
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical generation of aryl barium nucleophiles from unactivated barium metal†
Koji
Kubota
 *ab,
Sota
Kawamura
a,
Julong
Jiang
c,
Satoshi
Maeda
bc and
Hajime
Ito
*ab
*ab,
Sota
Kawamura
a,
Julong
Jiang
c,
Satoshi
Maeda
bc and
Hajime
Ito
*ab
aDivision of Applied Chemistry and Frontier Chemistry Center, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido, Japan. E-mail: kbt@eng.hokudai.ac.jp; hajito@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido, Japan
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan
First published on 1st October 2024
Abstract
Organobarium reagents are of interest as homologues of the Grignard reagents based on organomagnesium compounds due to their unique reactivity as well as regio- and stereoselectivity. However, reactions involving organobarium reagents are less developed in comparison to reactions involving Grignard reagents due to the lack of a simple and economical synthetic method and their high reactivity. To the best of our knowledge, there is no established method for the direct synthesis of organobarium compounds from commercially available bulk barium metal and organic halides. So far, the generation of organobarium compounds usually requires the use of activated barium (Rieke barium), which significantly reduces the practical utility of organobarium reagents and hinders the development of new organobarium-mediated transformations. Here, we present a mechanochemical strategy based on ball-milling that facilitates the direct generation of various aryl barium nucleophiles from commercially available unactivated barium metal and aryl halides without complicated pre-activation processes. Our simple mechanochemical protocol allows the rapid development of novel carbon–silicon-bond-forming reactions with hydrosilanes mediated by aryl barium nucleophiles; importantly, these reactions are difficult to achieve using other Grignard-type carbon nucleophiles. To the best of our knowledge, this is the first example of a nucleophilic substitution reaction involving an aryl barium species. Furthermore, this mechanochemical strategy established the first example of a nucleophilic addition to a carbonyl compound involving an aryl barium nucleophile. Preliminary theoretical calculations using the artificial force-induced reaction (AFIR) method to reveal the reaction mechanism of the hydrosilane arylation are also described.
Introduction
Since the pioneering study on the preparation of allyl barium species and their use in the α-selective allylation of carbonyl compounds by Yamamoto and Yanagisawa in 1991, organobarium nucleophiles have attracted substantial attention in synthetic chemistry and natural-product synthesis. Their unique reactivity as well as regio- and stereoselectivity offer several advantages over Grignard reagents prepared from magnesium, which, like barium, is an alkaline-earth-group element.1–3 Organobarium nucleophiles are typically prepared via the direct insertion of barium metal into a carbon–halogen bond (direct synthesis; Scheme 1A).2 However, due to the relatively high atomization energy of barium, such direct insertions are fundamentally challenging,4 and direct reactions with commercial barium metal are usually unsuccessful (Scheme 1A). Instead, the preparation of activated barium metal (Rieke barium) under harsh conditions using alkali metals is required (Scheme 1A).5 Unfortunately, this requirement greatly reduces the practical utility of organobarium nucleophiles. These synthetic difficulties have most likely hampered more extensive investigations into the reactivity of barium-based carbon nucleophiles, and, as a result, their basic reactivity may have remained underappreciated. Although the first synthesis of phenyl barium iodide was reported by Gilman and Schulze almost a century ago,6 systematic explorations of the reactivity of aryl barium nucleophiles toward electrophiles have not yet been reported.2,7 Therefore, the development of an operationally simple protocol using commercial barium metal would be highly desirable to promote its widespread synthetic applications and enable the rapid development of new reactions that involve organobarium nucleophiles. | ||
| Scheme 1 Generation of barium-based heavy Grignard reagents via the direct insertion of barium metal into carbon–halogen bonds. | ||
Recently, mechanochemical strategies using ball-milling techniques have attracted significant attention as alternatives to conventional solution-based approaches.8 The advantages of ball-milling include the reduced usage of harmful organic solvents, faster reaction kinetics, and significantly increased operational simplicity. Recent studies have also revealed that strong mechanical agitation during the ball-milling process can activate zero-valent bulk metals, thus facilitating the direct metalation of organic halides.9–15 In the area of the mechanical activation of alkaline earth metals, the groups of Harrowfield,11 Birke,12 Hanusa,13 Bolm,14 and Kananovich,15 as well as our own group,10c have independently reported the mechanochemical synthesis of Grignard reagents via the direct insertion of magnesium metal into organic halides. More recently, our group has demonstrated that ball-milling allows the generation of heavy Grignard reagents based on calcium from aryl halides and unactivated calcium metal, which is unreactive under solution conditions.10e Importantly, neither pre-activation processes nor the preparation of Rieke metals is required in these cases. These recent achievements inspired us to consider the mechanochemically driven generation of organobarium reagents via direct synthesis from unactivated commercial barium metal, which is sparingly reactive in solution (Scheme 1B). Based on the relative atomization energies of alkaline-earth metals, the activation of barium metal (atomization enthalpy ≈ 182 kJ mol−1) is more challenging than that of magnesium (∼146 kJ mol−1) or calcium (∼178 kJ mol−1) metal.4 However, we envisaged that the strong mechanical impact during ball-milling could allow sufficient surface activation to overcome the high atomization energy of barium metal and thus enable the direct generation of barium-based heavy Grignard reagents without complicated operating conditions (Scheme 1B).
