
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClosed-loop chemically recyclable covalent adaptive networks derived from elementary sulfur†
Chen-Yu
Shi
,
Xiao-Ping
Zhang
,
Qi
Zhang
,
Meng
Chen
,
He
Tian
 and
Da-Hui
Qu
*
and
Da-Hui
Qu
*
Key Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, P. R. China. E-mail: dahui_qu@ecust.edu.cn
First published on 26th September 2024
Abstract
The development of sulfur-rich polymers derived from elementary sulfur provides an innovative approach to industrial waste valorization. Despite significant advancements in polymerization techniques and promising applications beyond traditional polymers, polysulfide networks are still primarily stabilized by diene crosslinkers, forming robust C–S bonds that hinder the degradation of sulfur-based polymers. In this study, the anionic ring-opening copolymerization of chemically homologous S8 and cyclic disulfides was explored to yield robust sulfur-rich copolymers with high molecular weight. The incorporation of polysulfide segments not only efficiently activated the crosslinked networks for excellent reprocessability and mechanical adaptability but also endowed the resulting copolymer with high optical transparency in the near-infrared region. More importantly, the dynamic disulfide crosslinking sites promoted the chemical closed-loop recyclability of the polysulfide networks via reversible S–S cleavage. This innovative inverse vulcanization strategy utilizing dynamic disulfide crosslinkers offers a promising pathway for the advanced applications and upcycling of high-performance sulfur-rich polymers.
Introduction
To explore new sustainable strategies beyond traditional polymers synthesized from limited petrochemical resources, surplus elementary sulfur (S8) from petroleum refining has emerged as a feedstock monomer for the production of innovative sulfur-rich polymers with high value-added applications, such as infrared optics,1–6 energy storage,7,8 and heavy metal adsorption.9–11 This class of inorganic sulfur-based polymer chemistry exemplifies an innovative approach to waste valorization and has sparked significant research interest in polymerization strategies, material performance, and diverse applications across multiple fields in the last decade.12–15The initial application of S8 in polymer chemistry can be traced back to the vulcanization of natural rubber, where a small amount of S8 was added as a crosslinking agent to enhance the properties of the polymers.16 This example highlights the feasibility of ring-opening polymerization (ROP) of S8, leading to the formation of polymeric sulfur. However, due to the low ring strain of S8, linear polymeric sulfur is thermodynamically unstable and tends to depolymerize into cyclic S8 monomers via terminal sulfur radical backbite.17 Instead, “inverse vulcanization” has been widely used to produce stable and processable sulfur-based polymers by copolymerizing abundant amounts of S8 with minor diene crosslinkers (Fig. 1A).18
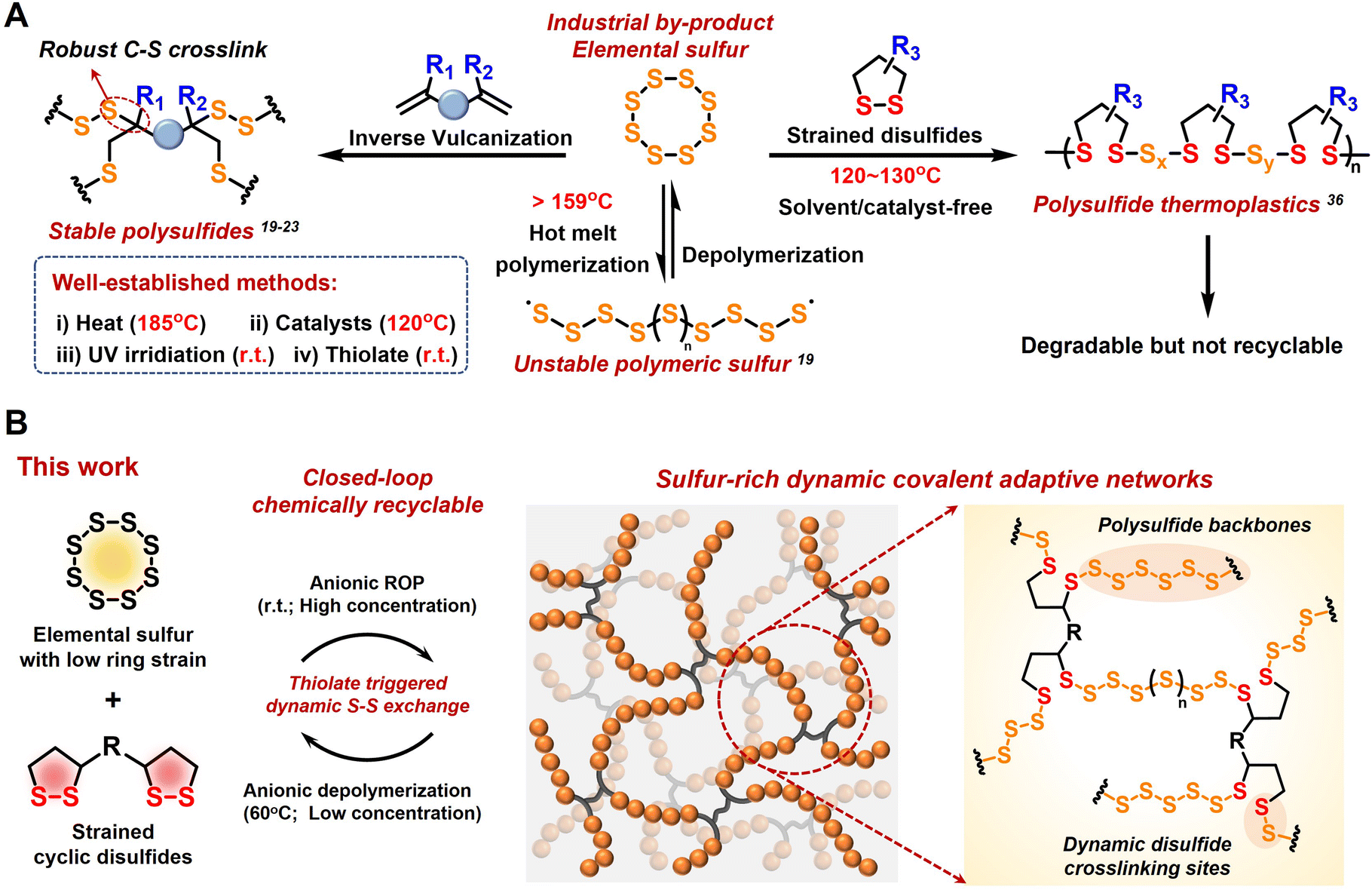 | ||
| Fig. 1 (A) Considerable advances in the “inverse vulcanization” strategies using S8 and dienes for innovative polymeric materials. (B) Schematic representation of the anionic copolymerization route of S8 and cyclic disulfides, yielding sulfur-rich dynamic CANs. The polysulfide backbones and dynamic disulfide crosslinking sites endow the resulting copolymer with reprocessability and chemical recyclability. | ||
Pyun and co-workers reported a groundbreaking method using liquid sulfur as a solventless reaction medium at 185 °C for its direct copolymerization with divinyl monomers, resulting in crosslinked polymeric materials with high sulfur content.19 The addition of catalysts and accelerators can reduce the generation of toxic H2S by lowering the reaction temperature to below 140 °C.20,21 Furthermore, photoinduced inverse vulcanization at room temperature has expanded the range of crosslinkers to low-boiling-point alkenes and alkynes.22 Zhang and co-workers achieved the room-temperature anionic hybrid copolymerization of S8 with acrylate to produce highly crosslinked copolymers containing short polysulfide segments.23 Moreover, thermomechanical and chemical S–S metathesis reactions endow the resulting polysulfide networks with reprocessability.24–26 Recently, Chalker and co-workers have achieved the electrochemical ROP and thermal depolymerization of norbornene-based cyclic trisulfide monomers.27 Besides inverse vulcanization, several approaches to the conversion of sulfur to functional polymers, including the multicomponent polymerization of S8, diamines, and diisocyanides to yield polythioureas and polythioamides,28,29 the polycondensation of S8 with dihalides and dithiols, and the ionic copolymerization of S8 with thiiranes and trithioles, have been reported to generate polysulfides with high sulfur content.30
