
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrowth of two-dimensional covalent organic frameworks on substrates: insight from microsecond atomistic simulations†
Zilin
Wang
 ab,
Hong
Du
ab,
Austin M.
Evans
*c,
Xiaojuan
Ni
d,
Jean-Luc
Bredas
*d and
Haoyuan
Li
*ab
ab,
Hong
Du
ab,
Austin M.
Evans
*c,
Xiaojuan
Ni
d,
Jean-Luc
Bredas
*d and
Haoyuan
Li
*ab
aSchool of Microelectronics, Shanghai University, Shanghai 201800, China. E-mail: lihaoyuan@shu.edu.cn
bDepartment of Chemistry, College of Sciences, Shanghai University, Shanghai 200444, China
cGeorge and Josephine Butler Polymer Laboratory, Department of Chemistry, University of Florida, Gainesville, Florida 32611-7200, USA. E-mail: austinevans@chem.ufl.edu
dDepartment of Chemistry and Biochemistry, The University of Arizona, Tucson, Arizona 85721-0041, USA. E-mail: jlbredas@arizona.edu
First published on 3rd October 2024
Abstract
While growing two-dimensional covalent organic frameworks (2D COFs) on substrates holds promise for producing functional monolayers, the presence of many defects in the resulting crystals often hinders their practical applications. Achieving structural order while suppressing defect formation necessitates a detailed atomic-level understanding. The key lies in understanding the polymerization process with high nano-scale accuracy, which presents significant challenges. Here, we perform microsecond atomistic molecular dynamics simulations to describe the deposition and polymerization of cyclohexa-m-phenylene on metal substrates, closely mimicking experimental conditions. Our improved approach highlights that 2D polymerization occurs through monomer addition and island coalescence, with a pre-bonding stage allowing monomers/oligomers to dynamically adjust their configurations to the expanding island structures. Our results elucidate the mechanisms underlying the formation of vacancy and dislocation defects during 2D polymerization as well as their healing processes. Overall, our findings underscore the significant roles that high surface mobility, effective monomer-substrate anchoring, high framework rigidity, moderate monomer coordination, and low bonding rate play in forming large, extended 2D crystals while suppressing vacancy and dislocation defects. We demonstrate how these factors can be tuned through substrate selection, deposition rate modulation, and temperature control, thereby offering valuable insight for strategically optimizing on-surface 2D polymerizations.
1. Introduction
Two-dimensional covalent organic framework (2D COF) monolayers are appealing for applications in 2D optoelectronic devices.1–3 Confining two-dimensional polymerization to an interface, which restricts the degrees of freedom, provides an effective means to fabricate thin-film or monolayer COFs.4–7 This is commonly achieved by depositing monomers onto a metallic substrate under vacuum and then thermally driving them to polymerize on this substrate.4,6–15 However, the inherent lack of 2D polymerization control generally results in small lateral crystalline domain sizes (100–10000 nm2) and high defect densities,1,12,13,16 which limits the relevance of the resulting monolayers in optoelectronic device applications.17,18 The complex interplay among monomer deposition, surface diffusion, and bond formation has made developing a holistic model of 2D polymerization at interfaces elusive.7,11,15 An in-depth understanding of the atomic-level dynamics associated with these processes is a prerequisite to achieving directed network control, increasing the monolayer lateral domain sizes, reducing the concentration of defects, and ultimately fabricating high-quality optoelectronic devices based on 2D COFs. Such insight is also beneficial to understanding 2D COF synthesis at other types of interfaces (e.g., liquid–liquid, liquid–solid, liquid–gas).19–26Experimentally probing 2D COF polymerization with high spatio-temporal resolution on substrates is challenging.27–29 One report that studied this process with high temporal resolution leveraged in situ scanning tunneling microscopy to track the polymerization of pyrene-2,7-diboronic acid at a solid–liquid interface of highly oriented pyrolytic graphite surface (solid–liquid interface).30 Initially, an amorphous layer is formed that begins to nucleate crystalline domains. These crystalline nuclei then begin to dynamically exchange as a result of the reversible boroxine bonds formed during this polymerization. These nuclei then undergo crystalline elongation with particle attachment characteristics. Also, separate investigations of confined polymerization under vacuum have shown substrate interactions greatly influence the rates of monomer diffusion and bond formation, which is empirically observed to modify the defect density and crystallinity of resulting monolayers.6,8,9,11,15 While these descriptions contribute to an empirical understanding of 2D COF polymerization on substrates, important questions remain regarding how the monomers dynamically assemble at the nanoscale, how defects are generated, and whether they can be healed in the course of polymerization. Addressing these questions calls for a molecular/atomic-level description of 2D COF polymerization on substrates.
Atomistic molecular modeling is one approach to interrogate the formation dynamics of 2D COFs.31–33 For example, Nguyen and Grünwald studied the microsecond-scale homogeneous crystallization dynamics of 2,3,6,7,10,11-hexahydroxytriphenylene and 1,4-phenylene bis(boronic acid) to form COF-5 and explored the formation of defect structures, which was enabled by using large time steps (5 fs) and neglecting explicit solvent molecules.32 Very recently, Hao et al. used 5 ns simulations to show how hydrogen bonds help direct the formation of a crystalline imine-linked COF on a graphene substrate in vacuum.33 While these studies provide valuable insight into COF formation, they are unable to capture the full complexity needed to derive a mechanistic model of COF formation at an interface.
The ability to describe experimentally relevant 2D COF formation conditions with long timescales and high atomistic accuracy exceeds the capabilities of common theoretical tools. Ab initio molecular dynamics, known for high accuracy, can typically consider processes only over a few picoseconds and involving a few hundred atoms,34 making their practical use in the context of 2D COF polymerization unfeasible. Methods such as reactive force fields reduce computational costs but are still limited to the nanosecond timescale and are bottlenecked by the limited availability of appropriate force–field parameters.35,36 Classical molecular dynamics, while significantly more computationally efficient, cannot directly describe the molecular reactions intrinsic to 2D COF growth. In the case of kinetic Monte Carlo models, experimental timescales can be matched with high levels of microscopic accuracy, which has been demonstrated for boronate ester-linked COFs.37–39 However, the development of kinetic Monte Carlo models relies on a good initial understanding of the microscopic processes involved and a substantial amount of rate data that must be validated,40,41 making its general deployment to a broad range of 2D COF systems and synthesis conditions challenging.
