
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroscale manipulation of bond exchange reactions in photocurable vitrimers with a covalently attachable photoacid generator†
Roman
Korotkov
a,
Walter
Alabiso
 a,
Alexander
Jelinek
c,
Max
Schmallegger
d,
Yang
Li
a,
Sandra
Schlögl
*a and
Elisabeth
Rossegger
*ab
a,
Alexander
Jelinek
c,
Max
Schmallegger
d,
Yang
Li
a,
Sandra
Schlögl
*a and
Elisabeth
Rossegger
*ab
aPolymer Competence Center Leoben GmbH, Sauraugasse 1, Leoben, 8700, Austria. E-mail: elisabeth.rossegger@pccl.at; sandra.schloegl@pccl.at
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9/V, Graz, 8010, Austria
cDepartment of Materials Science, Montanuniversität Leoben, Franz-Josef-Strasse 18, Leoben, 8700, Austria
dInstitute of Physical and Theoretical Chemistry, Graz University of Technology, Stremayrgasse 9/II, Graz, 8010, Austria
First published on 3rd September 2024
Abstract
Vitrimers are polymer networks with covalent bonds that undergo reversible exchange reactions and rearrange their topology in response to an external stimulus. The temperature-dependent change in viscoelastic properties is conveniently adjusted by selected catalysts. In these thermo-activated systems, the lack in spatial control can be overcome by using photolatent catalysts. Herein, we advance this concept to locally manipulate bond exchange reactions on a single digit microscale level. For this, we synthetize a linkable non-ionic photoacid generator, which is covalently attached to a thiol-click photopolymer. UV induced deprotection of the photoacid yields a strong immobilized sulfonic acid species, which is able to efficiently catalyze transesterification reactions. Covalent attachment of the formed acid prevents migration/leaching processes and enables a precise tuning of material properties. As proof of concept, positive toned microstructures with a resolution of 5 μm are inscribed in thin films using direct two-photon absorption laser writing and subsequent depolymerization. In addition, the possibility to locally reprogram bulk material properties is demonstrated by performing a post-modification reaction with ethylene glycol and carboxylic acids. The Young's modulus is varied between 3.3 MPa and 11.9 MPa giving rise to the versatility of the newly introduced catalysts for creating light processable and transformable materials.
1. Introduction
Covalent adaptable networks (CANs) are a unique class of functional polymers that are able to rearrange their network topology through dynamic exchange reactions of covalent bonds under certain stimuli.1–3 The bond exchange reactions follow either a dissociative or an associative pathway, which affects the viscoelastic properties of the related networks. In dissociative CANs, exchange reactions go through bond cleavage with subsequent bond formation leading to a reduction in crosslink density and a distinctive drop in viscosity. In contrast, associative CANs maintain their average crosslink degree during topology rearrangements as the first step in the bond exchange reactions involves the formation of a new bond.4 The viscosity follows an Arrhenius-type relationship and decreases with increasing temperature. At a sufficiently high bond exchange rate, the dynamic polymeric networks become malleable, weldable and recyclable.5,6Leibler and co-authors introduced the term vitrimer for this type of CANs by their investigation on dynamic epoxy-acid and epoxy-anhydride networks, which underwent thermo-activated transesterification reactions.7 Over the past years, transesterification-based vitrimers became one of the most reported class of CANs due to the commercial availability of monomers which contain ester and hydroxyl groups and/or form them during the polymerization/curing process.1,6 However, external or internal catalysts are typically required to accelerate the transesterification reactions and to facilitate a healing, welding or recycling of the materials at lower temperatures.8
The most commonly used catalysts for transesterification-based vitrimers are organic salts of zinc(II) such as acetate and acetylacetonate,9 Brønsted acids and organic bases.6,10 Bates and co-authors reported that strong Brønsted acids are more efficient in catalyzing bond exchange reactions than Zn complexes, which act as Lewis acids. Besides that, the strength of the acid significantly affects the kinetics and activation energy of the bond exchange reaction.11
Vitrimers mainly rely on thermoactivated dynamic bond exchange reactions, which lack spatial control.12 Several approaches have been described in the literature to achieve local control of topological rearrangements in vitrimers. One example is the exploitation of the photothermal effect of fillers such as carbon nanotubes, which convert light to thermal energy.13 Another approach involves the introduction of photosensitive chromophores (e.g. o-nitrobenzyl esters) into thermally curable vitrimers.14 Alternatively, catalysts with photosensitive protecting groups are used, which are locally activated by light exposure. Once the catalyst is released, the network is able to exclusively undergo topology rearrangements (at elevated temperature) in the irradiated parts of the polymer. In a pioneering study, Bowman et al. utilized a strong photolatent organic base to demonstrate controlled photoactivation of the catalyst in thiol–thioester vitrimers.15 This approach was adopted by our research group for transesterification-based vitrimers. We introduced a library of photolatent bases12,16 and acids17 to locally adjust topology rearrangements in various thermally and photochemically curable polymer networks. Whilst the concept is highly versatile for spatio-temporally controlling bond exchange reactions, the precision of the activation is limited as the released (low molecular weight) catalyst easily migrates over time or when treated with solvents. Common approaches to prevent catalyst migration are covalent attachment of the catalyst to the network18 or usage of polymeric catalysts.19
Herein, we synthesize a new photoacid generator comprising a functional vinyl group that enables its covalent attachment to a polymer matrix (Fig. 1). By following a semi-orthogonal curing protocol, the photoacid generator is immobilized in a thiol–ene photopolymer upon 405 nm light irradiation. In a second step, the photoacid is selectively activated by irradiation with 365 nm UV-light. However, the acidic species remains immobilized in the polymer network and thus, a manipulation of rearrangement reactions in the μm-level is applicable. Moreover, the prepared photoacid generator can be excited via two photon absorption enabling the writing of positive-tone micropatterns by two photon absorption lithography (780 nm). Along with solubility properties, we successfully prove that the mechanical properties of the dynamic photopolymer networks can be locally tailored by acid-mediated post-reactions with low-molecular weight carboxylic acids and alcohols.
