
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA homoleptic Fe(IV) ketimide complex with a low-lying excited state†
Phoebe R.
Hertler
a,
Arturo
Sauza-de la Vega
 b,
Andrea
Darù
b,
Arup
Sarkar
b,
Richard A.
Lewis
a,
Guang
Wu
a,
Laura
Gagliardi
*b and
Trevor W.
Hayton
*a
b,
Andrea
Darù
b,
Arup
Sarkar
b,
Richard A.
Lewis
a,
Guang
Wu
a,
Laura
Gagliardi
*b and
Trevor W.
Hayton
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106, USA. E-mail: hayton@chem.ucsb.edu
bDepartment of Chemistry, Pritzker School of Molecular Engineering, James Franck Institute, University of Chicago, Chicago, IL 60637, USA. E-mail: lgagliardi@uchicago.edu
First published on 10th September 2024
Abstract
The reaction of 4 equiv. of Li(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(tBu)Ph) with FeIICl2 results in isolation of [Li(Et2O)]2[FeII(NC(tBu)Ph)4] (1), in good yields. The reaction of 1 with 1 equiv. of I2 leads to formation of [FeIV(NC(tBu)Ph)4] (2), in moderate yields. 57Fe Mössbauer spectroscopy confirms the Fe(IV) oxidation state of 2, and X-ray crystallography reveals that 2 has a square planar coordination geometry along with several intramolecular H⋯C interactions. Furthermore, SQUID magnetometry indicates a small magnetic moment at room temperature, suggestive of an accessible S = 1 state. Both density functional theory and multiconfigurational calculations were done to elucidate the nature of the ground state. Consistent with the experimental results, the ground state was found to be an S = 0 state with an S = 1 excited state close in energy.
C(tBu)Ph) with FeIICl2 results in isolation of [Li(Et2O)]2[FeII(NC(tBu)Ph)4] (1), in good yields. The reaction of 1 with 1 equiv. of I2 leads to formation of [FeIV(NC(tBu)Ph)4] (2), in moderate yields. 57Fe Mössbauer spectroscopy confirms the Fe(IV) oxidation state of 2, and X-ray crystallography reveals that 2 has a square planar coordination geometry along with several intramolecular H⋯C interactions. Furthermore, SQUID magnetometry indicates a small magnetic moment at room temperature, suggestive of an accessible S = 1 state. Both density functional theory and multiconfigurational calculations were done to elucidate the nature of the ground state. Consistent with the experimental results, the ground state was found to be an S = 0 state with an S = 1 excited state close in energy.
Introduction
In comparison to the oxo, nitrido, and imido chemistry of Fe(IV),1–18 the coordination and organometallic chemistry of this oxidation state is relatively sparse.19–33 The paucity of examples is generally ascribed to the high oxidizing power of Fe4+, which can oxidize sensitive organometallic ligands.11,34–38 As a result, many of the Fe(IV) coordination complexes isolated thus far contain additional stabilizing features. For example, London dispersion forces are thought to play an important role in the stability of [FeIV(1-norbornyl)4] and [FeIV(cyclohexyl)4].39–42 Likewise, the solid-state structure of [FeIV(C5Me5)2][SbF6]2 displays multiple close contacts between the methyl groups of the cation and fluorine atoms of the anions.24 In contrast, the homoleptic Fe(IV) ketimide complex, [FeIV(NCtBu2)4],27 acquires its stability via the exceptionally strong σ- and π-donor properties of the ketimide ligand.43–45 In fact, the di-tert-butyl ketimide ligand can stabilize a large series of homoleptic M(IV) complexes of the type [MIV(NCtBu2)4] (M = Ti, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co).27,44–49 This ligand also stabilizes the U5+ ion in the homoleptic penta(ketimide) complex, [UV(NCtBu2)5],50 while the closely related phenyl-tert-butyl ketimide, [NC(tBu)Ph]–, can stabilize U(VI) and Ce(IV) – two ions that are also considered a challenge to isolate.50,51 Similarly, the stability of [FeIV(mnt)3]2– (mnt = maleonitriledithiolate) was thought to be due to highly covalent π-interaction between the Fe4+ centers and the [mnt]2– ligand.25
Given the paucity of Fe(IV) coordination complexes, we have continued to explore the chemistry of high-valent iron ketimides. Herein, we report the synthesis of unusual square planar, homoleptic Fe(IV) ketimide complex, [FeIV(NC(tBu)Ph)4], which features a singlet (S = 0) ground state. However, NMR spectral data, magnetism measurements, and density functional theory (DFT) and multiconfigurational calculations support the presence of a low-lying triplet state that is partially populated at room temperature.
