
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective total synthesis of (+)-cylindricine B†
Dallas M.
Dukes
,
Victor K.
Atanassov
and
Joel M.
Smith
 *
*
Florida State University, Department of Chemistry and Biochemistry, Laboratories of Molecular Recognition, 95 Chieftan Way, Tallahassee, FL 32306, USA. E-mail: jsmith17@fsu.edu
First published on 13th September 2024
Abstract
This article describes the first enantioselective synthesis of the Tasmanian marine alkaloid (+)-cylindricine B. The concise construction of the compound hinged on dearomative retrosynthetic logic combined with a tactical advance in the generation of congested, cyclic, alpha-tertiary amine centers. The scope of this key coupling reaction was explored in addition to providing a synthetic application for Cu-catalyzed enantioselective dearomatization of N-acyl-pyridiniums. The synthesis proceeds in five or six steps from commercially available starting materials.
Introduction
Marine natural products have been a unique source of leads for small molecule therapeutics for the past half-century.1,2 Despite their therapeutic promise, however, it can often be challenging to source these bioactive molecules in large quantities due to their relative paucity and/or arduous isolation. As a result, synthetic chemistry plays an elevated role in elucidating the medicinal indices of these natural products, as their necessity demands strategically and tactically practical designs for laboratory preparation.3 In particular, the total synthesis of marine alkaloids has benefitted the clinical advancement of several unique chemotypes, including tetrodotoxin,4,5 lurbinectedin (Zepzelca®),6,7 and salinosporamide A (Marizomib).8–10Isolated off the coast of Tasmania in southern Australia, the cylindricine natural product family is constituted of several structurally intricate and stereochemically dense marine alkaloids (see Fig. 1A). Topologically, they are characterized by a tricyclic scaffold with four stereogenic centres, a piperidone core, and most family members bear a C14 heteroatomic functionality that varies between a chloride (1, 9),11 alcohol (2), ether (3), acetate (4),12 thiocyanate (5–7), and isothiocyanate (8).13 Additionally, each family member has a chiral alpha-tertiary amine moiety that constitutes the primary synthetic hurdle, as these congested centres have been cited for being historically challenging for construction.14 Intriguingly, while many racemic15–21 and enantiospecific preparations of these alkaloids have been reported in the literature,22–33 only one approach towards cylindricine C (2) is enantioselective in nature.34 Herein, we report the inaugural enantioselective preparation of (+)-cylindricine B (9), a flagship isomer of this marine alkaloid family.