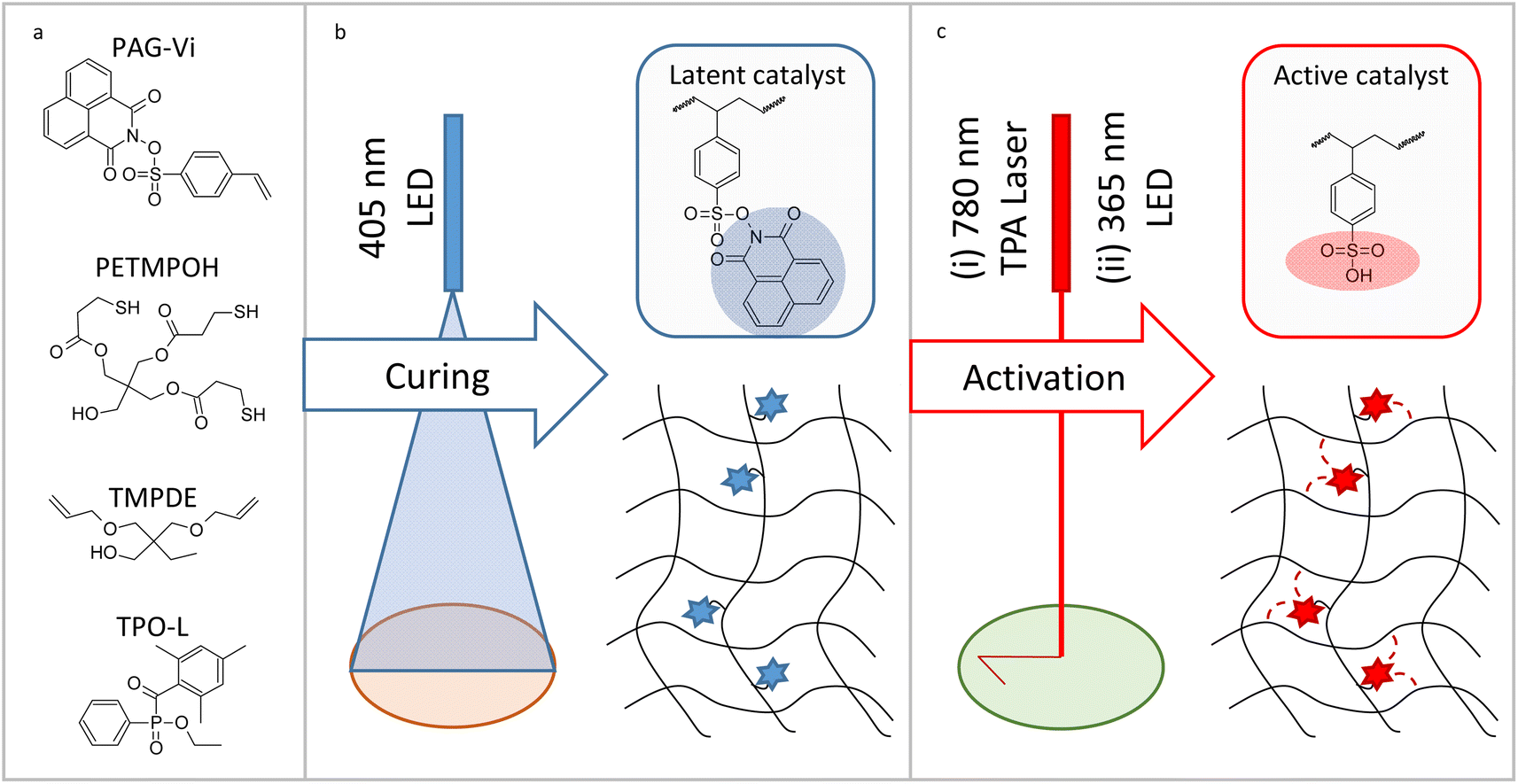 | ||
| Fig. 1 (a) Components used in the preparation of the thiol–ene formulations. (b) Preparation of dynamic photopolymers by visible light induced radical curing (405 nm) of the thiol–ene monomers and immobilization of the photoacid generator (PAG-Vi). (c) Selective activation of the immobilized PAG-Vi by exposure with (i) two photon absorption laser light at 780 nm or (ii) 365 nm LED light. | ||
2. Results and discussion
2.1. Synthesis and characterization of immobilizable photoacid generators
For preparing a photolatent transesterification catalyst bearing an immobilizable pendant group, a vinyl-functional aromatic sulfonic acid was equipped with a photocleavable naphthalimide protecting group (PAG-Vi). A non-ionic photoacid generator (PAG) was synthetized as this family is better soluble in organic solvents and resins in comparison to ionic PAGs.20 Naphtalimide was chosen as protective group due to the relatively clean photocleavage process of the N–O bond, which can be induced by UV-light21 as well as two photon absorption using 780 nm light of a femtosecond laser.22 Additionally, a derivative with a methyl group (PAG-Me) instead of the vinyl function was synthesized as non-linkable model compound. Both catalysts were prepared according to known synthesis protocols,23,24 following a substitution reaction between arylsulfonyl chloride and N-hydroxy-1,8-naphtalimide (Fig. 2a). The crude PAGs were purified by flash chromatography and white crystalline powders with 52.7% (PAG-Vi) and 74.4% (PAG-Me) yields were obtained. The structure and purity of the products was confirmed by 1H-, 13C-NMR and FTIR measurements (Fig. S1a–c and S2a–c†). The melting point of PAG-Vi and PAG-Me amounts to 192 °C and 236 °C, respectively (Fig. S3a and b†). According to TGA data (Fig. S4a and b†), the synthesized PAGs possess a high thermal stability in inert atmosphere with an onset of the mass loss curve at 324 °C (PAG-Vi) and 310 °C (PAG-Me). Despite their similar structure, PAG-Vi exhibited a higher residual mass at 900 °C (28.3%) than PAG-Me (21.0%), which can be explained by a polymerization across the vinyl groups that takes place during the heating ramp.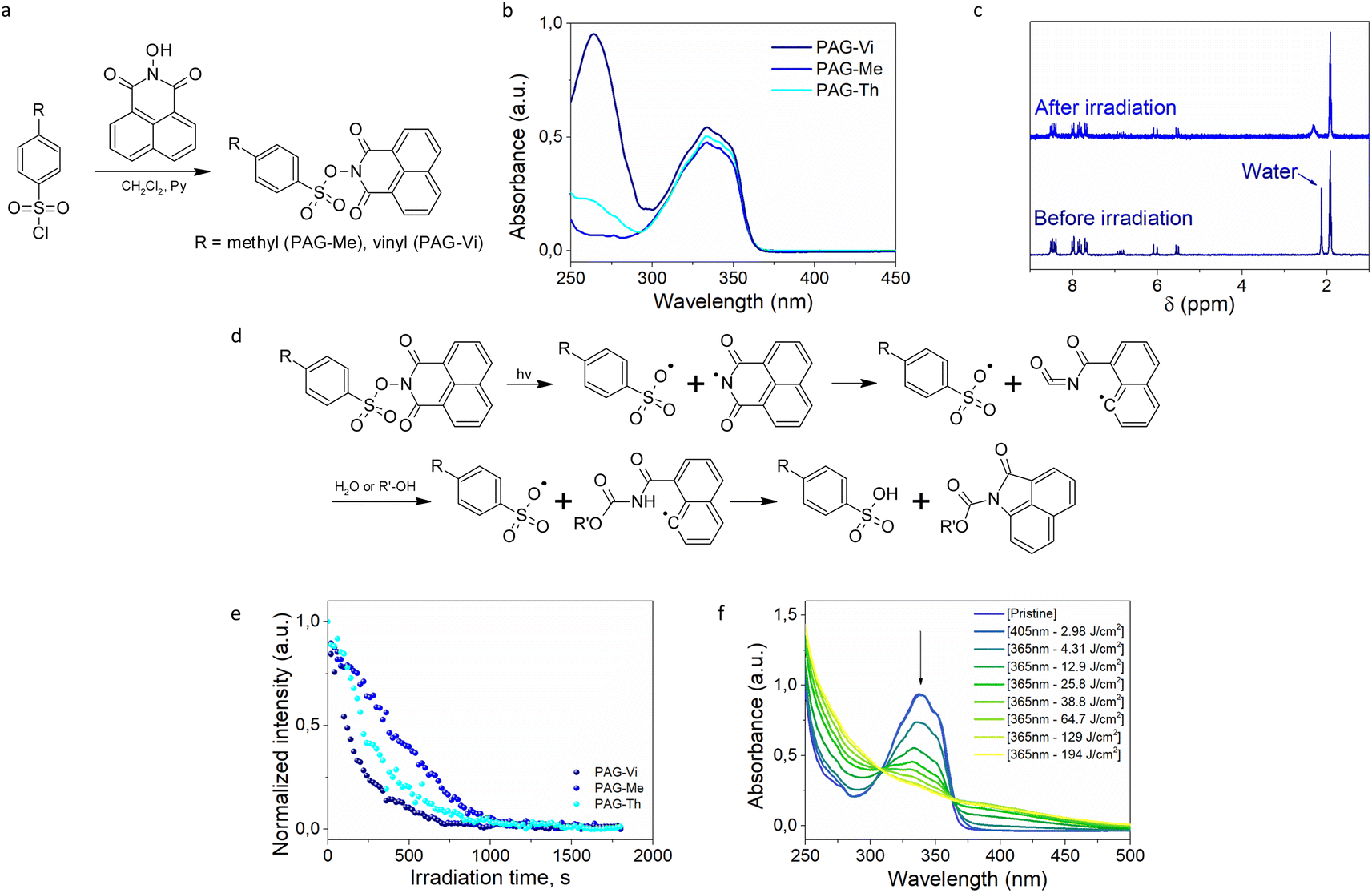 | ||
| Fig. 2 (a) Synthesis route for PAG-Vi and PAG-Me; (b) UV-vis absorption spectra of synthesized PAGs; (c) 1H NMR spectra of PAG-Vi dissolved in deuterated acetonitrile, prior to and after UV light (365 nm) irradiation; (d) photocleavage mechanism of naphtalimide-based PAGs;24 (e) normalized signal integral of the residue water peak obtained by 1H NMR spectroscopy in solution upon irradiation (365 nm), followed over time; (f) change of UV-vis absorption spectra upon photopolymerization (405 nm) and subsequent PAG activation (365 nm) of a formulation containing 5 mol% of PAG-Vi. | ||
To investigate the effect of the thiol–ene matrix on the photoreactivity of the naphtalimide-based PAGs, PAG-Th containing a thioether pendant group was synthesized as second model compound (Fig. S5†). For this, PAG-Vi (1 mmol) was stirred in an excess of 1,4-butanedithiol (20 mmol) at 90 °C for 2 hours until a clear solution was obtained. The product was precipitated in cyclohexane, subsequently filtered, and washed with cyclohexane three times. 1H-, 13C-NMR and FTIR spectroscopy data of PAG-Th support the proposed structure and are provided in ESI (Fig. S6a–c†).