Despite these considerable advancements in the up-conversion of elementary sulfur to polymeric materials, current strategies primarily focus on the formation of stable crosslinking sites by quenching active sulfur chains (radical or anionic terminals) with dienes.31,32 The formation of robust C–S bonds partially hinders the decrosslinking and depolymerization of sulfur-rich networks, which is theoretically possible due to dynamic S–S exchange and reformation in the polymeric backbones.33 Inspired by the anionic ROP of chemically homologous S8 and episulfides,34,35 cyclic disulfides show great potential as ideal comonomers considering their similar active species to those of S8. Recently, our group reported a novel class of H-crosslinked thermoplastics produced from the copolymerization of S8 and cyclic disulfides. Polysulfide was degraded into cyclic disulfides and sodium sulfide in a basic solution, but it was not recycled back to the original monomers.36 Therefore, new challenges arise in achieving the chemical closed-loop recycling of sulfur-based polymers without compromising the polymer stability or material performance.
Covalent adaptive networks (CANs) with exchangeable dynamic covalent bonds provide an interesting solution to the challenge of irreversible covalent crosslinking in sulfur-rich thermosets.37,38 In this work, we explored the efficient copolymerization of S8 and cyclic disulfides via disulfide-mediated ROP, providing sulfur-rich CANs with variable sulfur content (Fig. 1B). Moreover, the modifiable sidechain of cyclic disulfide, e.g., 1,2-dithiolane, provides a wide functionalization platform for improved performance and versatile applications.39–41 Compared with poly(disulfide) homo-polymeric networks, polysulfide copolymeric networks demonstrate superior dynamics while maintaining their mechanical properties, endowing the resulting material with enhanced machinability due to the highly dynamic S–S exchange within the polysulfide backbones. These sulfur-rich copolymers also have high optical transparency in the near-infrared (NIR) region, expanding their potential applicability as NIR transmissive materials. In addition, the dynamic disulfide crosslinking sites facilitate reversible S–S exchange and cleavage. Thiolate-triggered depolymerization drives the reaction equilibrium toward cyclic monomers, enabling the efficient separation and recovery of S8 and disulfide monomers. This chemical closed-loop recyclability under mild conditions highlights the promising trend of circular sulfur chemistry toward atomic economics and the upcycling pathways for high-performance polysulfide materials derived from S8.
Results and discussion
Structural characterization of linear polysulfide copolymers
First, we utilized the similar reactivities of S8 and cyclic disulfides in their anionic copolymerization, triggered by alkaline thiolate through the disulfide-mediated ROP mechanism.42 The methyl ester of thioctic acid (TAM) (Fig. S1†) served as the model cyclic disulfide comonomer, which was polymerized with S8 in the presence of 0.01 equiv. of phosphazene base (tBu-P1)/benzyl mercaptan (BnSH) (Fig. 2A). The poly(TAM) homopolymer was synthesized as the control sample. After the addition of the thiolate initiator, the reaction mixture quickly turned red and the inorganic S8 solid gradually dissolved in the poor solvent chloroform, which indicated the generation of a living thiolate anion via sulfur ring-opening and subsequent chain growth. The S8 monomer was fully converted when the TAM/S molar ratio x/y exceeded 1/2, yielding poly(TAMx–Sy) copolymers with a high molecular weight of over 10 kDa (Fig. S2†). The further addition of S8 led to large amounts of insoluble species, preventing the formation of longer polysulfide backbones with weak consecutive S–S bonds (Fig. 2B).43 The dramatically reduced molecular weight of the resultant poly(TAM1–S4) (Mn = 1237 Da) (Fig. S3†) was attributed to excess active polysulfide anions acting as initiators in living polymerization.44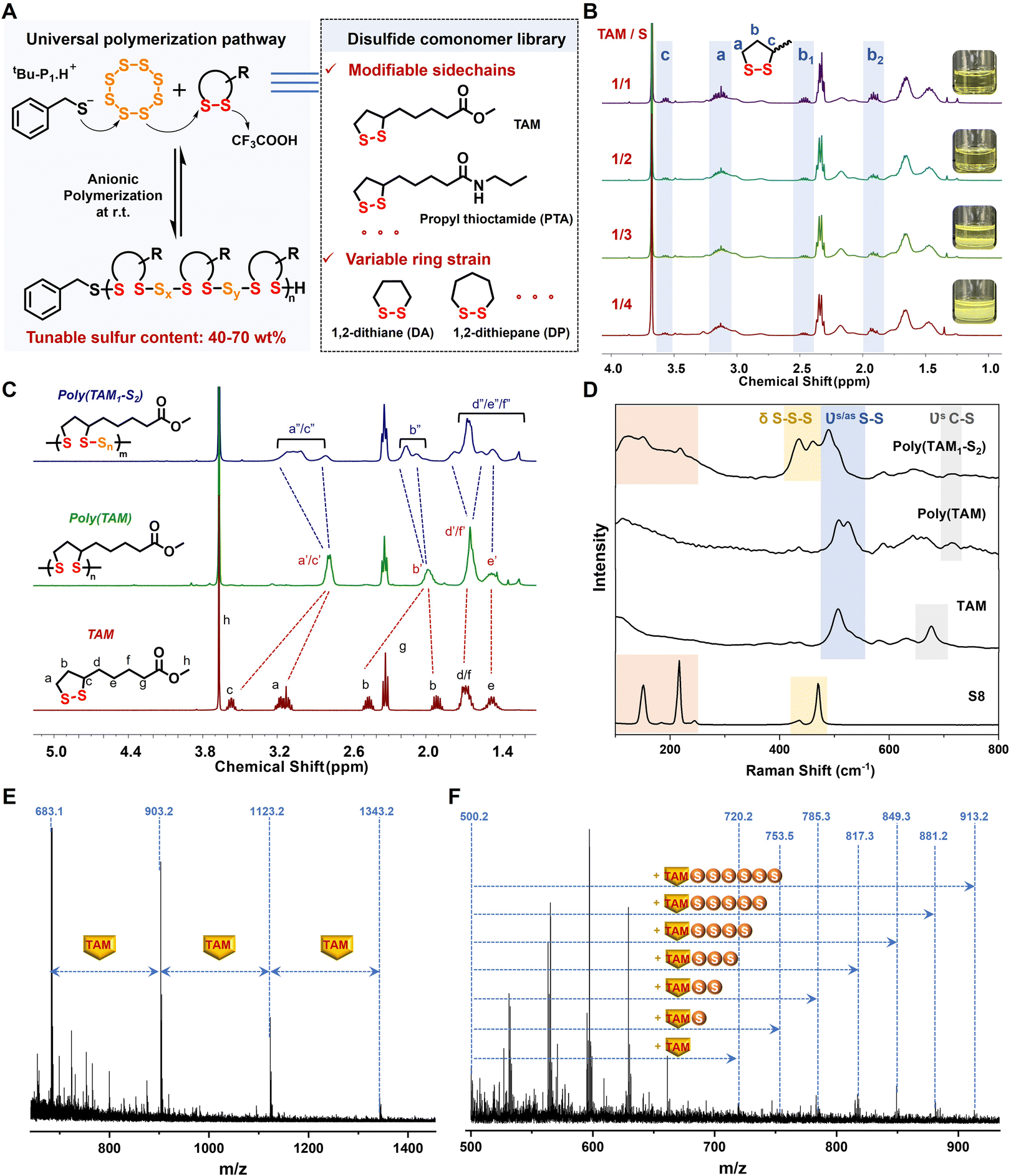 | ||