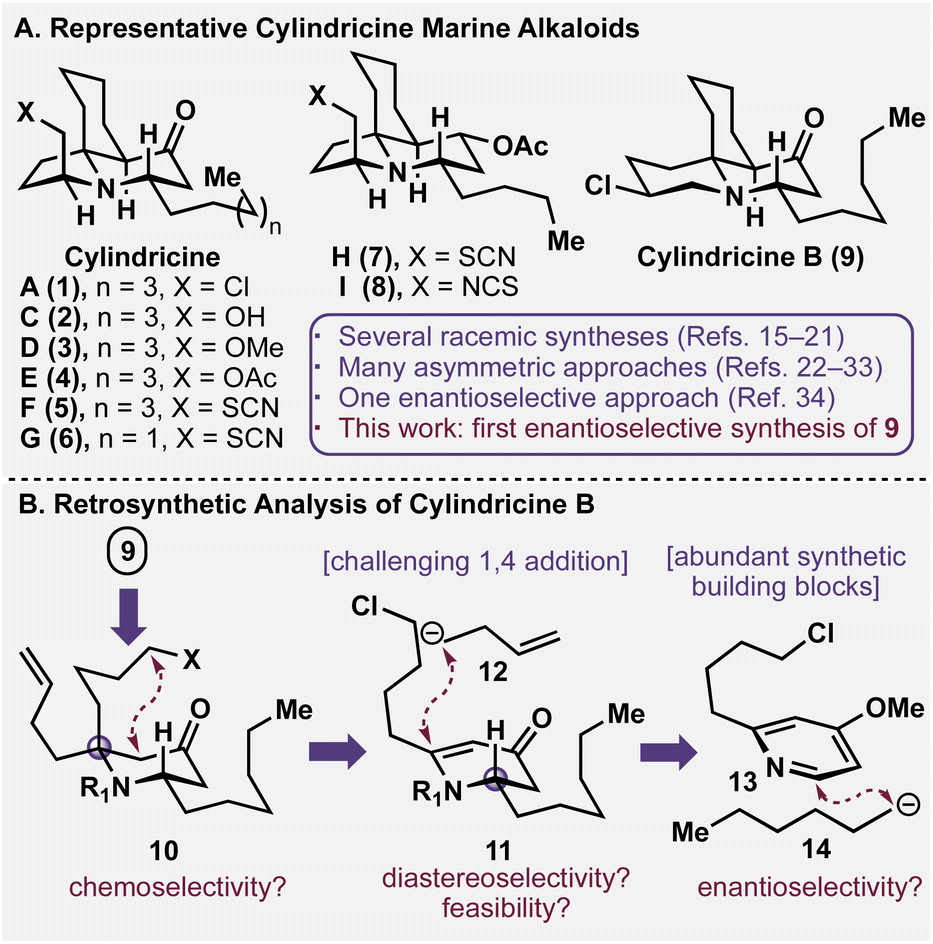 | ||
| Fig. 1 (A) Structures of various cylindricine natural products. (B) Dearomative retrosynthetic logic towards cylindricine B. | ||
A retrosynthetic analysis of cylindricine B (9) is depicted in Fig. 1B. It was envisioned that the chlorinated piperidine moiety could be forged at a late stage in the synthesis between the piperidine nitrogen and the adventitious butenyl side chain (see 10).15 Furthermore, intramolecular enolate trapping via nucleophilic substitution of a pendant halide would enable the generation of the cis-azadecalinone ring system.35 The planned introduction of the butenyl group in 10 would require the addition of a four-carbon nucleophile (12) in conjugate fashion to vinylogous amide 11. This prospective transformation is deceptively challenging, and while this type of addition is common for unsubstituted variants,36 the establishment of this fully substituted centre has little precedent in the chemical literature.37–39 However, we envisioned that its realization in this synthetic context would enable a direct access to the appropriately functionalized piperidone core. Vinylogous amide 11 was envisioned to arise from in situ activation and dearomatization of commercial pyridine 13 with anion 14 following formation of an intermediate pyridinium. It should be noted that Donohoe19 and Comins37 have described dearomative strategies towards the cylindricines. But, as articulated in their work, the establishment of the desired alpha tertiary amine either required circuitous downstream functional group interconversion or resulted in an undesired and/or unresolvable stereochemical outcome. By solving this difficult tactical requirement (i.e. conversion of 11 to 10), concise synthetic access would be realized through the leveraging of a feedstock pyridine and simple organometallic nucleophilic coupling partners as starting materials.
Results and discussion
The initial synthetic approach towards the cylindricines is depicted in Fig. 2. At the outset, a racemic construction of 9 was evaluated to assess the feasibility and selectivity of each step in the intended sequence. Commercially available pyridine 15 was selected as the starting point, and was treated with methyl chloroformate followed by n-hexylmagnesium bromide (16) at −78 °C to give piperidinone 17 in 84% yield. Next, the key construction of the fully substituted centre was evaluated. While many reaction parameters (e.g., metal salts, Lewis acids) and substrates (differing carbamoyl groups) were evaluated, it was determined that the use of superstoichiometric amounts of organocuprate in the presence of excess trimethylsilyl triflate (TMSOTf) resulted in a 67% yield of desired product 19a (see ESI†), following acidic hydrolysis of the intermediate silyl enol ether. Catalytic