To study the photosensitivity of the synthesized compounds, UV-vis spectroscopy measurements were performed at a concentration of 40 μM in acetonitrile (Fig. 2b). In solution, PAG-Vi possesses two main absorption bands with maxima at 333 nm and 264 nm, corresponding to the naphtalimide ring and the conjugated vinyl functionality, respectively. In contrast, PAG-Me and PAG-Th show only the absorption band of the naphtalimide ring (λmax = 333 nm). Comparing the width of the absorption bands, it is noted that PAG-Vi and PAG-Th feature a small red shift compared to PAG-Me. On the one hand, this can be attributed to the increased conjugation (PAG-Vi) and on the other hand, to the presence of electron-donating thioether functionalities (PAG-Th).25
In order to quantitively analyze the photoactivation kinetics of the synthesized PAGs and the formation of the acidic species, a 10 mM solution of the compounds in deuterated acetonitrile (CH3CN-d3) was irradiated directly inside an NMR probe head with UV-light and spectra were recorded simultaneously. 1H-NMR spectra of the PAG-Vi solution prior to and after one hour of irradiation are presented in Fig. 2c. Corresponding 1H-NMR spectra of PAG-Me and PAG-Th are presented in ESI (Fig. S7a and b†). For the three synthesized compounds, only minor changes in the aromatic region of the spectrum are observed. The most significant change is detected for the peak at 2.1 ppm, which is attributed to residual water in the deuterated solvent. Upon irradiation, this peak decreases in intensity and shifts to 2.6 ppm. This indicates the formation of a species bearing an exchangeable hydrogen atom interacting with the residual water or the change of the pH value in solution. We assume that this shift in the signal is caused by the formation of sulfonic acid as it is well established for similar compounds that this process requires the presence of water or an alcohol (Fig. 2d).24,26 The activation kinetics of the PAGs in the acetonitrile solution was evaluated based on the decrease of integral intensity of the peak at 2.1 ppm corresponding to water (Fig. 2e). In the case of the light activated PAG-Me, the acid generation is additionally evidenced by the shift in the signal of the methyl group from 2.55 to 2.45 ppm.
Thus, following the change of the residual water peak allows an estimation of the kinetics of sulfonic acid formation upon UV irradiation under our experimental conditions. For this, the normalized integral of the water signal was monitored over time, assuming that the change in integral is due to the formation of sulfonic acid. The results show that PAG-Vi and PAG-Th undergo the quickest photochemical transformation, which can be attributed to the increased electron density on the aromatic ring owing to the enhanced conjugation in PAG-Vi and electron donating thioether moieties in PAG-Th. Since PAG-Th reacts faster than PAG-Me, we assume that PAG-Vi will be also more reactive within a thiol–ene network than PAG-Me due to the presence of thioether links formed during the curing reaction. All three compounds reach a plateau after prolonged irradiation, indicating an exchange equilibrium between water and acid in solution.
