
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexible organic crystals with multi-stimuli-responsive CPL for broadband multicolor optical waveguides†
Xiuhong
Pan
,
Linfeng
Lan
and
Hongyu
Zhang
 *
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Qianjin Street, Changchun 130012, P. R. China. E-mail: hongyuzhang@jlu.edu.cn
First published on 27th September 2024
Abstract
Flexible organic crystals, capable of transmitting light and responding to various external stimuli, are emerging as a new frontier in optoelectronic materials. They hold immense potential for applications in molecular machines, sensors, displays, and intelligent devices. Here, we report on flexible organic crystals based on single-component enantiomeric organic compounds, demonstrating multi-stimuli-responsive circularly polarized light (CPL). These crystals exhibit remarkable elasticity, responsiveness to light and acid vapors, and tunable circularly polarized optical signals. Upon exposure to acid vapors, the fluorescence of the crystals shifts from initial yellow emission to green emission, attributable to the protonation-induced inhibition of excited-state intramolecular proton transfer. Under UV irradiation, the fluorescence emission undergoes a red-shift, resulting from the molecular transformation from an enol configuration to a ketone configuration. Notably, both processes are reversible and can be restored under daylight. The integration of reversible fluorescence changes under light and acid vapors stimuli, CPL signals, and flexible optical waveguides within a single crystal paves the way for the application of organic crystals as all-organic chiral functional materials.
Introduction
With the continuous development of optical information technology, human beings are endowed with the ability to process super-high-speed and large-capacity information, which makes humankind gradually move from the electronic age to the photonic age.1,2 In particular, in the process of optical information transmission and processing, an optical waveguide is one of the most significant and fundamental structures, in which light propagates in a specific direction through total reflection at the interface of an optical medium. Organic single crystals are expected to become the building blocks of next-generation miniature optoelectronic devices due to their anisotropic property, intense emission, ordered structure, and high carrier mobility.3–5 In the past two decades, significant progress has been made in the field of optical waveguides based on organic single crystals.6–9 The combination of the waveguide properties of organic crystals with a wide range of other properties, such as mechanical compliance,10–12 chiral optical signals,13 field effects,14 mechanical fluorescence chromatism,15 and heterogeneous photoluminescence structures,16 offers almost unlimited possibilities for the development of future advanced photonic devices. Dynamic crystalline materials have attracted significant attention in recent years for their potential in sensor and memory application.17–22 However, only few single-component organic crystals can rapidly switch circularly polarized light (CPL) output signals under multiple external stimuli,23–25 which severely limits direct implementation in multimode optoelectronic transmission devices. It is well-acknowledged that multicolor emissions especially red, green, and blue (RGB) colors are important in display technology.26,27 Furthermore, chiral functional materials with CPL properties have received widespread attention in multidisciplinary fields for their potential applications in 3D optical displays, encrypted transmission, optical storage devices, smart materials, etc.28–31 Therefore, it is of great significance to construct a single component CPL active material with tunable color emission to satisfy the requirements of multidimensional practical applications.32,33Significantly, there is an important class of organic materials (Schiff bases,34–36 diarylethenes,37 azobenzenes38) that can exhibit photochromism or acidichromism in the crystalline state, represented by the Schiff bases of salicylaldehydes and amines. In this work, comprehensively considering the optical properties (fluorescence and CPL signal), stimulus responsiveness, and mechanical properties, we successfully designed and synthesized a pair of single-component enantiomeric organic compounds, S-, R-4-methyl-2-(((1-phenylethyl)imino)methyl)phenol (S-, R-1) (Fig. 1a). Under acid stimulation, it was found that the color of S-, R-1 crystals changed from yellow to green under ultraviolet (UV) light at 365 nm (Fig. 1b and c). Based on the 1H NMR, infrared (IR), and ultraviolet-visible (UV-vis) absorption spectra, we speculate that protonation of the imine groups induced by acid fumigation inhibited excited-state intramolecular proton transfer (ESIPT), leading to a blue shift in the crystal fluorescence. In addition, the fluorescence color of S-, R-1 crystals can change from yellow to orange-red under UV light irradiation at 365 nm (Fig. 1b and c). Solid-state UV–vis absorption and IR spectra confirm that S-, R-1 undergoes reversible structural photoisomerization between the enol form and the trans-keto form. Subsequently, the mechanical and optical (optical waveguide and CPL) properties of the enantiomers before and after the stimulus were characterized. The results show that stimulus response has little effect on mechanical properties and CPL emission wavelength of crystals can also be switched by UV radiation or acid stimulation. Under remote-control photoirradiation, the optical waveguide and CPL emission wavelength can be reversibly switched via structural photoisomerization. To our knowledge, switchable multi-responsive flexible optical waveguides and CPL reported so far via photoisomerization-triggered configurational interconversion and acid stimulation are limited. The unique flexible organic crystals with CPL properties have the potential for optoelectronic devices with multimode output signals.
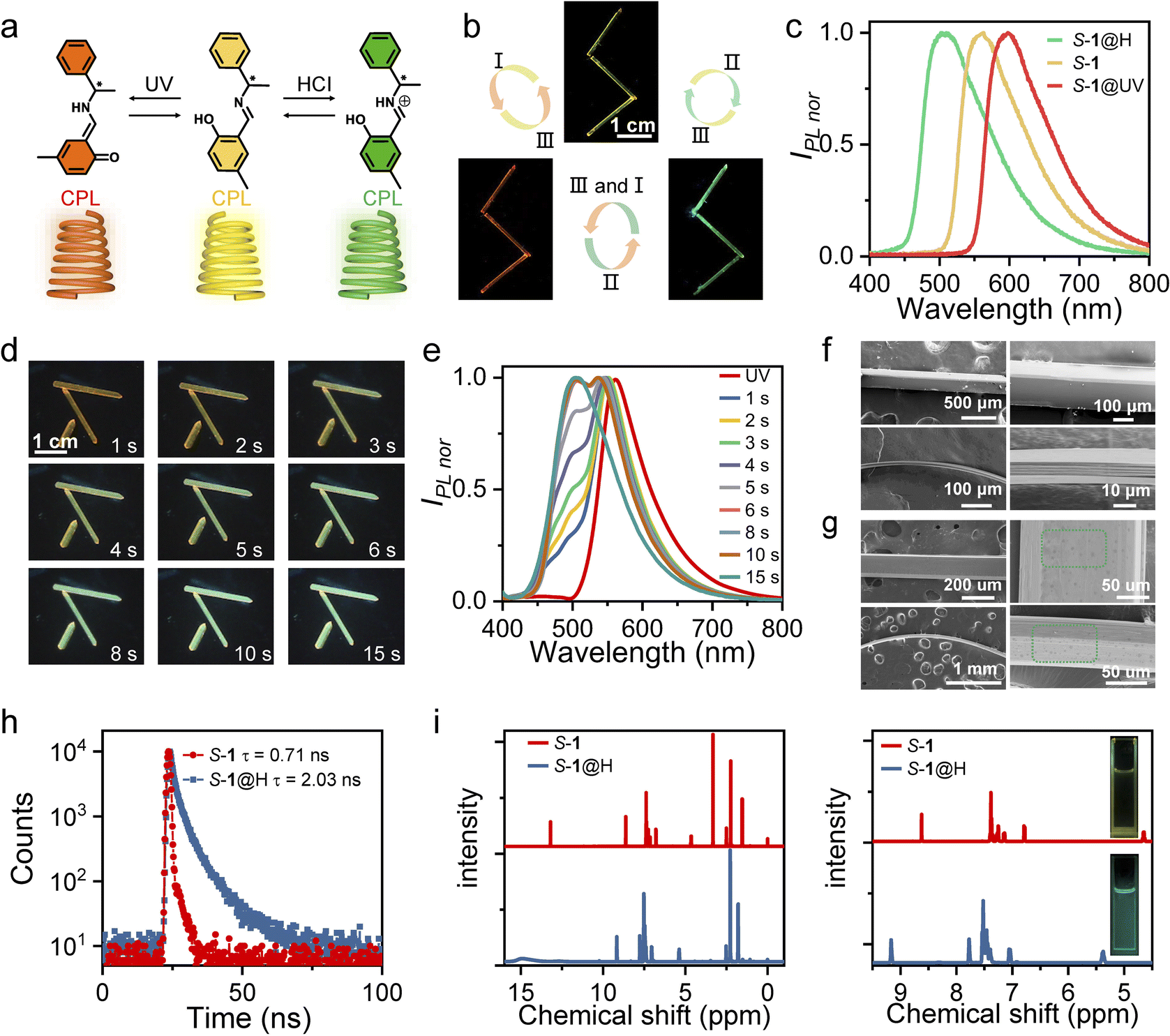 | ||