amounts (20 mol%) of Cu can be employed, but with lower yields and longer reaction times. At this stage of the synthesis, however, it was unknown whether this addition gave the desired diastereoselectivity needed to complete the synthesis of 9. Thus, as 2D NMR spectroscopic analysis was inconclusive as to the facial preference of the addition, it was decided to validate this through the synthesis of a more elaborated and putative precursor. In this vein, it was seen that targeting 22b (see Fig. 2) was operative, as its structure, if correct, had been previously reported by Snider and co-workers.15 With this goal in mind, chloropiperidine 19a was converted to its corresponding iodide (20) with NaI, and then treated with KOtBu in THF to promote cyclization to the cis azadecalinone 21a, while establishing the third stereocentre at C5 in 65% over the two steps. Cleavage of the methyl carbamate was accomplished with 2-mercaptoethanol in the presence of potassium phosphate at elevated temperature to cleanly deliver aminoketone 22a in 53% yield. This compound did not spectroscopically (1H NMR) match Snider's reported intermediate (22b), likely indicating that the conversion of 17 to 19a occurred in an anti fashion with respect to the established C2 stereocentre. This anti diastereoselectivity was significant because it provided evidence that conjugate additions to 2,6-disubstituted 4-piperidinones can proceed with the opposite facial preference compared with their 2-monosubstituted counterparts.36 As many alkaloid structures contain these alpha-tertiary amine substructures,14 this understanding of the feasibility and stereoselectivity will enable this transformation's use in downstream synthetic applications.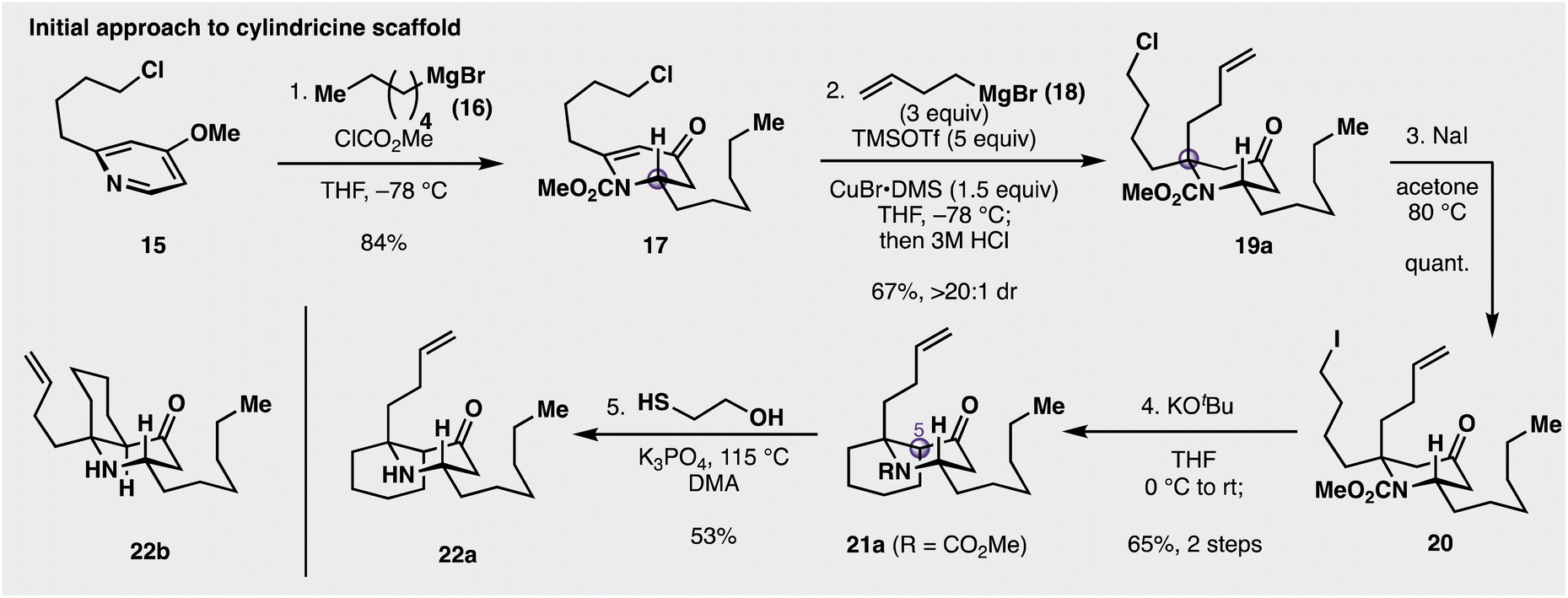 | ||
| Fig. 2 Original synthetic approach to the cylindricines. This approach resulted in the synthesis of 22a, which, based on comparison to past literature, is the 5,10-bis-epimeric diastereomer of the desired intermediate (structure is as depicted above). | ||
Given the success of this diastereoselective 1,4 addition to 17, we wanted to further evaluate the scope of nucleophiles competent for addition to this prochiral centre (see Fig. 3). In addition to butenylmagnesium bromide (16), other alkyl Grignard reagents added diastereoselectively to the 4-piperidinone including methyl (19b), isopropyl (19c), and even t-butylmagnesium bromide (19d) added in with moderate efficiency (22% yield). Utilization of a prochiral Grignard reagent derived from 2-bromohexane resulted in the formation of two new stereocentres with a 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio about the C7 stereocentre and >20:1 about the C6 stereocentre (see 19e). Another Grignard bearing an acetal functionality (19f), potentially useful