2.2. Curing kinetics and photoactivation of immobilized PAGs
PAG-Vi was then introduced in a dynamic thiol–ene photopolymer as linkable transesterification catalyst and its performance was compared to free PAG-Me.27 In thiol–ene chemistry, the radically induced step-growth polymerization between multifunctional thiols and alkenes proceeds under the formation of thioether bonds (Fig. S8a†). Since no free –OH and ester groups are formed during polymerization, it was necessary to select monomers bearing such functional groups to enable transesterification reactions at elevated temperatures in the resulting polymer network. As “ene” compound, trimethylolpropane diallyl ether 90 (TMPDE) was used and a pentaerythritol derivate partially esterified (75%) with 3-mercaptopropionic acid (PETMP-OH) was employed as thiol crosslinker. All resin formulations were prepared in a stoichiometric ratio of thiol groups to allyl moieties. To provide λ-orthogonality between the curing (405 nm) and activation (365 nm) reaction, a visible light absorbing radical photoinitiator (ethyl(2,4,6-trimethylbenzoyl)phenylphosphinate, TPO-L) was used to initiate the curing reaction.The influence of the synthetized PAGs on the curing kinetics was followed by FTIR-measurements on resin samples drop-cast between two CaF2 discs (Fig. S8b†). Upon exposure with 405 nm light, the characteristic thiol (2569 cm−1) and alkene (1645 cm−1) absorption bands disappeared simultaneously. In a thiol–ene resin without PAG, the final thiol as well as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conversion exceeded 95% at an exposure dose of 0.1 J cm−2. The addition of 5 mol% of each synthetized PAG significantly slowed down the photocuring kinetics. Although the PAGs are transparent at 405 nm, the chromophores seem to act as an internal filter. In a perfectly λ-orthogonal system, the photoacid should not be activated during network formation at irradiation with 405 nm light, to enable local control of material properties. However, it is known from the literature that a red shift in photoreactivity can often be observed, although no absorption in this region can be detected by UV-Vis spectroscopy. Barner-Kowollik's group investigated this phenomenon extensively by measuring so-called action plots.28 The activation of the PAGs was quantified by monitoring the decrease of the stretching band of the naphtalimide ring (1593 cm−1) upon light exposure with 405 and 365 nm. The results obtained at 405 nm irradiation clearly show that the systems are not fully orthogonal. The undesired conversion of PAG-Th, PAG-Vi and PAG-Me during the curing of the resin (405 nm) amounted to 11%, 12% and 27%, respectively (Fig. S8c†). The higher stability of PAG-Th and PAG-Vi might be related to steric hindrance based on the covalent binding of the PAGs to the polymeric network.29 However, the partial activation of PAG-Vi is not sufficient to catalyze transesterification reactions at elevated temperatures, as can be seen in stress relaxation experiments (Fig. 3a). The reactivity at 405 nm does not correlate with the cure kinetics, which slows down in the following order: PAG-Me ∼ PAG-Th > PAG-Vi. The particular slow conversion in the presence of PAG-Vi (85% final monomer conversion with an exposure dose of 2.5 J cm−2) can be attributed to the fact that the conjugated vinyl group of PAG-Vi is less reactive in the radical-mediated curing reaction than the terminal vinyl ether moieties of the alkene monomer.30 In contrast, the thiol moiety in PAG-Th possesses a comparable activity as the thiol group in PETMP-OH and thus, the cure kinetics is similar to thiol–ene formulations containing PAG-Me.31
C conversion exceeded 95% at an exposure dose of 0.1 J cm−2. The addition of 5 mol% of each synthetized PAG significantly slowed down the photocuring kinetics. Although the PAGs are transparent at 405 nm, the chromophores seem to act as an internal filter. In a perfectly λ-orthogonal system, the photoacid should not be activated during network formation at irradiation with 405 nm light, to enable local control of material properties. However, it is known from the literature that a red shift in photoreactivity can often be observed, although no absorption in this region can be detected by UV-Vis spectroscopy. Barner-Kowollik's group investigated this phenomenon extensively by measuring so-called action plots.28 The activation of the PAGs was quantified by monitoring the decrease of the stretching band of the naphtalimide ring (1593 cm−1) upon light exposure with 405 and 365 nm. The results obtained at 405 nm irradiation clearly show that the systems are not fully orthogonal. The undesired conversion of PAG-Th, PAG-Vi and PAG-Me during the curing of the resin (405 nm) amounted to 11%, 12% and 27%, respectively (Fig. S8c†). The higher stability of PAG-Th and PAG-Vi might be related to steric hindrance based on the covalent binding of the PAGs to the polymeric network.29 However, the partial activation of PAG-Vi is not sufficient to catalyze transesterification reactions at elevated temperatures, as can be seen in stress relaxation experiments (Fig. 3a). The reactivity at 405 nm does not correlate with the cure kinetics, which slows down in the following order: PAG-Me ∼ PAG-Th > PAG-Vi. The particular slow conversion in the presence of PAG-Vi (85% final monomer conversion with an exposure dose of 2.5 J cm−2) can be attributed to the fact that the conjugated vinyl group of PAG-Vi is less reactive in the radical-mediated curing reaction than the terminal vinyl ether moieties of the alkene monomer.30 In contrast, the thiol moiety in PAG-Th possesses a comparable activity as the thiol group in PETMP-OH and thus, the cure kinetics is similar to thiol–ene formulations containing PAG-Me.31
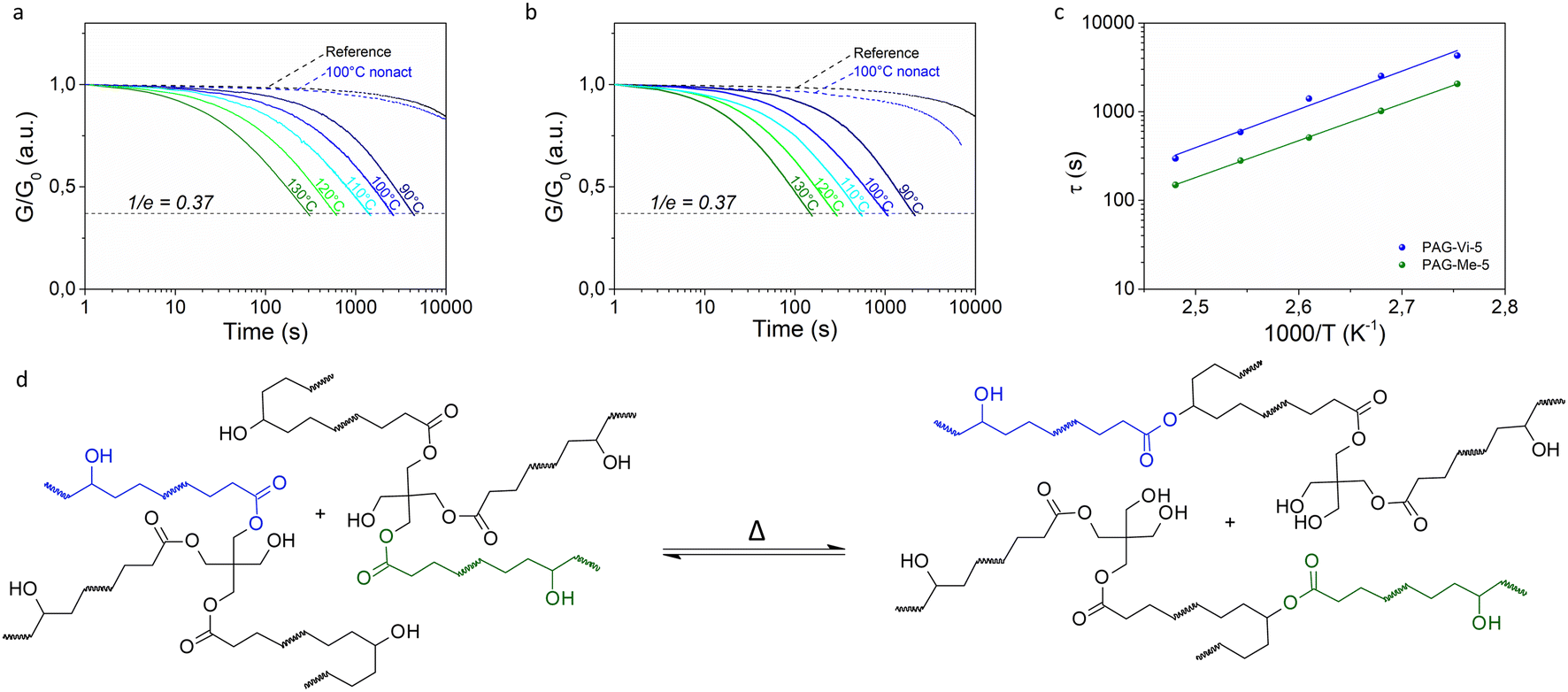 | ||
| Fig. 3 Stress relaxation curves of a UV-light activated thiol–ene photopolymer with (a) 5 mol% PAG-Vi or (b) 5 mol% PAG-Me measured at temperatures in the range 90–130 °C in comparison to the relaxation behavior of a non-activated (= only cured at 405 nm) and a reference sample (containing no PAG) at 100 °C; (c) Arrhenius plot of UV-activated formulations containing 5 mol% PAG-Vi or PAG-Me derived from measured relaxation times; (d) scheme of the thermo-activated transesterification in dynamic polymer networks. | ||
To activate the photoacid intentionally, the cured samples were subsequently exposed with UV-light (365 nm). UV irradiation led to a further increase in the curing degree of the monomers (97–99%) of PAG containing thiol–ene photopolymers as the radical photoinitiator has a higher reactivity in the UV region compared to 405 nm. Although the photocleavage of the PAGs required exposure doses of up to 25.9 J cm−2, final conversions higher than 90% could be reached for PAG-Vi and PAG-Th. In contrast, PAG-Me suffered from a lower reactivity and only a plateau around 80% conversion was achieved after prolonged UV exposure (129.4 J cm−2). The results obtained correlate well with the NMR photoactivation kinetics measurements performed in solution. The low activation rate of all introduced PAGs results from the lack of water and oxygen dissolved in the polymer matrix, which are required for the formation of sulfonic acid after photocleavage of the N–O group of naphthalimide-based PAGs (Fig. 2d).26 The higher photoactivity of PAG-Vi and PAG-Th are caused by the enhanced stability of the sulfonyl radical due to the stronger electron-donating effect of lone electron pairs of the sulfur atom in neighboring thioether groups in comparison to a methyl substituent.32 The incomplete activation of the PAG-Me under 365 nm light irradiation is likely caused by side reactions such as the photo-Fries rearrangement,33 which leads to the preservation of the naphthalimide ring structure. With respect to the immobilized PAG-Vi and PAG-Th, side reactions seem to play a minor role which is explained by the better stabilization of the benzene ring in the presence of the thioether substituent.