Here, we leverage a recently developed algorithm for chemical reactions in classical molecular dynamics with force field parameters carefully tuned to approach the accuracy of ab initio methods.42 This computational approach enabled us to carry out successful atomistic simulations of the deposition and polymerization of cyclohexa-m-phenylene (CHP) monomers43 (Fig. 1a and b) on metal (Ag and Cu) surfaces over microsecond timescales with high atomistic accuracy under conditions closely resembling experimental conditions. It has been shown experimentally15 that when a hexaiodo-substituted macrocycle CHP is deposited onto Ag or Cu, the iodine atoms cleave, leading to CHP radicals that polymerize and co-adsorbed iodine (Fig. 1a). CHP makes an ideal model system to study the surface polymerization mechanisms since the experimental data for its growth on Ag and Cu substrates are well-established15 and can be used to validate the calculations. Moreover, the molecular reaction pathways are well-understood, which enables us to provide an accurate modeling of the temperature dependence of the bond-formation rates in our simulations.15 Finally, CHP's simple D6h symmetry means that we do not have to contend with competing topological isomer formation.
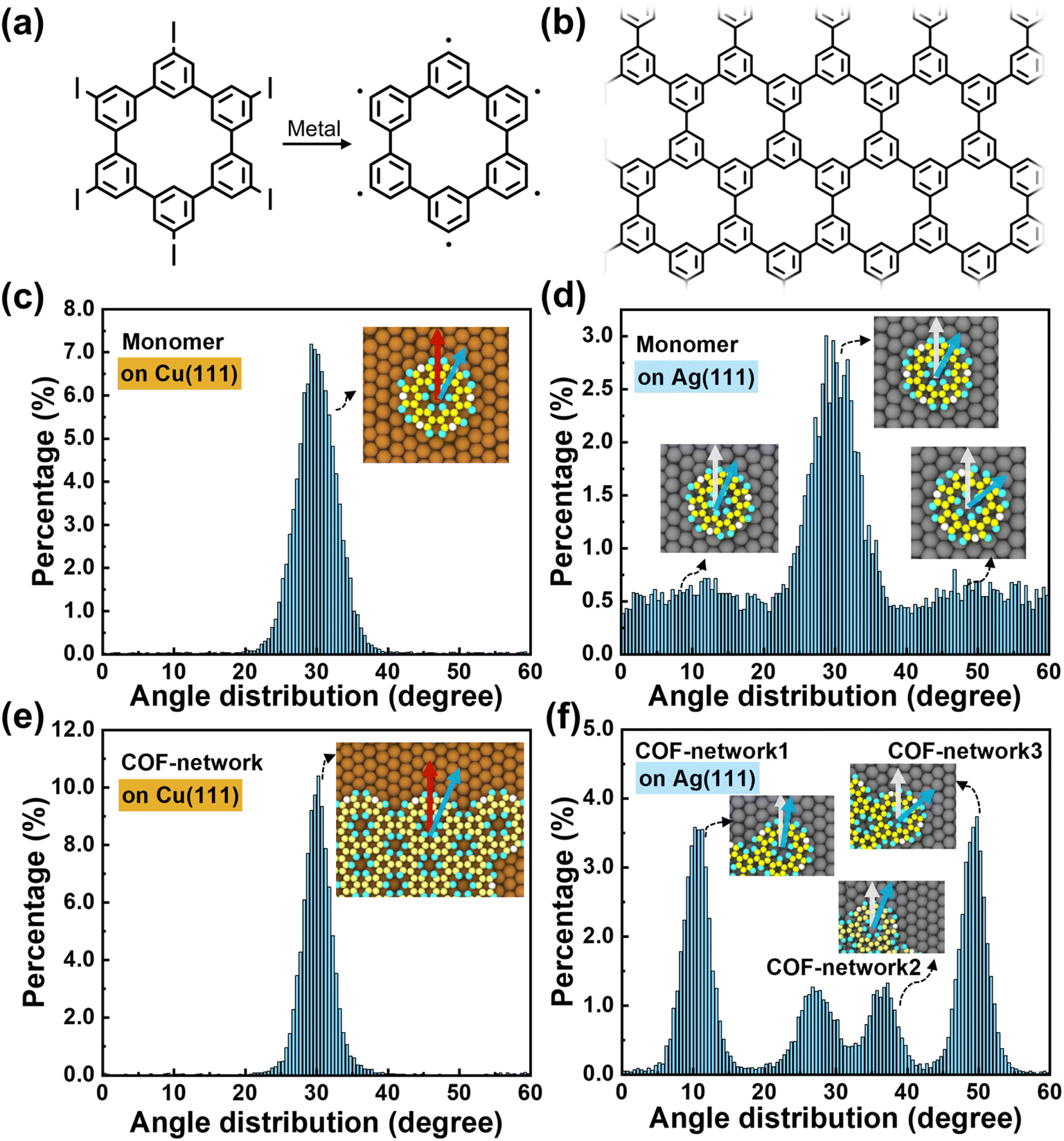 | ||
| Fig. 1 (a) Chemical structures of hexaiodo-substituted CHP and the generated CHP radical after being deposited on metal surfaces. (b) Chemical structure of the polyphenylene network. Distributions of the angles between the vector connecting the monomer center of mass and the radical atom (blue arrow) and the substrate (1, 0, −1) direction (red arrow) at 600 K on (c) Cu (111) and (d) Ag (111) substrates. Distributions of the angles of the monomer units of 2D islands on (e) Cu (111) and (f) Ag (111) substrates. Details of the calculation of the angular distributions and the values at other temperatures can be found in Fig. S1 and S2 in the ESI.† | ||
The improvements in our theoretical descriptions of 2D polymerization allowed us to uncover the mechanisms of monomer addition and island coalescence under experimentally relevant conditions. During 2D COF formation, monomers are evaporated onto a metallic substrate under vacuum and thermally annealed, which produces crystalline layers by reducing local stress.6,7,10,11 We mimic these two processes in our simulations. It has been empirically identified that the 2D COF quality depends on the substrate identity, reaction temperature, and monomer concentration.9,11,12,15 From this perspective, we deduce that 2D COF formation is the result of competition among monomer deposition, surface diffusion, and bond formation. Here, we aim to investigate the interplay among these factors using atomistic simulations. It is also important to acknowledge that, even with the efficient MD simulations applied here, achieving a complete match between experimental conditions and molecular simulations remains a difficult proposition. In this study, we modeled timescales in the microsecond range while maintaining high atomistic accuracy with the following conditions (see Methods section): (1) we consider a faster deposition rate to allow us to mimic the deposition and annealing stages in the simulations. (2) We use slightly elevated temperatures (570–620 K) compared to those used in experiments to enhance the rate of surface diffusion within the microsecond timescale.15 (3) To account for the appropriate temperature dependence of the reaction rates, we exploit the reaction energy barriers evaluated from density functional theory calculations.15 (4) We use large prefactors for the reaction rates to allow bond formation within microsecond timescales. As we will see in the following discussions, variations in deposition rate and temperature lead to distinct 2D polymer networks, indicating that the interplay among monomer deposition, monomer diffusion, and bond formation is successfully captured by our atomistic simulations. Consequently, the simulations preserve the interplay among deposition, surface diffusion, and bonding over longer timescales and the findings from our study can be extrapolated to typical experimental conditions.
