
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination engineering of single-atom ruthenium in 2D MoS2 for enhanced hydrogen evolution†
Dong
Guo‡
 a,
Xiong-Xiong
Xue‡
b,
Menggai
Jiao‡
c,
Jinhui
Liu‡
a,
Tian
Wu
a,
Xiandi
Ma
c,
Die
Lu
c,
Rui
Zhang
c,
Shaojun
Zhang
a,
Gonglei
Shao
*c and
Zhen
Zhou
*c
a,
Xiong-Xiong
Xue‡
b,
Menggai
Jiao‡
c,
Jinhui
Liu‡
a,
Tian
Wu
a,
Xiandi
Ma
c,
Die
Lu
c,
Rui
Zhang
c,
Shaojun
Zhang
a,
Gonglei
Shao
*c and
Zhen
Zhou
*c
aSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China
bSchool of Physics and Optoelectronics, Xiangtan University, Xiangtan 411105, P. R. China
cInterdisciplinary Research Center for Sustainable Energy Science and Engineering (IRC4SE2), School of Chemical Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: shaogonglei@zzu.edu.cn; zhenzhou@zzu.edu.cn
First published on 9th September 2024
Abstract
This study investigates the enhancement of catalytic activity in single-atom catalysts (SACs) through coordination engineering. By introducing non-metallic atoms (X = N, O, or F) into the basal plane of MoS2via defect engineering and subsequently anchoring hetero-metallic Ru atoms, we created 10 types of non-metal-coordinated Ru SACs (Ru–X–MoS2). Computations indicate that non-metal atom X significantly modifies the electronic structure of Ru, optimizing the hydrogen evolution reaction (HER). Across acidic, neutral, and alkaline electrolytes, Ru–X–MoS2 catalysts exhibit significantly improved HER performance compared with Ru–MoS2, even surpassing commercial Pt/C catalysts. Among these, the Ru–O–MoS2 catalyst, characterized by its asymmetrically coordinated O2–Ru–S1 active sites, demonstrates the most favorable electrocatalytic behavior and exceptional stability across all pH ranges. Consequently, single-atom coordination engineering presents a powerful strategy for enhancing SAC catalytic performance, with promising applications in various fields.
Introduction
Coordination engineering is emerging as a versatile approach in the field of catalysis. By deliberately designing and precisely controlling the coordination environment of heteroatoms, this method optimizes their properties and chemical reactivity. In single-atom catalysts (SACs), coordination engineering allows for the manipulation of the coordination environment of single-atom active sites, offering benefits such as tuned reactivity, optimized selectivity, and enhanced catalytic activity.1–3 Furthermore, SACs demonstrate remarkable stability and resistance to sintering or aggregation when their coordination structure is carefully designed,4,5 leading to extended catalyst lifetimes and improved catalytic performance. Additionally, coordination engineering enhances the utilization of metal atoms,6,7 particularly noble metals, promoting the economic and environmental sustainability of catalytic processes. Therefore, coordination engineering is a crucial scientific strategy for unlocking the catalytic potential of heteroatoms in SACs.In coordination engineering, the meticulous selection of coordination ligands is pivotal in tailoring catalytic properties. Ligands containing elements such as N,8–11 O,12–14 or S,15,16 can optimize the electron density of the metal atom, modulate the electronic structure, and enhance reactivity. For example, coordinating metal atoms with specific ligands on graphene-based carbon materials boosts catalytic activity and selectivity in various reactions.17–22 However, the regulatory mechanisms of hetero-metal atoms in SACs remain unclear, and the structure–activity relationship between specific coordination structures in SACs and their catalytic performance has yet to be established.
Transition metal dichalcogenides (TMDs) with well-defined two-dimensional (2D) layer structures offer ultra-high specific surface areas, rich surface properties, tunable chemical compositions, and S-rich terminal planes.23 These features enable the regulation of the coordination structures of hetero-metal atoms, making it feasible to elucidate the catalytic mechanisms of single metal active sites through coordination engineering in TMDs. Despite significant advancements, coordinating SACs in 2D TMDs remains challenging due to synthesis and stability issues. Single metal atoms on 2D surfaces tend to migrate, hindering their dispersion as individual atoms.24 Addressing these challenges is crucial for successful coordination engineering in both synthesis and application. Understanding the interactions between reactants and active sites at the atomic level is essential for optimizing catalytic performance. Thus, incorporating single-atom active sites in 2D TMDs through atomic-scale coordination engineering is advantageous for maximizing their catalytic potential and comprehending the mechanisms behind the optimized performance of 2D TMDs.
In this study, non-metal atoms X (X = N, O, or F) with varying electronegativities are introduced into 2D MoS2 nanosheets through defect engineering to coordinate Ru metal atoms. This atomic-scale manipulation technique allows for the precise modulation of the coordination environment of Ru atoms, ensuring their dispersion as individual atoms in the Ru–X–MoS2 catalysts. The catalytic activity of Ru, adjusted with different non-metal atoms, is systematically investigated through computational methods. These non-metal atoms optimize the d-band center of Ru atoms by varying the type and number of coordinating non-metal atoms, forming asymmetric Ru coordination structures. This unique configuration accelerates the kinetics of water dissociation, as confirmed by X-ray absorption fine structure (XAFS) spectroscopy. Among the Ru–X–MoS2 catalysts, the Ru–O–MoS2 catalyst exhibits outstanding hydrogen evolution reaction (HER) performance across the full pH range, surpassing both Ru–MoS2 and MoS2, and even outperforming commercial Pt/C catalysts.