| Fig. 2 Anionic ROP of linear polysulfides and structural characterization. (A) Schematic representation of thiolate induced polymerization routes of S8 and a series of cyclic disulfides with variable ring strain and sidechains. (B) 1H NMR spectra of poly(TAM–S) copolymers with different feed molar ratios of TAM and S. (C) 1H NMR spectra of the TAM monomer, poly(TAM) and poly(TAM1–S2). (D) Raman spectra of S8, TAM, poly(TAM) and poly(TAM1–S2). MALDI-TOF-MS spectra of (E) poly(TAM) and (F) poly(TAM1–S2). | ||
Spectroscopic characterization was performed to investigate the copolymerization structures, especially that of poly(TAM1–S2), which achieved an optimum monomer conversion of both TAM (∼79%) and S8 (∼100%) (Fig. S4†). As shown in the 1H NMR spectra (Fig. 2C), the broadening and shift of the characteristic proton peaks assigned to the disulfide five-membered ring signified that efficient copolymerization was achieved. Compared with the poly(TAM) homopolymer with discrete disulfide bonding, the further downfield shift of the characteristic proton peaks caused by electron-withdrawing polysulfide segments indicated the successful embedding of sulfur atoms between the ring-opening 1,2-dithiolane, whose intensity was positively correlated with the sulfur content (Fig. S5†). The presence of a linear sulfur-rich chain containing polysulfide blocks was also confirmed by the disappearance of the strong S8 Raman peaks at 151, 216, and 470 cm−1 and the broadened split S–S–S vibration band around 405–530 cm−1 (Fig. 2D and S6†).45 Moreover, the sulfur atom content was quantitatively analyzed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Specifically, the MS peaks of poly(TAM) showed a regular distribution with equal intervals of 220.0 m/z, which corresponded to the repeated TAM units linked by discrete disulfide bonds (Fig. 2E). For poly(TAM1–S2), six extra sulfur units of ca. 32.0 m/z were found, proving that the copolymerized backbones with polysulfide segments contained a maximum of eight sulfur atoms (Fig. 2F). The polysulfide segments lengthened as the feed ratio of S8 increased but shortened in the presence of excess S8 as a polysulfide anion initiator (Fig. S7†), which is in agreement with the low molecular weight of poly(TAM1–S4) (Fig. S3†).
Polymerization kinetics experiments were further conducted to investigate the underlying copolymerization mechanism. As shown in the time-dependent 1H NMR spectra, the TAM monomer content gradually decreased and was nearly consumed after 8 h (conversion >94%) (Fig. S8†). The consistent conversion rates of TAM and S8 indicated similar reactivity, as the characteristic monomer vibration peaks at 475 cm−1 in the corresponding Raman spectra disappeared after 8 h (Fig. S9†). Compared with the fast homopolymerization of TAM (<1.5 h), the relatively longer copolymerization period was ascribed to the low ring strain of S8, which limited the rate of chain propagation.17
To further expand the disulfide comonomer libraries, a series of cyclic disulfides with varied ring strains and side chains including propyl thioctamide (PTA), 1,2-dithiane (DA), and 1,2-dithiepane (DP) were successfully applied in the anion copolymerization reaction with S8 (Fig. 2A). The split peaks at 2.7–3.1 ppm in the 1H NMR spectra were assigned to methylene (a) protons bonded to the polysulfide blocks containing different numbers of sulfur atoms (Fig. S10–S12†). Compared with the five-membered monomer ring, the lower ring strain of six and seven-membered monomer rings caused the polymerization equilibrium to shift toward the monomer side, which may be related to the low molecular weights of the DA- and DP-based copolymers (2–5 kDa) (Fig. S13†).46,47 In summary, the universality of anionic copolymerization was confirmed to yield sulfur-rich copolymers with tunable sulfur content ranging from 40 to 70 wt% (Table S1†).