Importantly, we identified a crucial pre-bonding stage that significantly impacts the formation of extended 2D COF crystals, which is guided by an intricate interplay among monomer coordination, diffusion, and bond formation. Our models also describe the formation of vacancy and dislocation defects during polymerization. Overall, our results highlight the significance of high surface mobility, high framework rigidity, moderate monomer coordination, and low bonding rate in the formation of high-quality 2D COFs, which can be achieved by tuning the substrate, deposition rate, and temperature.
2. Results and discussion
2.1. Monomer conformation and motion on the metal substrate
In the isolated state, the dihedral angle between adjacent phenylene rings in CHP is approximately 150°. Upon deposition of the monomer on the substrate, its molecular conformation becomes coplanar due to van der Waals interactions with the metal surface (see Fig. S3†). Previous DFT calculations based on the local density approximation (LDA)44 have demonstrated that the CHP monomer exhibits its most stable binding configuration with the metal substrate when it resides at the Atop position (where the center of mass of the CHP radical is directly above a surface atom).15 This feature is also seen in our molecular dynamics simulations. For the Cu (111) substrate, ∼90% of the monomers are located at the Atop position, which is consistent with the LDA-DFT results that the Bridge and Hollow configurations have energies 1.8 eV and 2.0 eV higher, respectively. For the Ag (111) substrate, monomers are seen in our simulations to be located in both the Atop (∼45%) and Bridge (∼34%) positions, which agrees well with the LDA-DFT results that the energy difference between these configurations is small (0.1–0.2 eV).15At elevated temperatures, the CHP monomer does not keep fixed orientational angles (see Fig. 1) seen in the optimal configurations at 0 K (see ESI Video†). For the Cu substrate, we find that ∼90% CHP monomers have a 30° angle relative to the (1, 0, −1) direction of the metal surface at 600 K (see Fig. 1c), which is consistent with the potential energy profiles calculated based on the force field (Fig. S4†). This preferred orientation of the CHP monomer with respect to the Cu (111) surface originates from their lattice matching (Fig. S5†). However, the lattices are mismatched on the Ag (111) surface (Fig. S5†), leading to variations in orientations, as shown in Fig. 1d. As such, monomer diffusion on the Cu surface proceeds from Atop to Atop position with the initial and final configurations having the same orientation, while that on the Ag surface can switch among Atop and Bridge positions and with orientational changes. These results also demonstrate that the Cu surface has a stronger anchoring effect for CHP than the Ag surface.
2.2. Polymerization via monomer addition and island coalescence
We consider in the simulation a constant-rate deposition stage, followed by an annealing stage. There is a probability of bonding between deposited CHP monomers when they come into proximity on the surface, gradually forming an initial island consisting of two monomers, as shown in Fig. 2a. This island also diffuses on the metal surface, although at a slower rate. | ||
| Fig. 2 Illustrations of the CHP polymerization on the Ag (111) substrate: (a) monomer addition (red circles indicate bonding sites), (b) island coalescence, and (c) monomer-assisted island coalescence. | ||
Our MD simulations indicate that the polymerization of 2D COFs on the metal substrate occurs through two mechanisms: monomer addition and island coalescence. The process of monomer addition (Fig. 2a) resembles the classical crystal growth process, with monomers added sequentially to established nuclei. A small island and a monomer can diffuse and collide on the substrate, resulting in the extension of the island by one monomer unit. This repetitive process fosters the gradual formation of larger islands. Fig. 2a illustrates a typical monomer-addition process, where we take the example of the Ag (111) substrate: Monomers 1 and 2, upon being deposited on the metal substrate, collide and bond to form Island A (2aI → 2aII). Subsequently, Monomer 3 approaches this island, collides, and forms a larger Island B (2aII → 2aIII). This process continues, resulting in the formation of larger islands (2aIV and 2aV). We refer to the islands grown exclusively through monomer addition as the Initial Islands. As the islands expand, their diffusion rate on the metal surface decreases (see the mean square displacements in Fig. S6† and diffusion coefficients for monomers, dimers, and trimers in Table S1†). We observe that, on the Ag substrate, islands containing more than five monomers are virtually static within the simulated time frame, while on the Cu substrate those containing two or more monomer units exhibit minimal movement. As a result, the subsequent growth of the islands is primarily dependent on the diffusion of monomers, dimers, trimers, and tetramers on the substrate, along with their collisions. Thus, the locations of the initial islands heavily influence the formation of dense regions in the 2D network.
