
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium and copper co-catalyzed chloro-arylation of gem-difluorostyrenes – use of a nitrite additive to suppress β-F elimination†
Andrew J.
Intelli
 a,
Coriantumr Z.
Wayment
b,
Ryan T.
Lee
c,
Kedong
Yuan
d and
Ryan A.
Altman
*ab
a,
Coriantumr Z.
Wayment
b,
Ryan T.
Lee
c,
Kedong
Yuan
d and
Ryan A.
Altman
*ab
aBorch Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: raaltman@purdue.edu
bJames Tarpo Jr and Margaret Tarpo Department of Chemistry, Purdue University, West Lafayette, Indiana 47907, USA
cDepartment of Chemistry and Chemical Biology, Rutgers University, Piscataway, New Jersey 08854, USA
dGuangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target Clinical Pharmacology, Guangzhou Medical University, Guangzhou 511436, China
First published on 1st October 2024
Abstract
The installation of fluorine and fluorinated functional groups in organic molecules perturbs the physicochemical properties of those molecules and enables the development of new therapeutics, agrichemicals, biological probes and materials. However, current synthetic methodologies cannot access some fluorinated functional groups and fluorinated scaffolds. One such group, the gem-difluorobenzyl motif, might be convergently synthesized by reacting a nucleophilic aryl precursor and an electrophilic gem-difluoroalkene. Previous attempts have relied on forming unstable anionic or organometallic intermediates that rapidly decompose through a β-F elimination process to deliver monofluorovinyl products. In contrast, we report a fluorine-retentive palladium and copper co-catalyzed chloro-arylation of gem-difluorostyrenes that takes advantage of a nitrite (NO2−) additive to avoid the favorable β-F elimination pathway that forms monofluorinated products, instead delivering difluorinated products.
Introduction
The introduction of fluorine and fluorinated functional groups into organic molecules is a prominent strategy to perturb a molecule's physicochemical properties relevant to the development of therapeutics, agrichemicals, biological probes, and materials.1,2 One important functional group, the gem-difluorobenzyl motif, is typically used by medicinal chemists to block metabolically labile benzylic positions on drug candidates from benzylic oxidation by P450s, to reduce arene epoxidation by P450s by decreasing the electron density of the aromatic π system, or as a replacement for a labile oxygen atom.1,2 Such gem-difluoromethylene groups are most commonly accessed by deoxyfluorination reactions of carbonyl-containing molecules3–5 (Scheme 1A, Blue) that typically require reagents that can exhibit poor functional group compatibility (Deoxo-Fluor, XtalFluor, and Fluolead)3,5 or that, for many substrates, can competitively form monofluorovinyl side products (DAST and Deoxofluor).6,7 To complement this functional group interconversion strategy, alternative approaches to generate gem-difluoromethylene-containing compound might enable access to more elaborate substructures through convergent bond-forming processes.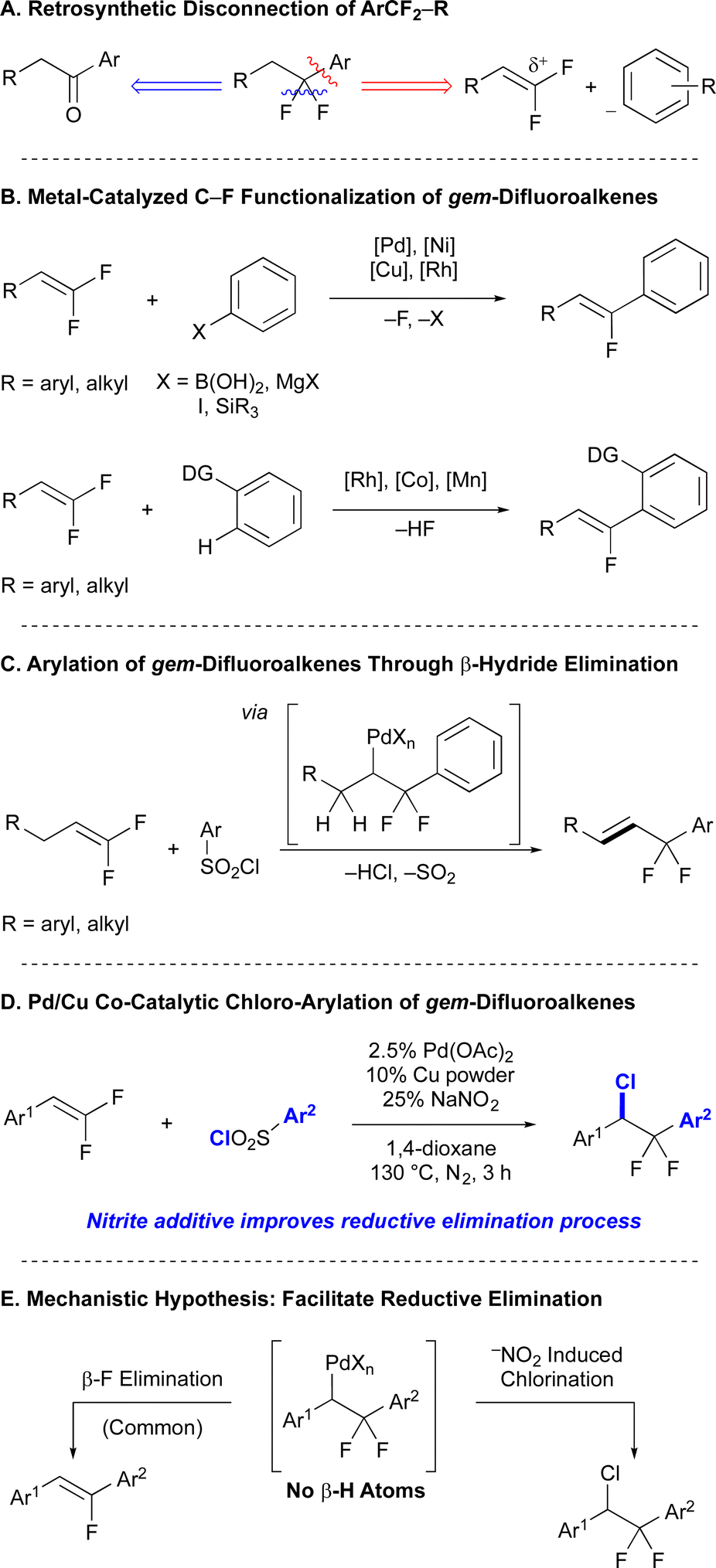 | ||
| Scheme 1 Metal catalyzed arylation reactions of gem-difluoroalkenes typically proceed through organometallic intermediates prone to undergo β-fluoride elimination, which can be overcome using a nitrite additive. | ||
As a complementary strategy to access gem-difluorobenzyl substructures, retrosynthetic disconnection of the CF2–Caryl bond would reveal a nucleophilic aryl species and a gem-difluoroalkene that bears electrophilic character at the terminal difluorinated carbon8 (Scheme 1A, Red). However, nucleophilic addition reactions of gem-difluoroalkenes that occur under basic conditions typically proceed through unstable anionic intermediates that decompose to form vinyl fluorides via β-F elimination pathways.9–12 Though some net nucleophilic hydrofunctionalization reactions have been developed using a variety of heteroatom nucleophiles,13–18 fluorine-retentive functionalization reactions of gem-difluorinated alkenes to generate new C–CF2 bonds remains limited.18–27 Instead, reactions of gem-difluoroalkenes with aryl nucleophiles typically proceed through unstable organometallic intermediates that also decompose via β-F elimination to form monofluorovinyl products (Scheme 1B).9–11,28–42