Bulk properties of crosslinked polysulfide covalent adaptive networks (CANs)
Having demonstrated the efficient anionic ROP of S8 and cyclic disulfide comonomers to yield linear sulfur-rich copolymers, we further designed an esterified bis(1,2-dithiolane) (DTA) (Fig. S14†) as a dynamic disulfide crosslinker to obtain S8-derived polysulfide crosslinked networks (Fig. 3A). The DTA/S8 feed molar ratio was set as 2–4 ([DTA]/[S] = 1/4 to 1/2) to ensure optimal monomer conversion. After the addition of 0.01 equiv. of thiolate initiator, the viscosity of the reaction system gradually increased within 30 min. Overnight curing accompanied by solvent evaporation resulted in yellow transparent polymer films. The copolymers were purified by TFA quenching and solvent immersion for further characterization. According to the elemental analysis (Fig. 3B), the sulfur content of the purified copolymers was 37.4 wt% for poly(DTA1–S2) and 40.2 wt% for poly(DTA1–S4), which was consistent with the theoretical values of 36.3 wt% for poly(DTA1–S2) and 43.0 wt% for poly(DTA1–S4). Furthermore, 24 h solubility tests in organic solvents, including acetone, DMSO, THF, DCM, DMF, CH3CN, and EtOH, demonstrated stable crosslinked networks without any evidence of dissolution or swelling (Fig. S15†). X-ray diffraction (XRD) analysis showed the disappearance of sharp S8 crystalline peaks after polymerization, indicating the amorphous nature of the resulting copolymer without residual monomers (Fig. S16†). In Raman spectra, the deformation S–S–S vibration band around 400–470 cm−1 was ascribed to the polysulfide backbones, whose intensity increased with increasing sulfur content. The C–S vibration band around 610–680 cm−1, corresponding to the disulfide comonomer, confirmed the formation of crosslinked sulfur-rich networks (Fig. S17†). Scanning electron microscopy showed a homogeneous surface morphology of the copolymer films without phase separation or S8 crystalline aggregates, proving the random copolymerization of S8 and DTA in the amorphous networks (Fig. S18†). Therefore, the decomposition temperature (Td) of thermally stable crosslinked poly(DTA1–S4) (Td = 234 °C) was higher than that of linear poly(TAM1–S2) (Td = 205 °C) at the same monomer feed ratio but was slightly lower than that of poly(DTA) (Td = 249 °C) due to the weaker polysulfide backbones (Fig. 3C).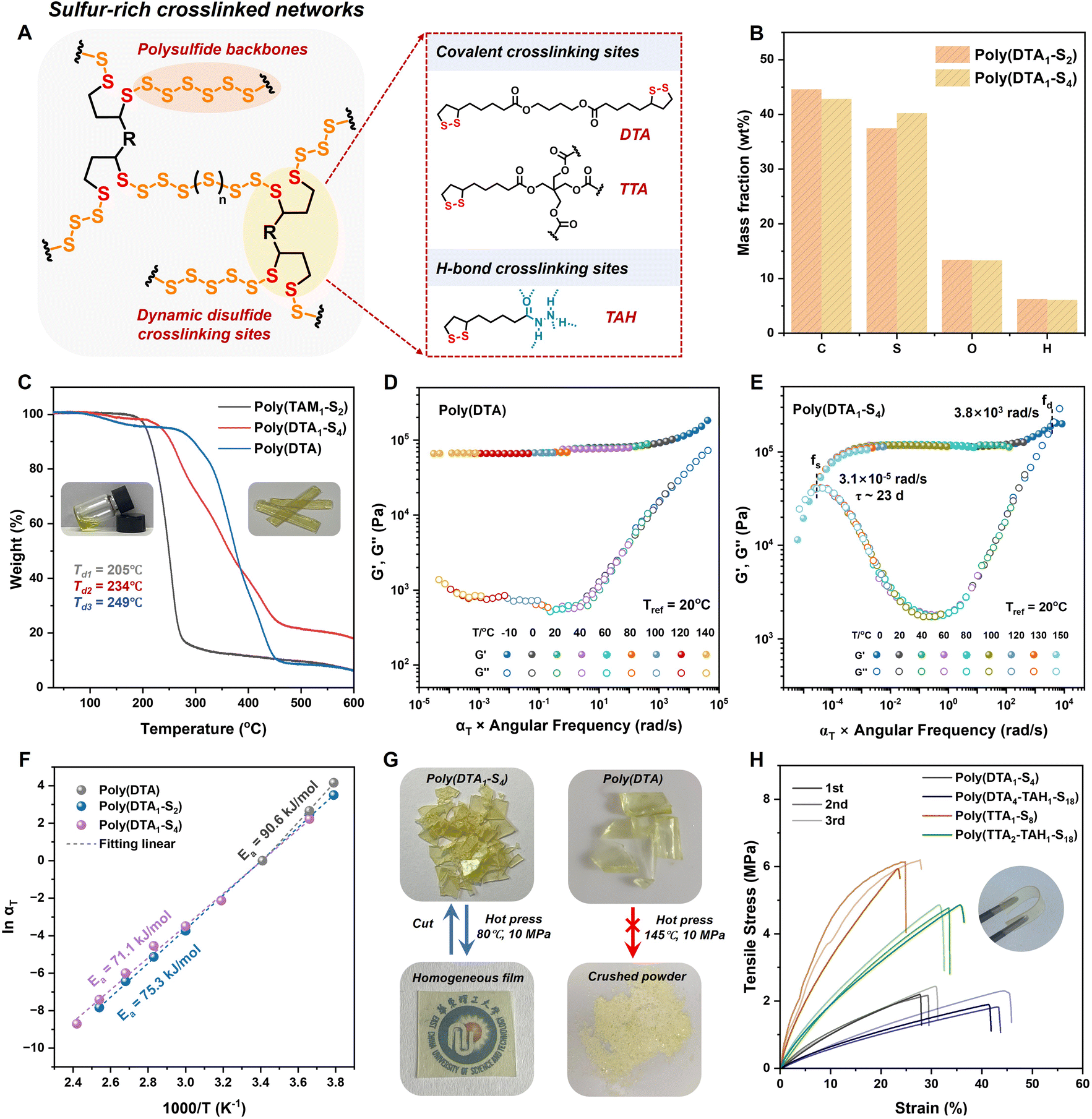 | ||
| Fig. 3 Structure, thermal and mechanical performance characterization of the polysulfide CANs. (A) Structural illustration of the polysulfide crosslinked networks and cyclic disulfide crosslinkers. (B) Elemental analysis of poly(DTA1–S2) and poly(DTA1–S4). (C) TGA of poly(DTA), poly(DTA1–S2) and poly(DTA1–S4). Time–temperature superposition for the frequency sweep plot of G′ and G′′ for (D) poly(DTA) and (E) poly(DTA1–S4). (F) Fitting of temperature-dependent αT for poly(DTA), poly(DTA1–S2) and poly(DTA1–S4) according to the Arrhenius equation. (G) Photographs of hot-press reprocessing of poly(DTA1–S4) and poly(DTA). (H) Strain–stress curves of sulfur-rich crosslinked copolymers. | ||
The bulk properties of sulfur-rich polymers obtained by anionic polymerization were further investigated, as they have been rarely explored despite the mature polymerization mechanism.48 The temperature-dependent rheology curves of the poly(disulfide) homopolymer and polysulfide copolymers showed broad elastic plateaus from 0 to 120 °C (Fig. S19†). This was consistent with the low glass transition temperature (Tg) below room temperature measured by differential scanning calorimetry (Fig. S20†), whose value decreased at higher sulfur content due to the reduced crosslinking density and the presence of flexible chain segments. The above observations proved the thermoset nature of the resulting copolymers with a stable rubbery state. It is worth noting that the loss modulus increased with increasing sulfur content at temperatures over 100 °C due to the motion of active segments and dynamic S–S exchange within the weak polysulfide backbones (Fig. S19†). The controllable network rearrangements were also confirmed by the slow stress relaxation above room temperature (Fig. S21†).