Results and discussion
As a proof-of-concept study, we focused on the mechanochemically driven generation of aryl barium species from 4-iodo-1,1′-biphenyl (1a) (1.0 equiv.) and commercial barium metal (Aldrich, >99.99% purity; 1.0 equiv.), using a Retch MM400 mixer mill (5 mL stainless-steel milling jar with two 10 mm diameter stainless-steel balls; Scheme 2A). The reaction of 1a was evaluated by monitoring the formation of the protonation product (2a) by treatment with aqueous HCl (1.0 M) (Scheme 2A). After an optimization study (for details, see the ESI†), we found that the addition of 2.0 equiv. of tetrahydrofuran (THF) was crucial to facilitate the formation of the organobarium nucleophile under mechanochemical conditions; the corresponding protonation product (2a) was obtained in 53% yield. Based on a previous study on the isolation of THF-coordinated aryl barium species by Westerhausen et al.,7a we anticipated that THF might enhance the thermodynamic stability of the organobarium compound by coordinating to the metal center. However, the fact that full conversion of 1a was observed despite the moderate yield of 2a suggests significant decomposition of the organobarium species during the reaction. We found that reactions with aryl bromides and chlorides resulted in poor conversion of the starting materials (for details, see the ESI†). To confirm the effectiveness of mechanical activation via ball-milling, we carried out a reaction between barium metal and 1a in THF at room temperature (Scheme 2B). Under solution conditions, the formation of 2a was not detected, and a large amount of unreacted 1a remained (>95% recovery) (Scheme 2B). This result indicates that the strong mechanical agitation provided by ball-milling is crucial for the activation of barium metal,9,10 to enable the subsequent generation of the corresponding aryl barium species. | ||
| Scheme 2 Mechanochemically driven generation of aryl barium nucleophiles. a For details of the reaction conditions, see the ESI.† | ||
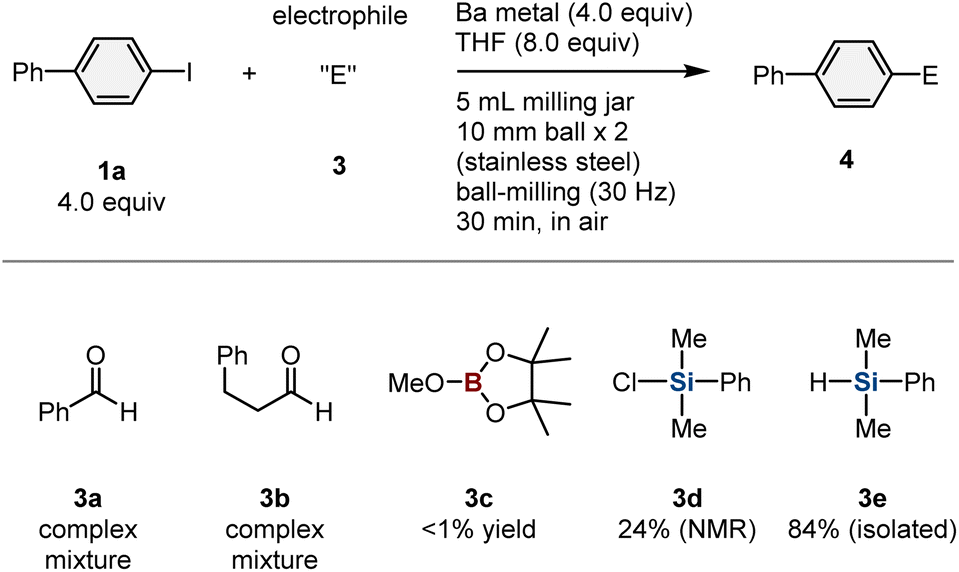
Having demonstrated the ability of this mechanochemical protocol to rapidly generate aryl barium nucleophiles from unactivated barium metal, we next investigated the development of new reactions using these nucleophiles. We hoped that the rapid screening approach enabled by the mechanochemical method would allow the discovery of new reactions with aryl barium nucleophiles.10e First, a series of mechanochemical reactions with various