Results and discussion
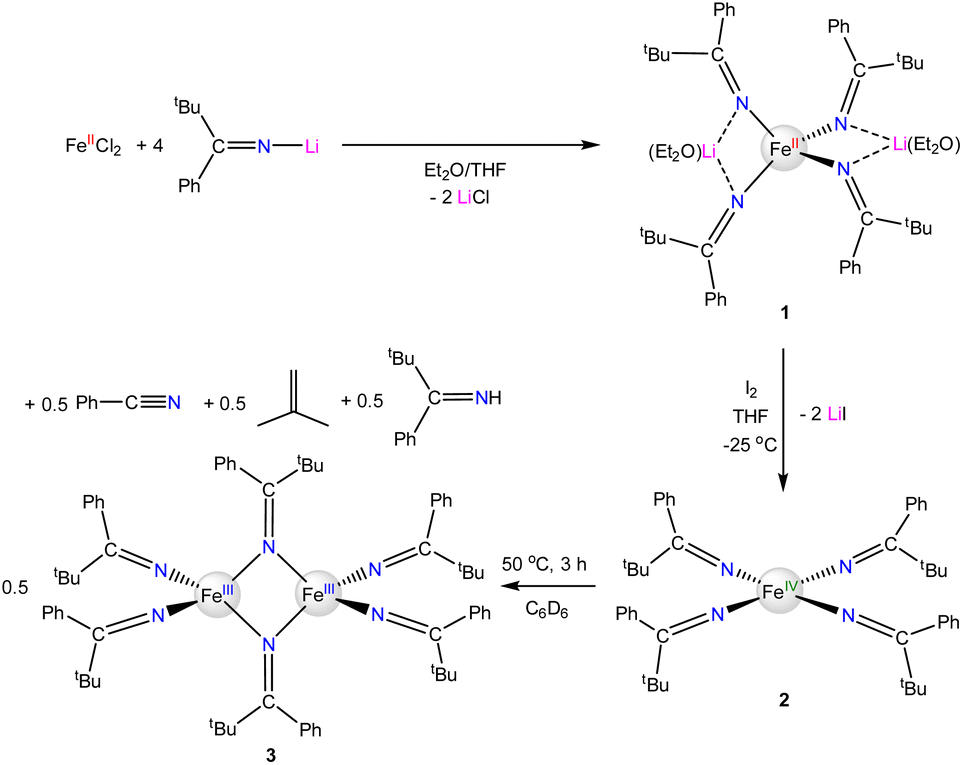
The reaction of FeIICl2 with 4 equiv. of Li(NC(tBu)Ph) results in formation of [Li(Et2O)]2[FeII(NC(tBu)Ph)4] (1), which crystallizes from a mixture of pentane and Et2O as large, orange blocks in 76% yield after work-up (Scheme 1). Complex 1 crystallizes in the monoclinic space group P21/c and features a distorted tetrahedral environment about the Fe center (Fig. 1). Additionally, the two Li+ cations are bound to two ketimide N atoms and one Et2O molecule. Similar structural features are found in the related homoleptic Fe(II) ketimide complexes, [Li(THF)]2[FeII(NCtBu2)4] and [Li(THF)2]2[FeII(NC13H8)4].27,52 The Fe–N distances in 1 range from 2.043(4) to 2.056(4) Å and are comparable to those observed for other Fe(II) ketimide complexes.27,52 Its 1H NMR spectrum in THF-d8 features a broad, paramagnetically shifted resonance at 16.0 ppm, assignable to the tBu environment, as well as broad resonances at 12.7 and 7.87 ppm, assignable to the o/m phenyl environments (Fig. S2†). Additionally, its 7Li{1H} NMR spectrum consists of an extremely broad, paramagnetically shifted resonance at 391 ppm (Fig. S3†), suggesting the presence of a contact-ion pair in solution. Similar behavior is observed in other [Li]2[MII(ketimide)4]-type complexes.27,44,47,52
 | ||
| Scheme 1 Synthesis of complexes 1–3. | ||
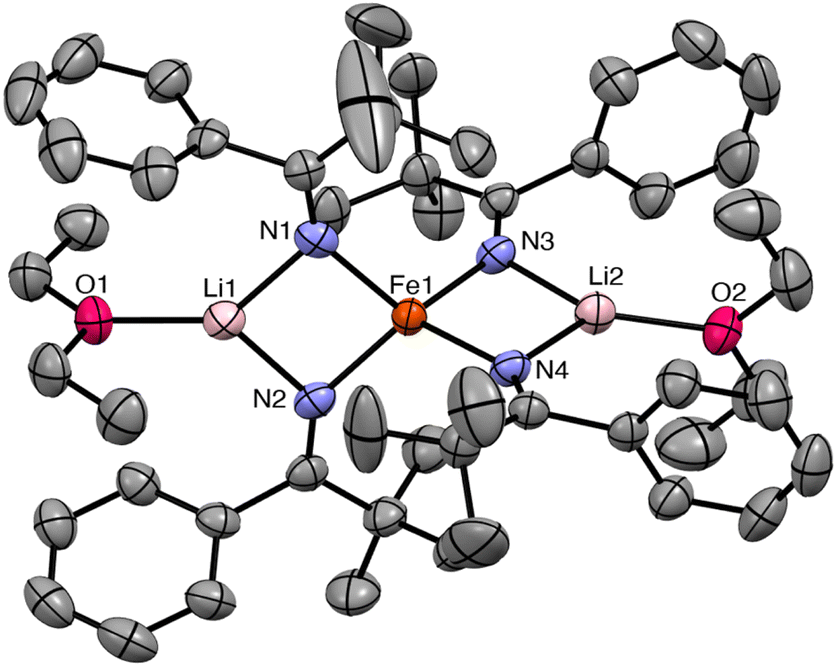 | ||
| Fig. 1 Solid-state structure of [Li(Et2O)]2[Fe(NC(tBu)Ph)4] (1) with 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–N1 = 2.051(4), Fe1–N2 = 2.056(4), Fe1–N3 = 2.045(4), Fe1–N4 = 2.043(4), N1–Fe1–N3 = 114.00(14), N1–Fe1–N2 = 93.21(14), and N1–Fe1–N4 = 121.09(14). | ||
The reaction of 1 with 1 equiv. of I2 results in formation of [FeIV(NC(tBu)Ph)4] (2), which can be isolated as black crystals in 32% yield after work-up (Scheme 1). Interestingly, we also observe formation of the previously reported Fe(III) complex, [FeIII2(NC(tBu)Ph)6] (3),52 during the oxidation of 1 to 2 (Fig. S10†), which is presumably formed by over-oxidation of 1 and which helps account for the low isolated yields of 2. Complex 2 can also be generated via oxidation of 1 with 1.7 equiv. of AgPF6 or 1.7 equiv. of [Cp2Fe][BF4]; however, neither reaction is synthetically useful. In the case of AgPF6, several byproducts are formed, including 3, despite the use of substoichiometric quantities of AgPF6. The oxidation of 1 with [Cp2Fe][BF4] is relatively clean, but complex 2 proved too difficult to separate from the [Cp2Fe] by-product, due to their similar solubilities.
Complex 2 crystallizes from Et2O and hexanes in the monoclinic space group C2/c (Fig. 2). In the solid state, 2 exhibits a distorted square planar geometry (τ4 = 0.19),53 wherein all four phenyl rings point up and all four tBu groups point down. Complex 2 features a nearly linear N1–Fe1–N1* angle (172.96(10)°). However, the N2–Fe1–N2* angle is notably smaller (159.70(10)°) and the Fe–N–C angle of these two ketimide ligands also deviates from linearity (Fe–N2–C12 = 149.24(14)°). This distortion is attributable to the presence of short inter-ligand H⋯C contacts. In particular, H22 is in close proximity to both C6 and C11 (H22⋯C6 = 2.726 Å and H22⋯C11 = 2.900 Å, Fig. 2), distances that are within the sum of their van der Waals radii (rHrC = 2.9 Å).41,54 These interactions may contribute to the overall stability of the complex, as has been noted for other Fe(IV) coordination and organometallic complexes,39–42 as well as the observed syn arrangement of the phenyl groups. Finally, the Fe–N distances in 2 are 1.7571(17) and 1.7859(15) Å. These values are similar to those previously reported for [FeIV(NCtBu2)4] (1.771(3) and 1.775(3) Å) and are somewhat shorter than Fe–N distances in [FeIV(TAML)(CN)2][PPh4]2 (1.807(9)–1.950(9) Å),32 [(N3N′)FeIV(CN)] (av. 1.82 Å),19 and [Fe(MesNCH2CH2NMes)2] (av. 1.89 Å).33 The latter complex also exhibits a distorted square planar geometry (τ4 = 0.22). Finally, the metrical parameters of a crystal of 2, collected at 287(2) K, are nearly identical to those collected at 100(2) K (Table S2†), demonstrating that the square planar geometry is maintained at room temperature in the solid state.