These findings were also corroborated by UV-Vis spectroscopy experiments on thin cured films. Samples containing PAG-Vi are lacking an absorption band at 264 nm (compared to the UV-Vis spectra of the pure compound in solution, see Fig. 2b) as the vinyl group is converted to a thioether bond during the thiol–ene reaction. During curing at 405 nm, almost no change in the absorbance profile was observed for compositions containing 5 mol% PAG-Vi or PAG-Th (Fig. S8d†). However, curing of the formulation containing PAG-Me resulted in a slight decrease in the main absorption peak at 333 nm, indicating partial activation as observed by FTIR spectroscopy. Subsequent irradiation with 365 nm light significantly reduced the intensity of the absorption peak (Fig. 2f) in all three resin formulations, again with PAG-Vi reacting faster than PAG-Th and PAG-Me. By considering NMR, FTIR and UV-Vis data, the efficiency of acid formation follows the order: PAG-Vi ∼ PAG-Th > PAG-Me; suggesting that thioether functionalities have a positive influence on the photo-release of the acidic species.
2.3. Polymer network properties and rheological investigations
Differential scanning calorimetry (DSC) measurements revealed that the addition of the PAGs do not significantly alter the thermal network properties. Compared to the reference network containing no PAG, the glass transition temperature (Tg) is slightly shifted to lower values in the presence of PAG-Me or PAG-Vi. This is mainly related to the lower cure degree obtained under irradiation with 405 nm, since subsequent activation of the photoacids by UV exposure increased the Tg of the polymer networks (Table S2†) due to additional post-curing reactions. Moreover, the lower functionality of PAG-Vi (monofunctional) and the plasticization character of PAG-Me (no reactive group) can lead to a reduction in the resulting Tg.For analyzing the influence and efficiency of the released photoacids (from PAG-Me and PAG-Vi) in catalyzing bond exchange reactions, stress-relaxation measurements were performed on photocured samples (containing 5 mol% of the respective PAG) and compared to a reference sample (containing no PAG) at 100 °C. The non-activated (= cured at 405 nm) sample with PAG-Vi showed nearly no relaxation and the curve was nearly identical to the reference sample (Fig. 3a). Although some acids are generated during curing at 405 nm, the amount seems to be too low to sufficiently accelerate the transesterification reaction at the applied temperature. In contrast, the non-activated sample containing PAG-Me started already to relax some stresses over time (Fig. 3b). This is in good agreement with the FTIR- and UV-Vis data, which had shown that PAG-Me has a higher reactivity than PAG-Vi upon 405 nm irradiation.
Subsequently, stress-relaxation measurements were performed in a temperature range from 90 to 130 °C on activated (= cured at 405 nm and irradiated at 365 nm with an exposure dose of 194 J cm−2) specimens. In general, the process of stress relaxation is accelerated at higher temperatures due to the faster bond exchange kinetics catalyzed by the photogenerated acid and the associated reorganization of the network structure against the applied deformation. To this effect, a temperature dependence of the characteristic relaxation time (τ) according to the Arrhenius model (eqn S1†) represents one of the key characteristics of vitrimers.2 Since the decrease in applied stress follows a general exponential trend, the relaxation times τ were assumed according to the Maxwell model, which defines τ as the time required to relax to 1/e (=37%) of the initial stress. Independent of the incorporated PAG, a linear dependency of τ on (1/T) is given in a semilogarithmic scale and thus an Arrhenius-type behavior is confirmed (Fig. 3c). Such properties imply a globally constant, temperature-independent network connectivity attributed to the associative bond exchange mechanism underlying dynamic transesterification (Fig. 3d).
The activation energies for the relaxation processes were 82.6 kJ mol−1 and 79.7 kJ mol−1 and the topology freezing temperatures, defined as the temperature at which the relaxation time is equal to 106 s, were 31 °C and 21 °C, for formulations containing PAG-Vi and PAG-Me, respectively. Benzenesulfonic acid, which is liberated by the activation of PAG-Me, is a very strong organic acid with pKa = −7.0.11 By activating the covalently linked PAG-Vi, a structurally analogous sulfonic acid (Fig. 2d) is formed, which is expected to have a similar pKa. However, the covalent immobilization of PAG-Vi in the polymer network restricts the mobility of the acid and leads to a reduced efficiency in catalyzing transesterification reactions. This results in longer relaxation times (∼factor of 3), a higher activation energy and topology freezing temperature compared to the free benzenesulfonic acid photo-released from PAG-Me.