for downstream manipulation, was diastereoselectively appended in 65% yield. Benzylmagnesium bromide (19g), vinylmagnesium bromide (19h), and isopropenylmagnesium bromide (19i) were also successfully added onto the piperidone scaffold in 65%, 43%, and 55% yields respectively. Furthermore, product 19j was synthesized in 79% from the addition of the corresponding phenyl Grignard reagent. While A1,3 strain between the 2-substituent and the carbamoyl group has proven a driving force for the diastereoselectivity of these additions in the past, we hypothesized the additional C6 alkyl chain complicated the traditionally accepted pathway for these additions (see Fig. 3). As Comins' has alluded to in prior work,37 the additional C6 substituent provided a complex stereoelectronic conundrum in which the axially disposed conformation of the C2 group in the substrate (A), in concert with the carbamoyl group and the C6 substituent, likely drives the observed addition to proceed through an initial twist-boat conformation (B) that results in the observed anti diastereoselectivity, that, upon hydrolysis, give the observed products (19a–k). Importantly, all these additions proceeded with complete diastereocontrol (>20:1) and provide a facile avenue for a diverse array of highly substituted 4-piperidone congeners.
:1 diastereomeric ratio about the C7 stereocentre and >20:1 about the C6 stereocentre (see 19e). Another Grignard bearing an acetal functionality (19f), potentially useful for downstream manipulation, was diastereoselectively appended in 65% yield. Benzylmagnesium bromide (19g), vinylmagnesium bromide (19h), and isopropenylmagnesium bromide (19i) were also successfully added onto the piperidone scaffold in 65%, 43%, and 55% yields respectively. Furthermore, product 19j was synthesized in 79% from the addition of the corresponding phenyl Grignard reagent. While A1,3 strain between the 2-substituent and the carbamoyl group has proven a driving force for the diastereoselectivity of these additions in the past, we hypothesized the additional C6 alkyl chain complicated the traditionally accepted pathway for these additions (see Fig. 3). As Comins' has alluded to in prior work,37 the additional C6 substituent provided a complex stereoelectronic conundrum in which the axially disposed conformation of the C2 group in the substrate (A), in concert with the carbamoyl group and the C6 substituent, likely drives the observed addition to proceed through an initial twist-boat conformation (B) that results in the observed anti diastereoselectivity, that, upon hydrolysis, give the observed products (19a–k). Importantly, all these additions proceeded with complete diastereocontrol (>20:1) and provide a facile avenue for a diverse array of highly substituted 4-piperidone congeners.