To better understand the room-temperature dynamic behaviors of sulfur-based CANs at different time scales, the rheological master curves (reference temperature, 20 °C) were fitted based on the time–temperature superposition.49 The storage modulus of poly(DTA1–S4) remained constant, ranging from 104 to 10−4 rad s−1, and was almost identical to that of the poly(DTA) homopolymer, demonstrating its satisfactory mechanical strength and creep resistance over long periods (Fig. 3D and E). With the increased sulfur rank in the polymeric backbones, polysulfide-mediated dynamic exchange and backbone cleavage led to an enhanced loss modulus and a final viscous flow state with governed terminal relaxation times, with the τ values of poly(DTA1–S2) and poly(DTA1–S4) reaching about 35 and 23 d, respectively (Fig. 3E and S22†). According to the Arrhenius equation, the fitted apparent activation energies (Ea) of poly(DTA), poly(DTA1–S2), and poly(DTA1–S4) were 90.6, 75.3, and 71.1 kJ mol−1, respectively (Fig. 3F). Considering the thermal and rheology measurements, we inferred that the embedding of weak polysulfide segments into the crosslinked networks efficiently activated the frozen backbones via dynamic bond exchange, endowing the resulting sulfur-rich CANs with synergic mechanical robustness and dynamic reconfigurability.
Reprocessability and mechanical properties of polysulfide CANs
The resulting copolymers exhibited good reprocessability, taking advantage of the dynamic reconfigurable sulfur-rich networks. Therefore, the purified polysulfide copolymer pieces could be reformed via hot-press molding at 80 °C to obtain homogeneous films for further uniaxial tensile tests (Fig. 3G). The mechanical properties of the remolded copolymer retained their original values even after three hot-press cycles (Fig. 3H). The consistent S–S–S vibration bands in the Raman spectra of the reshaped samples confirm their chemical stability and integrity after multiple reprocessing cycles (Fig. S23†). The rearrangement process was mediated by heat-induced sulfur radical exchange, during which the crosslinked networks maintained their integrity without depolymerization (Fig. S24†). Easily activated bond exchange under mild conditions was ascribed to the weak polysulfide backbones, which corresponded to the lower Ea determined using the Arrhenius equation. By contrast, the hot pressing of the brittle poly(DTA) homopolymer even at 145 °C provided only crushed powder due to the dominant covalent backbones with high bond energies (Fig. 3G). Further heating to 160 °C caused the depolymerization of poly(disulfide) networks driven by the thermodynamic equilibrium, resulting in a low-viscosity yellow liquid that failed to recure upon cooling (Fig. S25†).The modifiable sidechain of disulfide crosslinkers provided a wide regulation range for the mechanical properties of sulfur-rich copolymers. Thus, a series of copolymers with the same sulfur content but varied crosslinking sites were designed for uniaxial tensile tests. As shown in the stress–strain curves (Fig. 3H), two-arm disulfide crosslinked poly(DTA1–S4) exhibited a relatively low tensile strength of 2.3 MPa with an elongation of 30%. The use of four-arm disulfide crosslinkers increased the maximum mechanical strength of poly(TTA1–S8) to 6.1 MPa with a slightly lower elongation of 25% due to higher crosslinking density, whose value was comparable to those of diene-crosslinked sulfur-rich polymers.19,23 Thioctic acylhydrazine was further introduced as sacrificial H-bonding crosslinking sites (Fig. S26†),50 resulting in a maximum elongation of over 3000% (Fig. S27†), which expanded their applicability as soft materials.