| Fig. 1 (a) Chemical structures of S-, R-1, S-, R-1@UV and S-, R-1@H. (b) S-1 crystals with fluorescence switching under different stimuli: (I) UV irradiation for 30 s; (II) fuming with HCl acid for 15 s; (III) recovery at room temperature or heating (60 °C). Three crystals of similar size are arranged in a zig-zag shape for clear display. (c) PL spectra of S-1 crystals under different stimuli. (d) The color change process of acid fuming (HCl) at different times. (e) The fluorescence change process at different time of acid fuming (HCl). (f) Low (left) and high (right) magnification scanning electron microscope (SEM) images of a straight, bent original crystal S-1. (g) Low (left) and high (right) magnification SEM images of a straight, bent acid-fumigated crystal S-1. (h) Decay curves of crystals S-1 and S-1@H at room temperature. (i) Comparison of NMR (500 MHz, DMSO-d6) spectra of S-1 and S-1@H. On the right are expanded NMR spectra between 4.5–9.5 ppm (inset: photographic images of S-1 and S-1@H in DCM solution). | ||
Results and discussion
The general methods for preparing S-1 and R-1 are shown in Fig. S1–S6.† They were synthesized by the equimolar 2-hydroxy-5-methylbenzaldehyde and S-, R-1-phenylethan-1-amine, respectively. Single crystals of S-1 and R-1 were prepared via the solvent diffusion method at room temperature within 7–10 days. In general, the crystals are yellow needle-shaped (up to centimeters in length, tens to hundreds of microns in thickness, and hundreds of microns in width) with a maximum photoluminescent (PL) ≈ 550 nm (Fig. 1b and c). Interestingly, the color and emission of dichloromethane (DCM) solution S-, R-1 change dramatically upon adding hydrochloric (HCl) or trifluoroacetic acid (TFA) (Fig. 1i), and the formed protonated complex is defined as S-, R-1@H. The crystalline solids S-, R-1 also exhibit acidichromism upon fuming in HCl or TFA vapors. When the yellow needle-like crystal was placed in the center of the Petri dish which was surrounded by cotton absorbing hydrochloric acid, the maximum emission wavelength appeared blue-shifted, and finally, the entire crystal emits green fluorescence (Fig. 1d, e and Movie S1†). When the aforementioned S-, R-1@H samples were taken out from the Petri dish to air, the fluorescence of these crystals was restored as deprotonation occurred (Fig. S7†). The fluorescence transition process can be repeated multiple times, indicating that S-, R-1 has a reversible acid-responsive behavior. The crystals of compound S-1 and S-1@H are reversibly elastic; they can be bent without any visible damage, as it was confirmed by the scanning electron microscopic images of bent specimens that were fixed in their bent state for inspection (Fig. 1f and g). The lifetimes of crystals S-1 and S-1@H are determined to be 0.71 and 2.03 ns (Fig. 1h). The original crystals S-, R-1 and acid-treated crystals S-, R-1@H were dissolved in deuterated DMSO-d6 for H1 NMR testing (Fig. 1i) to investigate the mechanism of the acid reaction. To ensure only the hydrochloric acid that reacted with the crystal remained, we fumed the crystal with acid for 2 minutes and then left it in an open environment for several minutes to allow free hydrochloric acid vapor to dissipate. Compared to the initial state, all peaks in S-, R-1@H shifted to a lower field, with the two hydrogen peaks closest to the nitrogen atom showing significant changes, shifting from 4.67 and 8.63 ppm to 5.37 and 9.17 ppm. Simultaneously, IR absorption spectra and powder X-ray diffraction (PXRD) of the initial crystal 1 and the acid-fumed crystal 1@H were measured (Fig. S8 and S9†). The IR spectra show a shift of certain peaks to higher wavenumbers. Additionally, some PXRD peaks of the protonated crystal 1@H exhibit slight weakening, with the appearance of a weak new peak (Fig. S9†), indicating that the crystal phase after protonation remains largely consistent with the initial crystal 1. By comparing the 1H NMR, IR, and PXRD results, it was determined that partial protonation of crystal 1 occurred after fuming, resulting in the formation of the associated compound 1@H.UV-vis absorption and emission spectra of S-, R-1, S-, R-1@H in solution and crystalline states were obtained to further elucidate the mechanism of acid fumigation (Fig. S10 and S11†). S-, R-1 in DCM solution display a strong emission band peaked at 504 nm and an absorption band located at about 326 nm. The band at 504 nm with a huge Stokes shift of 178 nm is assigned to the emission of keto species produced through an ESIPT process (Fig. S10†).39–41 The wavelength absorption of the protonated compounds S-, R-1@H in DCM solution was shorter compared to S-, R-1 with an emission peak at 493 nm and a blue shift of 11 nm (Fig. S10†). The optical properties of S-, R-1 and S-, R-1@H solids show a similar trend as those of solution samples. As shown in Fig. S11,† the absorption bands of S-, R-1@H are obviously blue-shifted by about 35 nm compared with those of S-, R-1. In addition, crystals S-, R-1 display yellow fluorescence with an emission maximum at 550 nm, while crystals S-, R-1@H show green fluorescence at 500 nm which are significantly blue-shifted by 50 nm compared with crystals S-, R-1 (Fig. 1c and S11†). Under the stimulation of HCl or TFA, there are two different sources of hydrogen protons, one part from the phenolic hydroxyl group of S-, R-1, and the other part from HCI or TFA. Considering the size of PKa value, the nitrogen atom will preferentially interact with acid. The combination of hydrogen protons in the medium inhibits the process of excited state proton transfer and affects the emission of ketone isomers, thus an enol emission at short wavelengths is emitted in an acidic medium.42 Air or heating promotes the volatilization of the acid, and this deprotonation reaction directly promotes the emission of keto isomers, which is beneficial to the keto luminescence of ESIPT molecules at long wavelengths. Therefore, S-, R-1 in solid and solution states exhibits a blue shift (from yellow to green) of the luminescence spectrum upon acid stimulation. In this sense, the fluorescence of crystalline solids S-, R-1 can be conveniently modulated with a huge difference by protonic acid fuming.