Results and discussion
Comprehensive consideration of the coordination atom type and coordination number of non-metal atoms, the best combination for Ru atoms is when Ru atoms coordinated with two O atoms and one S atom to form an O2–Ru–S1 coordination structure, which worth own the best HER catalytic performance.Regulating the coordination structure of single atoms in a 2D plane is crucial for optimizing electrocatalytic performance. In the specific case of designing the coordination structure of Ru atoms in the basal plane of 2D MoS2, we focus on Ru coordinated with non-metal atoms of varying electronegativities (S = 2.58, N = 3.04, O = 3.44, F = 3.98). This deliberate coordination strategy aims to stimulate and optimize the catalytic activity of the Ru active site, thereby enhancing the hydrogen evolution performance of 2D MoS2 nanomaterials. To achieve this, density functional theory (DFT) was employed to calculate the catalytic activity of 2D MoS2 doped with Ru atoms bonded to different non-metal atoms. This approach allows for the design of a high-performance MoS2-based catalyst with a precisely engineered spatial coordination structure involving Ru atoms.
From a physical chemistry perspective, the HER activity can be evaluated by plotting the Gibbs free energy (ΔGH*) diagram for adsorbed hydrogen.25,26 An ideal HER catalyst should have a ΔGH* value close to zero, indicating a balanced compromise between the reaction barriers associated with both adsorption and desorption steps. The effects of non-metal atom type and coordination number on the HER activity of Ru–X–MoS2 were investigated theoretically by calculating ΔGH* using density functional theory (DFT).
Fig. 1a presents models of Ru–X–MoS2 with varying coordination numbers. The calculated ΔGH* values for Ru–X–MoS2 with different coordination numbers are summarized in Fig. 1b. The HER performance of Ru–X–MoS2 is significantly influenced by the non-metal atoms and coordination numbers over a wide range. The optimal coordination numbers for the non-metal atoms in F, N, and O coordination-modified catalysts are 3, 2, and 2, respectively, with ΔGH* values of 0.132, 0.124, and 0.046 eV. These findings indicate that O atoms are the most effective for enhancing the HER activity of Ru–X–MoS2, with the optimal coordination number being 2.
 | ||
| Fig. 1 DFT calculations of Ru–X–MoS2 for hydrogen evolution. (a) Ru–X–MoS2 model with X-atom coordination numbers 1, 2 and 3. (b) ΔGH* of Ru–X–MoS2 with different numbers of non-metal metal (N, O, and F) coordination. (c) Correlations between ΔGH* and the d-band center of the Ru atoms. Electron concentration distribution of S3–Ru (d), O2–Ru–S1 (e), N2–Ru–S1 (f) and F3–Ru (g). (h) Partial density of states for the Ru–X–MoS2. (i) Mutual equilibrium relation diagram of coordination atom type and coordination number on the HER catalytic performance. | ||
For the metal active center, the d-band model provides valuable insights into the nature of non-metal coordination-regulated HER activity. The ΔGH* values are plotted as a function of the Ru atom's d-band center in different coordination environments (Fig. 1c). Notably, a well-established volcanic relationship exists between the d-band center of the Ru atoms and the corresponding ΔGH* values. The d-band center values range from −2.404 eV to −1.650 eV, and excessively high or low values can lead to unfavorable catalytic activity. Near the summit of the volcano plot, the candidate structure O2–Ru–S1, with a d-band center value of −1.964 eV, exhibits an optimal d-band center for moderate H* adsorption, resulting in superior HER activity.
Additionally, the Ru atom forms a symmetric coordination structure (S3–Ru) when coordinated with three S atoms, displaying a uniform electron concentration distribution (Fig. 1d). When a non-metal atom X is bonded, the symmetric S3–Ru coordination structure is disrupted, forming an asymmetric coordination structure and resulting in electron aggregation at the S site (Fig. 1e–g). This local structural polarization creates an electron-rich environment at the S site, effectively reducing the energy barrier for hydrolysis dissociation.
Therefore, the modulation of non-metal coordination on the HER catalytic activity of Ru–X–MoS2 intrinsically stems from the tunability of the d-band center of the Ru atom and the asymmetric coordination structure formed by the non-metal atoms and Ru atoms. The partial density of states (PDOS) further indicates that non-metal coordination significantly impacts the distribution of d-orbital energy levels and the localization degree of the Ru atom, tuning the d-band center and affecting H* adsorption and HER catalytic activity (Fig. 1h).
Summarizing the relationship between the HER catalytic performance of Ru–X–MoS2 and the coordination atom type or coordination number in Fig. 1i, we find a critical value for the effect of different electronegative non-metal atoms and coordination numbers on HER performance. Comprehensive consideration of the coordination atom type and number reveals that the best combination for Ru atoms is when they are coordinated with two O atoms and one S atom, forming an O2–Ru–S1 coordination structure, which demonstrates the best HER catalytic performance.
Translating these theoretical insights into practical material synthesis, we accurately synthesized single-atom Ru coordinated with distinct non-metal atoms in 2D MoS2 electrocatalysts through a variety of manipulation techniques (Fig. 2a). Firstly, S-vacancies in 2D MoS2 (denoted as Vs-MoS2) were carefully created using a hydrogen peroxide solution at a moderate concentration.27 Subsequently, the Vs-MoS2 was treated with O2 plasma, forming O-containing functional groups near the vacancies (designated as O–MoS2). Next, the 2D MoS2 with oxygen-containing vacancies was immersed in a Ru solution, allowing Ru atoms to anchor at the adsorption sites of the oxygen-containing vacancies. Finally, the 2D MoS2 electrocatalyst containing Ru–O active sites (labeled as Ru–O–MoS2) was prepared by annealing the samples at low temperatures in an inert atmosphere within a tube furnace. Detailed synthesis steps for Ru–F–MoS2, Ru–N–MoS2, and Ru–O–MoS2 are provided in the Experimental methods section of the ESI. These procedures ensure the precise and controlled creation of single-atom Ru in 2D MoS2 electrocatalysts, each with tailored non-metal coordination.