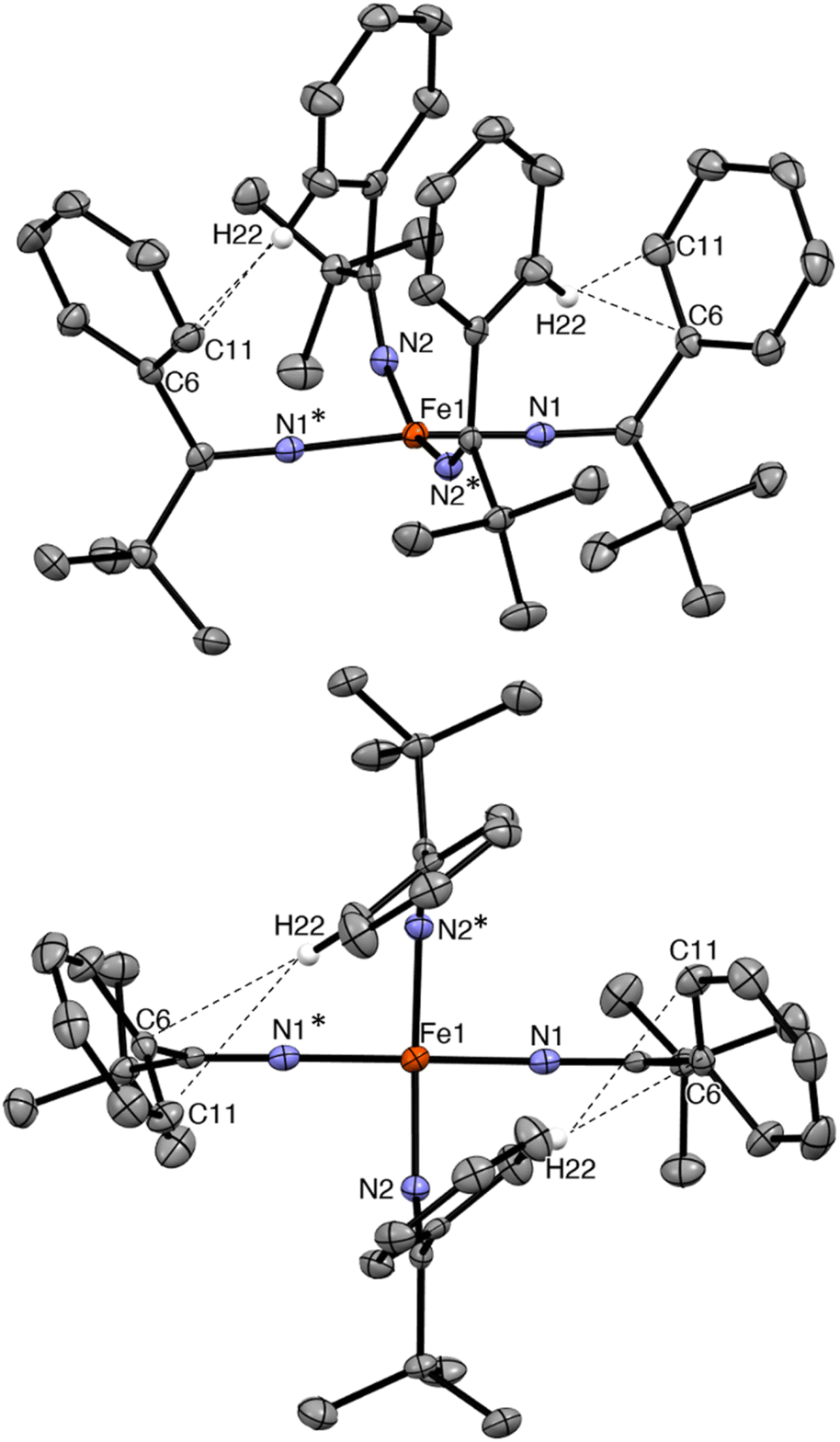 | ||
| Fig. 2 Two views of the solid-state molecular structure of [Fe(NC(tBu)Ph)4] (2) measured at 100 K, shown with 50% probability ellipsoids. Hydrogen atoms (except H22) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–N1 = 1.7571(17), Fe1–N2 = 1.7859(15), N1–Fe1–N2 = 90.26(7), N1–Fe–N2* = 90.99(7), N1–Fe1–N1* = 172.96(10), N2–Fe1–N2* = 159.70(10), Fe1–N1–C1 = 177.62(16), and Fe1–N2–C12 = 149.24(14). | ||
The 1H NMR spectrum of 2 in toluene-d8 exhibits four paramagnetically shifted resonances at 8.92, 7.70, 5.33, and 2.68 ppm in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2: 1:9 ratio, which are assignable to the m-Ar, o-Ar, p-Ar, and tBu protons, respectively. The o-Ar resonance is notably broad, likely because of the aforementioned H⋯C contacts, demonstrating that these interactions are probably maintained in solution. These resonances shift upon cooling. For example, at −60 °C in toluene-d8, the m-Ar resonance at 8.92 ppm shifts upfield to 7.37 ppm, whereas the p-Ar resonance at 5.33 ppm shifts downfield to 6.42 ppm (Fig. S6 and S7†). The shift of the 1H resonances upon cooling is not consistent with temperature-independent paramagnetism (TIP),55 but is instead the result of a thermally populated low-lying electronic excited state (see below).56,57 In contrast, [FeIV(N=CtBu2)4] is diamagnetic at room temperature, indicating the difference in the electronic structure imparted by the stronger donating bis(tert-butyl) ketimide ligand. For further comparison, the recently reported [Fe(MesNCH2CH2NMes)2] exhibits an S = 1 ground state at room temperature, suggesting that the bis(amide) ligand is a weaker donor than [NC(tBu)Ph]–.33
:2: 1:9 ratio, which are assignable to the m-Ar, o-Ar, p-Ar, and tBu protons, respectively. The o-Ar resonance is notably broad, likely because of the aforementioned H⋯C contacts, demonstrating that these interactions are probably maintained in solution. These resonances shift upon cooling. For example, at −60 °C in toluene-d8, the m-Ar resonance at 8.92 ppm shifts upfield to 7.37 ppm, whereas the p-Ar resonance at 5.33 ppm shifts downfield to 6.42 ppm (Fig. S6 and S7†). The shift of the 1H resonances upon cooling is not consistent with temperature-independent paramagnetism (TIP),55 but is instead the result of a thermally populated low-lying electronic excited state (see below).56,57 In contrast, [FeIV(N=CtBu2)4] is diamagnetic at room temperature, indicating the difference in the electronic structure imparted by the stronger donating bis(tert-butyl) ketimide ligand. For further comparison, the recently reported [Fe(MesNCH2CH2NMes)2] exhibits an S = 1 ground state at room temperature, suggesting that the bis(amide) ligand is a weaker donor than [NC(tBu)Ph]–.33