To have a deep insight into the electronic structure, we calculated the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) of S-1 and R-1 molecules (Fig. S12†). Due to the mirror symmetry of this pair of enantiomeric molecules, their HOMO and LUMO also exhibits a mirror–image relationship, respectively. From the HOMO of S-1 and R-1 molecules, the electron density was mainly distributed on the benzene ring of the salicylaldehyde moiety. Compared to the HOMO, the LUMO was significantly shifted in the C–N direction. The energy gap between HOMO and LUMO is calculated to be 0.16336 hartree, which corresponds to absorption wavelength of 279 nm.
Interestingly, S-, R-1 crystals also have a sensitive response under UV irradiation with good reproducibility (illumination crystals are defined as S-, R-1@UV). The fluorescent color of the crystals gradually changed from yellow to orange-red within 30 s under 365 nm UV light (Fig. 2a and Movie S1†), whereby the emission maximum shift from 550 nm to 598 nm (Fig. 2b). As the light source was removed, the emitted color of the crystals was restored to initial fluorescence at the time of 80 s (Fig. 2a and c). This stimulus response was observed in real-time and was also uniform and fully reversible. This photochromic phenomenon is associated with the photoisomerization of molecular configuration from enol to trans-keto.43 Unfortunately, the photogenerated structure could not be determined through single-crystal X-ray diffraction tests. Due to the low conversion ratio of photoreaction, the obtained crystal structure is still the enol form.
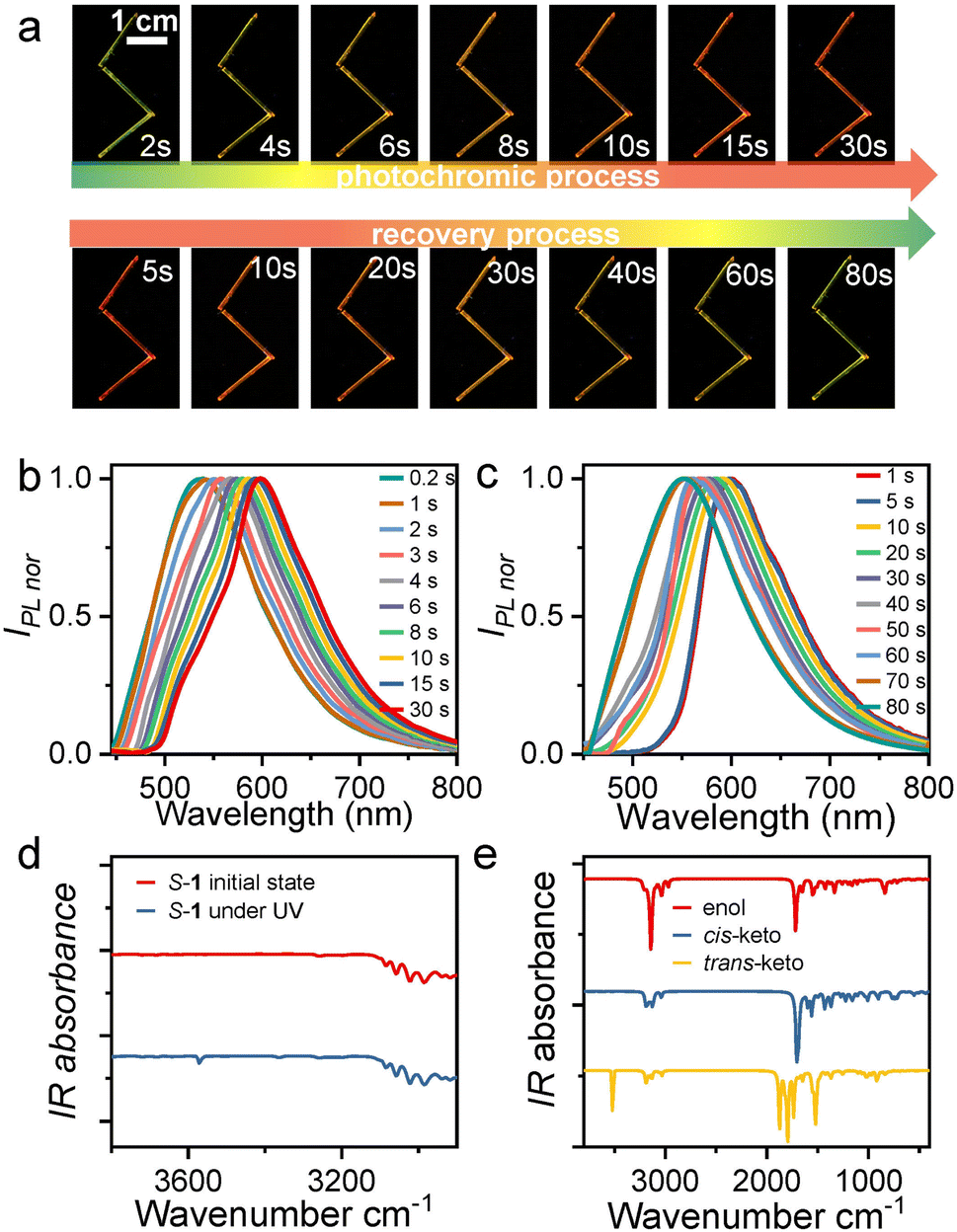 | ||
| Fig. 2 (a) Photographs of S-1 during the discoloration–recovery cycle under UV illumination at 365 nm. Three crystals of similar size are arranged in a zig-zag shape for clear display. (b) Solid-state maximum emission spectra of S-1 were recorded after 365 nm light irradiation for different illumination times. (c) Maximum emission spectra of S-1 crystals with different recovery times under light irradiation at 365 nm. (d) The experimental IR absorption spectra of S-1 at ambient conditions and under UV (365 nm) photoirradiation. (e) The calculated IR absorption spectra of S-1 with enol, cis-keto, and trans-keto forms, respectively. | ||
Photoisomerizations by UV light lead to photochromic behaviors. To further investigate the photochromism, we then recorded the solid-state UV–vis absorption spectrum for the crystal before and after UV photoirradiation (Fig. S11 and S13†). At ambient condition, both S-1 and R-1 absorb light of less than 500 nm, which agrees well with the yellow appearance. Upon UV photoirradiation, a new absorption band appears in the wavelength range from 500 to 600 nm, in good agreement with the orange-red appearance (Fig. S11†). Thus, the photochromic behaviors in R-1 and S-1 are associated with the occurrence of the UV photoirradiation induced new absorption band. We also performed the time-dependent solid-state UV–vis spectrum on a S-1 powder sample for monitoring the photoreaction process. Fig. S13† shows that the intensity of the 365 nm UV photoirradiation induced new absorption band increases with UV illumination time and becomes saturated within 30 s, which indicates that the photoreaction from the enol molecular configuration to the trans-keto one is complete. Under further daylight photoirradiation, the intensity of the new absorption band decease very quickly and the new absorption band vanishes within 100 s (Fig. S13†), indicating the recovery of the photoinduced trans-keto form to the initial enol one. The photoinduced trans-keto form recovers to the enol one even in the daylight due to the enol form is in the most stable ground electronic state while the photoinduced trans-keto form is in the excited electronic state,44 as also found in other salicylideneaniline-derived compounds.45S-1 showed no changes in color and absorption spectrum under hand-held UV irradiation for up to 1200 s (Fig. S13†) and exhibited a high fatigue resistance under multiple cycles of UV and daylight stimulation.