Infrared optical applications
In addition to enabling dynamic covalent networks with better mechanical adaptability and reprocessability, the incorporation of sulfur heteroatoms effectively reduced IR absorption via substituting C–C and C–H bonds, thus endowing sulfur-based polymers with higher IR transparency and refraction. As shown in the NIR transmittance spectra (Fig. 4A), poly(DTA1–S4) films with varied thicknesses (165–560 μm) all displayed high transmission in the range of 800–2200 nm, which was over 80% in the NIR region (800–1600 nm), comparable to that of previously reported sulfur-based polymer films with the same thickness.1,51 The unavoidable absorption bands around 1700 nm and above 2300 nm were ascribed to the C–H vibrations in disulfide comonomer units. The inherent yellow appearance of the poly(DTA1–S4) film resulted in the reduced transparency in the visible regions (<800 nm), which had no influence on its performance in IR optical applications. Considering the NIR transparency of the poly(DTA1–S4) film, we evaluated its imaging capability as lens materials for NIR cameras under the illumination of a 980 nm laser (Fig. 4B). The image resolution captured through the poly(DTA1–S4) film covered lens was comparable to that obtained through a bare lens, which decreased with increasing surface roughness of polymer films, thus proving its application potential as a candidate for reprocessable NIR optical devices.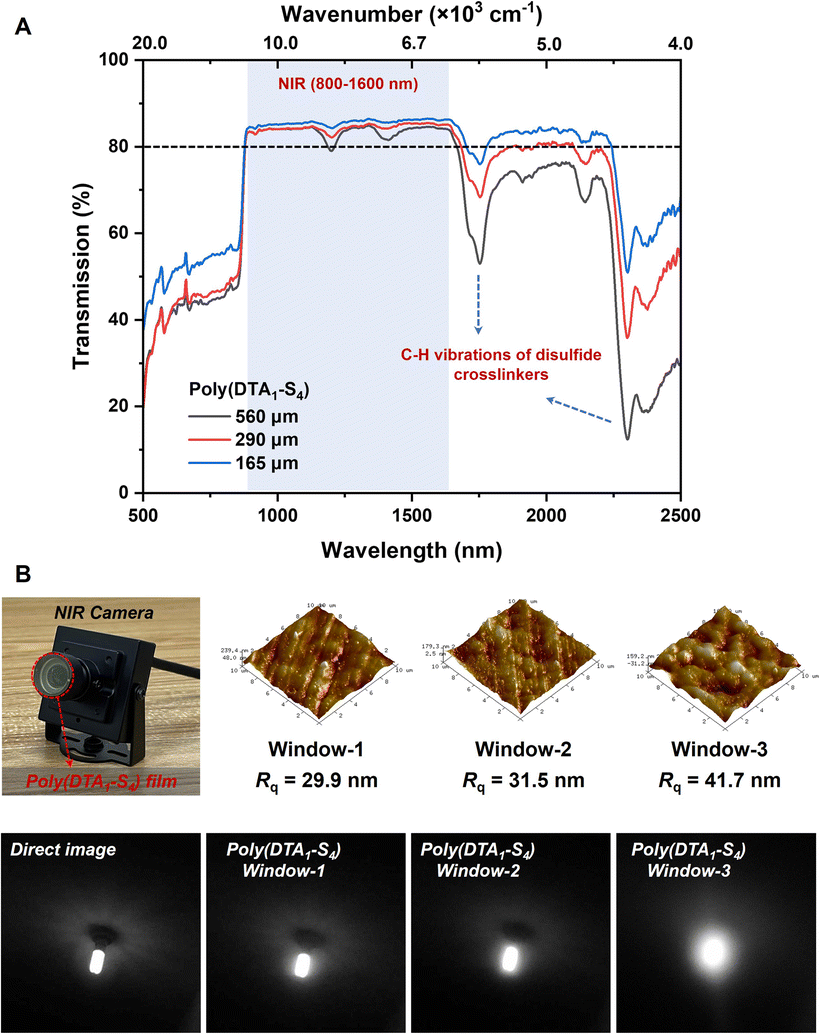 | ||
| Fig. 4 IR optical application of polysulfide CANs. (A) NIR transmittance spectra of poly(DTA1–S4) films with different thicknesses. (B) Optical images taken using a NIR camera with and without the poly(DTA1–S4) window. | ||
Chemical recyclability of polysulfide CANs
The abundance of dynamic covalent S–S bonds in the polysulfide backbones led to the further investigation of monomer recyclability, a chemically feasible process considering the thermodynamically favorable regeneration of S8 with low ring strain. However, this feat is challenging for most current S8-derived polysulfides stabilized by vinyl crosslinkers, as robust C–S bonding partly inhibits the decrosslinking and depolymerization of sulfur-rich networks. Herein, the cyclic disulfide comonomers offered reversible S–S crosslinking sites for efficient chemical closed-loop recyclability. After the linear poly(TAM1–S2) copolymer was diluted to a low concentration (0.5 M) in THF and heated to 60 °C, the polysulfides were degraded via thiolate–disulfide exchange using BnSH/tBu-P1 (0.01 equiv.) as the depolymerization catalyst.48 The colorless polymer solution gradually turned yellow after 10 h (Fig. S28†), as a high monomer conversion rate of 78% was achieved (Fig. S29 and S30†). Having proved the feasibility of thiolate-induced depolymerization, we further investigated the degradation of crosslinked polysulfides under the same conditions. The conversion of the insoluble poly(DTA1–S4) copolymer into a yellow solution indicated the reversible cleavage of the dynamic S–S crosslinking sites as well as active S–S exchange, thus generating oligomers and cyclic monomers (Fig. 5A and B). Two sulfur-containing comonomers could be efficiently purified and separated via simple extraction (see ESI Materials† for the detailed depolymerization procedure), yielding the recycled DTA monomer with a purity exceeding 95% and a recovery rate of 85% as well as the crystalline S8 solid with a recovery rate of 82%, which were confirmed using the consistent 1H NMR spectra and crystal XRD peaks when compared with the original samples (Fig. 5C and D). The conversion of the polysulfide segments back to cyclooctasulfur was guided by the thermodynamic equilibrium. Further repolymerization of the recycled DTA and S8 produced recycled poly(DTA1–S4), which exhibited almost identical network structures and mechanical properties compared with the original sample (Fig. S31–S33†). Therefore, the diverse dynamic disulfide crosslinkers provide an advanced paradigm for the circular production, degradation, and reuse of S8-derived polymers.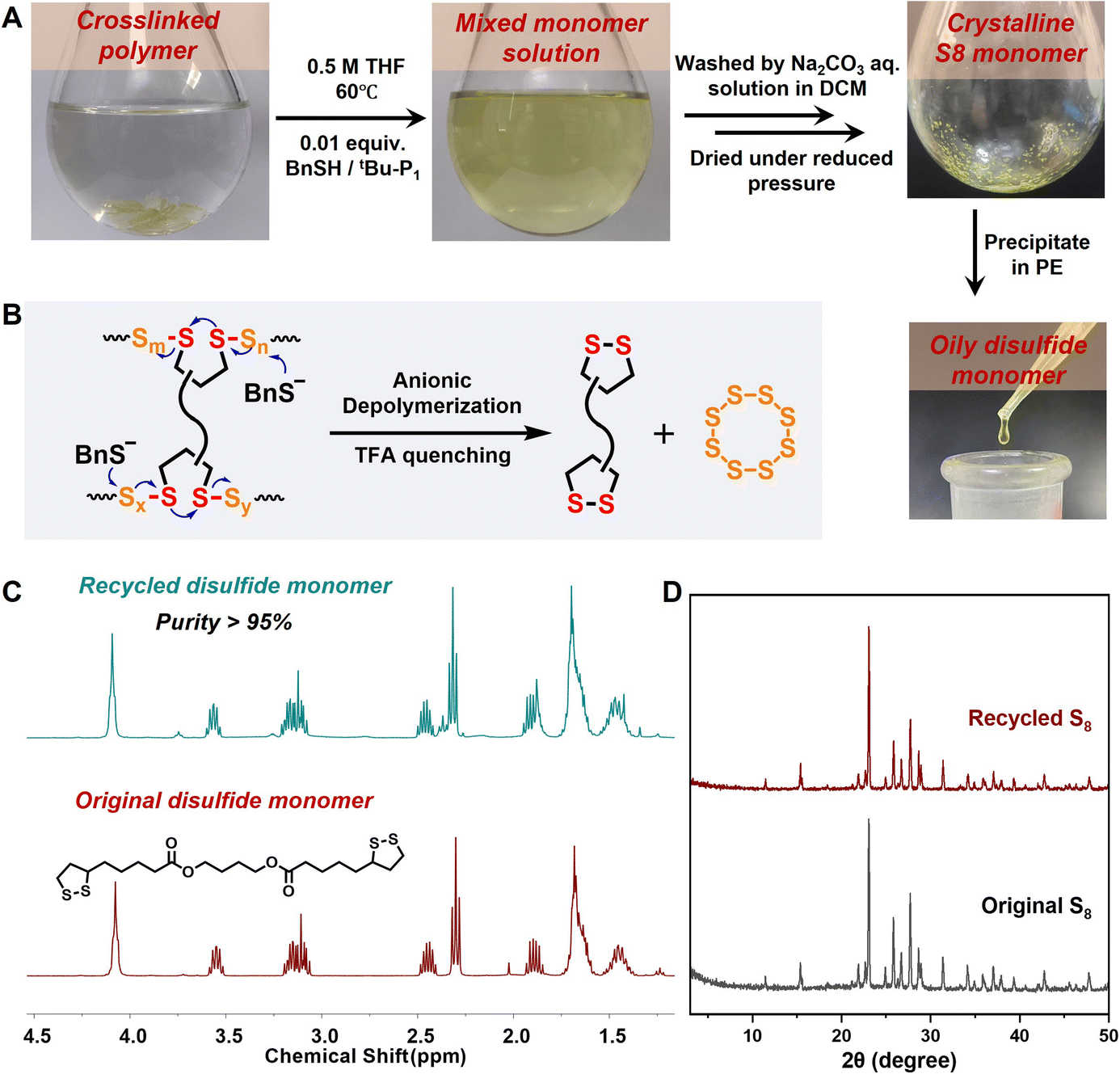 | ||