In contrast to these previous two-electron approaches, we have recently focused on functionalization reactions of gem-difluoroalkenes that proceed through radical intermediates,27,43–47 thus avoiding unstable anionic or organometallic intermediates that undergo β-fluoride elimination.9–11,20,30–39,41,42,48,49 With respect to C(Aryl)–CF2 bond-formation, a fluorine-retentive Pd/Cu co-catalyzed arylation of gem-difluoroalkenes with aryl sulfonyl chlorides proceeded through Pd-alkyl intermediates bearing β-F atoms, but avoided β-F elimination by offering the metal a kinetically favorable β-H elimination pathway (Scheme 1C).50 However, substrates lacking β-H atoms, such as gem-difluorostyrenes, readily underwent β-F elimination. Herein, we report a F-retentive Pd/Cu co-catalyzed regioselective difunctionalization reaction of gem-difluoroalkenes, which convergently generates new Calkyl–Cl and CF2–aryl bonds and increases complexity (Scheme 1D). Notably, the reaction exploits an uncommon nitrite (NO2−) additive to promote the reductive elimination of a Calkyl–Cl bond, thus avoiding the kinetically facile β-F elimination pathway (Scheme 1E).
Results and discussion
In early explorations, modification of our previously reported conditions27 delivered mixtures of desired chloro-arylated product (A), β-F elimination side products (B and C), and a chloro-sulfonylated product (D) in a substrate-dependent manner (Table 1). Reactions of electron-deficient gem-difluorostyrenes with electron-deficient aryl sulfonyl chlorides formed products A and B in equal quantities (entry 1); however, when the aryl ring of either substrate bore neutral or electron-donating substituents, side product B or D typically formed in 10–12% yield along with trace quantities of C (entries 2–5). To avoid competing β-F elimination processes, we initially explored conditions that facilitate reductive elimination of Pd complexes to form new C–X bonds by changing the properties of the ligand,51 using weaker coordinating solvents,52 exploiting additives that generate high-valent palladium species [Pd(II)/(IV) cycle] under anaerobic (e.g. PhICl2, PhI(OAc)2, Ph2ICl, Umemoto's reagent)53–56 or aerobic (e.g. PhICl2, PhI(OAc)2, −NO3, −NO2)57–62 conditions.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | Conv.b | A | B | C | D |
| a Unless otherwise stated, all reactions were carried out with gem-difluorostyrene (0.20 mmol), aryl sulfonyl chloride (0.40 mmol), 5% Pd(OAc)2, 10% CuCl2, NaCl (0.20 mmol) and heated for 1 h under an atmosphere of nitrogen. b Yields were determined by 19F NMR using α,α,α-trifluorotoluene as an internal standard. | |||||||
| 1 | 4-CN-Ph | 4-NO2-Ph | 60 | 29 | 28 | — | — |
| 2 | 3,4,5-tri-OMe-Ph | 4-NO2-Ph | >95 | 73 | 10 | 4 | — |
| 3 | 4-tBu-Ph | 4-NO2-Ph | 63 | 13 | 10 | 3 | — |
| 4 | 3,5-di-Me-Ph | 4-NO2-Ph | 40 | 15 | 12 | 1 | — |
| 5 | 4-OMe-Ph | 4-OMe-Ph | 91 | 17 | 1 | 2 | 11 |
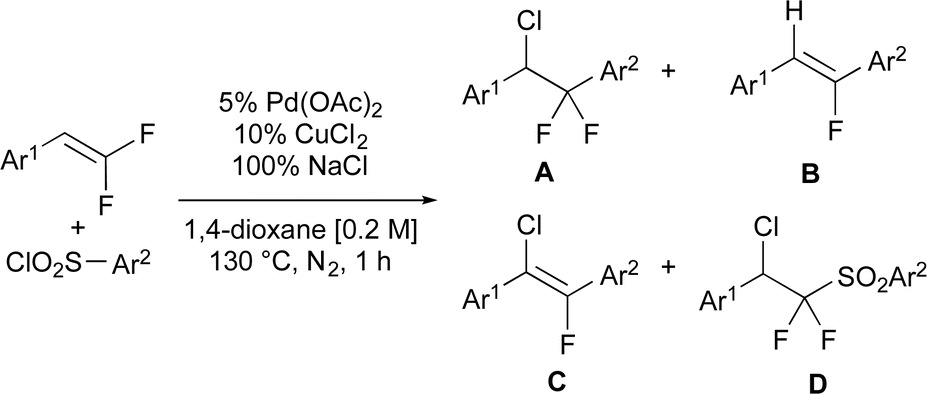
Eventually, the reaction of electron-deficient gem-difluoroalkene 1a with electron deficient aryl-sulfonyl chloride 2a to form chloro-arylated product 3aa was facilitated under anaerobic conditions in good selectivity in the presence of nitrite additives (Table 2, entries 1 and 2). With this improvement in hand, an additive screen demonstrated the unique ability of −NO2 to minimize formation of monofluoroalkene 3aa′. As a control experiment, the reaction run without any additives formed chloro-arylated product 3aa in modest yield and poor selectivity (entry 1). However, addition of 25% sodium nitrite [NaNO2] dramatically increased selectivity for 3aa over 3aa′ (entry 2), though the addition of sodium nitrate [NaNO3] only provided a more modest improvement in selectivity relative to NaNO2 (entry 3). The increase in selectivity was not dependent on Na+ and instead dependent on −NOx, as KNO2 and KNO3 provided comparable yield and selectivity to their Na+ counterparts (entries 4–5). Additionally, other Na-based additives, including halogens (entry 6), O-based anions (entry 7), non-coordinating ions (entry 8), anionic bases (entries 9 and 10) did not significantly increase selectivity for forming chloro-arylated product over β-F elimination product (for individual yields, see ESI Table S1†). In this screen, β-F elimination product C and chloro-sulfonylated product D were generally each detected in <2% quantities by 19F NMR of the crude reaction mixtures. Overall, exhaustive screening demonstrated the ability of −NO2 additives to facilitate formation of the C–Cl bonds and minimize β-fluoride elimination.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | % Additive | Conv.b | 3aa | 3aa′ | 3aa/3aa′ |