2.4. Extraction studies and micro-patterning
To prove the covalent binding of PAG-Vi to the polymer network during photopolymerization, extraction experiments were carried out. For this, fully cured samples containing 5 mol% of PAG-Me or PAG-Vi were immersed in dichloromethane at room temperature for three days and the extracts were then analyzed with GC-MS (Fig. 4a). The samples were fully cured upon 405 nm light exposure and no activation of the acid (at 365 nm irradiation) was performed.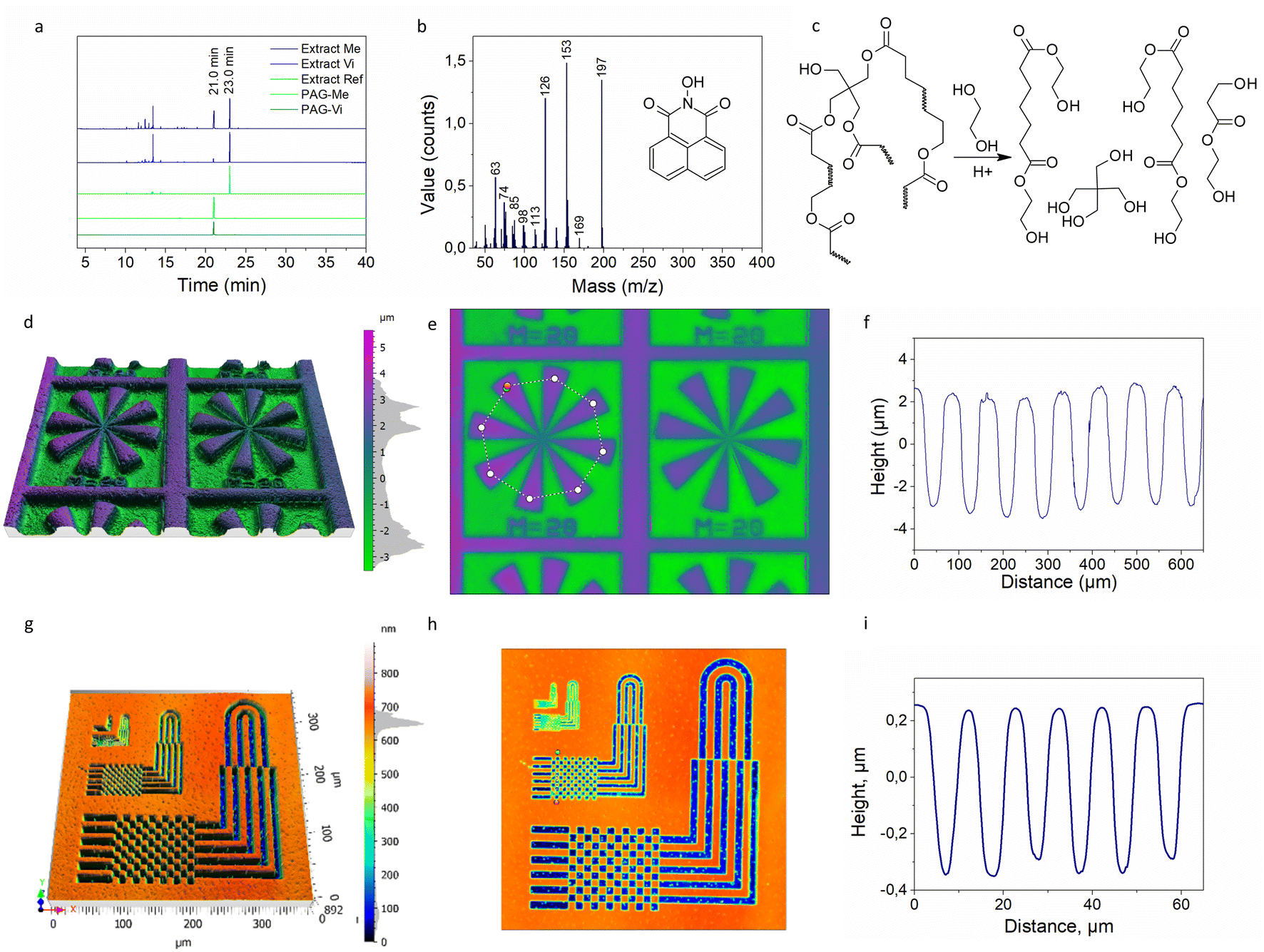 | ||
| Fig. 4 (a) Chromatograms of synthetized PAGs, and extracts from cured formulations; (b) mass-spectra of peak corresponding to PAGs (21 min retention time) in GC analysis; (c) reaction scheme of matrix depolymerization upon treatment with EG; analysis of a film with 5 mol% PAG-Vi patterned via photomask (365 nm) and subsequent development in EG: (d) 3D reconstruction of surface topography scans, (e) surface topography scans with marked profile pathway, (f) related surface profile; analysis of a film with 5 mol% PAG-Vi patterned via direct TPA laser writing (intensity: 20 mW, scanning speed: 500 μm s−1) and subsequent development in EG: (g) 3D reconstruction of surface topography scans; (h) surface topography scans with marked profile pathway; (i) related surface profile. | ||
GC-MS chromatograms of the pure PAGs in solution were used as reference. For both PAGs, a distinctive signal is observed at a retention time of 21 min (Fig. S9a and b†), which corresponds to 1,8-naphtalimide (Fig. 4b) as revealed by the related mass spectra (Fig. S9c and d†). The extract from the reference polymer sample, which contained no PAG, revealed only one significant peak at a retention time of 23 min, which corresponds to unreacted TPO-L (MS is presented in Fig. S9e†). The chromatograms of photopolymer extracts containing either PAG-Vi or PAG-Me show peaks with retention times between 10 and 15 min, whose mass spectra correspond to aliphatic fragments. These fragments are related to uncured monomers and oligomers present in the photopolymer network. In addition, both chromatograms display the signal related to TPO-L (23 min) with comparable intensity. While the naphtalimide peak (21 min) is visible in both extracts, its intensity is significantly lower for the sample containing PAG-Vi compared to PAG-Me. In particular, the ratio between the peak areas of naphtalimide and TPO-L was 0.11 and 0.87 for formulations containing PAG-Vi and PAG-Me, respectively. Thus, the results clearly evidence that the majority of the PAG-Vi molecules is immobilized in the photopolymer network.
To highlight the ability of controlling transesterification reactions on a microscale level, thin films of thiol–ene formulations containing 5 mol% of PAG-Vi and PAG-Me were prepared by spin-casting the resins on silicon wafers. After curing of the spin-casted films under 405 nm irradiation, positive-tone micropatterns were inscribed by photolithography exposing selected areas of the cured films with UV-light at 365 nm through a photomask. In the development step, the films were immersed in ethylene glycol (EG) at 100 °C for 1 h to selectively degrade the networks by acid-catalyzed transesterification reaction of the ester moieties within the network across the –OH groups of the applied solvent (Fig. 4c). After the treatment, the patterned films were washed with water and dried under vacuum at 80 °C to remove residual water and EG.
Topography scans of the developed samples containing either PAG-Vi or PAG-Me are shown in Fig. 4d, e, S10a and b,† respectively. In addition, profiles of the inscribed lines and spaces are presented in Fig. 4f (PAG-Vi) and Fig. S10c† (PAG-Me). For photopolymers containing the immobilized PAG-Vi, positive-tone patterns with an average depth of 7 μm and a lateral resolution of 30 μm were obtained by local depolymerization of the network. Films containing the free PAG-Me, exhibit shallow and blurry imprinted patterns. These were caused by the migration of the activated acid into the non-activated areas but also by leaching of the active catalyzing species in the applied solvent during the development step as p-toluene sulfonic acid is highly soluble in EG.
Advancing from photolithography, two-photon absorption (TPA) direct laser writing was further applied to inscribe even smaller patterns in thiol–ene photopolymers containing PAG-Vi as photolatent transesterification catalyst. This technique decreases the activation of “tails” and thus increases the resolution of the pattern. Direct writing was carried out with a femtosecond laser working at 780 nm. For sample preparation, the thiol–ene resins were spin-cast on silicon wafers, whose surface was chemically modified to ensure a high adhesion between the polymer film and the substrate (particularly during the development step).
The laser parameters were optimized by varying scanning speed (from 250 to 1500 μm s−1) and laser power (from 20 to 40 mW) to efficiently activate the acid catalyst without degrading the photopolymer during writing (Fig. S10d and e†). Fig. 4g and h show the resulting micro-patterns obtained after the development step in EG. Micro-patterns with a resolution of 5 μm with an average depth of 0.6 μm were inscribed in the photopolymer films by using a high scanning speed (500 μm s−1) and a low intensity (20 mW) of the femtosecond laser. Even resolution of 2 μm was achieved but without proper reliability (Fig. S10f–h†). The surface profile of the inscribed “chess-deck” pattern is shown in Fig. 4i and clearly evidences the possibility to adjust the properties of vitrimers on a microscale level.