As multiple islands grow simultaneously on the substrate, they may merge into a larger island (island coalescence) (2bI → 2bII in Fig. 2b). Small islands diffuse and collide: When Island E and Island F approach each other, they temporarily form a loose contact due to van der Waals forces (2bIII), with the smaller Island F continuing to move and rotate. This loosely bound configuration during the pre-bonding stage facilitates their subsequent bonding, which connects the two islands (2bIII → 2bIV). For large islands, diffusion on the substrate is restricted. However, small islands can diffuse, approach them, and coalesce. Additionally, we have observed that monomers can promote contact and bonding between islands, a process we refer to as monomer-assisted island coalescence. For instance, as illustrated in Fig. 2c, Monomer 4 is located between Island G and Island H (2cI), which also fosters the formation of a transient binding state (2cII). Monomer 4 collides with both Island G and Island H and, in this case, eventually bonds with Island G, forming Island J (2cIII). Finally, Island J and Island H collide and coalesce to form Island K (2cIII → 2cV).
The transient binding configurations described above represent a pre-bonding stage for island growth. This stage involves molecular collision leading to eventual bond formation, while allowing the monomers and small islands to adjust their positions and move towards a more thermodynamically favorable state. This corresponds to filling the concave structures surrounded by more monomer units to form a larger contact, thereby facilitating the formation of an integrated polymer network, as shown in Fig. 3. Based on this mechanism, it can be inferred that moderate coordination, high surface mobility, and low bonding rate are important to forming large, extended networks.
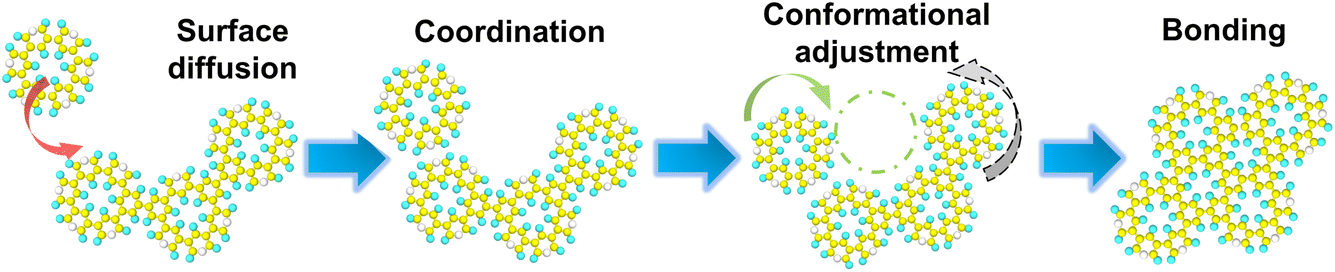 | ||
| Fig. 3 Proposed mechanism of polymerization on metal substrates. | ||
The type of substrate significantly influences the polymerization of 2D COFs, which manifests as monolayers with different sizes and defect densities. As shown in Fig. 4a and b, large and dense networks are formed on an Ag substrate, while on a Cu substrate, the network consists of numerous small and branched islands (see ESI Video†). These findings are consistent with previously reported experimental STM measurements.15 Compared to Ag, the interactions between monomers and the Cu substrate are stronger (the binding energies are calculated by the force field to be −35.6 kcal mol−1 (∼−1 kcal per mol per carbon) on Cu (111) and −32.5 kcal mol−1 (∼−0.9 kcal per mol per carbon) on Ag (111), respectively; see Table S2† for calculation details). The monomer mobility is also lower on Cu (111) than on Ag (111), with the diffusion barriers evaluated by LDA-DFT being 2.2 eV and 0.8 eV, respectively;15 the calculated monomer diffusivities given by our MD simulations are 0.335 and 4.734 cm2 s−1 on Cu (111) and Ag (111), respectively (see Table S1†). The restricted mobility on Cu not only leads to fewer bonding collisions, but also reduces the conformational adjustments in the pre-bonding stage, thereby limiting the formation of high-quality 2D COFs, as mentioned above.
 | ||
| Fig. 4 2D COFs formed at 1000 ns on (a) Ag (111) and (b) Cu (111) surfaces. (c) Evolution of the number of reactions during the simulation. Evolution of the island size on (d) Ag and (e) Cu substrates. Red and blue in panels (d) and (e) represent growth by monomer addition and island coalescence, respectively. During the deposition stage, a monomer is deposited every 1.25 ns while keeping the system at a temperature of 600 K; in the annealing stage, the system is maintained at 600 K without monomer deposition (see the Methods section for details). | ||
Interestingly, the formation of islands modifies the orientation of the monomers on the substrates. On the Cu substrate, the islands maintain the ∼30° angle seen for monomers (Fig. 1e). However, on the Ag substrate, the monomers, which originally have various orientations, become aligned within a given island due to constraints of covalent bonding. These islands prefer to orient around 10 or 50°, while those with dislocations can have values between these two limits, as illustrated in Fig. 1f. Overall, these results suggest that the monomer-substrate interactions should be tuned to balance increasing monomer mobility for extended crystal growth and anchoring monomer orientations to suppress dislocations.
Our quantitative analysis of bond formation during polymerization reveals distinct growth stages. Fig. 4c shows the number of bond formations occurring during the simulation. The initial step is an induction stage with low bonding occurrences, which is observed for polymerizations on both Ag and Cu substrates. This stage corresponds to diffusion-controlled nucleation, where the low monomer concentration results in few effective collisions and bond formations. Subsequently, on the Ag substrate, the system enters a regime of constant growth, where the growth rate of islands is primarily limited by the number of deposited monomers on the substrate. In comparison, the induction period is notably longer on the Cu substrate, and the subsequent growth rate increases gradually, indicating relatively restricted monomer and island diffusion during polymerization. As the concentration of monomers increases on the Cu substrate, the dependence of polymerization on diffusion decreases, leading to an increase in the growth rate. Overall, more bond formations are observed on the Ag substrate than on the Cu substrate within the same time frame, as a result of more rapid diffusion and formation of 2D COF crystals on its surface (Fig. S7†).