| a Unless otherwise stated, all reactions were carried out with gem-difluorostyrene 1a (0.20 mmol), aryl sulfonyl chloride 2a (0.40 mmol), 2.5% Pd(OAc)2, 10% Cu powder, XX% additive and heated for 3 h under an atmosphere of nitrogen. b Yields were determined by 19F NMR using α,α,α-trifluorotoluene as an internal standard. c All reported yields and selectivities represent an average of three independent runs. | ||||||
| 1c | None | 0 | >95 | 32 | 25 | 1.3 |
| 2 | NaNO2 | 25 | >95 | 56 | 6 | 9.3 |
| 3 | NaNO3 | 25 | >95 | 41 | 16 | 2.5 |
| 4 | KNO2 | 25 | >95 | 61 | 7 | 8.7 |
| 5 | KNO3 | 100 | >95 | 60 | 17 | 3.5 |
| 6 | NaCl, NaBr, NaI | 25 | 40–94 | 0–26 | 0–21 | 0–1.2 |
| 7 | NaOAc, NaTFA, NaOTf | 25 | 88–98 | 24–36 | 16–20 | 1.5–2 |
| 8 | NaPF6, NaBF4 | 25 | >95 | 34 | 17–19 | 1.8–2 |
| 9 | Na2CO3, NaHCO3 | 25 | >95 | 33–35 | 18–21 | 1.6–1.9 |
| 10 | Na3PO4, Na2HPO4 | 25 | >95 | 31–35 | 14–15 | 2.1–2.5 |
| 11c | NaNO2 | 2.5 | >95 | 41 | 18 | 2.2 |
| 12c | NaNO2 | 5 | >95 | 42 | 17 | 2.5 |
| 13c | NaNO2 | 10 | >95 | 52 | 12 | 4.4 |
| 14c | NaNO2 | 20 | >95 | 50 | 11 | 4.7 |
| 15c | NaNO2 | 50 | >95 | 53 | 8 | 6.3 |
| 16c | NaNO2 | 75 | >95 | 51 | 7 | 7.7 |
| 17c | NaNO2 | 100 | >95 | 51 | 5 | 11 |
Further studies more rigorously established the ability of NO2− to control the selectivity for promoting chlorination over β-F elimination. Specifically, increasing the equivalents of NaNO2 from 0 to 1.0 systematically increased the selectivity from 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 11:1 (entries 11–17), and 1.0 equiv. NaNO2 was selected for further studies. Similarly, the use of NaNO3 followed a similar trend, albeit with lower maximum selectivity (up to 3.9:1, ESI Table S2†). The use of NaNO2 also improved the yield and selectivity for a wide range of Pd-based (ESI Table S3†) and Cu-based (ESI Table S4†) catalyst systems, with the combination of Pd(OAc)2 and Cu powder performing best. Interestingly, on 1.0 and 5.0 mmol scales, the loading of NaNO2 could be lowered to 25% with no reduction in selectivity or yield; at these scales, the use of stoichiometric NaNO2 did not provide additional benefits to yield or selectivity. For this reaction, routine evaluation of solvent, temperature, and time indicated 130 °C/1,4-dioxane/3 h as optimal conditions (ESI Tables S5–S7†). In general, lower temperatures and shorter reaction times delivered less product, while hotter or longer reaction times provided no benefit (ESI Tables S6 and S7†).
:1 to 11:1 (entries 11–17), and 1.0 equiv. NaNO2 was selected for further studies. Similarly, the use of NaNO3 followed a similar trend, albeit with lower maximum selectivity (up to 3.9:1, ESI Table S2†). The use of NaNO2 also improved the yield and selectivity for a wide range of Pd-based (ESI Table S3†) and Cu-based (ESI Table S4†) catalyst systems, with the combination of Pd(OAc)2 and Cu powder performing best. Interestingly, on 1.0 and 5.0 mmol scales, the loading of NaNO2 could be lowered to 25% with no reduction in selectivity or yield; at these scales, the use of stoichiometric NaNO2 did not provide additional benefits to yield or selectivity. For this reaction, routine evaluation of solvent, temperature, and time indicated 130 °C/1,4-dioxane/3 h as optimal conditions (ESI Tables S5–S7†). In general, lower temperatures and shorter reaction times delivered less product, while hotter or longer reaction times provided no benefit (ESI Tables S6 and S7†).
The optimized conditions enabled coupling of a wide range of gem-difluorostyrenes with aryl sulfonyl chlorides to deliver chloro-difluorinated products (Fig. 1). Reactions of electron-rich gem-difluorostyrenes containing ethers and alkyl groups afforded products in moderate yields (3ba–3fa). Electron-deficient gem-difluorostyrenes containing O-benzyl, phenyl ester, trifluoromethyl, nitrile, tosylate and methyl ester groups (3aa, 3ga–3ka), and gem-difluorostyrenes bearing halogen atoms (3la–3na) also reacted efficiently to deliver the chloro-arylated products in moderate yields. Substrates containing heterocyclic moieties including N-benzenesulfonyl indole and benzofuran moieties were also compatible in this system (3oa–3pa). However, non-styrenyl gem-difluoroalkenes and gem-difluoroalkenes containing N–H and thioethers were not tolerated. Importantly, the selectivity for chloro-arylated products was not influenced by the electronic character of either aryl ring, with selectivities >9:1 [A:(B + C + D) as determined by 19F NMR of the crude reaction mixture], except for one outlier (entry 7, 5:1, ESI Table S8†). Moreover, the NaNO2 additive suppressed β-F elimination, with yields of B averaging at or below 4% (as determined by 19F NMR of the crude reaction mixture), except for one outlier with 6% (entry 7, ESI Table S8†).