To investigate the conformational changes of the photochromic molecules, the IR spectra of S-, R-1 before and under UV light irradiation were further analyzed (Fig. 2d, e and S14†). Given the structural and property similarities between R-1 and S-1, here we take S-1 as an example for discussion. The most significant change in the experimentally measured IR spectrum under UV irradiation is the appearance of a new absorption peak centered at 3571 cm−1 (Fig. 2d). Moreover, based on the single-crystal X-ray diffraction data, different molecular configurations of S-1, including the enol, cis-keto, and trans-keto forms, were constructed, and their IR spectra were further calculated (Fig. S15–17†). The calculations indicate that only the trans-keto form exhibits an IR absorption peak at 3582 cm−1 in the range above 3100 cm−1 (Fig. 2e). This IR absorption peak could be attributed to the free stretching vibration of the N–H bond in the trans-keto form, whereas in the cis-keto form, the N–H stretching vibration is restricted by N–H⋯O hydrogen bonding interactions. Therefore, it is reasonably to infer that the molecular configuration of S-1 molecules changes from the enol form to the trans-keto form upon UV irradiation.
The general mechanical properties have an important role in determining the mechanical compliance. The S-, R-1 crystals exhibit good elastic properties. By applying stress to the ends of the crystal with tweezers, the straight crystal could be bent into a semicircle without breaking or cracking (Fig. S18†). Once the tweezers were removed, the bent crystal quickly returned to its original shape without any deformation or crack. Interestingly, this reversible and repeatable process takes place under UV light, which means that S-1@UV crystals also have good flexibility. The needle-like S-1@H crystals also exhibit the same elastic bendable behavior (Fig. S18†). By the images of the scanning electron microscope (SEM), the bent crystal S-1@H maintains smooth except for some corroded spots on the surface as reflected (Fig. 1g). The linear correlation between stress and strain in the three-point bending experiments further confirms the elastic properties of the crystals (Fig. S18†). There is a slight larger in the elastic modulus of S-1@H (2.85 GPa) compared with that of S-1 (2.20 GPa), which probably be due to the fuming-caused defects as implied by the SEM image (Fig. 1f and g). The results show that acid fumigation had little effect on the mechanical properties of the crystals.
Single-crystal X-ray diffraction was performed on crystals S-, R-1 to identify the structural features responsible for the good elasticity. Crystals S-, R-1 belong to the monoclinic system and C2 space group. The growth direction of the S-, R-1 were confirmed to coincide with the crystallographic [010] direction and the bending surface was determined to be (100) by face indexing (Fig. S19 and S20†). Only the intermolecular interactions and stacking structure of S-1 are discussed for the sake of description (the corresponding data for R-1 are provided in Fig. S21†). The hydroxyl and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups are linked by intramolecular hydrogen bond interaction (O–H⋯N: 1.755 Å and 1.692 Å) (Fig. 3a and b). Each molecule connects with two neighbors through an intermolecular C–H⋯π hydrogen bond (2.643 Å), forming a chain structure along the crystallographic [010] direction (Fig. 3c). When the crystal was bent upon loading stress on the (100) plane, the molecular chains in the outer arc expanded by enlarging the C–H⋯π hydrogen bonds distance, and those in the inner arc contracted by shortening the C–H⋯π hydrogen bonds length (Fig. S22†). To further illustrate our hypothesis, the interaction energy frameworks of crystal 1 are analyzed by the CrystalExplorer software using the B3LYP hybrid functional with 6-31G (d, p) basis set, where semiempirical dispersion is included by using the D2 version of Grimme's dispersion.46 In the (010) and (001) planes, the thick blue cylinders with a largest total energy of 29.8 kJ mol−1 reflect the strongest interactions contributed by the π-stacked molecular columns that are responsible for layer contraction and expansion. As shown in Fig. 3d and e, the cylinder radius is proportional to the strength of the interaction energy. Other thinner cylinders perpendicular to the thicker ones with small total energies (e.g., 7.1 kJ mol−1) represent weak intermolecular interactions which promote short-range slippages thereby reducing the crystal brittleness.
N groups are linked by intramolecular hydrogen bond interaction (O–H⋯N: 1.755 Å and 1.692 Å) (Fig. 3a and b). Each molecule connects with two neighbors through an intermolecular C–H⋯π hydrogen bond (2.643 Å), forming a chain structure along the crystallographic [010] direction (Fig. 3c). When the crystal was bent upon loading stress on the (100) plane, the molecular chains in the outer arc expanded by enlarging the C–H⋯π hydrogen bonds distance, and those in the inner arc contracted by shortening the C–H⋯π hydrogen bonds length (Fig. S22†). To further illustrate our hypothesis, the interaction energy frameworks of crystal 1 are analyzed by the CrystalExplorer software using the B3LYP hybrid functional with 6-31G (d, p) basis set, where semiempirical dispersion is included by using the D2 version of Grimme's dispersion.46 In the (010) and (001) planes, the thick blue cylinders with a largest total energy of 29.8 kJ mol−1 reflect the strongest interactions contributed by the π-stacked molecular columns that are responsible for layer contraction and expansion. As shown in Fig. 3d and e, the cylinder radius is proportional to the strength of the interaction energy. Other thinner cylinders perpendicular to the thicker ones with small total energies (e.g., 7.1 kJ mol−1) represent weak intermolecular interactions which promote short-range slippages thereby reducing the crystal brittleness.