Complex 2 is soluble in hexanes, benzene, Et2O, and THF. It is stable as a solid for two weeks under an inert atmosphere at −25 °C; however, at longer storage times, it begins to decompose into the previously reported Fe(III) complex, 3.52 Complex 2 is also unstable in solution. Thermolysis of 2 for 3 h at 50 °C in C6D6, results in complete conversion to 3, according to 1H and 13C NMR spectroscopy (Scheme 1, Fig. S13 and S14†).52 Also present in these spectra are signals attributable to PhCN, isobutylene, and HNC(tBu)Ph, which are present in an approximately 1:1:1 ratio. These products are apparently derived from the one-electron oxidation of the ketimide ligand by the Fe(IV) center.47,48 For comparison, [FeIV(NCtBu2)4] also decomposes upon heating in C6D6 forming [Fe2(NCtBu2)5], tert-butyl cyanide, isobutane, and isobutylene;48 however, its complete conversion required 8 h at 50 °C, suggesting that the [NC(tBu)Ph]– ligand is more easily oxidized. Finally, the reaction of 2 with HNCPh2 in an attempt to form [FeIV(NCPh2)4] results in the formation of [FeIII2(NCPh2)6],52 along with isobutylene, PhCN, and HNCtBuPh (Scheme S1 and Fig. S15†). This observation indicates that the diphenyl ketimide ligand is likely unable to support the Fe(IV) state.
The cyclic voltammogram of 2 in THF reveals two reversible redox events, with E1/2 values of −1.40 and −0.36 V (vs. Fc/Fc+) (Fig. S20†). The former is attributed to the Fe(II)/Fe(III) redox couple, and the latter is attributed to the Fe(III)/Fe(IV) redox couple. For comparison, [FeIV(NCtBu2)4] features an Fe(II)/Fe(III) redox couple at −1.63 V and an Fe(III)/Fe(IV) redox couple at −0.53 V (vs. Fc/Fc+).27 The ca. 0.2 V shift in potential for 2 is unsurprising, given that phenyl ligands are less electron-donating than tert-butyl groups.58
The zero-field 57Fe Mössbauer spectrum for 2 at 90 K displays a doublet with δ = −0.162(2) mm s−1 and |ΔEQ| = 1.837(3) mm s−1 (Fig. 3 and Table 1). The isomer shift is very similar to those reported for [FeIV(NCtBu2)4] (δ = −0.15 mm s−1) and [Fe(MesNCH2CH2NMes)2] (δ = −0.15 mm s−1),33,48 as well as other Fe(IV) complexes,2,7,8,48,59 corroborating the proposed +4 oxidation state assignment. For further comparison, the Mössbauer spectrum of 1 exhibits a much larger isomer shift (δ = 0.924(2) mm s−1; Fig. S19†), consistent with its lower oxidation state. Interestingly, the zero-field 57Fe Mössbauer spectrum for 2, collected at 298 K, displays a similar isomer shift (−0.096(4) mm s−1)60 but much smaller quadrupolar splitting (|ΔEQ| = 1.172(7) mm s−1) (Fig. 3), suggesting a decrease in the electric field gradient with increasing temperature.61
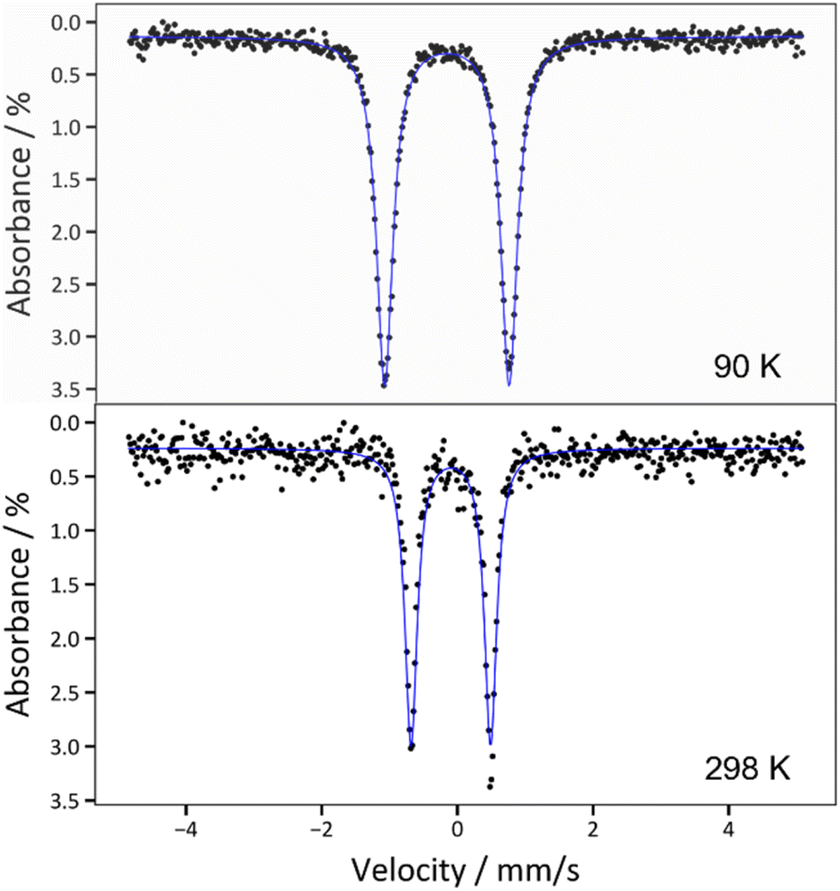 | ||
| Fig. 3 Zero-field 57Fe Mössbauer spectra of 2 collected at T = 90 K (top) and T = 298 K (bottom). The blue traces correspond to the overall fits, which are described in the main text. | ||
CtBu2)4], and [Fe(MesNCH2CH2NMes)2]. O. S. = Formal Oxidation State
Temperature-dependent dc magnetization data were collected for a crystalline sample of 2 at H = 1000 Oe, revealing a linear increase in χMT from 0.00 cm3 K mol−1 (μeff = 0.08 μB) at T = 2 K to 0.17 cm3 K mol−1 (μeff = 1.17 μB) at T = 300 K (Fig. 4). These results indicate that, at very low temperatures, 2 has a singlet spin ground state (S = 0). However, at room temperature, the moment is still well below that expected for an S = 1 system (1.00 cm3 K mol−1). To explain this data, we hypothesize that a low-lying excited state is partially populated as the temperature increases.