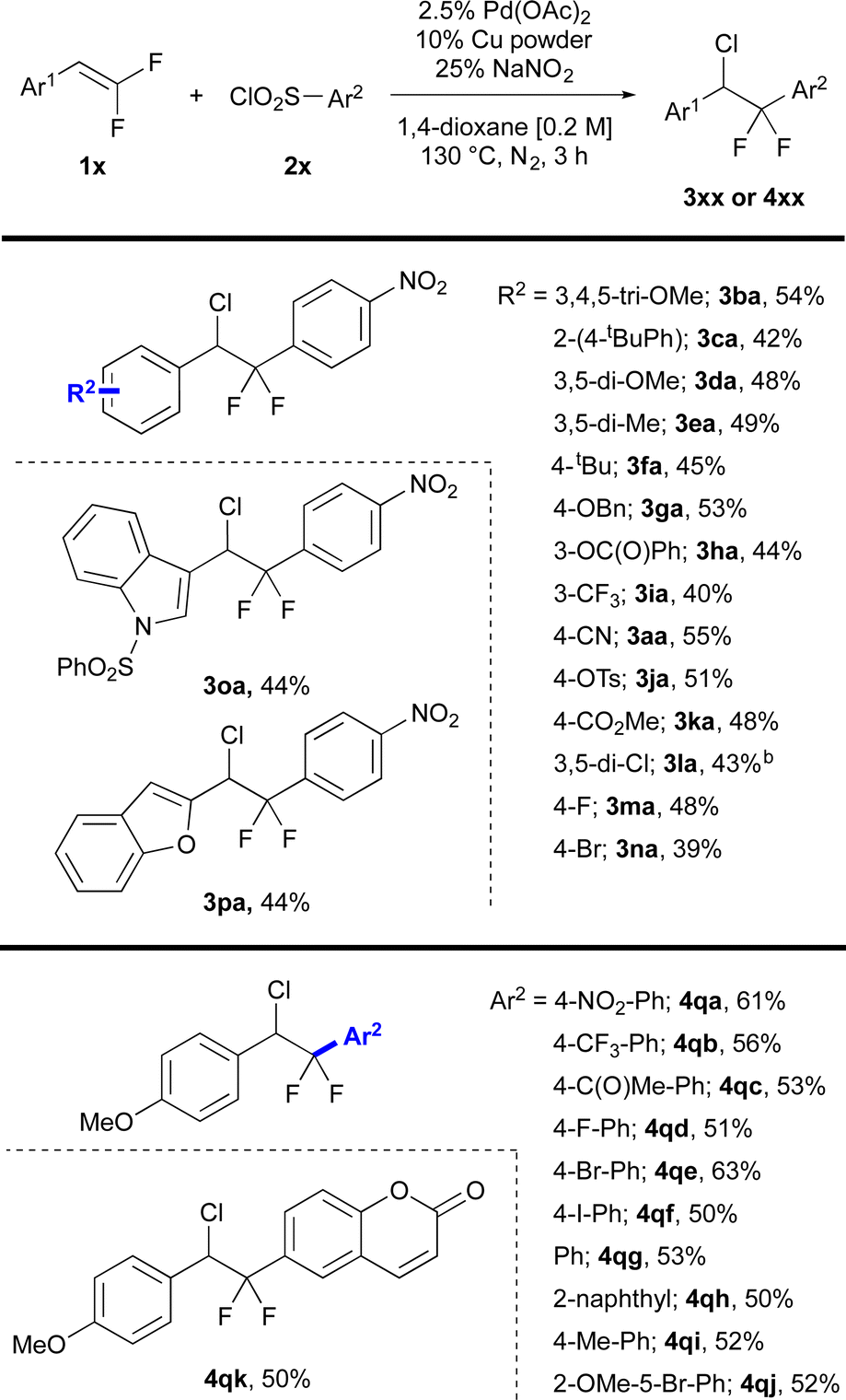 | ||
| Fig. 1 Scope of gem-difluorostyrenes and aryl sulfonyl chlorides. aUnless otherwise stated, all reactions were carried out with gem-difluorostyrene (1.0 mmol), aryl sulfonyl chloride (2.0 mmol), 2.5% Pd(OAc)2, 10% Cu powder, 25% NaNO2, 1,4-dioxane (5.0 mL) and heated at 130 °C for 3 h under an atmosphere of nitrogen. b5% mmol Pd(OAc)2. All reported yields and selectivities represent an average of two independent runs. | ||
A series of aryl sulfonyl chlorides bearing a range of electronic characters were coupled with gem-difluorostyrene 1q to deliver chloro-arylated products (Fig. 1). Aryl sulfonyl chlorides containing strong electron-withdrawing nitro, trifluoromethyl and ketone groups reacted smoothly to deliver chloro-arylated products in fair to good yields (4qa–4qb). Notably, an aryl sulfonyl chloride containing a ketone group successfully delivered the chloro-arylated product, which contrasts with deoxyfluorinating reagents that react preferentially with carbonyls (4qc). Additionally, aryl sulfonyl chlorides bearing halogens provided the corresponding chloro-arylated products in moderate to good yields (4qd–4qf). Moreover, such examples that tolerate aryl iodides and bromides suggest that the catalytic cycle does not involve Pd(0) intermediates. Aryl sulfonyl chlorides bearing electronically neutral phenyl and naphthyl groups (4qg–4qh) as well as electron-donating alkyl and ether groups also provided chloro-arylated products in moderate yields (4qi–4qj). Finally, a coumarin-derived aryl sulfonyl chloride reacted smoothly to deliver the chloro-arylated product in moderate yield (4qk). For this series, reactions of electronically neutral or rich aryl sulfonyl chloride proceeded in slightly lower selectivity 5 and 6:1 [A:(B + C + D) as determined by 19F NMR of the crude reaction mixture], solely due to an increase of product D, which likely derives from slow desulfination, not from an increase in β-F elimination (entries 24, 26 and 27, ESI Table S8†). Finally, aryl sulfonyl bromides were not compatible with this system, instead delivering aryl bromides as the predominant side product (detected via GC-MS).