electrophiles was evaluated (Table 1). When benzaldehyde (3a) or an aliphatic aldehyde (3b) was used as an in situ trapping reagent for the formed aryl barium nucleophile, the formation of the desired nucleophilic-addition products was not observed. Instead, a reduction of the carbonyl group or a Tishchenko reaction proceeded, probably triggered by reactions between the carbonyl compounds and barium metal. To prevent these undesired side reactions, we used reduction-resistant trialkoxyboron reagent 3c as the electrophile,16 albeit that the desired borylation product was not detected. However, we discovered that the use of chlorodimethylphenyl silane (3d) afforded the corresponding silylation product (4a) in 24% yield. The low yield of 4a is most likely due to the competing undesired reduction of a Cl–Si bond by barium metal.17 Therefore, we then tested dimethylphenylsilane (3e), which is significantly less reactive than the chlorosilane under reductive conditions, and 4a was obtained in 84% yield.18 Notably, this is the first example of a nucleophilic substitution reaction involving an aryl barium species.2,7
| a Conditions: 3 (0.25 mmol), 1a (1.0 mmol), Ba metal (1.0 mmol), and THF (2.0 mmol) in a stainless-steel milling jar (5 mL) with two stainless-steel balls (10 mm). |
|---|
|
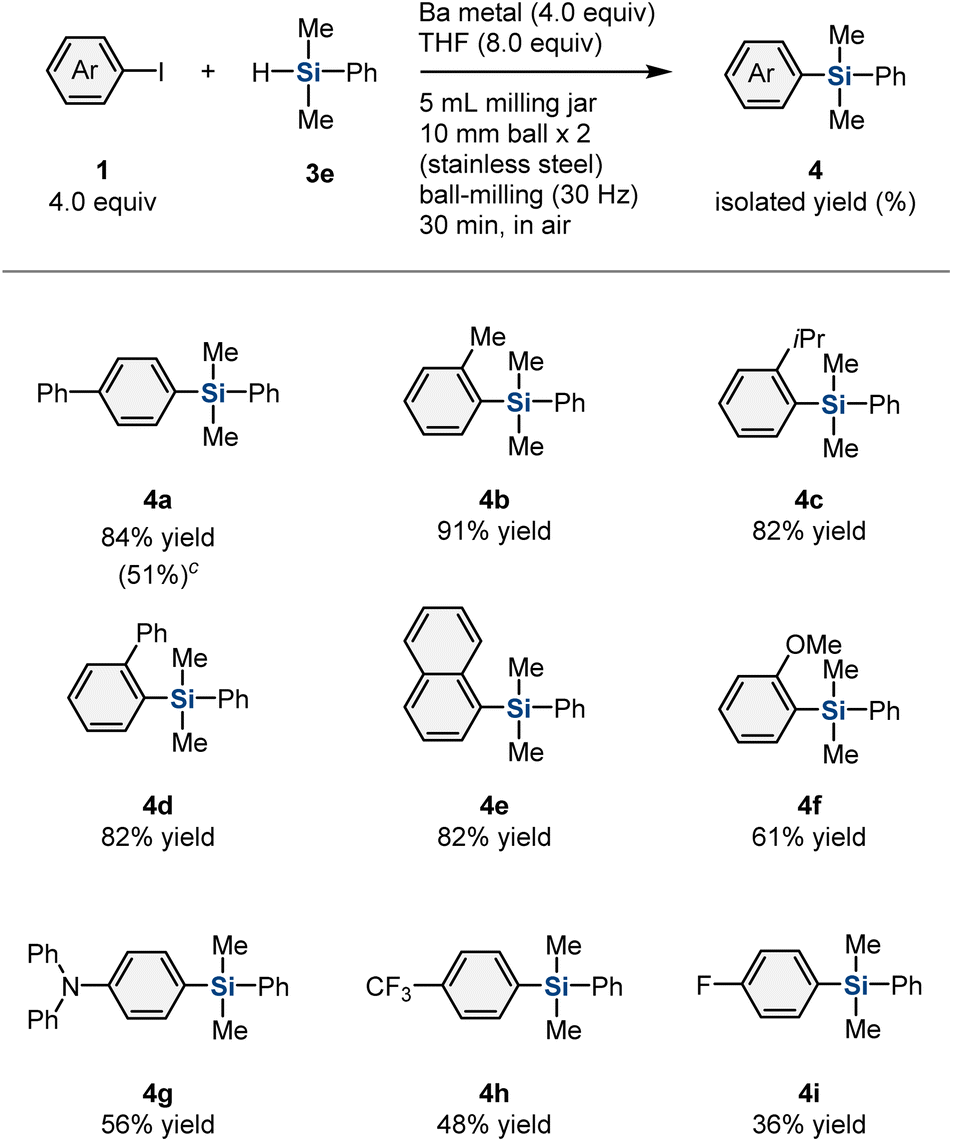
Next, the scope of aryl iodides for the mechanochemically driven generation of aryl barium nucleophiles was investigated (Table 2). Substrates with ortho-substituents, such as a methyl (1b), i-propyl (1c), or phenyl (1d) group, furnished the desired silylation products (4b–4d) in high yield (82–91%). 1-Iodonaphthalene (1e) was also converted into the corresponding product (4e) in good yield (82%). Substrates with an electron-donating group, such as a methoxy (1f) or amino (1g) group, were tolerated and furnished the desired products (4f and 4g) in moderate yield (61% and 56%, respectively). The reactions of aryl iodides with an electron-withdrawing group, such as trifluoromethyl (1h) or fluorine (1i), afforded the corresponding products (4h and 4i) in low-to-moderate yield (48% and 36%, respectively). Substrates 1h and 1i were fully consumed; the relatively low yield of 4h and 4i is probably due to the lower nucleophilicity of the corresponding aryl barium species. We also carried out the silylation reaction of 4-bromobiphenyl (1j) under mechanochemical conditions, which afforded the desired silylation product (4a) in moderate yield (51%).