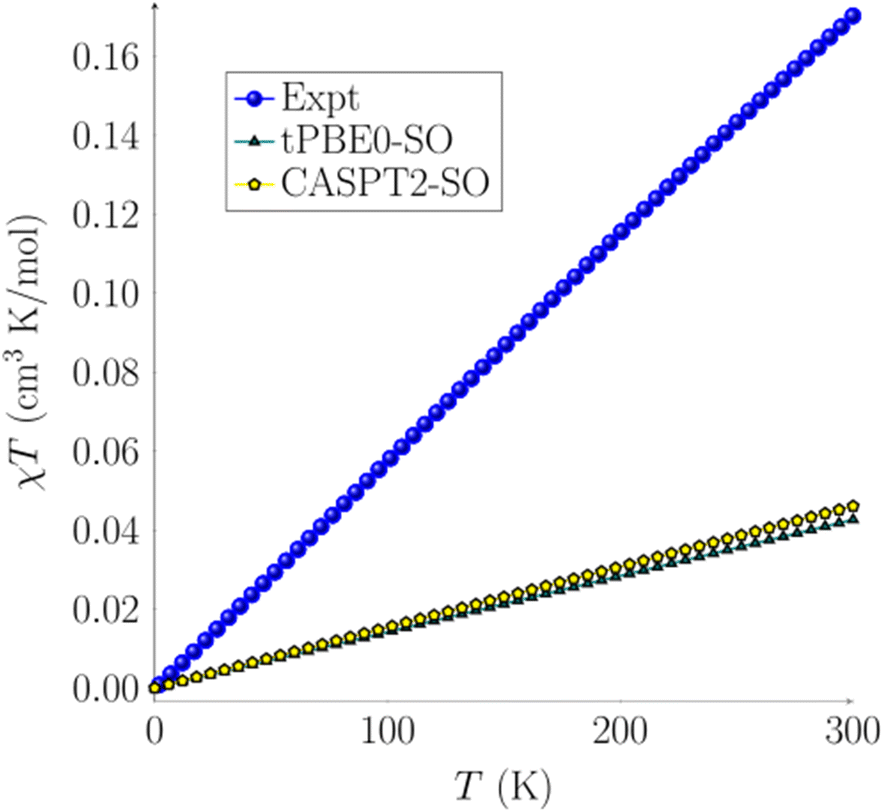 | ||
| Fig. 4 Magnetic susceptibility plot for complex 2 using TPSSh-D3BJ/def2-TZV optimized structures. Experimental data are shown in blue circles, CASPT2-SO (4, 10) yellow pentagons, and tPBE0-SO (4, 10) green triangles. Experimental data were collected under an applied magnetic field of H = 1k Oe from T = 2 to T = 300 K. | ||
In an effort to explain the spectroscopic and magnetic properties of complex 2, we performed an extensive computational investigation of its electronic structure using DFT and ab initio methods. Computational modeling was performed on the structure extracted from the crystal packing. Geometry optimization considering the singlet (S = 0), triplet (S = 1), and quintet (S = 2) spin states was performed with Kohn–Sham DFT using the functionals TPSSh-D3BJ,62–64 B3LYP-D3BJ,65–67 and M0668 together with def2-TZVP69 as the basis set for all the atoms. The Gaussian 16 rev. A03 software package70 was used for all DFT calculations (see the ESI† for full details). The functional TPSSh-D3BJ, which well describes the first–row transition metals including Fe-complexes,71 in agreement with the previous literature72,73 and with experiments, was chosen to carry out further DFT calculations (more details in the ESI†).
Structural optimization provides similar singlet and triplet geometries, which suggests that the structures have similar energy. For all the functionals tested here, the root means square deviation (RMSD) between the crystal structure and both the singlet and the triplet optimized geometries is minimal. The RMSD values are between 0.05 Å and 0.32 Å, and the RMDS between the singlet and triplet optimized structures is about 0.25 Å (see the ESI† for additional details).
Energetically, a singlet ground state was obtained using the selected functional TPSSh-D3BJ. The singlet–triplet gap is small with a value of 0.03 eV, which agrees with a low-lying triplet state as suggested experimentally. In contrast, the non-optimized geometry extracted from the crystal structure provides a singlet–triplet gap of about 0.5 eV, which cannot be referred to as a low-lying in character. Therefore, this structure is unsuitable for computing the net singlet or triplet state properties.
To better understand the variable temperature magnetism behavior of complex 2, complete active space self-consistent field (CASSCF)74 followed by perturbation theory (CASPT2)75 and pair-density functional theory (MC-PDFT with the tPBE and tPBE0 functionals)76,77 calculations were performed. These multireference calculations employed both the 100 K X-ray structure, in which only the hydrogen positions were optimized at the DFT level (discussed in the ESI†) and the TPSSh-D3BJ fully optimized structures previously discussed.