−NOx additives have served unique and modestly understood roles in many Pd-catalyzed processes,57,60,62–65 though the ability of the additives to influence β-elimination vs. reductive elimination processes show no precedent.66 With respect to the immediate reaction, the ability of −NO2 to influence the selectivity for difluorobenzyl vs. monofluorovinyl products (Avs.B) could result from multiple different phenomena. First, coordination of −NO2 to the putative Pd(Cl)(alkyl) intermediate 5 might block the binding site typically populated by the vicinal F atom, thus precluding the β-F elimination process (Fig. 2A). Second, binding of −NO2 might accelerate the reductive elimination process to form the C–Cl bond via intermediate 6, as the π-back bonding interaction between Pd(II) and −NO2 pulls electron density away from the metal,67 thus destabilizing the higher oxidation state form and reducing the energy barrier for reductive elimination (Fig. 2B), as has been demonstrated for reductive elimination of C–F bonds from Pd(IV) complexes.68 In contrast, other anions tested (e.g. halogens, −OTf, −OAc, −O2CCF3, −BF4, −PF6) do not engage in strong π-back bonding interactions that might help avoid β-F elimination. For a proposed mechanism and mechanistic studies, see the ESI (Fig. S1, Tables S9 and S10†.
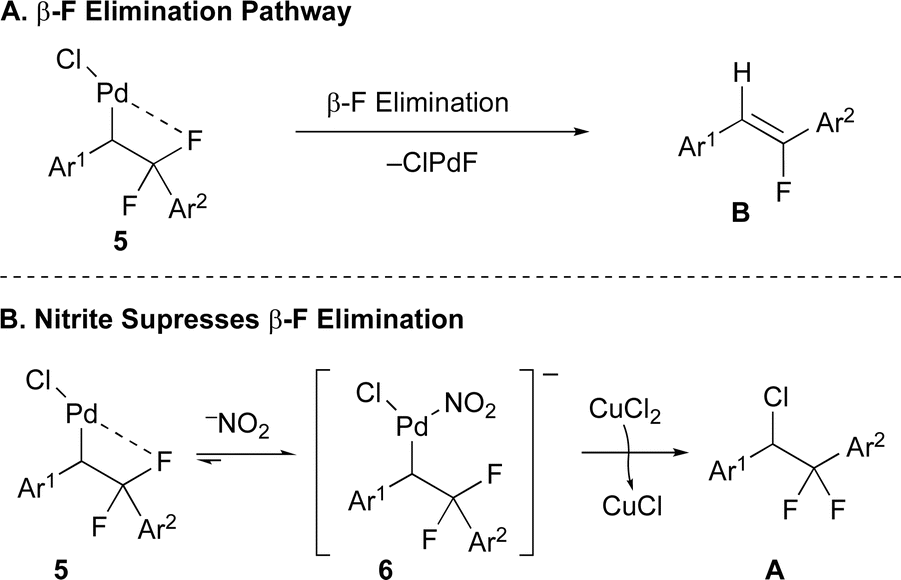 | ||
| Fig. 2 Proposed mechanisms by which −NO2 prevents β-F elimination. | ||
Conclusion
In conclusion, a palladium and copper co-catalyzed reaction utilizes aryl sulfonyl chlorides to add both aryl and chlorine groups across gem-difluoroalkenes in a regioselective difunctionalization reaction. Notably, this reaction exploited NaNO2 as a critical additive that enabled formation of the Calkyl–Cl bond and reduced the common β-F elimination pathway, thus improving selectivity for generating difluorobenzyl products over monofluorovinyl products, and we speculate that such additives might prove more generally useful at perturbing rates of reductive elimination and/or β-F elimination processes. Ongoing investigation to further understand the unique role of nitrite in this reaction will be reported in due time.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
K. Y. and R. A. conceived the project. A. J. I., C. Z. W., and R. T. L. carried out the experiments. A. J. I. and R. A. A. wrote the manuscript, and all authors revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support the National Institutes of Health (GM124661) and the donors of the Steve and Lee Ann Taglienti Endowment for supporting this project. The Purdue Interdepartmental NMR Facility is supported by the Institute for Cancer Research (P30 CA023168). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We appreciate early screening studies that were performed by Alonso Rodriguez and Suvajit Koley.Notes and references
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of Fluorine in Medicinal Chemistry, J. Med. Chem., 2015, 58(21), 8315–8359, DOI:10.1021/acs.jmedchem.5b00258.
- N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61(14), 5822–5880, DOI:10.1021/acs.jmedchem.7b01788.
- F. Beaulieu, L. P. Beauregard, G. Courchesne, M. Couturier, F. Laflamme and A. L'Heureux, Aminodifluorosulfinium Tetrafluoroborate Salts as Stable and Crystalline Deoxofluorinating Reagents, Org. Lett., 2009, 11(21), 5050–5053, DOI:10.1021/ol902039q.
- T. Umemoto, R. P. Singh, Y. Xu and N. Saito, Discovery of 4-Tert-Butyl-2,6-Dimethylphenylsulfur Trifluoride as a Deoxofluorinating Agent with High Thermal Stability as Well as Unusual Resistance to Aqueous Hydrolysis, and Its Diverse Fluorination Capabilities Including Deoxofluoro-Arylsulfinylation, J. Am. Chem. Soc., 2010, 132(51), 18199–18205, DOI:10.1021/ja106343h.
- G. S. Lal, G. P. Fez, R. J. Pesaresi and F. M. Prozonic, Bis(2-Methoxyethyl)Aminosulfur Trifluoride: A New Broad-Spectrum Deoxofluorinating Agent with Enhanced Thermal Stability, Chem. Commun., 1999,(2), 215–216, 10.1039/a808517j.
- M. Hudlicky, Fluorination with Diethylaminosulfur Trifluoride and Related Aminofluorosulfuranes, Org. React., 1988, 35, 515–633, DOI:10.1002/0471264180.or035.03.
- A. Lheureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. Laflamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, Aminodifluorosulfinium Salts: Selective Fluorination Reagents with Enhanced Thermal Stability and Ease of Handling, J. Org. Chem., 2010, 75(10), 3401–3411, DOI:10.1021/jo100504x.
- J. Zhang, J. D. Yang and J. P. Cheng, Chemoselective Catalytic Hydrodefluorination of Trifluoromethylalkenes towards Mono-/Gem-Di-Fluoroalkenes under Metal-Free Conditions, Nat. Commun., 2021, 12(1), 1–10, DOI:10.1038/s41467-021-23101-3.
- X. Zhang and S. Cao, Recent Advances in the Synthesis and C-F Functionalization of Gem-Difluoroalkenes, Tetrahedron Lett., 2017, 58(5), 375–392, DOI:10.1016/j.tetlet.2016.12.054.