 | ||
| Fig. 2 Synthesis and characterization of Ru–X–MoS2 catalysts. (a) Schematic representation of the synthesis process for Ru–O–MoS2. (b) Raman spectra of pure MoS2, Vs-MoS2, O–MoS2 and Ru–O–MoS2. (c) TGA data of pure MoS2 and Ru–O–MoS2. (d) Ru 3d peaks of pure MoS2, O–MoS2, Ru–F–MoS2, Ru–N–MoS2 and Ru–O–MoS2 according to XPS data. (e) O 1s peaks of pure MoS2, O–MoS2 and Ru–O–MoS2 according to XPS. | ||
Raman spectroscopy proves to be highly effective in tracking subtle changes in the microscopic structure of 2D MoS2. Fig. 2b clearly displays the Raman spectra of as-prepared pure MoS2, Vs-MoS2, O–MoS2 and Ru–O–MoS2 (Detailed Raman spectra of as-prepared pure MoS2, Vs-MoS2, F–MoS2, N–MoS2, O–MoS2, Ru–F–MoS2, Ru–N–MoS2 and Ru–O–MoS2 in Fig. S1†). Two distinct scattering characteristic peaks at 376.3 cm−1 and 402.3 cm−1 are attributed to the E12g and A1g vibrational modes of hexagonal MoS2. The formation of a vacancy in MoS2, as seen in Vs-MoS2, induces a shift of the main characteristic peak to a higher frequency. The introduction of the O atom in O–MoS2 results into a new Mo–O vibration peak at 818.7 cm−1. Subsequently, the Raman characteristic peak of E12g and A1g shift obviously to higher frequency in Ru–O–MoS2. Additionally, the Raman peak at 818.7 cm−1 disappears, indicating the incorporation of Ru–O into the MoS2 lattice.
Thermogravimetric analysis (TGA) was further employed to elucidate the structure of the synthetic Ru–O–MoS2 (Fig. 2c). Similarly, pure MoS2 and Ru–O–MoS2 exhibit a notable mass loss of S atom near 426.8 °C. The reported thermogravimetric results of oxygen-doped MoS2 show that the oxygen induced weight loss starts from 200 °C.28,29 Thus, the synthetic Ru–O–MoS2 displays an initial phase of weight loss from 187.2 °C to 319.8 °C, which can be attributed to the weight loss of the non-metal O atoms with 8.26%. Additionally, X-ray photoelectron spectroscopy (XPS) was utilized to further prove the presence of metal and non-metal elements (Fig. 2d). The Ru 3d peak of Ru–O–MoS2 shifted to a higher binding energy compared with that of Ru–F–MoS2 and Ru–N–MoS2. The higher binding energy of Ru–O bond predicts that the introduction of O atoms facilitates the formation of an asymmetric electron distribution, and the asymmetric electron distribution is favorable for the improvement of catalytic activity.30,31 The appearance of XPS in F 1s, O 1s and N 1s further confirms the successful bonding of non-precious metals into the MoS2 lattices (More XPS data in Fig. S2 and S3†). In the case of pure MoS2, only adsorbed oxygen (Oad) is observed at 532.3 eV, resulting from the adsorption of O2 from the air onto the surface of MoS2. Following the O2 plasma treatment, lattice oxygen (OL) appears at 531.0 eV for O–MoS2 in addition to Oad, indicating the entry of O atoms into the MoS2 lattice. In the target Ru–O–MoS2 sample, the proportion of OL significantly increases from 10.5% (O–MoS2) to 72.1% (Ru–O–MoS2). This shift in the O 1s peaks signifies the evolution of non-metal O atoms, corroborating the existence of the OL and the formation of Ru–O bonds in the Ru–O–MoS2 sample.
Subsequently, scanning electron microscope (SEM) images (Fig. S4†) reveal that MoS2, Ru–F–MoS2, Ru–N–MoS2 and Ru–O–MoS2 are in the shape of nanosheets. Scanning transmission electron microscopy (STEM) images of Ru–O–MoS2 at low magnifications (Fig. S5†) display the thin nanosheets, boosting the exposure of active sites. Atomically resolved STEM image reveals the atomic arrangement of Ru–O–MoS2 with the hexagonal crystal system (Fig. 3a). The lattice length of the (100) crystal faces is measured at 0.272 nm, and the lattice width of the (110) crystal faces is 0.165 nm, consistent with the theoretical cell structure of MoS2. This alignment is further validated by selected area electron diffraction (SAED) in set of Fig. 3a. Due to the thickness superposition of MoS2, each bright spot represents a superposition of Mo atoms and S atoms. Nevertheless, the Ru atoms can still be distinguished by the brightness of these atoms, as confirmed by the atomic height plane diagram (marked with a dotted red line in Fig. 3a). Meanwhile, the width of 2d(100) increases from 0.543 nm to 0.552 nm (Fig. 3b and c), indicating that Ru atoms with larger atomic radius are bonded into the 2D MoS2, which results in the increase in the width of the (100) crystal faces.