The CASSCF calculations were performed using the state average formalism (SA-CASSCF). From the optimized CASSCF wavefunction, CASPT2 and tPBE0 calculations were performed to obtain the singlet–triplet energy gap and the magnetic susceptibility in three selected active spaces (see the ESI† for computational details). Multireference calculations were also shown to be effective in rationalizing the electronic structure of the Fe(IV) nitride, [Cp′Fe(μ-N)]2 (Cp′ = 1,2,4-tBu3C5H2), which was not well described by DFT methods.78
The active spaces were chosen based on the occupation of the 3d frontier orbitals of the Fe(IV) center and the four N atoms surrounding it. Indeed, the Fe(IV) center of complex 2 has a 3d4 electron configuration; thus, the minimal active space considered is 4 electrons in 5 3d-orbitals (4, 5). To provide a more accurate electronic and chemical description, a second d-shell (4d) has been added to allow the correlation of the 4 electrons in 10 orbitals, active space (4, 10). This expansion of the active space is necessary to allow further delocalization of the 3d electrons into the 4d-shell. Furthermore, a (12, 9) active space was also chosen by including four Fe–N bonding orbitals to describe the high degree of covalency of metal–ketimide bonds (see ESI† for more details). Indeed, the Fe–N 3d-sp2 hybrid bonding orbitals allow a further expansion in the direction of the ketimide ligands.44,45,79 Therefore, such extended active spaces should suffice to explore the physical properties of the system in subsequent calculations. Consistent with the DFT results, CASPT2 predicts a singlet ground state followed by the triplet state, while tPBE0 almost agrees, with a singlet–triplet gap slightly negative; the quintet state lies significantly higher in all methods and is likely not populated to any meaningful extent at the temperatures studied. The singlet–triplet gaps at CASPT2 and tPBE0 are 0.11 eV and −0.02 eV respectively consistent with both states being thermally accessible (Table 2 and the ESI†).
| TPSSh-D3BJ | CASPT2 | tPBE0 | |
|---|---|---|---|
| Singlet | 0.00 | 0.00 | 0.02 |
| Triplet | 0.03 | 0.11 | 0.00 |
| Quintet | 0.27 | 2.26 | 1.84 |
After assessing the different spin states of complex 2, calculations of its magnetic susceptibility were performed. Spin–orbit coupling (SOC) was computed with the restricted active space state interaction method (RASSI-SO)80–82 on top of the previously described spin states at both CASPT2-SO and tPBE-SO levels. The spin–orbit energies are reported and discussed in the ESI† (see Tables S12–S14†). The effective spin Hamiltonian for the zero-field parameters and magnetic susceptibility were computed with the SINGLE_ANISO module using OpenMolcas.83 CASPT2-SO and tPBE0-SO with the (4, 10) active space magnetic susceptibility values are reported in Fig. 4. A total of 5 quintets, 35 triplets, and 22 singlets roots were calculated for all active spaces. These states were chosen based on an energy cut-off of 40000 cm−1. We observe a linear behavior of χT over the temperature range. The linear plot confirms a singlet ground state with a low-lying triplet excited state. A pure singlet state (diamagnetic) would have resulted in a horizontal line at χT = 0, whereas a pure triplet state (paramagnetic) would have resulted in a plateauing curve. The mix of such states provides a straight line with a slope (see the ESI† for more details). The computed χT vs. T using tPBE0-SO and CASPT2-SO agree with each other (Fig. 4). However, the computed χcalc values only account for the spin–orbit coupling without thermal contributions; thus they are still below the experimental data but within an acceptable deviation, with a difference of about 0.0004 cm3 mol−1. The error in χcalc is a consequence of performing the geometry optimization calculations at a temperature of 0 K rather than in the 2–300 K range, thus in the absence of any phonon or thermal bath where a deformation of the geometrical structure and subsequent spin-phonon coupling may take place.
Conclusions
In summary, we report the synthesis of [FeIV(NC(tBu)Ph)4], a rare example of a nearly square planar, Fe(IV) coordination complex. Its +4 oxidation state was confirmed via57Fe Mössbauer spectroscopy. In addition, SQUID magnetometry and VT-NMR spectroscopic studies indicate that this complex features a low-lying excited state. The calculated magnetic susceptibility data also show that a low-lying triplet excited state contributes to the paramagnetic behavior observed for 2. These properties set 2 apart from its di-tert-butyl analogue, [FeIV(NCtBu2)4]. The latter complex is diamagnetic at all temperatures studied, likely due to the better electron donating ability of the di-tert-butyl ketimide ligand, which results in a stronger ligand field and better separated ground state. The better donating ability of di-tert-butyl ketimide is also supported by the cyclic voltammetry data of both complexes. Overall, this work expands our understanding of Fe(IV) coordination chemistry and better defines the conditions under which ketimides can be used to stabilize high oxidation states.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1 and 2 have been deposited at the Cambridge Structural Database under 1: CCDC 2254712; 2 (100 K): 2254713; 2 (287 K): 2363649.Author contributions
PRH and RAL performed the synthesis and characterization. GW oversaw the crystallographic analysis. ASdV, AD, and AS performed the computational analysis. LG and TWH supervised the work. PRH, ASdV, AD, AS, LG, and TWH contributed to the writing and editing of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
PRH, RAL, GW, and TWH thank the National Science Foundation (CHE 2055063) for financial support of this work. The computational work (ASdV, AD, AS, and LG) was supported by the Air Force Office Scientific Research by Grant FA9550-20-1-0360. This research made use of a 400 MHz NMR spectrometer supported in part by the NIH Shared Instrumentation Grant, S10OD012077. The MRL Shared Experimental Facilities are supported by the MRSEC Program of the National Science Foundation under award NSF DMR 1720256, a member of the NSF-funded Materials Research Facilities Network. The authors acknowledge the University of Chicago Research Computing Center (RCC) for providing computational resources.Notes and references
- J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 10515–10517 CrossRef CAS PubMed.
- J. J. Scepaniak, C. S. Vogel, M. M. Khusniyarov, F. W. Heinemann, K. Meyer and J. M. Smith, Science, 2011, 331, 1049–1052 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- M. P. Mehn and J. C. Peters, J. Inorg. Biochem., 2006, 100, 634–643 CrossRef CAS PubMed.
- C. M. Thomas, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2006, 128, 4956–4957 CrossRef CAS PubMed.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que, S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed.
- C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon and K. Meyer, Angew. Chem., Int. Ed., 2008, 47, 2681–2684 CrossRef CAS PubMed.
- T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 6252–6254 CrossRef CAS PubMed.
- M. S. Vad, A. Lennartson, A. Nielsen, J. Harmer, J. E. McGrady, C. Frandsen, S. Mørup and C. J. McKenzie, Chem. Commun., 2012, 48, 10880–10882 RSC.
- J. England, J. O. Bigelow, K. M. Van Heuvelen, E. R. Farquhar, M. Martinho, K. K. Meier, J. R. Frisch, E. Münck and L. Que, Chem. Sci., 2014, 5, 1204–1215 RSC.