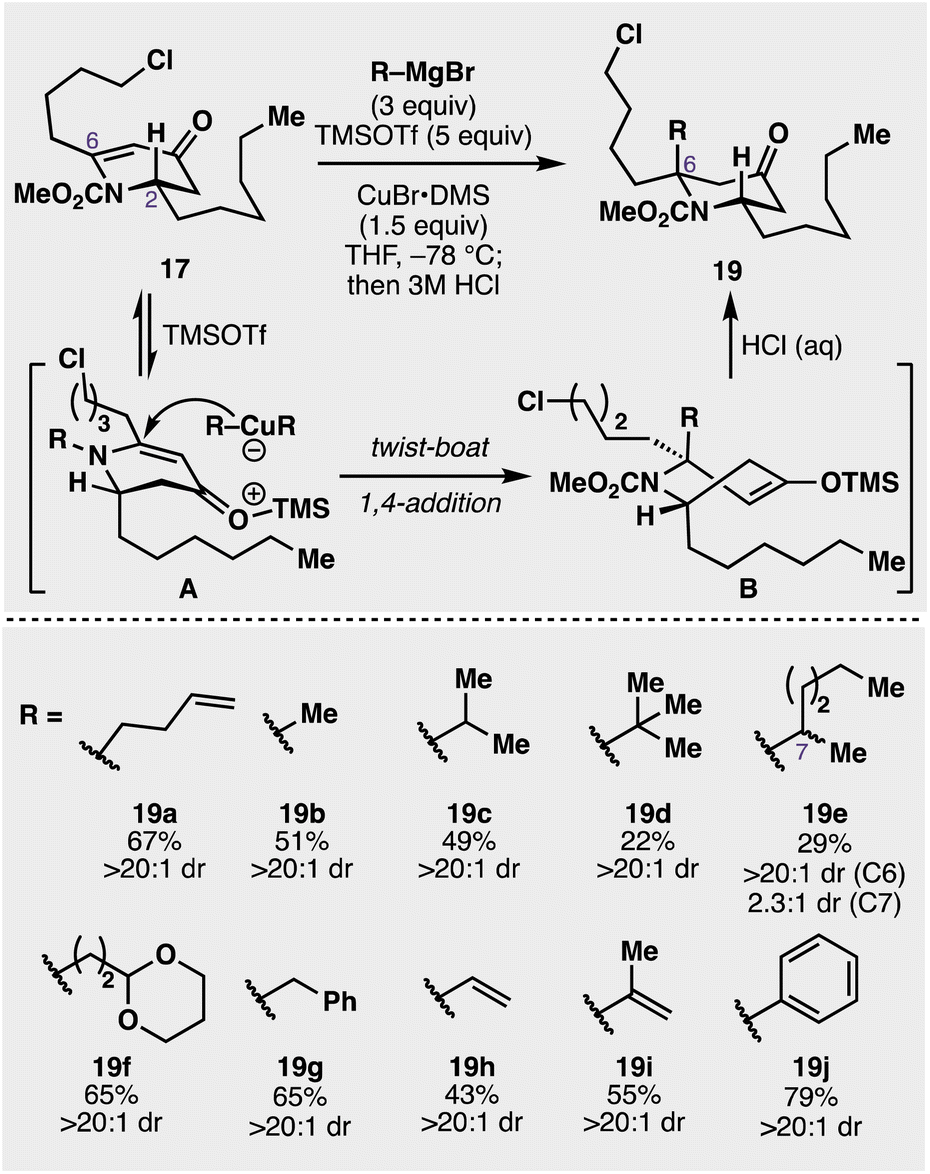 | ||
| Fig. 3 Investigation of the scope for the 1,4 addition to vinylogous amide 17 to furnish multiply substituted piperidinones of structure 19. | ||
Having understood the diastereoselective nature for synthesizing this fully substituted centre, a modified approach to the synthesis of cylindricine B (9) was undertaken (Fig. 4). In addition to a strategic change in the starting material, it was also surmised that the synthesis could be rendered enantioselective. To this end, butenylpyridine 23 was employed as the starting material for the enantioselective generation of piperidinone 24. Based on precedent from Harutyunyan and co-workers,40 treatment of 23 with 3 equivalents of methyl chloroformate and Grignard 16 (3 equiv.) in the presence of the CuBr complexed with (R,R)-Ph-BPE in CH2Cl2 gave 24 in 67% yield and with 80.3% ee. Notably, a higher asymmetric induction was observed when only 2 equivalents of the chloroformate and the nucleophile (16) were employed, but the yield of 24 decreased to only 32%, but with higher enantioselectivity (91.4% ee). We attribute the lower enantiomeric excess in the first iteration was due to a slightly higher rate for the racemic background reaction of the Grignard reagent adding the pyridinium electrophile. Additionally, the preparation of 16 in ether was essential for the enantioselectivity as utilizing THF as a cosolvent resulted in poor asymmetric induction (17% ee). With 24 in hand, diastereoselective addition of chlorobutyl Grignard 25 in the presence of CuBr(DMS) generated piperidone 19k in good yield and with high diastereoselectivity. Next, deprotonation of 19k with excess KOtBu facilitated the intramolecular displacement of the pendant chloride to synthesize the azadecalinone 21b,35 which, upon further treatment with 2-mercaptoethanol and base gave a modest yield (28%) of amine 22b.41 Conveniently, separating the cyclization and carbamate cleavage events resulted in a much higher two-step yield of 66% percent (72% and 91% for each step, respectively). Finally, chlorination of the amine was cleanly accomplished with NCS, and subsequent treatment with a CuCl/CuCl2 mixture promoted