 | ||
| Fig. 3 Atomic structure characterization of Ru–O–MoS2 catalysts. (a) STEM image of Ru–O–MoS2. Inset: the SAED image of Ru–O–MoS2. The Ru atoms are marked with red circle dotted line. Atomic height cross sections of (b) Ru-coordinated and (c) pure MoS2, respectively. d(100) denotes the spacing of the (100) crystal planes in the MoS2 lattice. (d) The content of Ru atoms in each sample based on ICP and XPS data. (e) EDS mapping image of Ru–O–MoS2. | ||
Inductively coupled plasma optical emission spectrometry (ICP-OES) and XPS were utilized to determine the atomic content of Ru (Fig. 3d). According to the ICP data, the content of Ru is 3.51 wt% in the Ru–O–MoS2 sample, 4.35 wt% in Ru–N–MoS2, and 4.32 wt% in Ru–F–MoS2, which closely match the values based on XPS (3.73 wt%). XPS reveals that the contents of N, O, and F in Ru–N–MoS2, Ru–O–MoS2, and Ru–F–MoS2 are 3.85 wt%, 3.62 wt%, and 2.80 wt%, respectively (Fig. S6†). STEM-energy dispersive spectroscopy (EDS) of Ru–O–MoS2 illustrates the characteristic peaks of S, Mo, Ru, and O elements (Fig. S7†), which is consistent with the XPS results. EDS mapping also shows that S, Mo, Ru, and O elements are uniformly distributed on Ru–O–MoS2 (Fig. 3e), indicating that Ru atoms do not aggregate to form clusters/particles, and this uniform distribution facilitates the improvement of the proton transport capacity.32 These findings provide strong evidence for the formation of Ru–O active sites in 2D MoS2.
To precisely assess the catalytic performance of single-atom Ru combined with different non-metal atoms X (X = N, O, or F) in the MoS2 base plane, the Ru–X–MoS2 electrocatalysts were systematically evaluated on the electrocatalytic hydrogen evolution performance across the full pH range (Fig. 4). The linear scanning voltammetry (LSV) curves in acidic, neutral, and alkaline electrolytes are depicted in Fig. 4a–c, respectively. Ru–MoS2 catalyst exhibits lower overpotentials of 58, 116 and 123 mV in acidic, neutral, and alkaline electrolytes, respectively, to drive a current density of 10 mA cm−2 compared with MoS2, and still larger than the commercial Pt/C catalyst (overpotentials of 53, 80 and 122 mV in acidic, neutral, and alkaline electrolytes, respectively). Interestingly, the catalytic performance of Ru–X–MoS2 demonstrates significant improvement compared with Ru–MoS2 and even outperformed the commercial Pt/C catalyst. Conversely, the X–MoS2 (X = N, O, or F) catalysts, which are solely bonded with non-metal atoms X, reveal almost negligible performance in comparison to Ru–X–MoS2 (Fig. S8a–c†). These results imply that the optimization of the coordination environment for single-atom Ru by non-metal atoms plays a key role in enhancing the catalytic activity of Ru–X–MoS2. Among the Ru–X–MoS2 catalysts, it is noteworthy that Ru–O–MoS2 displays outstanding catalytic performance, exhibiting remarkably low overpotentials of 16, 54, and 50 mV at 10 mA cm−2 in acidic, neutral, and alkaline electrolytes, respectively (Fig. 4d–f and Tables S1–S3†). As a result, the coordination environment modification of Ru atoms with moderately electronegative oxygen atoms is more conducive to the progression of the HER across the full pH range.
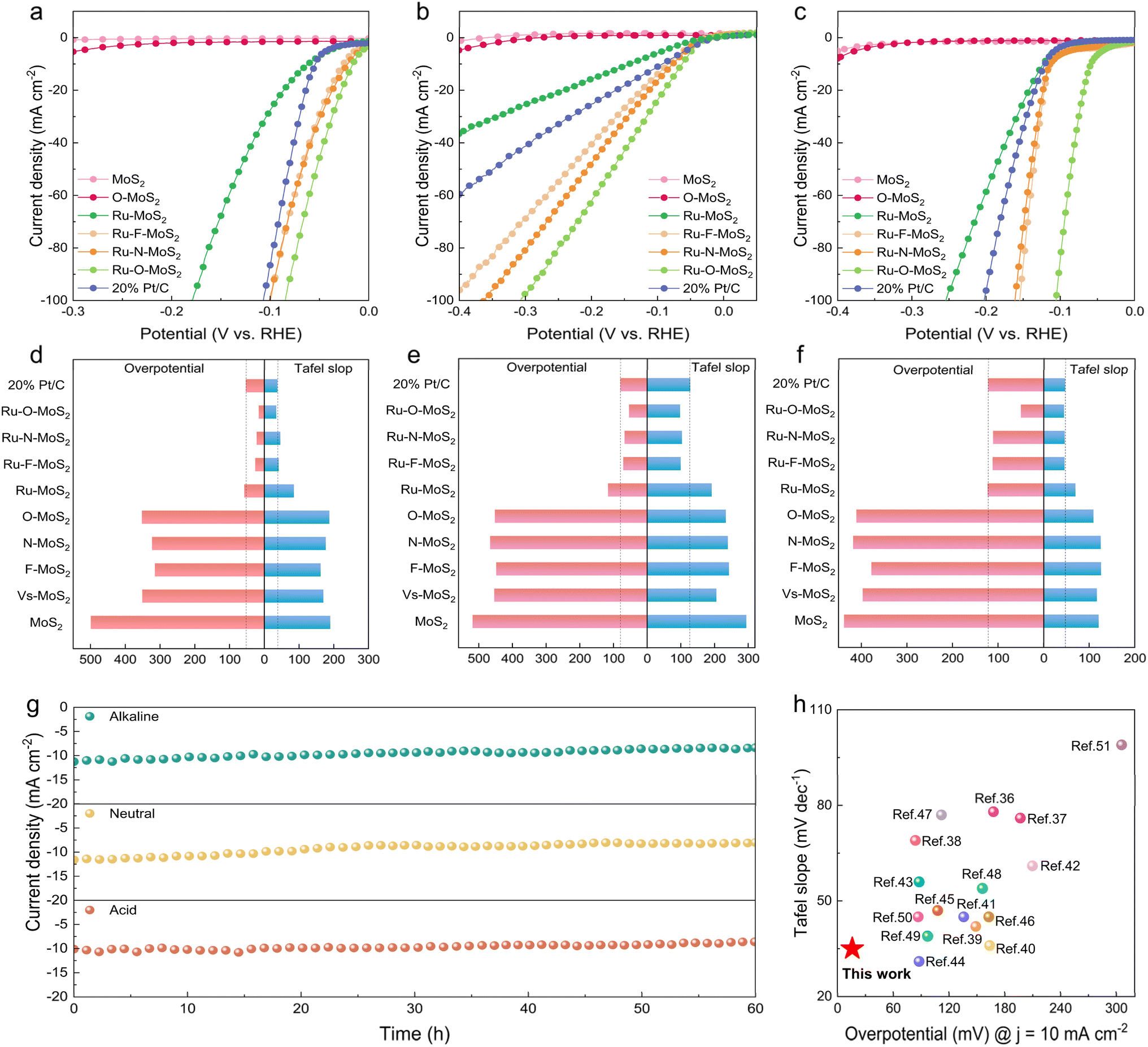 | ||