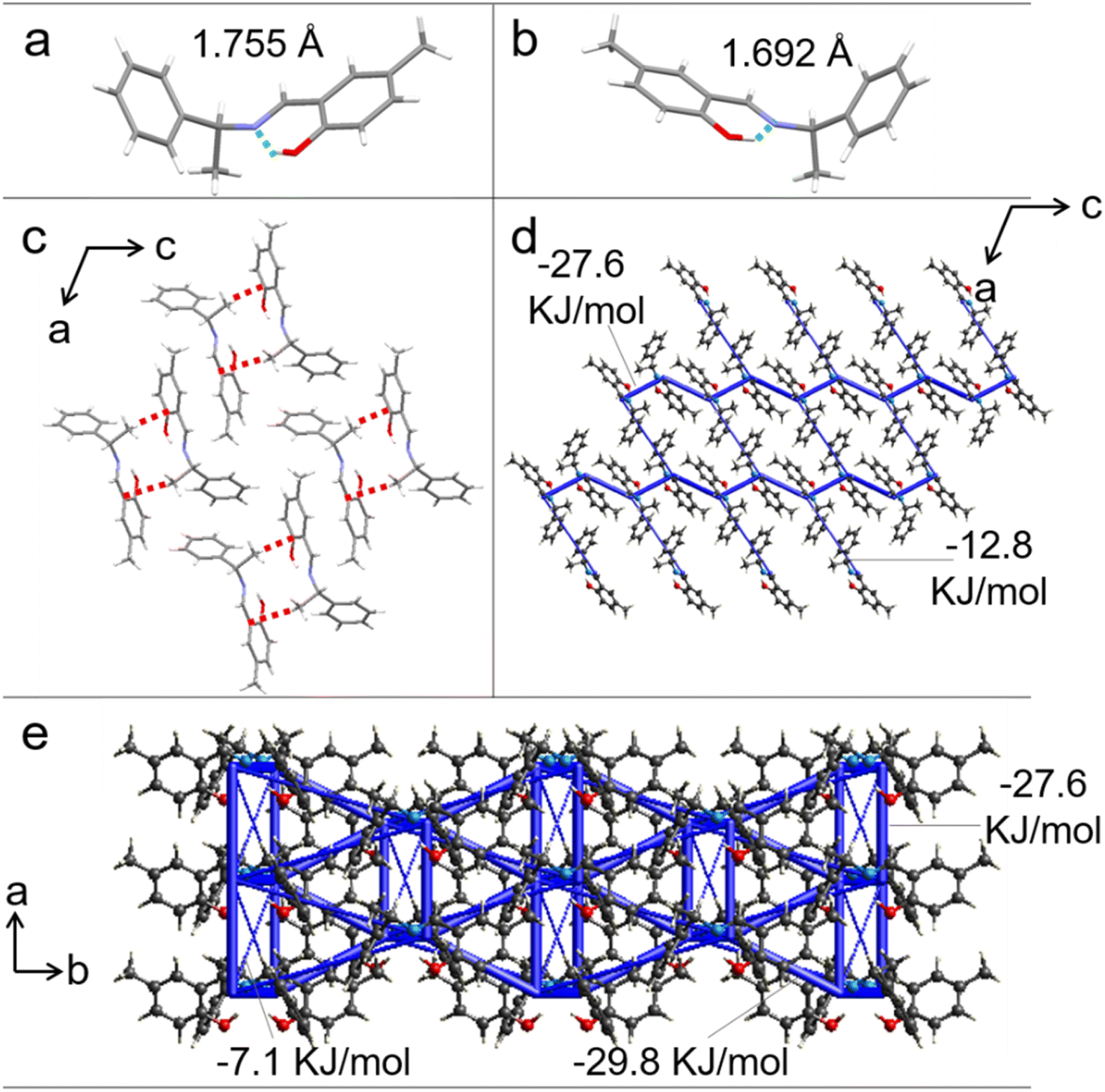 | ||
| Fig. 3 (a and b) Molecular configurations in the crystal structures of S-1 (a) and R-1 (b). (c) Intermolecular interactions in S-1 along the b-axis (red dotted line for C–H⋯π). (d and e) Energy frameworks of S-1 crystal viewed down b (d) and c (e) axis. The blue cylinders represent the interaction energy between molecules. | ||
Considering the chiral structures of S-1 and R-1, their chiral characteristics were investigated by recording the circular dichroism (CD) and CPL spectra. The mirror-symmetry CD absorption spectra of both the DCM solution (Fig. 4a) and the powders (Fig. 4b) of S-1 and R-1 displayed a significant Cotton effect, with S-1 and R-1, exhibiting a positive and negative Cotton effect, respectively. Interestingly, S-1@H and R-1@H in DCM solution show intense CD signal peaks, which is exactly a mirror-image symmetry relation (Fig. 4c), consistent with the absorption spectra. Additionally, when the solid samples of S-1 and R-1 were irradiated by 365 nm UV light or acid-fumigated, the CD spectra changed significantly (Fig. S23†), and consistent with the absorption spectra, indicating chirality in the discrete and aggregated states. When the S-1 and R-1 solid samples were fumigated in a concentrated HCl atmosphere, the CD spectra were blue-shifted by about 50 nm. This is attributed to the corresponding changes in chiral molecules caused by acid-fuming protonation. For S-1@UV and R-1@UV, a new broad CD peaks emerge from 500 to 580 nm, which is attributed to the remarkable change of molecular configuration after UV photoirradiation, resulting in the corresponding variation of the electronic transition of the chiral molecule. The chiral structures of S-1 and R-1 were further explored by CPL techniques to probe the chiroptical properties in the excited state. When excited at 365 nm, the powders of S-1 and R-1 displayed a significant Cotton effect, with S-1 and R-1, exhibiting a positive and negative Cotton effect, respectively. The corresponding DC curves consistent with the photoluminescence spectrum of S-1 and R-1. Then, we prepared powders with different stimulus responses to explore whether acid stimulation and illumination could achieve multi-color CPL. The prepared S-, R-1@H and S-, R-1@UV powders also showed obvious cotton effect and were consistent with the photoluminescence spectra (Fig. S24†), and the green, yellow and orange-red CPL signals were successfully obtained by acid/light stimulation. The glum factor, commonly used to evaluate the magnitude of CPL, is defined as glum = 2(IL − IR)/(IL + IR), where IL and IR refer to the intensity of left-and right-handed CPL, respectively.47–49 The maximum glum values of S-1 and R-1 are about ±3 × 10−3 (Fig. S24†). As the material is crystalline and large enough, the prepared single crystals of S-, R-1, S-, R-1@H and S-, R-1@UV were picked for CPL test. The CPL spectra of the enantiomers in the single-crystal state are similar to those in the powders state, but surprisingly, the CPL intensities are slightly increased (Fig. 4d–f and S25†). The difference of the CPL intensities in the powder state and the single-crystal state can be explained by the destruction of the defects in the structure.50 The single-crystals maintain intact supramolecular chirality leading to enhanced CPL signals. According to the above data, multi-color CPL emission of green, yellow and orange-red can be easily achieved with acid stimulation and light response, providing a convenient method for the preparation of multi-color CPL materials.