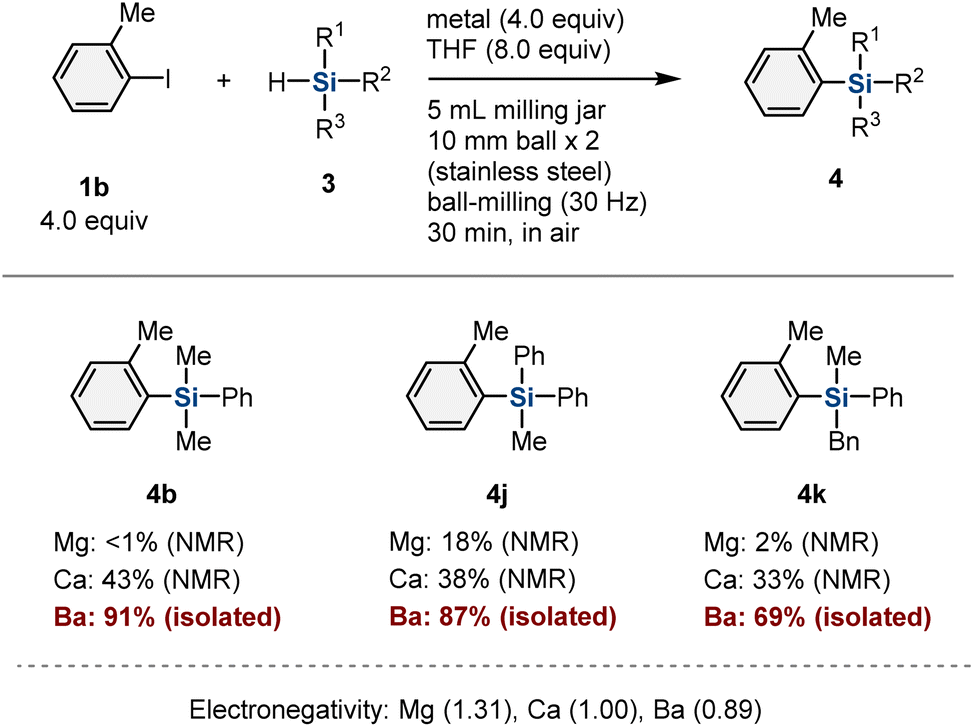
Next, we turned our attention to the scope of applicable hydrosilane reagents (Table 3). We found that trialkyl-substituted hydrosilanes (3g–3i) are applicable to this mechanochemical protocol, and the desired products (4k–4m) were obtained in moderate-to-good yield (69%, 43%, and 46% yield, respectively). Trimethylsilyl-substituted hydrosilane (3j) furnished the corresponding product (4n) in 17% yield.

To confirm that this reactivity is unique to organobarium species, we conducted the mechanochemical reaction using other alkaline-earth metals (Table 4). The use of magnesium instead of barium showed poor results (4b: <1%; 4j: 18%; 4k: 2%).10c,11–15 The reactions in the presence of calcium metal provided the products in much lower yield (4b: 43%; 4j: 38%; 4k: 33%) than those in the presence of barium metal (4b: 88%; 4j: 87%; 4k: 69%).10e Considering the relative electronegativity values of these metals (Table 4), these results suggest that the higher nucleophilicity of the aryl barium nucleophiles compared to those of the other Grignard-type reagents facilitates the substitution of hydrosilanes to form silylation products.
A reaction using Rieke barium under solution-based conditions was also performed for comparison with the mechanochemical method (Scheme 3).5 The Rieke method using lithium biphenylide was applied to generate the corresponding aryl barium species from 1b, and the desired silylation product (4b) was obtained in 56% yield (Scheme 3). This result suggests that barium-based heavy Grignard reagents (Ar–BaX) similar to those present in solution are likely generated under mechanochemical conditions and subsequently react with hydrosilanes to form the silylation products.