| Fig. 4 Hydrogen evolution performance evaluation of Ru–X–MoS2 catalysts. HER polarization curves in (a) acidic, (b) neutral and (c) alkaline electrolytes. The corresponding Tafel slop and overpotential at 10 mA cm−2 in (d) acidic, (e) neutral and (f) alkaline electrolytes. (g) Durability test of Ru–O–MoS2 in acidic, neutral, and alkaline electrolytes. (h) HER activity comparison using the Tafel slope (mV dec−1) vs. overpotential at the current density of 10 mA cm−2 in acidic electrolyte. | ||
Tafel slopes are used to examine the kinetics of HER in acidic, neutral, and alkaline electrolytes (Fig. 4d–f). The Tafel slope values for Ru–MoS2 in acidic, neutral, and alkaline electrolytes are 85, 192 and 70 mV dec−1, respectively. These values are lower than that of the reference samples MoS2 and O–MoS2, which are 190/187 mV dec−1 in acidic electrolytes, 295/234 mV dec−1 in neutral electrolytes, and 121/109 mV dec−1 in alkaline electrolytes. Nevertheless, after modifying the coordination environment of single-atom Ru by non-metal atoms, the Tafel slop values of Ru–X–MoS2 are significantly reduced compared with that of Ru–MoS2. Among them, the Tafel slop values of Ru–O–MoS2 are 35, 98 and 45 mV dec−1 in acidic, neutral, and alkaline electrolytes, respectively, which are even lower than the commercial Pt/C catalyst (38, 128 and 47 mV dec−1 in acidic, neutral, and alkaline electrolytes, respectively). In contrast, the Tafel slop of X–MoS2 catalysts bonded only with non-metal atoms X is very large compared with Ru–X–MoS2 (Fig. S8d–f†). The smaller Tafel slope value of Ru–X–MoS2 indicates the better hydrogen-evolving kinetics. Overall, the coordination environment modification of the single-atom Ru by non-metal atoms on the 2D MoS2 platform promotes HER across the full pH range.
Additionally, cyclic voltammetry (CV) was performed at various scan speeds (v) (Fig. S9–S11†). The double-layer capacitance (Cdl) values for each sample were then calculated by using Δj vs. ν plots.33,34 Among them, the Cdl values of Ru–X–MoS2 are significantly higher than those of other samples. Ru–O–MoS2 possesses the highest Cdl values in acidic, neutral, and alkaline electrolytes, measuring 27.3, 28.3, and 38.5 mF cm−2, respectively (Fig. S12a–c†). The enhanced Cdl in Ru–O–MoS2 should derive from the enlarged d(100) spacing and efficient catalytic activity after bonding with Ru and O atoms. This indicates that Ru–O–MoS2 possesses a larger electrochemically active surface area and increased charge storage capacity, which are favorable for the electrocatalytic HER. The electrochemical impedance spectra (EIS) are presented in Fig. S12d–f† for all samples. Ru–X–MoS2 exhibited smaller semicircular diameters compared with MoS2 and X–MoS2, indicating that the coordination environment modification of the single-atom Ru with non-metal atoms reduces charge-transfer resistance (Rct) and facilitates efficient charge transfer from the catalysts to the electrodes. The Rct of Ru–O–MoS2 in acidic, neutral, and alkaline electrolytes is 47.35 Ω, 55.49 Ω and 23.7 Ω, respectively, and the minimal Rct value of Ru–O–MoS2 may be attributed to the modulation of the electronic structure, which is consistent with the PDOS analysis described above. The turn over frequency (TOF) and mass activity of Ru–X–MoS2 catalysts far exceeded that of the commercial Pt/C, as shown in Fig. S13.† Among them, the Ru–F–MoS2/Ru–N–MoS2/Ru–O–MoS2 catalysts have a large electrochemically active surface area (ECSA), which makes their specific activities very similar. The larger ECSA promotes the mass transport process.35 The Faraday efficiency of Ru–O–MoS2 catalysts in acidic, neutral and alkaline electrolytes was 98.9%, 98.8% and 99.3%, respectively (Fig. S14†).