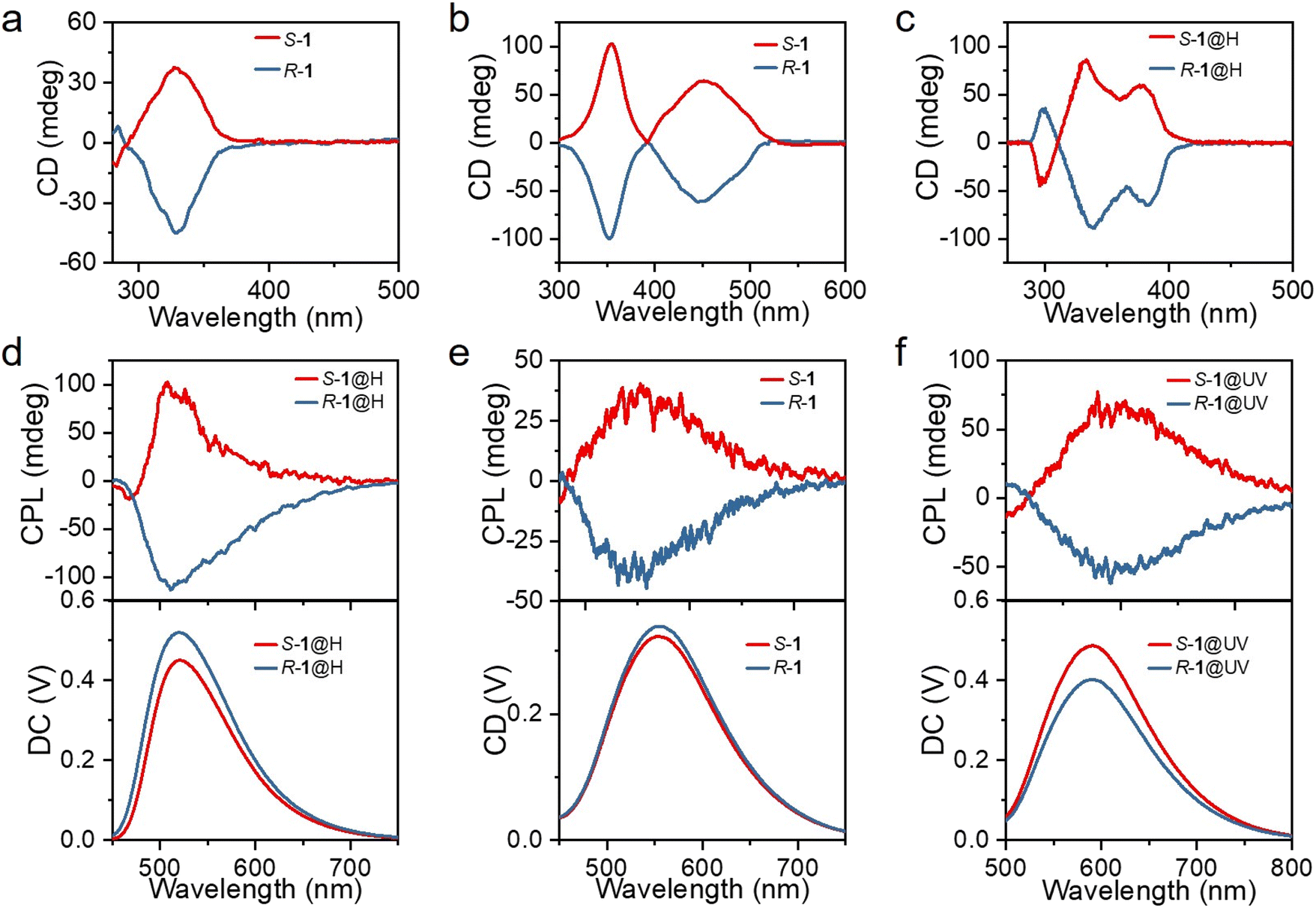 | ||
| Fig. 4 (a) CD spectra of S-1 and R-1 compounds in DCM. (b) CD spectra of S-1 and R-1 powder samples. (c) CD spectra of S-1@H and R-1@H compounds in DCM. (d) CPL emission spectra of crystals S-1@H and R-1@H. (e) CPL emission spectra of crystals S-1 and R-1. (f) CPL emission spectra of crystals S-1@UV and R-1@UV. | ||
Taking advantage of the integrated elastic-bending and photochromic as well as acidichormic behaviors of crystals S-, R-1, a flexible multi-mode color tunable crystalline optical waveguides is designed by a standard experiment.51,52 The active optical waveguide capabilities of straight and bent crystal S-1 in the initial state were first tested. When different positions of the crystal were excited by a pulsed laser (Nd: YAG, 355 nm), the emission was generated and propagated (optical waveguides) to both ends of the crystal (Fig. 5a and b). By changing the excitation position from left to right along the crystal, six emission spectra were recorded at the left end of the crystal. The fluorescence color is slightly red-shifted due to photochromism and blue-shifted due to acid discoloration. Similarly, the optical waveguide tests for S-1@UV and S-1@H are also carried out under the same conditions (Fig. 5c–f). The intensity of the emission recorded in all measurements decreases as the laser beam moves away from the left end. By fitting the data in Fig. 5g, the light loss coefficients (OLCs) calculated by the reported method53,54 have been determined to be 0.197 dB mm−1, 0.216 dB mm−1, 0.186 dB mm−1, 0.207 dB mm−1, 0.190 dB mm−1 and 0.226 dB mm−1 for the initial straight, curved, photo-stimulated straight, curved, and acid smoked straight and curved crystals, respectively (Fig. 5h). It can be seen that acid/light stimulation has minimal impact on the light transmission ability of single crystals. Based on acidichormic properties of the crystal, a heterojunction sample 1 with only one-half fumed in HCl vapor was prepared (Fig. 5i). When the sample was excited with a 355 nm laser at seven sites from 0 to 6, the fluorescence emission of the output signals gradually changes from green (500 nm) to yellow (556 nm) (Fig. 5k). Furthermore, another heterojunction sample with one-half fumed in HCl vapor and one-half irradiated with UV light was prepared (Fig. 5j). The samples were also excited with a 355 nm laser at seven sites from 0 to 6, and the outputs recorded at the tip of the crystal are outlined (Fig. 5l). The optical waveguide kept a constant orange-red emission profile for the first three tests, then changed into a dual-band of yellow emission, and finally a single green emission band. The CIE coordinates of the optical waveguide outputs as plotted in Fig. 5m localize in the region of green, yellow and orange-red. These results show that the signal output of crystal waveguides can be adjusted flexibly by ultraviolet light and acid stimulation, which opens up a new application for organic crystals, namely the flexible optical tuner of crystalline optical waveguides.