- C. Liu, H. Zeng, C. Zhu and H. Jiang, Recent Advances in Three-Component Difunctionalization of Gem -Difluoroalkenes, Chem. Commun., 2020, 56(72), 10442–10452, 10.1039/D0CC04318D.
- S. Koley and R. A. Altman, Recent Advances in Transition Metal-Catalyzed Functionalization of Gem-Difluoroalkenes, Isr. J. Chem., 2020, 60(3–4), 313–339, DOI:10.1002/ijch.201900173.
- T. Fujita, K. Fuchibe and J. Ichikawa, Transition-Metal-Mediated and -Catalyzed C-F Bond Activation by Fluorine Elimination, Angew. Chem., Int. Ed., 2019, 131(2), 396–408, DOI:10.1002/ange.201805292.
- J. K. Jin, W. X. Zheng, H. M. Xia, F. L. Zhang and Y. F. Wang, Regioselective Radical Hydroboration of Gem-Difluoroalkenes: Synthesis of α-Borylated Organofluorines, Org. Lett., 2019, 21(20), 8414–8418, DOI:10.1021/acs.orglett.9b03173.
- X. Liu, E. E. Lin, G. Chen, J. L. Li, P. Liu and H. Wang, Radical Hydroboration and Hydrosilylation of Gem-Difluoroalkenes: Synthesis of α-Difluorinated Alkylborons and Alkylsilanes, Org. Lett., 2019, 21(20), 8454–8458, DOI:10.1021/acs.orglett.9b03218.
- J. P. Sorrentino, D. L. Orsi and R. A. Altman, Acid-Catalyzed Hydrothiolation of Gem-Difluorostyrenes to Access α,α-Difluoroalkylthioethers, J. Org. Chem., 2021, 86(3), 2297–2311, DOI:10.1021/acs.joc.0c02440.
- D. L. Orsi, M. R. Yadav and R. A. Altman, Organocatalytic Strategy for Hydrophenolation of Gem-Difluoroalkenes, Tetrahedron, 2019, 75(32), 4325–4336, DOI:10.1016/j.tet.2019.04.016.
- D. L. Orsi, B. J. Easley, A. M. Lick and R. A. Altman, Base Catalysis Enables Access to α,α-Difluoroalkylthioethers, Org. Lett., 2017, 19(7), 1570–1573, DOI:10.1021/acs.orglett.7b00386.
- J. P. Sorrentino and R. A. Altman, Fluorine-Retentive Strategies for the Functionalization of Gem -Difluoroalkenes, Synthesis, 2021, 53(21), 3935–3950, DOI:10.1055/a-1547-9270.
- X. T. Feng, J. X. Ren, X. Gao, Q. Q. Min and X. Zhang, 3,3-Difluoroallyl Sulfonium Salts: Practical and Bench-Stable Reagents for Highly Regioselective Gem-Difluoroallylations, Angew. Chem., Int. Ed., 2022, 61(42), e202210103, DOI:10.1002/anie.202210103.
- F. Liu, Z. Zhuang, Q. Qian, X. Zhang and C. Yang, Ru-Catalyzed Defluorinative Alkylation or Catalyst-Free Hydroalkylation of Gem-Difluoroalkenes Enabled by Visible Light, J. Org. Chem., 2022, 87(5), 2730–2739, DOI:10.1021/acs.joc.1c02662.
- T. Kawashima, M. Ohashi and S. Ogoshi, Nickel-Catalyzed Formation of 1,3-Dienes via a Highly Selective Cross-Tetramerization of Tetrafluoroethylene, Styrenes, Alkynes, and Ethylene, J. Am. Chem. Soc., 2017, 139(49), 17795–17798, DOI:10.1021/jacs.7b12007.
- S. Shibutani, K. Nagao and H. Ohmiya, Organophotoredox-Catalyzed Three-Component Coupling of Heteroatom Nucleophiles, Alkenes, and Aliphatic Redox Active Esters, Org. Lett., 2021, 23(5), 1798–1803, DOI:10.1021/acs.orglett.1c00211.
- T. De Tan, J. M. I. Serviano, X. Luo, P. C. Qian, P. L. Holland, X. Zhang and M. J. Koh, Congested C(Sp3)-Rich Architectures Enabled by Iron-Catalysed Conjunctive Alkylation, Nat. Catal., 2024, 7(3), 321–329, DOI:10.1038/s41929-024-01113-8.
- T. Narita, T. Hagiwara, H. Hamana, K. Tomooka, Y.-Z. Liu and T. Nakai, Unique Radical Addition Reactions onto Perfluoro-Enol Esters, Tetrahedron Lett., 1995, 36(34), 6091–6094 CrossRef CAS.
- T. F. Herpin, W. B. Motherwell and M. J. Tozer, The Synthesis of Difluoromethylene-Linked C-Glycosides and C-Disaccharides, Tetrahedron, 1994, 5(11), 2269–2282 CrossRef CAS.
- G. Kachkovskyi, M. Cieślak, P. Graczyk, P. Zawadzki, J. Kalinowska-Tłuścik and M. Werłos, Photocatalytic Approach to α,α-Difluoroalkyl, Synthesis, 2022, 54(19), 4294–4303, DOI:10.1055/s-0041-1737546.
- K. Yuan, T. Feoktistova, P. H. Y. Cheong and R. A. Altman, Arylation of Gem-Difluoroalkenes Using a Pd/Cu Co-Catalytic System That Avoids β-Fluoride Elimination, Chem. Sci., 2021, 12(4), 1363–1367, 10.1039/d0sc05192f.
- X. Lu, Y. Wang, B. Zhang, J. J. Pi, X. X. Wang, T. J. Gong, B. Xiao and Y. Fu, Nickel-Catalyzed Defluorinative Reductive Cross-Coupling of Gem-Difluoroalkenes with Unactivated Secondary and Tertiary Alkyl Halides, J. Am. Chem. Soc., 2017, 139(36), 12632–12637, DOI:10.1021/jacs.7b06469.
- W. Dai, H. Shi, X. Zhao and S. Cao, Sterically Controlled Cu-Catalyzed or Transition-Metal-Free Cross-Coupling of Gem-Difluoroalkenes with Tertiary, Secondary, and Primary Alkyl Grignard Reagents, Org. Lett., 2016, 18(17), 4284–4287, DOI:10.1021/acs.orglett.6b02026.