 | ||
| Scheme 3 Solution-based reaction using Rieke barium. a For details of the reaction conditions, see the ESI.† | ||
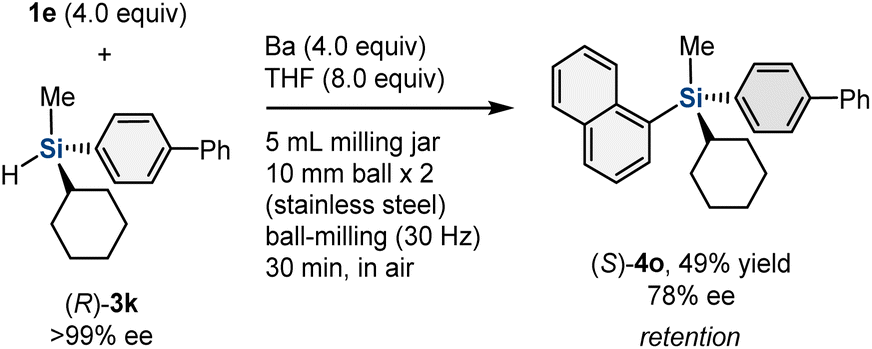
To obtain mechanistic insight into the mechanochemically driven organobarium-mediated silylation, we investigated the reaction with chiral hydrosilane (R)-3k (>99% ee) (Scheme 4). (R)-3k was prepared via preparative chiral high-performance liquid chromatography (HPLC) separation.19 The reaction between 1e and (R)-3k in the presence of barium metal under mechanochemical conditions afforded (S)-4o with 78% ee. Although the stereospecificity is not perfect, the reaction mainly proceeded with retention of the configuration of the silicon stereogenic center, excluding a simple SN2 type mechanism.
 | ||
| Scheme 4 Mechanochemically driven hydrosilane arylation with chiral hydrosilane 3k. a For details of the reaction conditions, see the ESI.† | ||
To gain deeper insight into the reaction mechanism, we conducted a preliminary computational study using the artificial force-induced reaction (AFIR) method.20 Unlike conventional density-functional-theory (DFT) calculations, in which proposed structures and mechanisms are required, the AFIR method allows the comprehensive and unbiased exploration of chemical structures and reaction pathways.20 Since organobarium compounds often form a dimeric complex,21 we first studied the monomeric and dimeric Ar–BaI (Ar = o-tolyl) species, as well as their reaction with hydrosilane. For the monomeric Ar–BaI(thf)2 species, the sampling calculation using the SC-AFIR method revealed 1856 possible structures, while 3729 possible structures were suggested for the dimeric species. Further calculations confirmed that the dimeric species is significantly more stable than the monomeric one (Fig. 1A). The most stable dimeric Ar–BaI geometry was then subjected to reaction with Me2PhSiH. In the calculated mechanism, when the silane approaches the dimeric Ar–BaI species, it immediately becomes a ligand on the barium species (Fig. 1A). In the optimized structure of intermediate I, Me2PhSiH coordinates to two barium atoms simultaneously, with its phenyl ring acting as a π-ligand to form a half-sandwich complex and its hydride forming a hydrogen-bridged bond with another barium center.21 Moreover, the hydride is extracted in a unique way via a five-coordinate silicon species (cf. the structures of II and TS1). This is indeed not an SN2-type reaction, and retention of the configuration of the silicon stereogenic center is assured. A further NBO analysis clearly indicated that the 3d-orbital of the Si atom plays a critical role in forming a penta-coordinated Si complex and therefore allows this unique substitution pathway (for details, see the ESI†). Importantly, these results are consistent with the experimental observations discussed in Scheme 4. In addition, when the silicon–hydrogen bond is completely cleaved viaTS1, the hydride becomes an H-bridge connecting both barium atoms, and the iodide-bridged bonds no longer exist. Interestingly, once the new silicon–carbon bond is formed, the tolyl moiety then becomes another π-ligand to form a half-sandwich complex with a barium center (cf. the structure of III), suggesting very unique bonding behavior of the barium atom. It is noteworthy that similar single-crystal X-ray structures have already been reported.22