amino chlorination of the butenyl side chain. Initially, this reaction formed a mixture of cylindricine A, cylindricine B, and their corresponding epimers. As precedented by Snider15 and Heathcock,16 this atom-transfer reaction was not diastereoselective, to which, following chromatographic separation of the reaction mixture, rendered pure samples of (+)-cylindricine B (9) and a 9:1 mixture of 13-epi-cylindricine B and 13-epi-cylindricine A. The successful synthesis of the natural product further confirmed the diastereoselectivity of the key 1,4-addition to establish the chiral alpha-tertiary amine centre. Lastly, it is worth noting that while earlier reports claimed that carbamate cleavage for substrates such as 21b were not feasible,16 the syntheses above disprove any previous claims of inertness about these protected, multiply substituted piperidone scaffolds.
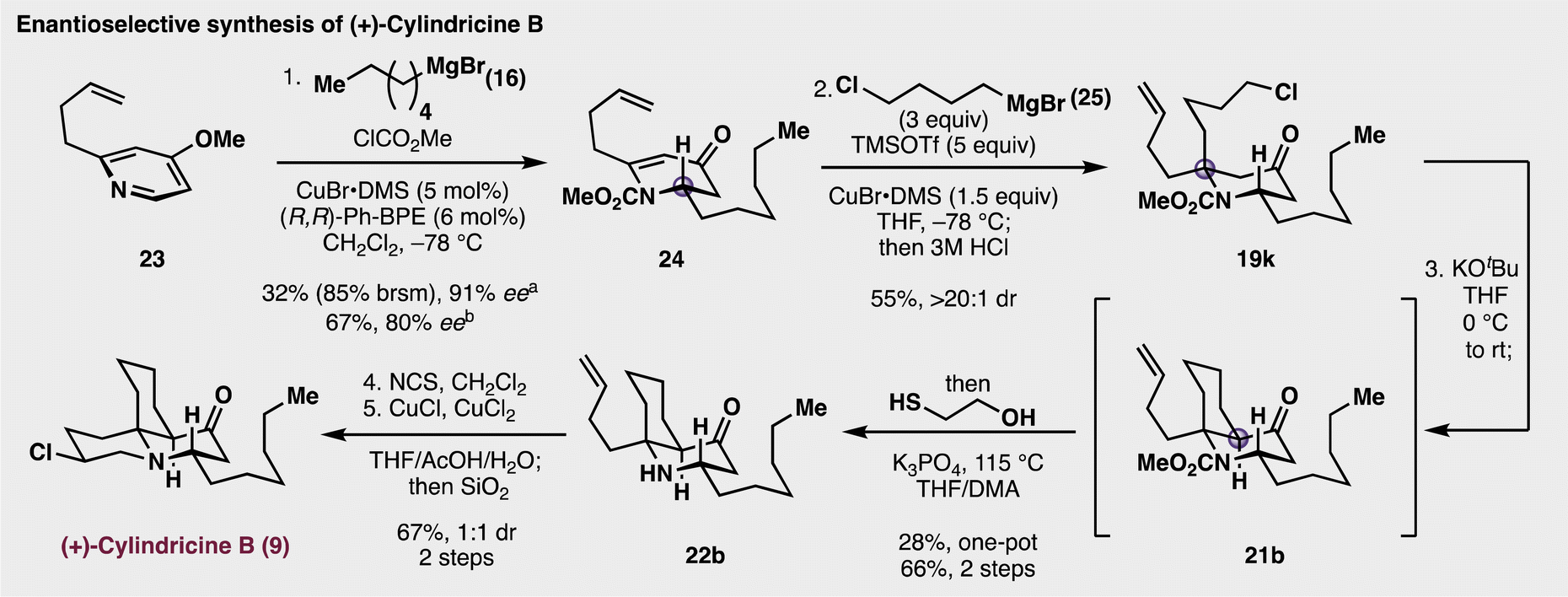 | ||
| Fig. 4 Modified and successful synthetic approach to cylindricine B. aReaction performed with 2 equiv. of C6H13MgBr and 2 equiv. of ClCO2Me. bReaction performed with 3 equiv. of C6H13MgBr and 3 equiv. of ClCO2Me. | ||
Conclusion
In conclusion, we have accomplished the first reported enantioselective synthesis of (+)-cylindricine B (9) in 5 or 6 steps from commercially available starting materials. This also constitutes only the second enantioselective route to the cylindricine family of marine alkaloids for which, to date, asymmetric syntheses have mainly hinged on using the chiral pool as synthetic starting points. A rare, but tactically important 1,4-addition to a 4-piperidinone (e.g., 24) was utilized and further evaluated for its broader synthetic utility. This new protocol for the synthesis of (+)-9 proceeds with a maximum 16% overall yield constituting one of the most concise routes reported for this synthetic target. Overall, the brevity of the approach was enabled by overcoming tactical challenges associated with the construction of multiply substituted piperidines in addition to the strategic leveraging of robust heteroaromatic starting materials. As dearomative retrosynthetic logic continues to inspire efficient access to numerous alkaloid scaffolds, it is contended that the future is bright for its ability to inspire novel tactical and strategic disconnections in heterocyclic synthesis.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