| Fig. 5 Chemical recyclability of polysulfide CANs. (A) Photographs of the depolymerization of crosslinked polysulfides and monomer regeneration process. (B) Schematic representation of the thiolate induced depolymerization process. (C) 1H NMR spectra of original and recycled DTA monomers. (D) XRD patterns of original and recycled S8 monomers. | ||
Conclusions
In summary, we proposed a universal strategy for the anionic copolymerization of S8 and cyclic disulfides with varying ring strains and sidechains to produce sulfur-rich copolymers with tunable sulfur content (40–70 wt%). The active segment motion and dynamic S–S exchange in crosslinked networks with high sulfur rank provided polysulfide CANs with excellent adaptability and reprocessability without compromising mechanical performance. Notably, the dynamic S–S crosslinking sites facilitated the depolymerization of polysulfide networks. Unlike the dark opaque appearance of commonly reported sulfur-based polymers, the free-standing transparent polymer films exhibited high optical transmission in the NIR range, extending their potential application in NIR transmissive optical devices. This facile and general copolymerization approach presents new opportunities for the upcycling of S8 towards sustainable polysulfide materials and advanced NIR imaging applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors discussed the results and commented on the manuscript. Design of the study: C.-Y. S., H. T., and D.-H. Q.; acquisition of funding: C.-Y. S., H. T., and D.-H. Q.; project supervision: Q. Z. and D.-H. Q.; design and chemical synthesis: C.-Y. S. and X.-P. Z.; writing original draft: C.-Y. S. and X.-P. Z.; writing – review and editing: Q. Z., M. C., and D.-H. Q., with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22220102004, 22025503, and 22405090), the Shanghai Municipal Science and Technology Major Project (grant no. 2018SHZDZX03), the Innovation Program of Shanghai Municipal Education Commission (2023ZKZD40), the Fundamental Research Funds for the Central Universities, the Programme of Introducing Talents of Discipline to Universities (grant no. B16017), the Science and Technology Commission of Shanghai Municipality (grant no. 21JC1401700), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (grant no. SN-ZJU-SIAS-006), the Postdoctoral Fellowship Program of CPSF (GZB20230210), and the China Postdoctoral Science Foundation (2023TQ0114 and 2024M750900). We thank LetPub (http://www.letpub.com.cn) for their linguistic assistance during the preparation of this manuscript.Notes and references
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. v. d. Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed
.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef CAS PubMed
- T. S. Kleine, T. Lee, K. J. Carothers, M. O. Hamilton, L. E. Anderson, L. R. Diaz, N. P. Lyons, K. R. Coasey, W. O. Parker Jr, L. Borghi, M. E. Mackay, K. Char, R. S. Glass, D. L. Lichtenberger, R. A. Norwood and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 17656–17660 CrossRef CAS PubMed
- J. Molineux, T. Lee, K. J. Kim, K.-S. Kang, N. P. Lyons, A. Nishant, T. S. Kleine, S. W. Durfee, J. Pyun and R. A. Norwood, Adv. Opt. Mater., 2024, 12, 2301971 CrossRef CAS
- S. J. Tonkin, L. N. Pham, J. R. Gascooke, M. R. Johnston, M. L. Coote, C. T. Gibson and J. M. Chalker, Adv. Opt. Mater., 2023, 11, 2300058 CrossRef CAS
- J. Pyun and R. A. Norwood, Prog. Polym. Sci., 2024, 156, 101865 CrossRef CAS
- F. Zhao, J. Xue, W. Shao, H. Yu, W. Huang and J. Xiao, J. Energy Chem., 2023, 80, 625–657 CrossRef CAS
- X. Zhang, K. Chen, Z. Sun, G. Hu, R. Xiao, H.-M. Cheng and F. Li, Energy Environ. Sci., 2020, 13, 1076–1095 RSC
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed
- A. Nayeem, M. F. Ali and J. H. Shariffuddin, Environ. Res., 2023, 216, 114306 CrossRef CAS PubMed