Moreover, electrochemical stability is also an important index to strictly evaluate whether the catalyst has commercial value. Ru–O–MoS2 was performed for 60 h electrocatalytic HER in acidic, neutral, and alkaline electrolytes, and no significant attenuation of the current density was observed (Fig. 4g), and the Ru–O–MoS2 catalyst after stability tests was nearly unchanged based on a result of XRD, TEM, and XPS characterization (Fig. S15–S19†). In addition, the Ru ion concentrations in the neutral and alkaline electrolytes were only 30.1 and 24.6 µg L−1 (Fig. S20†). All these results indicate that Ru single atoms and O atoms form a stable active site structure in the MoS2 plane. The stable catalytic activity at the full pH range is highly advantageous for practical applications, as it allows the catalyst to adapt to the complex and variable industrial production environment. Furthermore, the electrocatalytic activity of Ru–O–MoS2 is compared with that of previously reported TMDs-based electrocatalysts for HER in acidic solutions (Fig. 4h and Table S4†). The catalytic activity of Ru–O–MoS2 in acidic electrolytes with an overpotential of 16 mV and a Tafel slop of 35 mV dec−1, exceeds that of metal-coordinated TMDs catalysts already reported in the literature.36–51 The excellent catalytic activity of Ru–O–MoS2 indicates that the O atom plays a pivotal role in further stimulating the Ru atom, changing the electron density distribution around the Ru atom owing to the moderate electronegativity of O atom in comparison to N, F and S atom.
To further understand the catalytic mechanism of Ru–O–MoS2 in the process of hydrogen evolution, the essence of the O2–Ru–S1 active site in improving catalytic performance is revealed from a microscopic perspective. First, X-ray Diffraction (XRD) was utilized to analyze the structural changes of MoS2, with particular focus on the (002) and (100) crystal face (Fig. 5a). Comprehensive XRD data for pure MoS2, Vs-MoS2, F–MoS2, N–MoS2, O–MoS2, Ru–F–MoS2, Ru–N–MoS2, and Ru–O–MoS2 composites are provided in Fig. S21.† Upon comparison, the crystal faces (002) and (100) initially shift to lower angles and then to higher angles (Fig. 5b and c). By using the Bragg equation: 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ for analysis, the corresponding interplanar spacing d(002) and d(100) significantly increase in the presence of defect structure and F, N or O bonding. When Ru atoms and non-metal atoms bond to form X–Ru active centers into the MoS2 lattice, the interplanar spacing d(002) and d(100) becomes smaller, but remain greater than that of the pure MoS2 (Fig. 5d). Taking Ru–O–MoS2 as an example, d(002) increases from 0.604 nm of pure MoS2 to 0.616 nm of O–MoS2 and then to 0.609 nm of Ru–O–MoS2. Similarly, d(100) increases from 0.272 nm of pure MoS2 to 0.274 nm of O–MoS2 and then to 0.273 nm of Ru–O–MoS2. This indicates that the alteration in the Ru–O–MoS2 lattice results in more active sites and increased physical space, which is highly conducive to the HER.
sinθ = nλ for analysis, the corresponding interplanar spacing d(002) and d(100) significantly increase in the presence of defect structure and F, N or O bonding. When Ru atoms and non-metal atoms bond to form X–Ru active centers into the MoS2 lattice, the interplanar spacing d(002) and d(100) becomes smaller, but remain greater than that of the pure MoS2 (Fig. 5d). Taking Ru–O–MoS2 as an example, d(002) increases from 0.604 nm of pure MoS2 to 0.616 nm of O–MoS2 and then to 0.609 nm of Ru–O–MoS2. Similarly, d(100) increases from 0.272 nm of pure MoS2 to 0.274 nm of O–MoS2 and then to 0.273 nm of Ru–O–MoS2. This indicates that the alteration in the Ru–O–MoS2 lattice results in more active sites and increased physical space, which is highly conducive to the HER.
 | ||
| Fig. 5 Catalytic mechanism analysis of Ru–O–MoS2 catalysts. (a) XRD data of pure MoS2, Vs-MoS2, O–MoS2 and Ru–O–MoS2. Local XRD data detail in (b) (002) and (c) (100) lattice plane of pure MoS2, Vs-MoS2, O–MoS2 and Ru–O–MoS2. (d) The interplanar spacing d(002) and d(100) of pure MoS2, Vs-MoS2, F–MoS2, N–MoS2, O–MoS2, Ru–F–MoS2, Ru–N–MoS2 and Ru–O–MoS2. (e) Ru K-edge XANES spectra and (f) EXAFS spectra for Ru K-edge of Ru–O–MoS2. RuO2 and Ru foil as a reference. Corresponding EXAFS fitting curves of Ru–O–MoS2 nanosheets at (g) k space and (h) R space, respectively, inset showing the schematic model. The WT for the Ru atom based on EXAFS signals of (i) 2D Ru–O–MoS2 nanosheets, (j) RuO2 and (k) Ru foil. | ||
For further clarifying the coordination structure and local electronic environment of the Ru active site, XAFS was adopted to prove that O-modified Ru atoms carry with the best catalytic activity. The electronic state of the Ru species at the K-edge is investigated using X-ray absorption near-edge structure (XANES), which reveals that the positions of Ru–O–MoS2 situated between the Ru foil and RuO2, reflecting the ionic features of these Ru–O–MoS2 species (Fig. 5e). The absorption edge position of Ru–O–MoS2 closely approaches that of RuO2, indicating that the valence state of Ru in Ru–O–MoS2 is similar to that of Ru in RuO2. The local structure of Ru atom in Ru–O–MoS2 is unveiled by the Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectrum (Fig. 5f). The Ru atom in Ru–O–MoS2 manifests a distinctive characteristic peak at 1.51 Å, which differs from the peak value of Ru foil at 2.42 Å, and almost coincides with the peak value of RuO2 at 1.47 Å. This indicates that the Ru atoms did not aggregate to form a Ru–Ru bond, which is consistent with the fact that no Ru clusters or particles were found by STEM.