- B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145–12148 CrossRef CAS PubMed.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que, Science, 2003, 299, 1037–1039 CrossRef CAS PubMed.
- M. R. Bukowski, K. D. Koehntop, A. Stubna, E. L. Bominaar, J. A. Halfen, E. Münck, W. Nam and L. Que, Science, 2005, 310, 1000–1002 CrossRef CAS PubMed.
- J. England, Y. Guo, E. R. Farquhar, V. G. Young Jr, E. Münck and L. Que Jr, J. Am. Chem. Soc., 2010, 132, 8635–8644 CrossRef CAS PubMed.
- V. F. Oswald, J. L. Lee, S. Biswas, A. C. Weitz, K. Mittra, R. Fan, J. Li, J. Zhao, M. Y. Hu, E. E. Alp, E. L. Bominaar, Y. Guo, M. T. Green, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2020, 142, 11804–11817 CrossRef CAS PubMed.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS PubMed.
- J. P. Bigi, W. H. Harman, B. Lassalle-Kaiser, D. M. Robles, T. A. Stich, J. Yano, R. D. Britt and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 1536–1542 CrossRef CAS PubMed.
- A. K. Maity, J. Murillo, A. J. Metta-Magaña, B. Pinter and S. Fortier, J. Am. Chem. Soc., 2017, 139, 15691–15700 CrossRef CAS PubMed.
- C. Souilah, S. A. V. Jannuzzi, D. Demirbas, S. Ivlev, M. Swart, S. DeBeer and A. Casitas, Angew. Chem., Int. Ed., 2022, 61, e202201699 CrossRef CAS PubMed.
- O. Prakash, P. Chábera, N. W. Rosemann, P. Huang, L. Häggström, T. Ericsson, D. Strand, P. Persson, J. Bendix, R. Lomoth and K. Wärnmark, Chem.–Eur. J., 2020, 26, 12728–12732 CrossRef CAS PubMed.
- A. Casitas, J. A. Rees, R. Goddard, E. Bill, S. DeBeer and A. Fürstner, Angew. Chem., Int. Ed., 2017, 56, 10108–10113 CrossRef CAS PubMed.
- M. Malischewski, K. Seppelt, J. Sutter, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2018, 57, 14597–14601 CrossRef CAS PubMed.
- M. Malischewski, K. Seppelt, J. Sutter, F. W. Heinemann, B. Dittrich and K. Meyer, Angew. Chem., Int. Ed., 2017, 56, 13372–13376 CrossRef CAS PubMed.
- M. Malischewski, M. Adelhardt, J. Sutter, K. Meyer and K. Seppelt, Science, 2016, 353, 678–682 CrossRef CAS PubMed.
- C. Milsmann, S. Sproules, E. Bill, T. Weyhermüller, S. D. George and K. Wieghardt, Chem.–Eur. J., 2010, 16, 3628–3645 CrossRef CAS PubMed.
- M. Atanasov, P. Surawatanawong, K. Wieghardt and F. Neese, Coord. Chem. Rev., 2013, 257, 27–41 CrossRef CAS.
- R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 12814–12816 CrossRef CAS PubMed.
- B. K. Bower and H. G. Tennent, J. Am. Chem. Soc., 1972, 94, 2512–2514 CrossRef CAS.
- C. C. Cummins and R. R. Schrock, Inorg. Chem., 1994, 33, 395–396 CrossRef CAS.
- A. Chanda, D.-L. Popescu, F. T. de Oliveira, E. L. Bominaar, A. D. Ryabov, E. Münck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606–619 CrossRef CAS PubMed.
- J. England, E. R. Farquhar, Y. Guo, M. A. Cranswick, K. Ray, E. Münck and L. Que Jr, Inorg. Chem., 2011, 50, 2885–2896 CrossRef CAS PubMed.
- J. H. Lim, X. Engelmann, S. Corby, R. Ganguly, K. Ray and H. S. Soo, Chem. Sci., 2018, 9, 3992–4002 RSC.
- B. Zhang, J. P. Joyce, N. Wolford, W. W. Brennessel, S. DeBeer and M. Neidig, Angew. Chem., Int. Ed., 2024, 63, e202405113 CrossRef CAS PubMed.
- T. J. Collins, K. L. Kostka, E. S. Uffelman and T. L. Weinberger, Inorg. Chem., 1991, 30, 4204–4210 CrossRef CAS.
- T. J. Collins, Acc. Chem. Res., 1994, 27, 279–285 CrossRef CAS.
- T. Kojima, F. Ogishima, T. Nishibu, H. Kotani, T. Ishizuka, T. Okajima, S. Nozawa, Y. Shiota, K. Yoshizawa, H. Ohtsu, M. Kawano, T. Shiga and H. Oshio, Inorg. Chem., 2018, 57, 9683–9695 CrossRef CAS PubMed.
- A. Ikezaki, M. Takahashi and M. Nakamura, Chem. Commun., 2013, 49, 3098–3100 RSC.
- Y. Morimoto, Y. Shimaoka, Y. Ishimizu, H. Fujii and S. Itoh, Angew. Chem., Int. Ed., 2019, 58, 10863–10866 CrossRef CAS PubMed.
- H. Li, L. Wang, Y. Hu, Z. Zhang, D. Wan, Q. Fan, R. B. King and H. F. Schaefer III, J. Phys. Chem. A, 2020, 124, 6867–6876 CrossRef CAS PubMed.
- D. J. Liptrot, J.-D. Guo, S. Nagase and P. P. Power, Angew. Chem., Int. Ed., 2016, 55, 14766–14769 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- H. Li, Y. Hu, D. Wan, Z. Zhang, Q. Fan, R. B. King and H. F. Schaefer III, J. Phys. Chem. A, 2019, 123, 9514–9519 CrossRef CAS PubMed.
- C. R. Graves, A. E. Vaughn, E. J. Schelter, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2008, 47, 11879–11891 CrossRef CAS PubMed.
- R. A. Lewis, S. P. George, A. Chapovetsky, G. Wu, J. S. Figueroa and T. W. Hayton, Chem. Commun., 2013, 49, 2888–2890 RSC.
- P. L. Damon, C. J. Liss, R. A. Lewis, S. Morochnik, D. E. Szpunar, J. Telser and T. W. Hayton, Inorg. Chem., 2015, 54, 10081–10095 CrossRef CAS PubMed.
- A. M. Martins, M. M. Marques, J. R. Ascenso, A. R. Dias, M. T. Duarte, A. C. Fernandes, S. Fernandes, M. J. Ferreira, I. Matos, M. Conceição Oliveira, S. S. Rodrigues and C. Wilson, J. Organomet. Chem., 2005, 690, 874–884 CrossRef CAS.