Fig. 4d and e show the evolution of the largest and average island sizes over time. We found that both monomer addition and island coalescence contribute to forming 2D crystals on the Ag substrate, as indicated by their alternating stages in Fig. 4d. In contrast, on the Cu substrate, constrained diffusion leads to the generation of localized small islands, and the development of the main 2D crystal proceeds in a step-wise manner (see Fig. 4e), primarily through the merging of islands. The change in island sizes in the annealing stage indicates ongoing island growth without new monomer deposition; it is unreacted monomers on the substrate that continue to grow existing islands by monomer addition. Island coalescence can also facilitate the growth of crystals, which is particularly noticeable in the case of Ag where this process rapidly increases the average island size.
2.3. Defect formation and healing
Our simulations demonstrate that monovacancy, divacancy, multivacancy, and dislocation defects emerge during monomer addition and island coalescence. We now illustrate representative examples of the appearance of such defects. As shown in Fig. 5a, when Monomer 1 diffuses close to Island A – which has a concave structure formed when a connecting monomer is not adjusted to the thermodynamically most favorable configuration in its pre-bonding stage, see 5aI—, entering the cavity is difficult in the pre-bonding stage due to steric hindrance; however, frequent collisions due to van der Waals interactions can lead to bonding and eventual closure of the cavity, resulting in a vacancy (5aII → 5aIV). During the subsequent growth, it is difficult for such a vacancy to get in contact with monomers and heal. Depending on the shape of the islands and the bonding positions, divacancy and multivacancy defects can also occur. Fig. 5b illustrates the successive formation of a multivacancy defect and a divacancy. Island B formed through island coalescence possesses multiple semi-closed regions. Monomers can diffuse and connect with the semi-closed ring structure, gradually closing the ring and resulting, for instance, in a five-vacancy defect (5bI → 5bV). Subsequently, another semi-closed region of this island can collide and bond with Monomer 3, forming a divacancy defect (5bVI → 5bVIII). Since the edges of islands usually do not match perfectly, the coalescence of islands also tends to produce vacancies, as illustrated in Fig. 5c. Compared to monomers, adjustment of the conformations of islands during the pre-bonding stage is more difficult due to their reduced mobilities. Based on the mechanisms of vacancy formation, a low bonding rate and a high surface mobility, which facilitate the adjustment of monomer and island configurations in the pre-bonding stage to prevent the formation of concave structures, also suppress the formation of vacancies. Lattice elongation by monomer addition is also preferred to reduce vacancy. | ||
| Fig. 5 Illustrations of the formation of (a) monovacancy and (b and c) divacancy and multivacancy defects. See text for details. | ||
Dislocation structures can also accompany island coalescence and monomer addition, a feature mostly observed on the Ag surface. To illustrate this point, consider Fig. 6a where Island A and Island B first diffuse and come in proximity (6aI). Collisions lead to bond formation on one end, connecting the two islands and forming a semi-closed structure (6aII). At this point, maintaining contact on the other end due to van der Waals forces results in a deformation along the branch of the covalent network from its crystal lattice. When a bond forms at the contact, a dislocation with multivacancy is formed (6aII–6aIII). Subsequent monomers during deposition and diffusion may partially fill the vacancies (6aIV); however, the dislocation is difficult to eliminate.
 | ||
| Fig. 6 Illustrations of the formation and partial filling of (a) a dislocation and (b) a seven-membered ring. See text for details. | ||
Fig. 6b illustrates the formation of a seven-membered ring through monomer addition. Initially, Island C and Island D combine to form a semi-closed ring structure (6bI → 6bII). Then, Monomer 1 collides and eventually bonds to one side of the semi-closed structure during its diffusion, forming a near-closed structure (6bII → 6bIV). If it further bonds, the ring would close with a single vacancy defect. However, we found that it is possible for a second monomer, Monomer 2, to diffuse into the middle of the semi-closed region, pushing away the earlier connected monomer unit, and then to bond to the other side of the semi-closed structure (6bV → 6bVIII). Finally, the ring completely closes, forming a seven-membered ring structure (6bIX). The vacancy in this defect can still be filled by a monomer; indeed, we observed the diffusion of Monomer 3 into the interior of the heptagon and its subsequent bonding (6bX → 6bXII).
On the Cu surface, the monomers tend to have fixed orientation owing to the strong monomer-substrate anchoring and the good lattice match we mentioned above. This phenomenon counteracts the deformation of the COF framework induced by van der Waals interactions in the pre-bonding stage, thereby reducing the likelihood of dislocation formation. However, suppressing dislocation formation through strong monomer-substrate anchoring and good lattice match may increase the diffusion barriers, thereby restricting the monomer mobility that is necessary for collision and conformational adjustment during the pre-bonding stage. This restricted mobility leads to the formation of more branched islands instead of large, dense crystals. In other words, there is a tradeoff between suppressing dislocation defects and forming dense 2D structures. Consequently, fine-tuning the monomer-substrate interactions becomes imperative to strike a balance between achieving an extended COF crystal and suppressing dislocation formation. Based on these aspects, a high framework rigidity reduces lateral structural deformations induced by monomer coordination or from thermal vibrations, thereby improving the quality of the 2D crystal.
2.4. Impact of the deposition rate and the temperature
The above results illustrate the intricate relationship among monomer coordination, surface diffusion, and bonding reactions during the polymerization of 2D polymers. These characteristics are expected to vary across different chemical structures and substrates and are markedly influenced by the experimental conditions. For a specific combination of monomer chemical structure and substrate, it is possible to fine-tune the polymerization process towards higher-quality crystals by adjusting factors such as the deposition rate and temperature. Next, we theoretically investigate their impacts to gain insight into an optimization of such experimental conditions. The tuning of these factors has so far been only explored empirically.10–12Fig. 7a–f show the distributions of bond connections between the same absolute number of monomers at different deposition rates. A slower deposition rate (which corresponds to a longer deposition time) leads to more connected bonds at the end of the deposition stage, as shown in Fig. 7a and d. This is understandable as the monomers have more time to diffuse and react. When the simulations proceed into the annealing stage, the monomers in the fast-deposition cases reacted with each other, leading to smaller differences in the bond density (Fig. 7b and e). However, the impact of the deposition rate cannot be fully eliminated via an annealing process. After a simulation time of 1000 ns (including deposition and annealing), the framework with the slowest deposition rate still has the densest network. This difference is more pronounced on the Cu surface given its lower monomer mobility than on the Ag surface; this feature is also illustrated in Fig. S8.†
 | ||
| Fig. 7 Ratios of monomers with different numbers of connections at different deposition rates on the Ag (111) substrate: (a) at the end of the deposition stage, (b) at 200 ns after the deposition stage, and (c) at a total time of 1000 ns. Ratios of monomers with different numbers of connections at different deposition rates on the Cu (111) substrate: (d) at the end of the deposition stage, (e) at 200 ns after the deposition stage, and (f) at a total time of 1000 ns. A bar plot representation of the ratios of bond connections is provided in Fig. S9.† | ||
Thus, modifying the deposition rate can tune the relative rates between surface diffusion and bond reaction. A slower deposition rate means a stronger impact of monomer mobility, particularly when bond formation is diffusion-limited as on the Cu surface. At a faster deposition rate, only a limited number of monomers have reacted at the end of the deposition stage, resulting in more monomers reacting during annealing, albeit to a lesser degree.