This project included a joint conceptualization (JMS and DMD) while its experimental implementation was also carried out jointly (DMD and VKA). The original draft of the manuscript was composed by the corresponding author (JMS), and all authors were involved in its review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge and are grateful generous support from Florida State University (FSU), and the National Science Foundation (CHE-2154662) for funding. We also thank Prof. James Frederich for thoughtful discussions.Notes and references
- K.-H. Altmann, Chimia, 2017, 71, 646 CrossRef CAS.
- C. Jiménez, ACS Med. Chem. Lett., 2018, 9, 959–961 CrossRef PubMed.
- C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC.
- R. González-Cano, M. C. Ruiz-Cantero, M. Santos-Caballero, C. Gómez-Navas, M. Á. Tejada and F. R. Nieto, Toxins, 2021, 13, 483 CrossRef PubMed.
- Y. Kishi, T. Fukuyama, M. Aratani, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura and H. Kakoi, J. Am. Chem. Soc., 1972, 94, 9219–9221 CrossRef CAS PubMed.
- A. Desai, C. J. Smith, Y. Ashara, J. J. Orme, S. Zanwar, A. Potter, C. Hocum, J. N. Moffett, A. J. Schwecke, R. Manochakian, Y. Lou, Y. Zhao, V. Ernani, P. Savvides, J. Molina, A. Dimou, A. S. Mansfield, K. Parikh and K. Leventakos, Clin. Lung Cancer, 2023, 24, 689–695e1 CrossRef CAS PubMed.
- E. J. Corey, D. Y. Gin and R. S. Kania, J. Am. Chem. Soc., 1996, 118, 9202–9203 CrossRef CAS.
- B. C. Potts and K. S. Lam, Mar. Drugs, 2010, 8, 835–880 CrossRef CAS PubMed.
- A. Endo and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 8298–8299 CrossRef CAS PubMed.
- L. R. Reddy, P. Saravanan and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 6230–6231 CrossRef CAS PubMed.
- A. J. Blackman, C. Li, D. C. R. Hockless, B. W. Skelton and A. H. White, Tetrahedron, 1993, 49, 8645–8656 CrossRef CAS.
- C. P. Li and A. J. Blackman, Aust. J. Chem., 1994, 47, 1355–1361 CrossRef CAS.
- C. P. Li and A. J. Blackman, Aust. J. Chem., 1995, 48, 955–965 CrossRef CAS.