Furthermore, the model-based EXAFS fitting results are shown in Fig. 5g–h and Table S5 of the ESI,† which further confirm that each Ru atom is mainly coordinated with O atoms (with a coordination number of 1.9 ± 0.2, corresponding to a Ru–O bond length of 1.99 Å), and is slightly coordinated with S atoms (with a coordination number of 1.1 ± 0.3, corresponding to a Ru–S bond length of 2.21 Å). These results can infer that the coordination structure of Ru atom in MoS2 is O2–Ru–S1 as shown in set of Fig. 4h, where the Ru atom is located directly above the Mo atom, which is consistent with STEM observations. Additionally, Wavelet Transforms (WT) results further validate this coordination pattern, as the O2–Ru–S1 bond with a lower k value about 3.7 for the Ru–O–MoS2, compared with k = 3.71 for RuO2 and k = 10.4 for Ru foil (Fig. 5i–k and S22†). These findings provide robust evidence that Ru primarily coordinates with two O atoms and one S atom (O2–Ru–S1) in the Ru–O–MoS2 nanosheets, supporting its unique coordination environment. This coordination structure formed by two O atoms and a S atom with a Ru atom results in local electric field polarization in the base plane of MoS2, with electrons aggregated in the S sites, and the electron-rich S sites effectively promote HER. Combined with the increased active sites and distensible physical space in the structure of Ru–O–MoS2, the catalysts with unique O2–Ru–S1 active sites exhibit the best HER performance and excellent electrocatalytic stability in acidic, neutral, and alkaline electrolytes.
Conclusions
In summary, we have successfully developed a series of Ru–X–MoS2 catalysts (where X = N, O, or F) with non-metal atoms asymmetrically coordinated Ru atom active sites. The innovative approach of introducing non-metal atoms to optimize the d-band center of Ru atoms and reduce the ΔGH* of Ru active sites has significantly enhanced the electrocatalytic hydrogen evolution performance. Theoretical calculations and experimental results demonstrated that the non-metal atoms not only form asymmetric coordination structures with Ru atoms, leading to electron-enriched active sites, but also promote an optimal catalytic environment. Among the synthesized catalysts, Ru–O–MoS2, with its O2–Ru–S1 coordination structure, exhibited the highest catalytic activity and excellent stability across the full pH range, outperforming even commercial Pt/C catalysts. This coordination engineering strategy thus represents a crucial method for unlocking the full potential of SACs in HER.Future investigations could focus on further exploring the coordination environment of SACs, potentially incorporating other non-metal elements to tune the electronic properties and catalytic performance. Additionally, scaling up the synthesis process and integrating these catalysts into practical applications will be essential steps toward commercial viability.
Data availability
The data that supporting the findings of this study are available within the article and its ESI.†Author contributions
Guo D. and Shao G. conceived the research and performed the experiments; Jiao M. and Xue X. contributed to the DFT simulations; Guo D., Shao G., Wu T., Lu D. and Zhang R. performed data analysis; Guo D., Shao G., Jiao M., Zhang S., Liu J., Xue X. and Zhou Z. contributed to manuscript editing; all authors contributed to the general discussion.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22205209 and U21A20281), China Postdoctoral Science Foundation (2022M722867), Key Research Project of Higher Education Institutions in Henan Province (23A530001 and 24A530009), Henan Province Key Research and Promotion Project – Scientific and Technological Breakthroughs (232102230088), and Special Fund for Young Teachers from the Zhengzhou University (JC23257011). The computations were performed at the National Supercomputing Center in Zhengzhou, China.References
- K. Fujisawa, B. R. Carvalho, T. Zhang, N. Perea-López, Z. Lin, V. Carozo, S. L. L. M. Ramos, E. Kahn, A. Bolotsky, H. Liu, A. L. Elías and M. Terrones, ACS Nano, 2021, 15, 9658–9669 CrossRef CAS
.
- W. Tan, S. Xie, D. Le, W. Diao, M. Wang, K.-B. Low, D. Austin, S. Hong, F. Gao, L. Dong, L. Ma, S. N. Ehrlich, T. S. Rahman and F. Liu, Nat. Commun., 2022, 13, 7070 CrossRef CAS PubMed
- J. Yang, Y. Huang, H. Qi, C. Zeng, Q. Jiang, Y. Cui, Y. Su, X. Du, X. Pan, X. Liu, W. Li, B. Qiao, A. Wang and T. Zhang, Nat. Commun., 2022, 13, 4244 CrossRef CAS PubMed
- Z. Qi, Y. Zhou, R. Guan, Y. Fu and J.-B. Baek, Adv. Mater., 2023, 35, 2210575 CrossRef CAS
- X. Ma, H. Liu, W. Yang, G. Mao, L. Zheng and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 12220–12229 CrossRef CAS PubMed
- L. Gloag, S. V. Somerville, J. J. Gooding and R. D. Tilley, Nat. Rev. Mater., 2024, 9, 173–189 CrossRef CAS