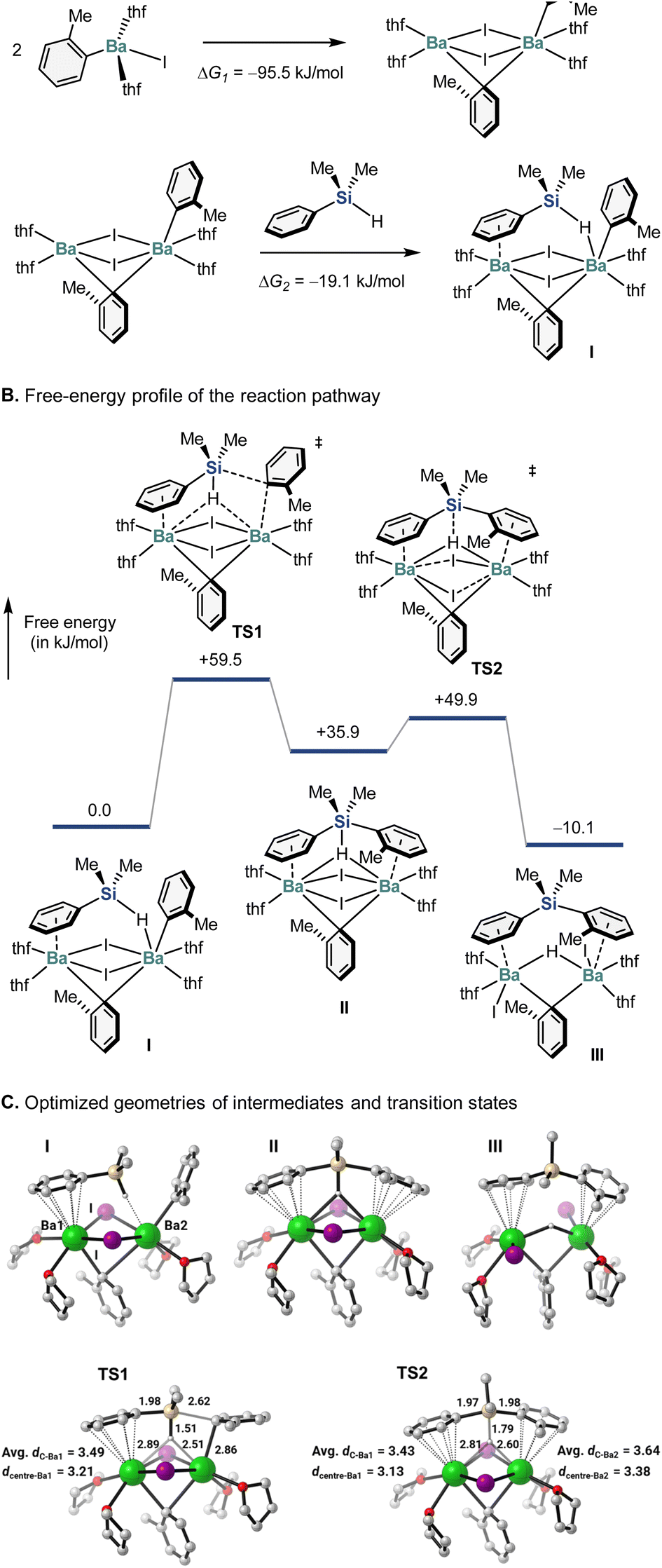 | ||
| Fig. 1 Theoretical calculations (B3LYP-D3/6-311G(d,p)-SDD) of the aryl-barium-mediated hydrosilane arylation. Bond lengths are given in Å. | ||
Encouraged by the success of the silylation reactions, we furthermore explored the development of new carbon–carbon-bond-forming reactions using the mechanochemically generated aryl barium species. After a quick re-screening of electrophiles, we discovered that the reaction of 1a with diaryl ketone 5a proceeded smoothly to give the desired tertiary alcohol 6a in 83% isolated yield (>99% 1H NMR yield) (Scheme 5). To the best of our knowledge, this is the first example of a nucleophilic addition to a carbonyl compound involving an aryl barium nucleophile. Notably, other possible byproducts such as a pinacol coupling product 7a and the simple ketone-reduction product 8a, which can be generated through a single-electron reduction with barium metal, were not detected in the crude reaction mixture. This Barbier-type arylation of diaryl ketones is a difficult transformation when magnesium metal is used as a reductant due to the formation of these competing byproducts. Indeed, we found that the use of magnesium metal instead of barium metal under mechanochemical conditions resulted in the formation of a mixture of these products (Scheme 5). This result highlights the unique and distinctive reactivity of barium metal under the applied mechanochemical conditions.
 | ||
| Scheme 5 Barbier-type ketone arylation involving mechanochemically generated aryl barium species. a For details of the reaction conditions, see the ESI.† | ||
Next, the substrate scope regarding the ketone arylation was explored (Table 5). The mechanochemically generated aryl barium nucleophile reacted with various diaryl ketones (5a–5e) to deliver the desired alcohols (6a–6e) in good to excellent yield (69–94%). Reactions using sterically hindered aryl iodides (1b and 1c) proceeded smoothly to give the corresponding addition products (6f and 6g) in good yield (72% and 66% yield, respectively). It is important to note that the reaction of sterically hindered 1c in the presence of magnesium metal afforded the desired product 6g in poor yield (31%) and significant amounts of other byproducts were formed (pinacol coupling byproduct 7g: 24%; reduction byproduct 8g: 22%; for details, see the ESI†).

To demonstrate the practical utility of this method, we also investigated a scaled-up reaction (Scheme 6). For that purpose, a ketone-arylartion reaction was conducted on the 2.5 mmol scale in a 10 mL stainless-steel jar with two 10 mm stainless-steel balls. The desired product (6a) was obtained in 77% isolated yield, which is comparable to the yield obtained from the reactions on the smaller scale. This result emphasizes the practical utility of this mechanochemical protocol.