- M. Li and G. C. Tsui, Stereoselective Palladium-Catalyzed Hiyama Cross-Coupling Reaction of Tetrasubstituted Gem-Difluoroalkenes, Org. Lett., 2024, 26(1), 376–379, DOI:10.1021/acs.orglett.3c04037.
- R. T. Thornbury and F. D. Toste, Palladium-Catalyzed Defluorinative Coupling of 1-Aryl-2,2-Difluoroalkenes and Boronic Acids: Stereoselective Synthesis of Monofluorostilbenes, Angew. Chem., Int. Ed., 2016, 128(38), 11801–11804, DOI:10.1002/ange.201605651.
- P. Tian, C. Feng and T. P. Loh, Rhodium-Catalysed C(Sp2)-C(Sp2) Bond Formation via C–H/C–F Activation, Nat. Commun., 2015, 6, 7472, DOI:10.1038/ncomms8472.
- W. Dai, J. Xiao, G. Jin, J. Wu and S. Cao, Palladium- and Nickel-Catalyzed Kumada Cross-Coupling Reactions of Gem -Difluoroalkenes and Monofluoroalkenes with Grignard Reagents, J. Org. Chem., 2014, 79(21), 10537–10546, DOI:10.1021/jo5022234.
- L. Kong, X. Zhou and X. Li, Cobalt(III)-Catalyzed Regio- and Stereoselective α-Fluoroalkenylation of Arenes with Gem-Difluorostyrenes, Org. Lett., 2016, 18(24), 6320–6323, DOI:10.1021/acs.orglett.6b03203.
- S. Porey, Y. Bairagi, S. Guin, X. Zhang and D. Maiti, Nondirected C-H/C-F Coupling for the Synthesis of α-Fluoro Olefinated Arenes, ACS Catal., 2023, 13(21), 14000–14011, DOI:10.1021/acscatal.3c02975.
- R. Doi, K. Kajiwara, T. Negoro, K. Koh and S. Ogoshi, Regioselective C-F Bond Transformations of Silyl Difluoroenolates, Org. Lett., 2023, 25(29), 5542–5547, DOI:10.1021/acs.orglett.3c02057.
- H. Tan, Y. Zong, Y. Tang and G. C. Tsui, Stereoselective Rhodium(I)-Catalyzed C-F Bond Arylation of Tri- and Tetrasubstituted Gem-Difluoroalkenes with Boronic Acids, Org. Lett., 2023, 25(5), 877–882, DOI:10.1021/acs.orglett.3c00108.
- Y. Wang, Y. Tang, Y. Zong and G. C. Tsui, Highly Selective C-F Bond Functionalization of Tetrasubstituted Gem-Difluoroalkenes and Trisubstituted Monofluoroalkenes Using Grignard Reagents, Org. Lett., 2022, 24(22), 4087–4092, DOI:10.1021/acs.orglett.2c01639.
- B. Pang, Y. Wang, L. Hao, G. Wu, Z. Ma and Y. Ji, Tandem C-C/C-N Bond Formation via Rh(III)-Catalyzed α-Fluoroalkenylation and Sequential Annulation of 2-Arylquinazolinones and Gem-Difluorostyrenes, J. Org. Chem., 2023, 88(1), 143–153, DOI:10.1021/acs.joc.2c02006.
- Y. Xiao, W. Huang and Q. Shen, Stereoselective Formation of Z-Monofluoroalkenes by Nickel-Catalyzed Defluorinative Coupling of Gem-Difluoroalkenes with Lithium Organoborates, Chin. Chem. Lett., 2022, 33(9), 4277–4280, DOI:10.1016/j.cclet.2022.01.020.
- Z. Dong, P. Li, X. Li and B. Liu, Rh(III)-Catalyzed Diverse C—H Functionalization of Iminopyridinium Ylides, Chin. J. Chem., 2021, 39(9), 2489–2494, DOI:10.1002/cjoc.202100203.
- W. Y. Xu, Z. Y. Xu, Z. K. Zhang, T. J. Gong and Y. Fu, Tunable Synthesis of Monofluoroalkenes and Gem-Difluoroalkenes via Solvent-Controlled Rhodium-Catalyzed Arylation of 1-Bromo-2,2-Difluoroethylene, Angew. Chem., Int. Ed., 2023, 62(40), e202310125, DOI:10.1002/anie.202310125.
- A. J. Intelli, R. T. Lee and R. A. Altman, Peroxide-Initiated Hydrophosphinylation of Gem-Difluoroalkenes, J. Org. Chem., 2023, 88(19), 14012–14021, DOI:10.1021/acs.joc.3c01562.
- D. L. Orsi, J. T. Douglas, J. P. Sorrentino and R. A. Altman, Cobalt-Catalyzed Selective Unsymmetrical Dioxidation of Gem-Difluoroalkenes, J. Org. Chem., 2020, 85(16), 10451–10465, DOI:10.1021/acs.joc.0c00415.
- R. M. Herrick, M. K. Abd El-Gaber, G. Coy and R. A. Altman, A Diselenide Additive Enables Photocatalytic Hydroalkoxylation of Gem-Difluoroalkenes, Chem. Commun., 2023, 59(37), 5623–5626, 10.1039/d3cc01012k.
- J. P. Sorrentino, R. M. Herrick, M. K. Abd El-Gaber, A. Z. Abdelazem, A. Kumar and R. A. Altman, General Co-Catalytic Hydrothiolation of Gem-Difluoroalkenes, J. Org. Chem., 2022, 87(24), 16676–16690, DOI:10.1021/acs.joc.2c02343.
- S. Koley, K. T. Cayton, G. A. González-Montiel, M. R. Yadav, D. L. Orsi, A. J. Intelli, P. H. Y. Cheong and R. A. Altman, Cu(II)-Catalyzed Unsymmetrical Dioxidation of Gem-Difluoroalkenes to Generate α,α-Difluorinated-α-Phenoxyketones, J. Org. Chem., 2022, 87(16), 10710–10725, DOI:10.1021/acs.joc.2c00925.
- X. Lu, Y. Wang, B. Zhang, J. J. Pi, X. X. Wang, T. J. Gong, B. Xiao and Y. Fu, Nickel-Catalyzed Defluorinative Reductive Cross-Coupling of Gem-Difluoroalkenes with Unactivated Secondary and Tertiary Alkyl Halides, J. Am. Chem. Soc., 2017, 139(36), 12632–12637, DOI:10.1021/jacs.7b06469.