Finally, we investigated the impact of substrate temperature. A higher temperature helps overcome the surface diffusion and reaction energy barriers, simultaneously increasing the monomer mobility and the bond formation rate. However, their relative accelerations can differ as the process with a higher energy barrier will increase to a larger extent. Our calculations suggest that the bond formation rates increase faster than the monomer surface mobility as the temperature goes up (Fig. S7†). At a higher temperature, the monomers have more kinetic energy, making the coordination process more difficult but allowing for easier monomer conformational adjustment in the pre-bonding stage. The combined effect of these factors is thus expected to be complicated by multiple interacting features. Our results suggest that the network density increases with temperature over the investigated range of 570–620 K for the Ag surface, albeit to different degrees at different deposition rates (Fig. 8a and b). However, the network density on the Cu surface begins to drop above 610 K for the two considered deposition rates (Fig. 8c and d). As such, a careful tuning of the temperature might be needed depending on the type of substrate and the deposition rate.
 | ||
| Fig. 8 Ratios of monomers with different numbers of connections at different temperatures after a total simulation time of 1000 ns on the Ag (111) substrate for deposition steps of (a) 250 ns and (b) 500 ns. Ratios of monomers with different numbers of connections at different temperatures after a total simulation time of 1000 ns on the Cu (111) substrate for deposition steps of (c) 250 ns and (d) 500 ns. | ||
3. Conclusions
In summary, we have leveraged efficient molecular dynamics simulations with parameters approaching density functional theory-level accuracy to investigate the two-dimensional polymerization of cyclohexa-m-phenylene on two metal substrates. Our greatly improved theoretical approach enabled us to simulate 2D COF polymerization over microsecond timescales with high atomistic accuracy under conditions closely resembling those used experimentally.The polymerization of 2D COFs on metal substrates exhibits an induction period followed by growth, both of which are significantly impacted by the diffusion of the monomers on the substrate. Induction corresponds to the formation of initial islands and is limited by surface diffusion. The growth process is driven by both monomer addition and island coalescence. On the Ag substrate (high monomer surface mobility), the crystal forms mainly via monomer addition and also involves island coalescence, while on the Cu substrate (low monomer surface mobility), it progresses mainly step-wise through the merging of islands. These results are intriguing because they resemble chain-growth and step-growth linear polymerizations, respectively, where growth is dominated by either monomer addition or chain–chain coupling. From this analogy, we derive that high monomer surface mobility may be desirable for achieving high-quality 2D COFs.
We identified a pre-bonding stage critical to forming large 2D crystals. The monomers, loosely coordinated by van der Waals interactions, can dynamically adjust their configurations with respect to the island structure towards the thermodynamically more favorable stage, i.e., filling concave structures. The simulations reveal that vacancy and dislocation defects arise during island coalescence and monomer addition processes. Surface diffusion, bonding, and closure of specific concave regions can lead to the formation of vacancy defects, while subsequent growth may reduce the extent of vacancies through partial or complete filling. Framework deformation due to low diffusion barriers and thermal vibrations leads to lattice mismatch situations (in particular for islands with long branches) where bonding can result in difficult-to-eliminate dislocations. Deformation of the COF framework in this pre-bonding stage must be suppressed because it yields dislocations that are challenging to anneal. These findings suggest that utilizing moderate monomer coordination strength (beneficial to entering the pre-bonding stage), minimizing the deformation of the 2D COF network (e.g., via tuning monomer-substrate interactions or increasing its structural rigidity), maintaining high surface mobility, and lowering the bond formation rate can reduce the occurrence of dislocation defects and promote the formation of large 2D crystals.45,46 These factors can be modified by tuning substrate type, deposition rate, and temperature, as we demonstrated in the simulations and as has been previously observed experimentally. Based on this understanding, it is possible that high-quality 2D crystals can be generated without the need for reversible defect annealing if these kinetic factors are appropriately balanced. Going forward, it will be important to consider how this complex interplay of factors influences the growth of 2D COFs at other interfaces (e.g., liquid–liquid, liquid–solid, liquid–gas).
Our observations underscore the intricate nature of 2D COF formation on substrates. While the mechanisms elucidated in this study are expected to apply to the formation processes of other types of 2D COFs, factors such as the COF chemical structure and substrate interactions will significantly influence the growth profiles. Understanding this complex interplay of factors as a function of interface and COF structure warrants further investigation.
4. Methods
For simplicity, we directly model the deposition of CHP radicals on metal substrates. Three atomic layers of Cu (111) or Ag (111) of a lateral dimension of 38 nm × 32 nm are used as the substrate, with the bottom layer fixed in the simulations. The General Amber Force Field47 is used to describe the CHP molecules with the restrained electrostatic potential (RESP)48 charges, an approach that has shown good accuracy for small molecules.49–52 The embedded-atom method (EAM)53 is used to describe the metal substrate54 and the interactions between CHP and the metal substrates are described via a Lennard-Jones potential with parameters taken from Heinz.55 We benchmarked the monomer-substrate interactions of the force field in comparison to those obtained at the DFT level (PBE,56 RPBE,57 and PBEsol58) and found that they can be reasonably described around the equilibrium position (see Fig. S10†).The monomers were deposited from a distance of 35–40 Å to the substrate, with an initial velocity of 0.001 Å fs−1 towards the substrate. A total of 400 monomers were deposited over a period of 5 ns, 250 ns, and 500 ns, after which the system was further simulated for another 995 ns, 750 ns, and 500 ns, resulting in a total simulation time of 1000 ns. The final system has 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 and 84840 atoms for simulations on the Ag and Cu substrates, respectively.