2.5. Reprogramming of bulk properties
Along with the solubility, the introduction of the linkable transesterification catalyst PAG-Vi paves the way towards the reprogramming of mechanical properties in a localized manner. In a post-modification step, the networks are immersed in solutions containing selected alcohols or carboxylic acids, which are then selectively incorporated by carrying out transesterification reactions in the previously light activated areas of the film. To demonstrate the potential of the latent catalyst PAG-Vi for adjusting bulk properties in a controlled way, free-standing thiol–ene films with and without PAG-Vi were prepared.First, they were immersed in EG at 100 °C for 30 min and tensile properties of the treated test specimen were characterized by uniaxial tensile tests. The related stress–strain curves are presented in Fig. S11a–i.† The sample containing the activated PAG-Vi (365 nm, exposure dose of 194 J cm−2) underwent transesterification reactions between the ester groups of the polymer matrix and the hydroxyl groups of the solvent (Fig. 5a). As a result of the decreasing crosslinking density, the Young's modulus was reduced by 65% and a softer network with a higher elongation at break (134%) was obtained (Fig. 5b and c). Prolonged treatment with EG led to dissolution of the polymer network, similar as observed by micropatterning experiments. The cured sample with the non-activated PAG-Vi showed similar tensile properties as the reference network (without PAG-Vi). In both cases a slight increase in the modulus of elasticity and a decrease in elongation at break were observed. We assume that this slight embrittlement of the materials is caused by the extraction of non-reacted monomers (acting as plasticizers) upon swelling of the material in EG.
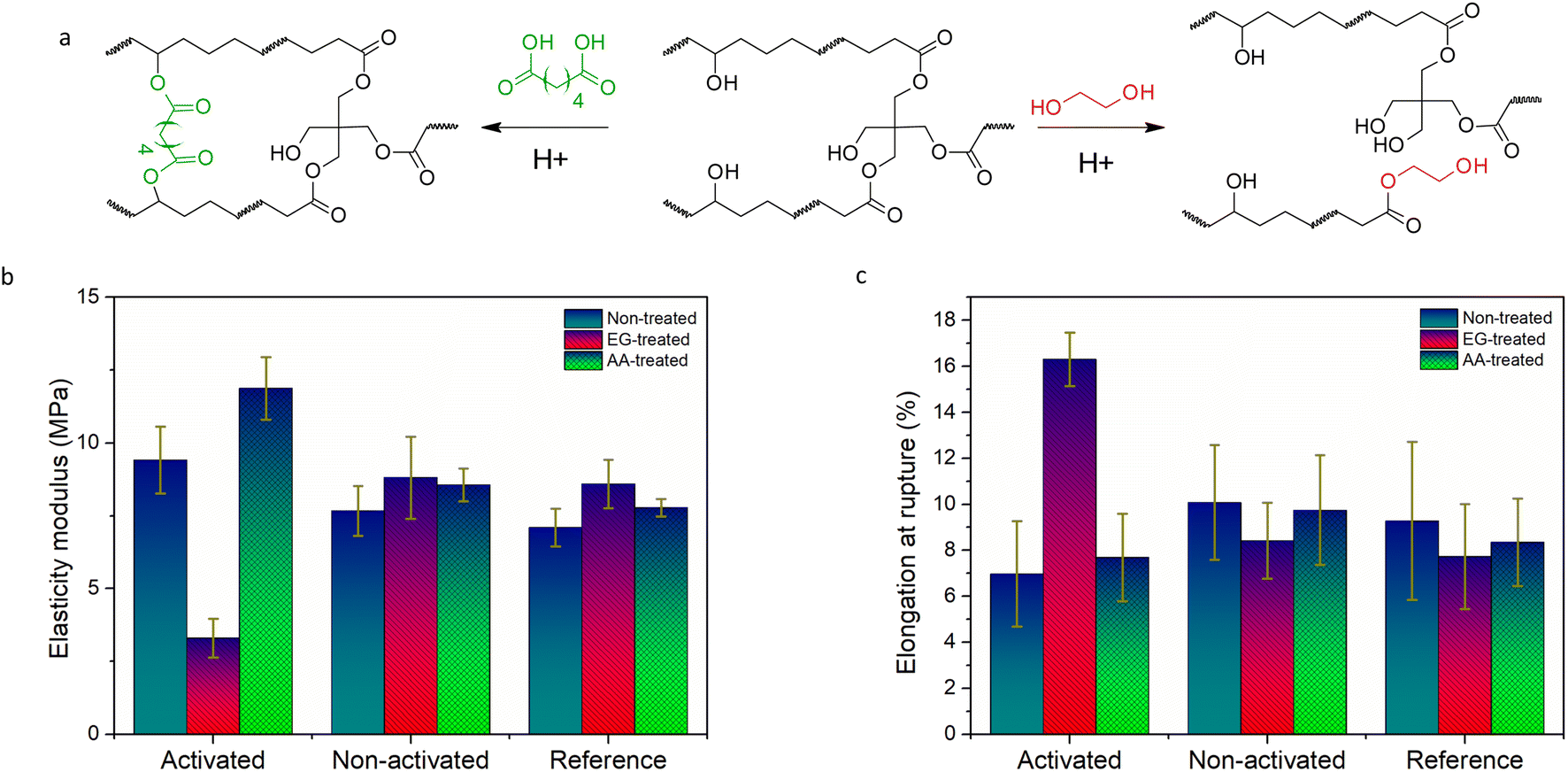 | ||
| Fig. 5 (a) Reaction schemes of an additional crosslinking of the polymer network upon treatment with adipic acid and a decrease of the crosslinking degree upon treatment with EG; comparison of the (b) Young's modulus and (c) elongation at rupture of activated (cured at 405 nm and exposed at 365 nm) and non-activated (cured at 405 nm) thiol–ene photopolymers with 5 mol% PAG-Vi in comparison to a reference network containing no PAG after post-curing, and treatment with either EG or adipic acid. | ||
To increase the Young's modulus of the material, adipic acid was incorporated into the polymer network as an additional crosslinking agent by swelling the photopolymer in a saturated adipic acid solution in acetone, followed by a drying step. The samples were post-baked at 150 °C for 4 h in vacuum in order to enable esterification reactions between the dicarboxylic acid and free hydroxyl groups of the matrix. These reactions led to additional cross-linking of the polymer network, resulting in a 26% increase in the Young's modulus. In contrast, the effect on the ultimate elongation is less pronounced, which is likely attributable to the flexible structure of adipic acid. Furthermore, the mechanical properties of the reference sample and the non-activated one are nearly constant again. In both cases, only a slight increase in the modulus of elasticity and an even smaller decrease in elongation at break were observed. This proves, that transesterification reactions lead to the change in mechanical properties (in case of activated samples), which do not occur in the reference samples and the non-activated ones and consequently don't reveal any change in the mechanical properties.