- W. Dai, H. Shi, X. Zhao and S. Cao, Sterically Controlled Cu-Catalyzed or Transition-Metal-Free Cross-Coupling of Gem-Difluoroalkenes with Tertiary, Secondary, and Primary Alkyl Grignard Reagents, Org. Lett., 2016, 18(17), 4284–4287, DOI:10.1021/acs.orglett.6b02026.
- K. Yuan, T. Feoktistova, P. H. Y. Cheong and R. A. Altman, Arylation of Gem-Difluoroalkenes Using a Pd/Cu Co-Catalytic System That Avoids β-Fluoride Elimination, Chem. Sci., 2021, 12(4), 1363–1367, 10.1039/d0sc05192f.
- J. F. Hartwig, Electronic Effects on Reductive Elimination to Form Carbon-Carbon and Carbon-Heteroatom Bonds from Palladium(Ll) Complexes, Inorg. Chem., 2007, 46(6), 1936–1947, DOI:10.1021/ic061926w.
- Y. P. Budiman, A. Jayaraman, A. Friedrich, F. Kerner, U. Radius and T. B. Marder, Palladium-Catalyzed Homocoupling of Highly Fluorinated Arylboronates: Studies of the Influence of Strongly vs. Weakly Coordinating Solvents on the Reductive Elimination Process, J. Am. Chem. Soc., 2020, 142(13), 6036–6050, DOI:10.1021/jacs.9b11871.
- P. Sehnal, R. J. K. Taylor and L. J. S. Fairlamb, Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis, Chem. Rev., 2010, 110(2), 824–889, DOI:10.1021/cr9003242.
- G. Yin, X. Mu and G. Liu, Palladium(II)-Catalyzed Oxidative Difunctionalization of Alkenes: Bond Forming at a High-Valent Palladium Center, Acc. Chem. Res., 2016, 49(11), 2413–2423, DOI:10.1021/acs.accounts.6b00328.
- A. J. Hickman and M. S. Sanford, High-Valent Organometallic Copper and Palladium in Catalysis, Nature, 2012, 484(7393), 177–185, DOI:10.1038/nature11008.
- D. C. Powers and T. Ritter, Palladium(III) in Synthesis and Catalysis, in Higher Oxidation State Organopalladium and Platinum Chemistry, ed. A. J. Canty, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 129–156, DOI:10.1007/978-3-642-17429-2_6.
- K. J. Stowers, A. Kubota and M. S. Sanford, Nitrate as a Redox Co-Catalyst for the Aerobic Pd-Catalyzed Oxidation of Unactivated Sp 3-C-H Bonds, Chem. Sci., 2012, 3(11), 3192–3195, 10.1039/c2sc20800h.
- D. Kalyani, A. D. Satterfield and M. S. Sanford, Palladium-Catalyzed Oxidative Arylhalogenation of Alkenes: Synthetic Scope and Mechanistic Insights, J. Am. Chem. Soc., 2010, 132(24), 8419–8427, DOI:10.1021/ja101851v.
- D. Kalyani and M. S. Sanford, Oxidatively Intercepting Heck Intermediates: Pd-Catalyzed 1,2- and 1,1-Arylhalogenation of Alkenes, J. Am. Chem. Soc., 2008, 130(7), 2150–2151, DOI:10.1021/ja0782798.
- J. Li, R. H. Grubbs and B. M. Stoltz, Palladium-Catalyzed Aerobic Intramolecular Aminoacetoxylation of Alkenes Enabled by Catalytic Nitrate, Org. Lett., 2016, 18(21), 5449–5451, DOI:10.1021/acs.orglett.6b02722.
- M. N. Wenzel, P. K. Owens, J. T. W. Bray, J. M. Lynam, P. M. Aguiar, C. Reed, J. D. Lee, J. F. Hamilton, A. C. Whitwood and I. J. S. Fairlamb, Redox Couple Involving NOx in Aerobic Pd-Catalyzed Oxidation of Sp3-C–H Bonds: Direct Evidence for Pd-NO3-/NO2- Interactions Involved in Oxidation and Reductive Elimination, J. Am. Chem. Soc., 2017, 139(3), 1177–1190, DOI:10.1021/jacs.6b10853.
- Z. K. Wickens, P. E. Guzmán and R. H. Grubbs, Aerobic Palladium-Catalyzed Dioxygenation of Alkenes Enabled by Catalytic Nitrite, Angew. Chem., Int. Ed., 2015, 54(1), 236–240, DOI:10.1002/anie.201408650.
- J. E. Báckvall, J. E. Nordberg, R. E. Wiberg, K. B. Saegebarth, K. A. Yoshimura, N. Tamura, M. Tamura, M. Yasui, T. Berzofsky, J. A. Peisach and J. Horecker, A Comment on the Recently Proposed Mechanism for the Oxidation of Olefins with PdCl(NO2)(CH3CN)2, J. Am. Chem. Soc., 1986, 108(16), 7107–7108 CrossRef.
- N. I. Kuznetsova, V. A. Likholobov, M. A. Fedotov and Y. I. Yermakov, The Mechanism of Formation of Ethylene Glycol Monoacetate from Ethylene in the System MeCO2H + LiNO3 + Pd(OAc)2, J. Chem. Soc., Chem. Commun., 1982, 2(170), 973–974 RSC.
- S. J. Lou, D. Q. Xu and Z. Y. Xu, Mild and Versatile Nitrate-Promoted C-H Bond Fluorination, Angew. Chem., Int. Ed., 2014, 53(39), 10330–10335, DOI:10.1002/anie.201404423.
- I. J. S. Fairlamb, Redox Active NOx Ligands in Palladium-Mediated Processes, Angew. Chem., Int. Ed., 2015, 127(36), 10558–10570, DOI:10.1002/ange.201411487.
- J. L. Burmeister and R. C. Timmer, The Pi-Bonding Effects of Other Ligands on Coordinated Nitrite Ion in Palladium (Il) and Platinum (II) Complexes, J. Inorg. Nucl. Chem., 1966, 28, 1973–1978 CrossRef CAS.
- Y. J. Mao, G. Luo, H. Y. Hao, Z. Y. Xu, S. J. Lou and D. Q. Xu, Anion Ligand Promoted Selective C-F Bond Reductive Elimination Enables C(Sp2)-H Fluorination, Chem. Commun., 2019, 55(96), 14458–14461, 10.1039/c9cc07726j.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04939j |
| This journal is © The Royal Society of Chemistry 2024 |