100 and 84840 atoms for simulations on the Ag and Cu substrates, respectively.
A parallel algorithm (REACTER)42 is used to perform predefined bond formations among CHP radicals during the MD simulations (see Fig. S11†). Radicals within 3 Å of each other were allowed to form a bond with a probability based on the reaction energy barriers derived from DFT calculations by Bieri et al.15 and prefactors tuned to allow feasible bond formation during the simulation timeframe (see Fig. S12†).59 Reversible reactions were not considered due to their high reaction energy barriers.15
The Nosé-Hoover60 thermostat was used to control the temperature. We considered temperatures between 570 and 620 K. The combination of a temperature higher than those reported experimentally,15 a faster deposition rate, and a faster bonding rate preserves the competition among deposition, diffusion, and bond formation found in actual experimental timescales. The time step was set to 1 fs. The velocity Verlet61 integrator was used for time integration of the potential energy equation.
The Gaussian 16 (version C.02)62 code and Vienna ab initio simulation package (VASP, version 6.3.0)63,64 were used for the density functional theory calculations aimed at comparing the energy profiles from the force field. The Gaussian smearing width was set to 0.1 eV. A plane wave cut-off of 500 eV and a 3 × 5 × 1 k-point mesh were used. All MD simulations were performed with the Large-scale Atomic Molecular Massively Parallel Simulator (LAMMPS, version 2 Aug 2023 update 1).65 The RESP charges of the system was calculated with the Multiwfn66 software (see Fig. S13 and Table S3†). The LJ cutoff was set to 10 Å, and the long-range Coulomb interactions were calculated using the particle–particle/particle-mesh (PPPM)67 method.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
Z. L. Wang conducted molecular dynamics calculations, analyzed the data, and contributed to writing the manuscript. H. Du carried out quantum chemistry calculations and contributed to data visualization. A. M. Evans contributed to mechanistic analysis and manuscript preparation. X. J. Ni contributed to analyzing the monomer-substrate interactions and writing the manuscript. J.-L. Bredas contributed to analyzing the mechanisms and writing the manuscript. H. Y. Li conceived the idea, supervised the project, and contributed to writing the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work at Shanghai University was supported by the National Natural Science Foundation of China (grant number: 22103053) and the Shanghai Technical Service Center of Science and Engineering Computing at Shanghai University. The work at the University of Arizona was supported by the UofA College of Science and the Office of Naval Research under Award no. N00014-24-1-2114.References
- A. M. Evans, M. J. Strauss, A. R. Corcos, Z. Hirani, W. Ji, L. S. Hamachi, X. Aguilar-Enriquez, A. D. Chavez, B. J. Smith and W. R. Dichtel, Two-Dimensional Polymers and Polymerizations, Chem. Rev., 2022, 122(1), 442–564 CrossRef CAS.
- D. D. Medina, M. L. Petrus, A. N. Jumabekov, J. T. Margraf, S. Weinberger, J. M. Rotter, T. Clark and T. Bein, Directional Charge-Carrier Transport in Oriented Benzodithiophene Covalent Organic Framework Thin Films, ACS Nano, 2017, 11(3), 2706–2713 CrossRef CAS PubMed.
- M. S. Lohse and T. Bein, Covalent Organic Frameworks: Structures, Synthesis, and Applications, Adv. Funct. Mater., 2018, 28(33), 1705553 CrossRef.
- H. Gao, H. Wagner, D. Zhong, J. Franke, A. Studer and H. Fuchs, Glaser Coupling at Metal Surfaces, Angew. Chem., Int. Ed., 2013, 52(14), 4024–4028 CrossRef CAS.
- H. Zhou, J. Liu, S. Du, L. Zhang, G. Li, Y. Zhang, B. Z. Tang and H.-J. Gao, Direct Visualization of Surface-Assisted Two-Dimensional Diyne Polycyclotrimerization, J. Am. Chem. Soc., 2014, 136(15), 5567–5570 CrossRef CAS.
- C. Chen, T. Joshi, H. Li, A. D. Chavez, Z. Pedramrazi, P.-N. Liu, H. Li, W. R. Dichtel, J.-L. Bredas and M. F. Crommie, Local Electronic Structure of a Single-Layer Porphyrin-Containing Covalent Organic Framework, ACS Nano, 2018, 12(1), 385–391 CrossRef CAS PubMed.
- J. P. Daum, A. Ajnsztajn, S. A. Iyengar, J. Lowenstein, S. Roy, G. Gao, E. H. Tsai, P. M. Ajayan and R. Verduzco, Solutions Are the Problem: Ordered Two-Dimensional Covalent Organic Framework Films by Chemical Vapor Deposition, ACS Nano, 2023, 17(21), 21411–21419 CrossRef PubMed.
- S. Zint, D. Ebeling, T. Schlöder, S. Ahles, D. Mollenhauer, H. A. Wegner and A. Schirmeisen, Imaging Successive Intermediate States of the On-Surface Ullmann Reaction on Cu (111): Role of the Metal Coordination, ACS Nano, 2017, 11(4), 4183–4190 CrossRef CAS PubMed.
- D. Zhang, G. Wang, C. Chen, T. Joshi, X.-K. Chen, A. M. Evans, M. Matsumoto, W. R. Dichtel, H. Li, M. F. Crommie and J.-L. Brédas, Mechanism of Formation of Benzotrithiophene-Based Covalent Organic Framework Monolayers on Coinage-Metal Surfaces: C–C Coupling Selectivity and Monomer–Metal Interactions, Chem. Mater., 2020, 32(24), 10688–10696 CrossRef CAS.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Controlling On-Surface Polymerization by Hierarchical and Substrate-Directed Growth, Nat. Chem., 2012, 4(3), 215–220 CrossRef CAS PubMed.