- A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491–522 RSC.
- B. B. Snider and T. Liu, J. Org. Chem., 1997, 62, 5630–5633 CrossRef CAS.
- J. F. Liu and C. H. Heathcock, J. Org. Chem., 1999, 64, 8263–8266 CrossRef CAS.
- A. C. Flick, M. J. A. Caballero and A. Padwa, Org. Lett., 2008, 10, 1871–1874 CrossRef CAS.
- A. C. Flick, M. J. Arevalo Caballero and A. Padwa, Tetrahedron, 2010, 66, 3643–3650 CrossRef CAS.
- T. J. Donohoe, P. M. Brian, G. C. Hargaden and T. J. C. O'Riordan, Tetrahedron, 2010, 66, 6411–6420 CrossRef CAS.
- G. Lapointe, K. Schenk and P. Renaud, Org. Lett., 2011, 13, 4774–4777 CrossRef CAS.
- A. Pastor, R. Prinsloo, K. N. Burford, A. R. Macdonald, M. Parvez, C. Gendy and T. G. Back, J. Org. Chem., 2023, 88, 13813–13824 CrossRef CAS.
- G. A. Molander and M. Rönn, J. Org. Chem., 1999, 64, 5183–5187 CrossRef CAS.
- B. M. Trost and M. T. Rudd, Org. Lett., 2003, 5, 4599–4602 CrossRef CAS.
- R. Hanzawa and H. Fuwa, Org. Lett., 2023, 25, 1984–1988 CrossRef CAS.
- M. Piccichè, A. Pinto, R. Griera, J. Bosch and M. Amat, Org. Lett., 2022, 24, 5356–5360 CrossRef.
- T. Arai, H. Abe, S. Aoyagi and C. Kibayashi, Tetrahedron Lett., 2004, 45, 5921–5924 CrossRef CAS.
- S. Canesi, D. Bouchu and M. A. Ciufolini, Angew. Chem., Int. Ed., 2004, 43, 4336–4338 CrossRef CAS.
- J. Liu, R. P. Hsung and S. D. Peters, Org. Lett., 2004, 6, 3989–3992 CrossRef CAS.
- J. Liu, J. J. Swidorski, S. D. Peters and R. P. Hsung, J. Org. Chem., 2005, 70, 3898–3902 CrossRef CAS.
- J. J. Swidorski, J. Wang and R. P. Hsung, Org. Lett., 2006, 8, 777–780 CrossRef CAS.
- H. Abe, S. Aoyagi and C. Kibayashi, J. Am. Chem. Soc., 2005, 127, 1473–1480 CrossRef CAS.
- G. Pandey and V. Janakiram, Chem.–Eur. J., 2015, 21, 13120–13126 CrossRef CAS PubMed.
- Y.-H. Huang, Z.-J. Liu and P.-Q. Huang, Org. Chem. Front., 2022, 9, 58–63 RSC.
- T. Shibuguchi, H. Mihara, A. Kuramochi, S. Sakuraba, T. Ohshima and M. Shibasaki, Angew. Chem., Int. Ed., 2006, 45, 4635–4637 CrossRef CAS PubMed.
- D. M. Dalton and T. Rovis, Org. Lett., 2013, 15, 2346–2349 CrossRef CAS PubMed.
- J. D. Brown, M. A. Foley and D. L. Comins, J. Am. Chem. Soc., 1988, 110, 7445–7447 CrossRef CAS.
- S. V Tsukanov, L. R. Marks and D. L. Comins, J. Org. Chem., 2016, 81, 10433–10443 CrossRef PubMed.
- D. B. Gotchev and D. L. Comins, J. Org. Chem., 2006, 71, 9393–9402 CrossRef CAS PubMed.
- Y. Yang, RSC Adv., 2015, 5, 18894–18908 RSC.
- Y. Guo, M. Castiñeira Reis, J. Kootstra and S. R. Harutyunyan, ACS Catal., 2021, 11, 8476–8483 CrossRef CAS PubMed.
- T. Scattolin, T. Gharbaoui and C. Chen, Org. Lett., 2022, 24, 3736–3740 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04910a |
| This journal is © The Royal Society of Chemistry 2024 |