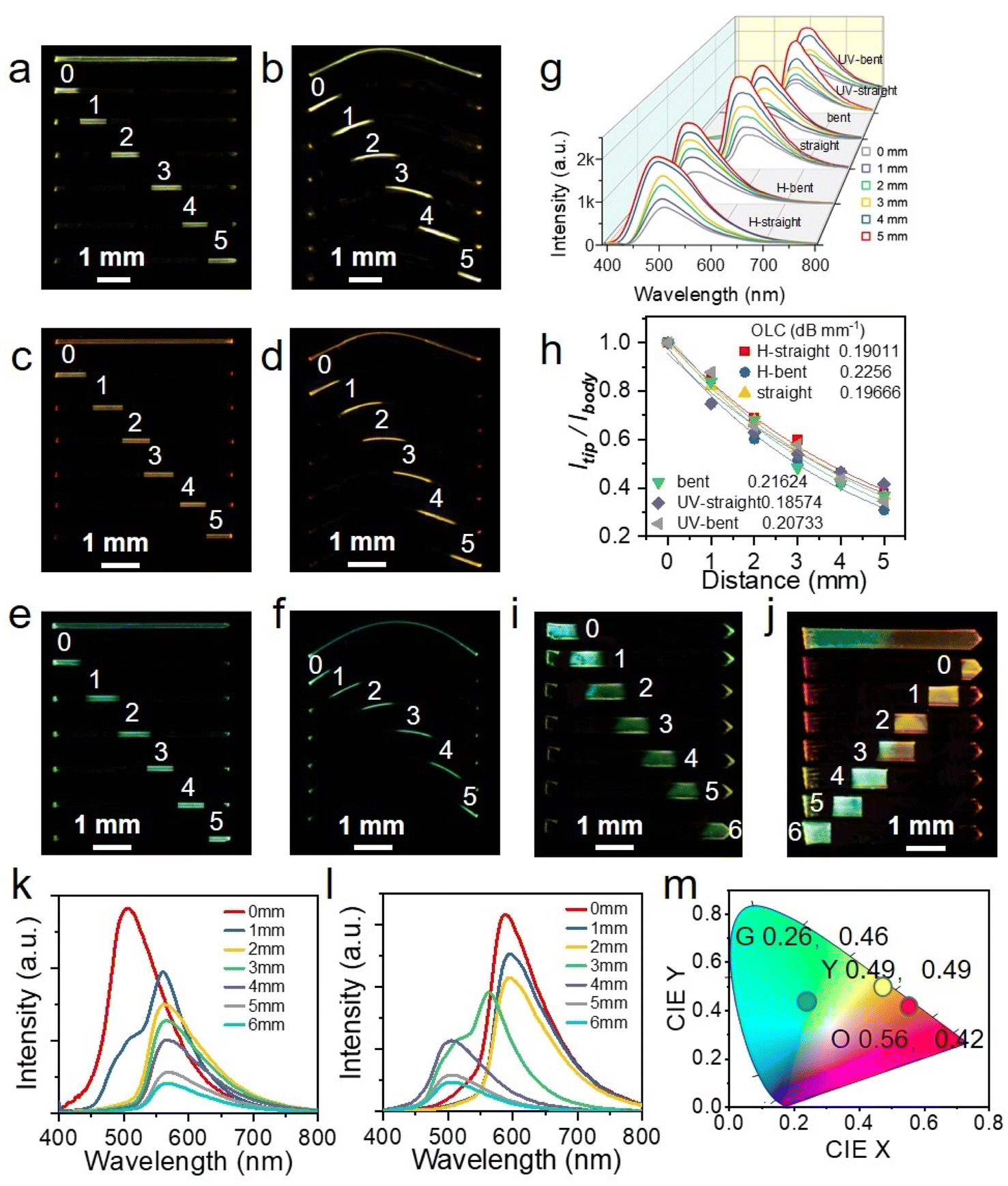 | ||
Fig. 5 (a–f) Fluorescence photographs of S-1 (a and b), S-1@UV (c and d) and S-1@H (e and f) in straight and bent states excited with a UV lamp (uppermost line) and a 355 nm laser-focused at different positions. (g) Fluorescence spectra collected at the tip of the crystal S-1 in different states and their dependence on the distance between the tip and the excitation site. (h) Comparison of the optical loss coefficients for the crystal S-1 in different states. The light loss coefficient (α) is obtained by a single exponential fit to the function Itip/Ibody = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−αD), where Itip and Ibody are the fluorescence intensity of the externally coupled and incident light, respectively, and D is the distance between the excitation site and the tip where the emission is collected. (i and j) Optical waveguide properties of a heterojunction sample 1 with only one-half fumed with HCl vapor (i) and another heterojunction sample 2 with one-half fumed in HCl vapor and another one-half irradiated with UV light (j). (k and l) Fluorescence spectra collected at the tip of the heterojunction sample 1 (k) and 2 (l), and their dependence on the distance between the tip and the excitation site. (m) CIE coordinates of optical waveguide emission spectra of S-1@H (G), S-1 (Y), S-1@UV (O) crystals. exp(−αD), where Itip and Ibody are the fluorescence intensity of the externally coupled and incident light, respectively, and D is the distance between the excitation site and the tip where the emission is collected. (i and j) Optical waveguide properties of a heterojunction sample 1 with only one-half fumed with HCl vapor (i) and another heterojunction sample 2 with one-half fumed in HCl vapor and another one-half irradiated with UV light (j). (k and l) Fluorescence spectra collected at the tip of the heterojunction sample 1 (k) and 2 (l), and their dependence on the distance between the tip and the excitation site. (m) CIE coordinates of optical waveguide emission spectra of S-1@H (G), S-1 (Y), S-1@UV (O) crystals. | ||
Conclusions
In summary, we developed multimode stimulus-responsive flexible organic crystals based on chiral Schiff base molecules. These crystalline materials exhibit reversible acid-responsive behavior, characterized by protonation in the presence of hydrochloric acid vapor, a blue shift in fluorescence emission, and recovery to the initial state under daylight, with heating accelerating this process. Additionally, the crystals display a reversible photochromic phenomenon, where fluorescence emission is red-shifted under UV irradiation due to molecular configurational changes. Remarkably, the crystals retain their elasticity and multi-color (green, yellow, orange-red) CPL signals under both acid fumigation and light conditions, underscoring their potential for applications in flexible CPL devices. The observed fluorescence emission changes under multimode stimulation enable the creation of a flexible optical tuner for crystal optical waveguides. These findings lay the groundwork for future research on multifunctional flexible organic molecular crystal materials and are anticipated to stimulate further exploration into the use of flexible crystals as stimulus-responsive waveguide materials in the CPL domain.Data availability
The data supporting this article have been included as part of the ESI.† The data that support the findings of this study are available from the corresponding author, Hongyu Zhang, upon reasonable request.Author contributions
Xiuhong Pan, performed the experiment and data analyses, wrote the manuscript draft. Linfeng Lan, revised the manuscript. Hongyu Zhang, conceived the ideas and designed the research, revised the manuscript and provided funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52373181 and 52173164), and the Natural Science Foundation of Jilin Province (20230101038JC).Notes and references
- G. Loke, W. Yan, T. Khudiyev, G. Noel and Y. Fink, Adv. Mater., 2020, 32, 1904911 CrossRef CAS PubMed.
- M. Alexander Schmidt, A. Argyros and F. Sorin, Adv. Opt. Mater., 2016, 4, 13–36 CrossRef CAS.
- A. L. Briseno, J. Aizenberg, Y. Han, R. A. Penkala, H. Moon, A. J. Lovinger, C. Kloc and Z. Bao, J. Am. Chem. Soc., 2005, 127, 12164–12165 CrossRef CAS.
- S. Hayashi and T. Koizumi, Chem.–Euro. J., 2018, 24, 8507–8512 CrossRef CAS.
- K. S. Park, J. Baek, Y. Park, L. Lee, J. Hyon, Y.-E. Koo Lee, N. K. Shrestha, Y. Kang and M. Mo Sung, Adv. Mater., 2017, 29, 1603285 CrossRef.
- H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 CrossRef CAS PubMed.
- A. V. Kumar, M. Godumala, J. Ravi and R. Chandrasekar, Angew. Chem., Int. Ed., 2022, 61, e202212382 CrossRef CAS PubMed.
- N. Mitetelo, D. Venkatakrishnarao, J. Ravi, M. Popov, E. Mamonov, T. V. Murzina and R. Chandrasekar, Adv. Opt. Mater., 2019, 7, 1801775 CrossRef.
- D. Barman, M. Annadhasan, R. Chandrasekar and P. Krishnan Iyer, Chem. Sci., 2022, 13, 9004–9015 RSC.
- W. M. Awad, D. W. Davies, D. Kitagawa, J. M. Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Zhang, C. Calvin Sun, C. Bardeen, R. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 RSC.