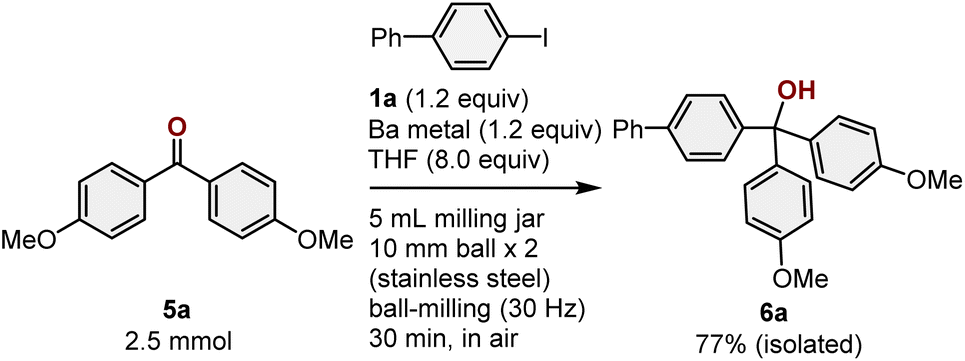 | ||
| Scheme 6 Scaled-up reaction of 5a. a For details on the reaction conditions, see the ESI.† | ||
Conclusions
In summary, we have demonstrated a mechanochemical strategy using ball-milling for the direct generation of aryl barium nucleophiles via the reaction between aryl halides and commercial unactivated barium metal. Importantly, this method does not require complicated pre-activation processes. Using this simple protocol, we discovered a carbon–silicon-bond-forming reaction mediated by an aryl barium species with hydrosilanes; such reactions are rather difficult to achieve using conventional Grignard reagents. To the best of our knowledge, this is the first example of a nucleophilic substitution reaction involving an aryl barium species. In addition, we developed a barium-mediated Barbier-type arylation of diaryl ketones, which is not an easy transformation when magnesium metal is used due to the competing formation of byproducts such as a pinacol coupling product and the simple ketone-reduction product. The details of the reaction mechanism for the mechanochemically driven hydrosilane arylation were discussed based on theoretical calculations using the artificial force-induced reaction (AFIR) method. Further studies to apply this mechanochemical strategy to the generation of allyl-, propargylic-, benzyl-, and simple alkyl barium species are currently in progress in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. K. and H. I. conceived and designed the study. K. K., J. J., S. M., and H. I. co-wrote the paper. S. K. performed the chemical experiments and analyzed the data. J. J. and S. M. conducted the theoretical study. All authors discussed the results and the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) via KAKENHI grants 24H00453, 24H01050, 24H01832, 22H00318, and 22K18333; by the JST via CREST grant JPMJCR19R1; by FOREST grant JPMJFR201I; as well as by the Institute for Chemical Reaction Design and Discovery (ICReDD), which was established by the World Premier International Research Initiative (WPI), MEXT, Japan. We thank Mr Keisuke Kondo for his help in cross-checking experiments. We sincerely thank the reviewers for their many constructive comments in revising this paper.Notes and references
- For pioneering studies on the preparation of allyl barium species and their use in organic synthesis, see: (a) A. Yanagisawa, S. Habaue and H. Yamamoto, J. Am. Chem. Soc., 1991, 113, 8955 CrossRef CAS; (b) A. Yanagisawa, S. Habaue, K. Yasue and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 6130 CrossRef CAS; (c) A. Yanagisawa, K. Ogasawara, K. Yasue and H. Yamamoto, Chem. Commun., 1996, 367 RSC; (d) K. Yasue, A. Yanagisawa and H. Yamamoto, Bull. Chem. Soc. Jpn., 1997, 70, 493 CrossRef CAS; (e) A. Yanagisawa, Y. Yamada and H. Yamamoto, Synlett, 1997, 9, 1090 CrossRef; (f) F. Raeppel and D. Heissler, Synlett, 2005, 16, 2534 Search PubMed; (g) A. Yanagisawa, S. Yamafuji and T. Sawae, Synlett, 2016, 27, 2019 CrossRef CAS; (h) A. Yanagisawa, T. Jitsukawa and K. Yoshida, Synlett, 2013, 24, 635 CrossRef CAS.
- For a comprehensive review on organic reactions promoted by barium reagents, see: A. Yanagisawa and K. Yoshida, Synlett, 2011, 2929 CrossRef CAS.
- For selected studies on propargylation and benzylation with barium reagents, see: (a) A. Yanagisawa, S. Okitsu and T. Arai, Synlett, 2005, 1679 CrossRef CAS; (b) A. Yanagisawa, T. Suzuki, T. Koide, S. Okitsu and T. Arai, Chem.–Asian J., 2008, 3, 1793 CrossRef CAS PubMed; (c) A. Yanagisawa, T. Sawae, S. Yamafuji, T. Heima and K. Yoshida, Synlett, 2015, 26, 1073 CrossRef CAS; (d) A. Yanagisawa, T. Koide and K. Yoshida, Synlett, 2010, 1515 CrossRef CAS.
- (a) J. S. Alexander and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2002, 2002, 2761 CrossRef; (b) M. Westerhausen, M. Gärtner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem.–Eur. J., 2007, 13, 6292 CrossRef CAS PubMed.