- R. A. Lewis, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 4660–4668 CrossRef CAS PubMed.
- R. A. Lewis, D. E. Smiles, J. M. Darmon, S. C. E. Stieber, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 8218–8227 CrossRef CAS PubMed.
- R. A. D. Soriaga, J. M. Nguyen, T. A. Albright and D. M. Hoffman, J. Am. Chem. Soc., 2010, 132, 18014–18016 CrossRef CAS PubMed.
- L. A. Seaman, G. Wu, N. Edelstein, W. W. Lukens, N. Magnani and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 4931–4940 CrossRef CAS PubMed.
- M. K. Assefa, D.-C. Sergentu, L. A. Seaman, G. Wu, J. Autschbach and T. W. Hayton, Inorg. Chem., 2019, 58, 12654–12661 CrossRef CAS PubMed.
- P. R. Hertler, R. A. Lewis, G. Wu and T. W. Hayton, Inorg. Chem., 2023, 62, 11829–11836 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. C. Bart, K. Chłopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901–13912 CrossRef CAS PubMed.
- L. A. Abriata, G. N. Ledesma, R. Pierattelli and A. J. Vila, J. Am. Chem. Soc., 2009, 131, 1939–1946 CrossRef CAS PubMed.
- N. V. Shokhirev and F. A. Walker, J. Phys. Chem., 1995, 99, 17795–17804 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. P. Hendrich, W. Gunderson, R. K. Behan, M. T. Green, M. P. Mehn, T. A. Betley, C. C. Lu and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17107–17112 CrossRef CAS PubMed.
- P. Gütlich, E. Bill and A. X. Trautwein, in Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, ed. P. Gütlich, E. Bill and A. X. Trautwein, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 73–135 Search PubMed.
- J. Li, B. C. Noll, A. G. Oliver, C. E. Schulz and W. R. Scheidt, J. Am. Chem. Soc., 2013, 135, 15627–15641 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comp. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Wallingford, CT, 2016 Search PubMed.
- K. P. Jensen and J. Cirera, J. Phys. Chem. A, 2009, 113, 10033–10039 CrossRef CAS PubMed.
- A. Darù, C. Martín-Fernández and J. N. Harvey, ACS Catal., 2022, 12, 12678–12688 CrossRef.
- N. Jiang, A. Darù, Š. Kunstelj, J. G. Vitillo, M. E. Czaikowski, A. S. Filatov, A. Wuttig, L. Gagliardi and J. S. Anderson, J. Am. Chem. Soc., 2024, 146, 12243–12252 CrossRef CAS PubMed.
- B. O. Roos, in Advances in Chemical Physics, 1987, pp. 399–445 Search PubMed.
- K. Andersson, P. Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- G. Li Manni, R. K. Carlson, S. Luo, D. Ma, J. Olsen, D. G. Truhlar and L. Gagliardi, J. Chem. Theory Comput., 2014, 10, 3669–3680 CrossRef CAS PubMed.
- R. Pandharkar, M. R. Hermes, D. G. Truhlar and L. Gagliardi, J. Phys. Chem. Lett., 2020, 11, 10158–10163 CrossRef CAS PubMed.
- M. Reiners, M. Maekawa, C. G. Daniliuc, M. Freytag, P. G. Jones, P. S. White, J. Hohenberger, J. Sutter, K. Meyer, L. Maron and M. D. Walter, Chem. Sci., 2017, 8, 4108–4122 RSC.
- A. W. Cook, P. Hrobárik, P. L. Damon, D. Najera, B. Horváth, G. Wu and T. W. Hayton, Inorg. Chem., 2019, 58, 15927–15935 CrossRef CAS PubMed.
- B. O. Roos and P.-Å. Malmqvist, Phys. Chem. Chem. Phys., 2004, 6, 2919–2927 RSC.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- G. Li Manni, I. Fdez. Galván, A. Alavi, F. Aleotti, F. Aquilante, J. Autschbach, D. Avagliano, A. Baiardi, J. J. Bao, S. Battaglia, L. Birnoschi, A. Blanco-González, S. I. Bokarev, R. Broer, R. Cacciari, P. B. Calio, R. K. Carlson, R. Carvalho Couto, L. Cerdán, L. F. Chibotaru, N. F. Chilton, J. R. Church, I. Conti, S. Coriani, J. Cuéllar-Zuquin, R. E. Daoud, N. Dattani, P. Decleva, C. de Graaf, M. G. Delcey, L. De Vico, W. Dobrautz, S. S. Dong, R. Feng, N. Ferré, M. Filatov, L. Gagliardi, M. Garavelli, L. González, Y. Guan, M. Guo, M. R. Hennefarth, M. R. Hermes, C. E. Hoyer, M. Huix-Rotllant, V. K. Jaiswal, A. Kaiser, D. S. Kaliakin, M. Khamesian, D. S. King, V. Kochetov, M. Krośnicki, A. A. Kumaar, E. D. Larsson, S. Lehtola, M.-B. Lepetit, H. Lischka, P. López Ríos, M. Lundberg, D. Ma, S. Mai, P. Marquetand, I. C. D. Merritt, F. Montorsi, M. Mörchen, A. Nenov, V. H. A. Nguyen, Y. Nishimoto, M. S. Oakley, M. Olivucci, M. Oppel, D. Padula, R. Pandharkar, Q. M. Phung, F. Plasser, G. Raggi, E. Rebolini, M. Reiher, I. Rivalta, D. Roca-Sanjuán, T. Romig, A. A. Safari, A. Sánchez-Mansilla, A. M. Sand, I. Schapiro, T. R. Scott, J. Segarra-Martí, F. Segatta, D.-C. Sergentu, P. Sharma, R. Shepard, Y. Shu, J. K. Staab, T. P. Straatsma, L. K. Sørensen, B. N. C. Tenorio, D. G. Truhlar, L. Ungur, M. Vacher, V. Veryazov, T. A. Voß, O. Weser, D. Wu, X. Yang, D. Yarkony, C. Zhou, J. P. Zobel and R. Lindh, J. Chem. Theory Comput., 2023, 19, 6933–6991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and crystallographic, spectroscopic, electrochemical, and magnetic characterization details for 1 and 2 (PDF and CIF files). CCDC 2254712, 2254713 and 2363649. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04880f |
| This journal is © The Royal Society of Chemistry 2024 |