- L. Catalano, D. P. Karothu, S. Schramm, E. Ahmed, R. Rezgui, T. J. Barber, A. Famulari and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 17254–17258 CrossRef CAS PubMed.
- J. M. Halabi, E. Ahmed, L. Catalano, D. P. Karothu, R. Rezgui and P. Naumov, J. Am. Chem. Soc., 2019, 141, 14966–14970 CrossRef CAS PubMed.
- N. Mitetelo, D. Venkatakrishnarao, J. Ravi, M. Popov, E. Mamonov, T. V. Murzina and R. Chandrasekar, Adv. Opt. Mater., 2019, 7, 1801775 CrossRef.
- G. Zhao, H. Dong, Q. Liao, J. Jiang, Y. Luo, H. Fu and W. Hu, Nat. Commun., 2018, 9, 4790 CrossRef PubMed.
- Y. Li, Z. Ma, A. Li, W. Xu, Y. Wang, H. Jiang, K. Wang, Y. Zhao and X. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 8910–8918 CrossRef CAS PubMed.
- Z. Li, J.-J. Wu, X.-D. Wang, K.-L. Wang, S. Zhang, W.-F. Xie and L.-S. Liao, Adv. Opt. Mater., 2019, 7, 1900373 CrossRef CAS.
- S. Hayashi, S.-y. Yamamoto, D. Takeuchi, Y. Ie and K. Takagi, Angew. Chem., Int. Ed., 2018, 57, 17002–17008 CrossRef CAS PubMed.
- S. Zhao, H. Yamagishi, O. Oki, Y. Ihara, N. Ichiji, A. Kubo, S. Hayashi and Y. Yamamoto, Adv. Opt. Mater., 2022, 10, 2101808 CrossRef CAS.
- L. Lan, L. Li, Q. Di, X. Yang, X. Liu, P. Naumov and H. Zhang, Adv. Mater., 2022, 34, 2200471 CrossRef CAS PubMed.
- R. Huang, C. Wang, Y. Wang and H. Zhang, Adv. Mater., 2018, 30, 1800814 CrossRef PubMed.
- X. Pan, L. Lan, L. Li, P. Naumov and H. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202320173 CrossRef CAS PubMed.
- X. Pan, L. Lan, Q. Di, X. Yang and H. Zhang, Wearable Electron., 2024, 1, 111–118 CrossRef.
- W. Wu, K. Chen, H. Yu, J. Zhu, Y. Feng, J. Wang, X. Huang, L. Li, H. Hao, T. Wang and N. W. P. Naumov, Chem. Sci., 2024, 15, 9287–9297 RSC.
- H. Murata, S. Suzuki, K. Terakubo, Y. Imai and S. Ito, Chem.–Asian J., 2024, 19, e202400293 CrossRef CAS PubMed.
- J. Song, Y. Zhou, Z. Pan, Y. Hu, Z. He, H. Tian and X. Ma, Matter, 2023, 6, 2005–2018 CrossRef CAS.
- B. Zhao, X. Gao, N. Lu and J. Deng, Adv. Opt. Mater., 2020, 8, 2000858 CrossRef CAS.
- C. Wei, L. Bai, X. An, M. Xu, W. Liu, W. Zhang, M. Singh, K. Shen, Y. Han, L. Sun, J. Lin, Q. Zhao, Y. Zhang, Y. Yang, M. Yu, Y. Li, N. Sun, Y. Han, L. Xie, C. Ou, B. Sun, X. Ding, C. Xu, Z. An, R. Chen, H. Ling, W. Li, J. Wang and W. Huang, Chem, 2022, 8, 1427–1441 CAS.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- Z.-G. Wu, H.-B. Han, Z.-P. Yan, X.-F. Luo, Y. Wang, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Adv. Mater., 2019, 31, 1900524 CrossRef PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- L. Song, Y. Dong, B. Zhao, Y. Wu and J. Deng, Adv. Opt. Mater., 2024, 12, 2400215 CrossRef CAS.
- W. Wu, K. Chen, X. Zhang, T. Wang, S. Li, H. Zhao, L. Zhou, X. Huang and H. Hao, Chem.–Euro. J., 2023, 29, e202202598 CrossRef CAS.
- W. Zhong, J. Zhang, Y. Lin, S. Li, Y. Yang, W.-J. Wang, C. Si, F. E. Kühn, Z. Zhao, X.-M. Cai and B. Z. Tang, Chem. Sci., 2024, 15, 3920–3927 RSC.
- Y.-Y. Tang, J.-C. Liu, Y.-L. Zeng, H. Peng, X.-Q. Huang, M.-J. Yang and R.-G. Xiong, J. Am. Chem. Soc., 2021, 143, 13816–13823 CrossRef CAS PubMed.
- P. Jin, J. Xue, L. Sun, J. Huang, J. Zhang and H. Tian, Nat. Commun., 2019, 10, 4232 CrossRef PubMed.
- M. Yamauchi, K. Yokoyama, N. Aratani, H. Yamada and S. Masuo, Angew. Chem., Int. Ed., 2019, 58, 14173–14178 CrossRef CAS.
- C. Azarias, Š. Budzák, A. D. Laurent, G. Ulrichb and D. Jacquemin, Chem. Sci., 2016, 7, 3763–3774 RSC.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- A. C. Sedgwick, L. L. Wu, H. H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880 RSC.
- W.-Q. Liao, Y.-L. Zeng, Y.-Y. Tang, H. Peng, J.-C. Liu and R.-G. Xiong, J. Am. Chem. Soc., 2021, 143, 21685–21693 CrossRef CAS PubMed.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- W.-Q. Liao, B.-B. Deng, Z.-X. Wang, T.-T. Cheng, Y.-T. Hu, S.-P. Cheng and R.-G. Xiong, Adv. Sci., 2021, 8, 2102614 CrossRef CAS.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, Perth, 2017 Search PubMed.
- X. Wang, S. Ma, B. Zhao and J. Deng, Adv. Funct. Mater., 2023, 33, 2214364 CrossRef CAS.
- X. Yang, X. Gao, Y.-X. Zheng, H. Kuang, C.-F. Chen, M. Liu, P. Duan and Z. Tang, CCS Chem., 2023, 5, 2760–2789 CrossRef CAS.
- H. Shang, Z. Ding, Y. Shen, B. Yang, M. Liu and S. Jiang, Chem. Sci., 2020, 11, 2169–2174 RSC.
- M.-X. Yu, C.-P. Liu, Y.-F. Zhao, S.-C. Li, Y.-L. Yu, J.-Q. Lv, L. Chen, F.-L. Jiang and M.-C. Hong, Angew. Chem., Int. Ed., 2022, 61, e202201590 CrossRef CAS PubMed.
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed.
- V. Gude, P. S. Choubey, S. Das, S. Bhaktha B. N., C. M. Reddy and K. Biradha, J. Mater. Chem. C, 2021, 9, 9465–9472 RSC.
- M. Annadhasan, S. Basak, N. Chandrasekhar and R. Chandrasekar, Adv. Opt. Mater., 2020, 8, 2000959 CrossRef CAS.
- Z. Lu, Y. Zhang, H. Liu, K. Ye, W. Liu and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4299–4303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2373505 (for R-1) and 2373506 (for S-1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05005c |
| This journal is © The Royal Society of Chemistry 2024 |