- T. C. Wu, H. Xiong and R. D. Rieke, J. Org. Chem., 1990, 55, 5045 CrossRef CAS.
- H. Gilman and F. Schulze, Bull. Soc. Chim., 1927, 41, 1333 CAS.
- (a) J. Langer, M. Gärtner, R. Fischer, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2007, 10, 1001 CrossRef CAS; (b) S.-O. Hauber, F. Lissner, G. B. Deacon and M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 5871 CrossRef CAS PubMed.
- For selected reviews on the use of ball-milling for organic synthesis, see: (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (b) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (c) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13 CrossRef CAS PubMed; (d) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007 CrossRef PubMed; (e) T.-X. Métro, J. Martinez and F. Lamaty, ACS Sustainable Chem. Eng., 2017, 5, 9599 CrossRef; (f) T. K. Achar, A. Bose and P. Mal, Beilstein J. Org. Chem., 2017, 13, 1907 CrossRef CAS PubMed; (g) O. Eguaogie, J. S. Vyle, P. F. Conlon, M. A. Gîlea and Y. Liang, Beilstein J. Org. Chem., 2018, 14, 955 CrossRef CAS PubMed; (h) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC; (i) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435 RSC; (j) C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285 CrossRef CAS PubMed; (k) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018 CrossRef PubMed; (l) K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066 CrossRef CAS; (m) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344 CrossRef CAS; (n) J. A. Leitch and D. L. Browne, Chem.–Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed; (o) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246 CrossRef CAS; (p) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145 CrossRef CAS PubMed; (q) V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51 CrossRef CAS PubMed.
- For a tutorial review on the mechanical activation of zero-valent metals, see: A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763 CrossRef.
- For selected examples of mechanical activation of zero-valent metals, see: (a) Q. Cao, J. L. Howard, E. Wheatley and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 11339 CrossRef CAS PubMed; (b) W. I. Nicholson, J. L. Howard, G. Magri, A. C. Seastram, A. Khan, R. R. A. Bolt, L. C. Morrill, E. Richards and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 23128 CrossRef CAS PubMed; (c) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed; (d) V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed; (e) P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 CrossRef CAS PubMed; (f) R. Takahashi, P. Gao, K. Kubota and H. Ito, Chem. Sci., 2023, 14, 499 RSC; (g) N. Davison, J. A. Quirk, F. Tuna, D. Collison, C. L. McMullin, H. Michaels, G. H. Morritt, P. G. Waddell, J. A. Gould, M. Freitag, J. A. Dawson and E. Lu, Chem, 2023, 9, 576 CrossRef CAS; (h) K. Fujishiro, Y. Morinka, Y. Ono, T. Tanaka, L. T. Scott, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 8163 CrossRef CAS PubMed.
- J. M. Harrowfield, R. J. Hart and C. R. Whitaker, Aust. J. Chem., 2001, 54, 423 CrossRef CAS.
- V. Birke, C. Schütt, H. Burmeier and W. K. L. Ruck, Fresenius Environ. Bull., 2011, 20, 2794 CAS.
- I. R. Speight and T. P. Hanusa, Molecules, 2020, 25, 570 CrossRef CAS PubMed.
- V. S. Pfennig, R. C. Villella, J. Nikodemus and C. Bolm, Angew. Chem., Int. Ed., 2022, 61, e202116514 CrossRef CAS PubMed.
- J. V. Nallaparaju, T. Nikonovich, T. Jarg, D. Merzhyievski, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2023, 62, e202305775 CrossRef PubMed.
- M. Fukuzawa, F. Takahashi, K. Nogi, T. Sasamori and H. Yorimitsu, Org. Lett., 2020, 22, 2303 CrossRef PubMed.
- The generation of the corresponding hydrosilane was confirmed by NMR analysis; for details, see the ESI†.
- H.-J. Lee, C. Kwak, D.-P. Kim and H. Kim, Green Chem., 2021, 23, 1193 RSC.
- X. Wang, C. Feng, J. Jiang, S. Maeda, K. Kubota and H. Ito, Nat. Commun., 2023, 14, 5561 CrossRef CAS PubMed.
- (a) S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC; (b) S. Maeda, Y. Harabuchi, M. Takagi, T. Taketsugu and K. Morokuma, Chem. Rec., 2016, 16, 2232 CrossRef CAS PubMed.
- M. Wiesinger, B. Maitland, C. Färber, G. Ballmann, C. Fischer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 16654 CrossRef CAS PubMed.
- (a) A. Uresk, N. J. O'Neil, Z. Zhou, Z. Wei and M. A. Petrukhina, J. Organomet. Chem., 2019, 897, 57 CrossRef CAS; (b) P. M. Chapple and Y. Sarazin, Eur. J. Inorg. Chem., 2020, 2020, 3321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05361c |
| This journal is © The Royal Society of Chemistry 2024 |