3. Conclusions
A covalent attachable photoacid generator was successfully synthesized, which enabled local control of dynamic transesterification reactions in photocurable materials on a microscale level. Across its vinyl group, PAG-Vi was immobilized in a thiol–ene photopolymer during the radical curing reaction initiated by 405 nm light irradiation. Subsequent photocleavage of the naphtalimide-based PAG yielded strong sulfonic acid species, which was exploited to locally catalyze thermo-activated transesterification reactions. The performance of PAG-Vi was compared to reference compounds (PAG-Me and PAG-Th). FTIR-, UV-Vis and NMR-spectroscopy measurements revealed that neighboring thioether functionalities accelerate the photo-release of the acidic species (365 nm) whilst they stabilize the compound under visible light exposure (405 nm). This improves the λ-orthogonality between the curing and activation reaction, which is also confirmed by stress relaxation experiments. The immobilization of the catalyst was confirmed by extraction studies followed by GC-MS analysis and was exploited to manipulate material properties on a microscale. As proof of concept, positive-tone micropatterns with a resolution of 5 μm were inscribed in the thiol–ene photopolymer by direct TPA laser patterning. Here, the deprotected acid was used to catalyze a depolymerization reaction of the network in ethylene glycol at 100 °C. The selective adjustment of crosslink density in a post-modification step was further applied to change mechanical (bulk) properties of free-standing thiol–ene films. Depending on the applied low molecular weight alcohol or carboxylic acid, the material's Young's modulus was varied between 3.3 and 11.9 MPa. The introduced concept is highly versatile as the bulk properties are expected to be programmed with numerous compounds bearing free alcohol, ester or carboxylic acid moieties in a local manner. By being able to locally tune bulk properties of vitrimers simply by light activation and a post-modification step, the approach paves the way towards the design of multi-material structures and gradient materials, which additionally benefit from the features of vitrimers. On a microscale level, this is interesting for biomimetic structures, (micro)robotics, electronics, smart surfaces or microfluidics.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. K. (formal analysis: lead; investigation: lead; validation: equal; visualization: lead; writing – original draft: lead; writing – review & editing: equal) W. A. (investigation: supporting; validation: supporting; writing – review & editing: supporting) A. J. (investigation: supporting; validation: supporting; writing – review & editing: supporting) M. S. (investigation: supporting; validation: supporting; writing – review & editing: supporting) Y. L. (investigation: supporting; validation: supporting; writing – review & editing: supporting) S. S. (funding acquisition: lead; project administration: supporting; supervision: supporting; validation: supporting; visualization: supporting; writing – review & editing: equal) E. R. (conceptualization: lead; project administration: lead; supervision: lead; validation: equal; visualization: supporting; writing – original draft: supporting; writing – review & editing: equal).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Part of the research work was performed with the “SMART” project. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 8400088. Part of the research was also carried out within the COMET-Module project “Repairtecture” (project no.: 904927) at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Climate Action, Environment, Energy, Mobility, Innovation and Technology and the Federal Ministry of Labour and Economy. The PCCL is funded by the Austrian Government and the State Governments of Styria, Upper and Lower Austria.References
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS
, https://www.sciencedirect.com/science/article/pii/S0032386120302299..
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC
-
(a) B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, Chem. Eng. J., 2020, 385, 123820 CrossRef CAS
-
(a) Y. Yang, Y. Xu, Y. Ji and Y. Wei, Prog. Mater. Sci., 2021, 120, 100710 CrossRef CAS
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS
- S. Kaiser, J. Jandl, P. Novak and S. Schlögl, Soft Matter, 2020, 16, 8577–8590 RSC
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed
-
(a) W. Alabiso and S. Schlögl, Polymers, 2020, 12, 1160 CrossRef
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS PubMed
- D. Reisinger, S. Kaiser, E. Rossegger, W. Alabiso, B. Rieger and S. Schlögl, Angew. Chem., Int. Ed., 2021, 60, 14302–14306 CrossRef CAS
-
(a) Y. Yang, Z. Pei, X. Zhang, L. Tao, Y. Wei and Y. Ji, Chem. Sci., 2014, 5, 3486–3492 RSC
- M. Giebler, W. Alabiso, V. Wieser, S. Radl and S. Schlögl, Macromol. Rapid Commun., 2021, 42, e2000466, DOI:10.1002/marc.202000466
- B. T. Worrell, M. K. McBride, G. B. Lyon, L. M. Cox, C. Wang, S. Mavila, C.-H. Lim, H. M. Coley, C. B. Musgrave, Y. Ding and C. N. Bowman, Nat. Commun., 2018, 9, 2804 CrossRef
- D. Reisinger, A. Hellmayr, M. Paris, M. Haas, T. Griesser and S. Schlögl, Polym. Chem., 2023, 14, 3082–3090 RSC
-
(a) E. Rossegger, K. Moazzen, M. Fleisch and S. Schlögl, Polym. Chem., 2021, 12, 3077–3083 RSC
-
(a) F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2019, 113, 297–304 CrossRef CAS
-
(a) X. Niu, F. Wang, X. Li, R. Zhang, Q. Wu and P. Sun, Ind. Eng. Chem. Res., 2019, 58, 5698–5706 CrossRef CAS
- W. Xu, T. Li, G. Li, Y. Wu and T. Miyashita, J. Photochem. Photobiol., A, 2011, 219, 50–57 CrossRef CAS
- M. Ikbal, R. Banerjee, S. Atta, D. Dhara, A. Anoop and N. D. P. Singh, J. Org. Chem., 2012, 77, 10557–10567 CrossRef CAS
- G.-J. Ye, T.-T. Zhao, Z.-N. Jin, P.-Y. Gu, J.-Y. Mao, Q.-H. Xu, Q.-F. Xu, J.-M. Lu, N.-J. Li and Y.-L. Song, Dyes Pigm., 2012, 94, 271–277 CrossRef CAS
-
(a) Q. Sun, B. Feng, Z. Sun, R. Liu, H. Ding and Y. Jin, React. Chem. Eng., 2024, 9, 630–641 RSC
- J.-P. Malval, S. Suzuki, F. Morlet-Savary, X. Allonas, J.-P. Fouassier, S. Takahara and T. Yamaoka, J. Phys. Chem. A, 2008, 112, 3879–3885 CrossRef CAS PubMed
-
(a) S. Yordanova-Tomova, D. Cheshmedzhieva, S. Stoyanov, T. Dudev and I. Grabchev, Sensors, 2020, 20, 3892 CrossRef CAS PubMed
- J.-P. Malval, F. Morlet-Savary, X. Allonas, J.-P. Fouassier, S. Suzuki, S. Takahara and T. Yamaoka, Chem. Phys. Lett., 2007, 443, 323–327 CrossRef CAS
-
(a) M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2020, 59, 6832–6838, DOI:10.1002/anie.201915132
- S. L. Walden, J. A. Carroll, A.-N. Unterreiner and C. Barner-Kowollik, Adv. Sci., 2024, 11, e2306014, DOI:10.1002/advs.202306014
- S. Enomoto, D. T. Nguyen and S. Tagawa, Jpn. J. Appl. Phys., 2013, 52, 06GC03 CrossRef
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS PubMed
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573, DOI:10.1002/anie.200903924
-
(a) G. Noirbent and F. Dumur, Eur. Polym. J., 2020, 132, 109702 CrossRef CAS
- E. Torti, G. Della Giustina, S. Protti, D. Merli, G. Brusatin and M. Fagnoni, RSC Adv., 2015, 5, 33239–33248 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04932b |
| This journal is © The Royal Society of Chemistry 2024 |