- F. G. Müller, L. S. Lisboa and J. M. Chalker, Adv. Sustainable Syst., 2023, 7, 2300010 CrossRef
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed
- M. M. Coleman, J. R. Shelton and J. L. Koenig, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 154–166 CrossRef CAS
- J.-Y. Chao, T.-J. Yue, B.-H. Ren, G.-G. Gu, X.-B. Lu and W.-M. Ren, Angew. Chem., Int. Ed., 2022, 61, e202115950 CrossRef CAS PubMed
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Top. Curr. Chem., 2019, 377, 16 CrossRef PubMed
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed
- L. J. Dodd, Ö. Omar, X. Wu and T. Hasell, ACS Catal., 2021, 11, 4441–4455 CrossRef CAS
- J. Jia, J. Liu, Z.-Q. Wang, T. Liu, P. Yan, X.-Q. Gong, C. Zhao, L. Chen, C. Miao, W. Zhao, S. Cai, X.-C. Wang, A. I. Cooper, X. Wu, T. Hasell and Z.-J. Quan, Nat. Chem., 2022, 14, 1249–1257 CrossRef CAS PubMed
- H. Yang, J. Huang, Y. Song, H. Yao, W. Huang, X. Xue, L. Jiang, Q. Jiang, B. Jiang and G. Zhang, J. Am. Chem. Soc., 2023, 145, 14539–14547 CrossRef CAS PubMed
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC
- P. Yan, W. Zhao, S. J. Tonkin, J. M. Chalker, T. L. Schiller and T. Hasell, Chem. Mater., 2022, 34, 1167–1178 CrossRef CAS
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem.–Eur. J., 2020, 26, 10035–10044 CrossRef CAS PubMed
- J. M. M. Pople, T. P. Nicholls, L. N. Pham, W. M. Bloch, L. S. Lisboa, M. V. Perkins, C. T. Gibson, M. L. Coote, Z. Jia and J. M. Chalker, J. Am. Chem. Soc., 2023, 145, 11798–11810 CrossRef CAS PubMed
- T. Tian, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS PubMed
- N. P. Tarasova, A. A. Zanin, E. G. Krivoborodov and Y. O. Mezhuev, RSC Adv., 2021, 11, 9008–9020 RSC
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS
- J. Bao, K. P. Martin, E. Cho, K.-S. Kang, R. S. Glass, V. Coropceanu, J.-L. Bredas, W. O. Parker, Jr., J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2023, 145, 12386–12397 CrossRef CAS PubMed
- T. Sato, M. Abe and T. Otsu, Makromol. Chem., 1979, 180, 1165–1174 CrossRef CAS
- R. W. Clarke, T. Sandmeier, K. A. Franklin, D. Reich, X. Zhang, N. Vengallur, T. K. Patra, R. J. Tannenbaum, S. Adhikari, S. K. Kumar, T. Rovis and E. Y.-X. Chen, Nature, 2023, 616, 731–739 CrossRef CAS PubMed
- S. Penczek, R. Ślazak and A. Duda, Nature, 1978, 273, 738–739 CrossRef CAS
- A. Duda and S. Penczek, Macromolecules, 1982, 15, 36–40 CrossRef CAS
- Y. Deng, Z. Huang, B. L. Feringa, H. Tian, Q. Zhang and D.-H. Qu, Nat. Commun., 2024, 15, 3855 CrossRef CAS PubMed
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed
- C. J. Kloxina and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC
- C.-Y. Shi, Q. Zhang, H. Tian and D.-H. Qu, SmartMat, 2020, 1, e1012 CrossRef
- C.-Y. Shi, Q. Zhang, B.-S. Wang, D.-D. He, H. Tian and D.-H. Qu, CCS Chem., 2023, 5, 1422–1432 CrossRef CAS
- C.-Y. Shi, D.-D. He, Q. Zhang, F. Tong, Z.-T. Shi, H. Tian and D.-H. Qu, Natl. Sci. Rev., 2023, 10, nwac139 CrossRef CAS
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed
- J. J. Griebel, N. A. Nguyen, A. V. Astashkin, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258–1261 CrossRef CAS
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS PubMed
- J. Wreczycki, D. M. Bielinski, M. Kozanecki, P. Maczugowska and G. Mloston, Materials, 2020, 13, 2597 CrossRef CAS PubMed
- M. Haghdadi and M. Hamzehluiyan, Chin. J. Chem., 2008, 26, 471–479 CrossRef CAS
- S. M. Bachrach, J. T. Woody and D. C. Mulhearn, J. Org. Chem., 2002, 67, 8983–8990 CrossRef CAS PubMed
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed
- W. Cui, W. You and W. Yu, Macromolecules, 2021, 54, 824–834 CrossRef CAS
- Y. Deng, Q. Zhang, C. Shi, R. Toyoda, D.-H. Qu, H. Tian and B. L. Feringa, Sci. Adv., 2022, 8, eabk3286 CrossRef CAS PubMed
- C. Cui, F. Wang, X. Chen, T. Xu, Z. Li, K. Chen, Y. Guo, Y. Cheng, Z. Ge and Y. Zhang, Adv. Funct. Mater., 2024, 34, 2315469 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05031b |
| This journal is © The Royal Society of Chemistry 2024 |