- T. Cui, L. Li, C. Ye, X. Li, C. Liu, S. Zhu, W. Chen and D. Wang, Adv. Funct. Mater., 2022, 32, 2108381 CrossRef CAS
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W.-C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1800396 CrossRef
- Y. Guo, M. Wang, Q. Zhu, D. Xiao and D. Ma, Nat. Catal., 2022, 5, 766–776 CrossRef CAS
- B. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. Chen, Nat. Commun., 2019, 10, 631 CrossRef CAS PubMed
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- Y. Xu, M. Chu, F. Liu, X. Wang, Y. Liu, M. Cao, J. Gong, J. Luo, H. Lin, Y. Li and Q. Zhang, Nano Lett., 2020, 20, 6865–6872 CrossRef CAS PubMed
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019, 18, 1215–1221 CrossRef CAS PubMed
- C. Tang, Y. Jiao, B. Shi, J. Liu, Z. Xie, X. Chen, Q. Zhang and S. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS
- J. Qin, H. Liu, P. Zou, R. Zhang, C. Wang and H. L. Xin, J. Am. Chem. Soc., 2022, 144, 2197–2207 CrossRef CAS
- L. Bai, C. Hsu, D. T. L. Alexander, H. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef
- Q. Liu, Y. Li, L. Zheng, J. Shang, X. Liu, R. Yu and J. Shui, Adv. Energy Mater., 2020, 10, 2000689 CrossRef CAS
- X. X. Wang, D. A. Cullen, Y.-T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef
- Y. Guo, B. Dai, J. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 723–732 CrossRef CAS PubMed
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed
- X. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed
- Y. Chen, Y. Li, Y. Liang, X. Chen and J. Cui, Micro Nano Lett., 2021, 16, 274–279 CrossRef CAS
- J. Guo, F. Huo, Y. Cheng and Z. Xiang, Catal. Today, 2020, 347, 56–62 CrossRef CAS
- J. Xu, X. Xue, G. Shao, C. Jing, S. Dai, K. He, P. Jia, S. Wang, Y. Yuan, J. Luo and J. Lu, Nat. Commun., 2023, 14, 7849 CrossRef CAS PubMed
- D. Li, B. Wang, X. Long, W. Xu, Y. Xia, D. Yang and X. Yao, Angew. Chem., Int. Ed., 2021, 60, 26483–26488 CrossRef CAS PubMed
- Q. Zhang and B. Wang, Trans. Tianjin Univ., 2023, 29, 360–386 CrossRef CAS
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T.-p. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS PubMed
- Y. Li, Z. Kan, L. Jia, D. Zhang, Y. Hong, J. Liu, H. Huang, S. Li and S. Liu, Trans. Tianjin Univ., 2023, 29, 313–320 CrossRef CAS
- Z. Lai, Q. He, T. H. Tran, D. V. M. Repaka, D.-D. Zhou, Y. Sun, S. Xi, Y. Li, A. Chaturvedi, C. Tan, B. Chen, G.-H. Nam, B. Li, C. Ling, W. Zhai, Z. Shi, D. Hu, V. Sharma, Z. Hu, Y. Chen, Z. Zhang, Y. Yu, X. Renshaw Wang, R. V. Ramanujan, Y. Ma, K. Hippalgaonkar and H. Zhang, Nat. Mater., 2021, 20, 1113–1120 CrossRef CAS
- Y. Zhang, T. Yang, J. Li, Q. Zhang, B. Li and M. Gao, Adv. Funct. Mater., 2023, 33, 2210939 CrossRef CAS
- G. Shao, J. Xu, S. Gao, Z. Zhang, S. Liu, X. Zhang and Z. Zhou, Carbon Energy, 2024, 6, e417 CrossRef CAS
- Z. Liu, K. Nie, X. Qu, X. Li, B. Li, Y. Yuan, S. Chong, P. Liu, Y. Li, Z. Yin and W. Huang, J. Am. Chem. Soc., 2022, 144, 4863–4873 CrossRef CAS
- J. Xu, G. Shao, X. Tang, F. Lv, H. Xiang, C. Jing, S. Liu, S. Dai, Y. Li, J. Luo and Z. Zhou, Nat. Commun., 2022, 13, 2193 CrossRef CAS PubMed
- J. Li, A. Listwan, J. Liang, F. Shi, K. Li and J. Jia, Chem. Eng. J., 2021, 422, 130100 CrossRef CAS
- W. Zhan, X. Zhai, Y. Li, M. Wang, H. Wang, L. Wu, X. Tang, H. Zhang, B. Ye, K. Tang, G. Wang and M. Zhou, ACS Nano, 2024, 18, 10312–10323 CrossRef CAS PubMed
- Y. Li, Q. Gu, B. Johannessen, Z. Zheng, C. Li, Y. Luo, Z. Zhang, Q. Zhang, H. Fan, W. Luo, B. Liu, S. Dou and H. Liu, Nano Energy, 2021, 84, 105898 CrossRef CAS
- S. Bolar, S. Shit, P. Samanta, N. Chandra Murmu, H. Kolya, C.-W. Kang and T. Kuila, Composites, Part B, 2022, 230, 109489 CrossRef CAS
- L. Jiang, Y. Zhang, X. Luo, L. Yu, H. Li and Y. Li, Chem. Eng. J., 2021, 425, 130611 CrossRef CAS
- X. Li, X. Lv, X. Sun, C. Yang, Y.-Z. Zheng, L. Yang, S. Li and X. Tao, Appl. Catal. B Environ., 2021, 284, 119708 CrossRef CAS
- J. Dong, X. Zhang, J. Huang, J. Hu, Z. Chen and Y. Lai, Chem. Eng. J., 2021, 412, 128556 CrossRef CAS
- M. Li, B. Cai, R. Tian, X. Yu, M. B. H. Breese, X. Chu, Z. Han, S. Li, R. Joshi, A. Vinu, T. Wan, Z. Ao, J. Yi and D. Chu, Chem. Eng. J., 2021, 409, 128158 CrossRef CAS
- L. Ma, L. Jiang, X. Li, P. Zuo, C. Xu, Z. Cheng, M. Tian, Y. Yuan, X. Zhang, Y. Lu, Y. Zhao and L. Qu, Chem. Eng. J., 2022, 445, 136618 CrossRef CAS
- Q. Zhou, Z. Wang, H. Yuan, J. Wang and H. Hu, Appl. Catal. B Environ., 2023, 332, 122750 CrossRef CAS
- R. Chen, M. Ma, Y. Luo, L. Qian, S. Wan, S. Xu and X. She, Trans. Tianjin Univ., 2022, 28, 440–445 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04905e |
| ‡ These authors contributed equally: Dong Guo, Xiong-Xiong Xue, Menggai Jiao and Jinhui Liu. |
| This journal is © The Royal Society of Chemistry 2024 |