- O. Ourdjini, R. Pawlak, M. Abel, S. Clair, L. Chen, N. Bergeon, M. Sassi, V. Oison, J.-M. Debierre, R. Coratger and L. Porte, Substrate-Mediated Ordering and Defect Analysis of a Surface Covalent Organic Framework, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84(12), 125421 CrossRef.
- J. Eichhorn, D. Nieckarz, O. Ochs, D. Samanta, M. Schmittel, P. J. Szabelski and M. Lackinger, On-Surface Ullmann Coupling: The Influence of Kinetic Reaction Parameters on the Morphology and Quality of Covalent Networks, ACS Nano, 2014, 8(8), 7880–7889 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Surface Mediated Synthesis of 2D Covalent Organic Frameworks: 1,3,5-Tris(4-Bromophenyl)Benzene on Graphite(001), Cu(111), and Ag(110), Chem. Commun., 2009,(29), 4456 RSC.
- S. Schlögl, T. Sirtl, J. Eichhorn, W. M. Heckl and M. Lackinger, Synthesis of Two-Dimensional Phenylene–Boroxine Networks through in Vacuo Condensation and on-Surface Radical Addition, Chem. Commun., 2011, 47(45), 12355 RSC.
- M. Bieri, M.-T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, Two-Dimensional Polymer Formation on Surfaces: Insight into the Roles of Precursor Mobility and Reactivity, J. Am. Chem. Soc., 2010, 132(46), 16669–16676 CrossRef CAS.
- R. Dong, T. Zhang and X. Feng, Interface-Assisted Synthesis of 2D Materials: Trend and Challenges, Chem. Rev., 2018, 118(13), 6189–6235 CrossRef CAS.
- N. Keller and T. Bein, Optoelectronic Processes in Covalent Organic Frameworks, Chem. Soc. Rev., 2021, 50(3), 1813–1845 RSC.
- K. Du, L. Xiong, C. Fu, X. Ni, J.-L. Bredas and H. Li, Impact of Structural Defects on the Electronic Properties of Two-Dimensional Covalent Organic Frameworks, ACS Mater. Lett., 2023, 6(2), 335–344 CrossRef.
- H. Sahabudeen, H. Qi, M. Ballabio, M. Položij, S. Olthof, R. Shivhare, Y. Jing, S. Park, K. Liu and T. Zhang, Highly Crystalline and Semiconducting Imine-based Two-dimensional Polymers Enabled by Interfacial Synthesis, Angew. Chem., Int. Ed., 2020, 59(15), 6028–6036 CrossRef CAS.
- I. Castano, A. M. Evans, R. dos Reis, V. P. Dravid, N. C. Gianneschi and W. R. Dichtel, Mapping Grains, Boundaries, and Defects in 2D Covalent Organic Framework Thin Films, Chem. Mater., 2021, 33(4), 1341–1352 CrossRef CAS PubMed.
- R. P. Bisbey, C. R. DeBlase, B. J. Smith and W. R. Dichtel, Two-Dimensional Covalent Organic Framework Thin Films Grown in Flow, J. Am. Chem. Soc., 2016, 138(36), 11433–11436 CrossRef CAS.
- H. B. Balch, A. M. Evans, R. R. Dasari, H. Li, R. Li, S. Thomas, D. Wang, R. P. Bisbey, K. Slicker, I. Castano, S. Xun, L. Jiang, C. Zhu, N. Gianneschi, D. C. Ralph, J.-L. Brédas, S. R. Marder, W. R. Dichtel and F. Wang, Electronically Coupled 2D Polymer/MoS2 Heterostructures, J. Am. Chem. Soc., 2020, 142(50), 21131–21139 CrossRef CAS PubMed.
- Z. Jiang, R. Dong, A. M. Evans, N. Biere, M. A. Ebrahim, S. Li, D. Anselmetti, W. R. Dichtel and A. G. Livingston, Aligned Macrocycle Pores in Ultrathin Films for Accurate Molecular Sieving, Nature, 2022, 609(7925), 58–64 CrossRef CAS.
- F. Yu, W. Liu, S.-W. Ke, M. Kurmoo, J.-L. Zuo and Q. Zhang, Electrochromic Two-Dimensional Covalent Organic Framework with a Reversible Dark-to-Transparent Switch, Nat. Commun., 2020, 11(1), 5534 CrossRef CAS PubMed.
- A. M. Evans, A. Giri, V. K. Sangwan, S. Xun, M. Bartnof, C. G. Torres-Castanedo, H. B. Balch, M. S. Rahn, N. P. Bradshaw, E. Vitaku, D. W. Burke, H. Li, M. J. Bedzyk, F. Wang, J.-L. Brédas, J. A. Malen, A. J. H. McGaughey, M. C. Hersam, W. R. Dichtel and P. E. Hopkins, Thermally Conductive Ultra-Low-k Dielectric Layers Based on Two-Dimensional Covalent Organic Frameworks, Nat. Mater., 2021, 20(8), 1142–1148 CrossRef CAS PubMed.
- L. Wang, X. Wang, Z.-L. Zhao, L.-J. Wan and D. Wang, Stranski–Krastanov Growth of Two-Dimensional Covalent Organic Framework Films, J. Am. Chem. Soc., 2024, 146(20), 14079–14085 CrossRef CAS PubMed.
- Z. Ou, B. Liang, Z. Liang, F. Tan, X. Dong, L. Gong, P. Zhao, H. Wang, Y. Zou, Y. Xia, X. Chen, W. Liu, H. Qi, U. Kaiser and Z. Zheng, Oriented Growth of Thin Films of Covalent Organic Frameworks with Large Single-Crystalline Domains on the Water Surface, J. Am. Chem. Soc., 2022, 144(7), 3233–3241 CrossRef CAS.
- K. Sun, O. J. Silveira, Y. Ma, Y. Hasegawa, M. Matsumoto, S. Kera, O. Krejčí, A. S. Foster and S. Kawai, On-Surface Synthesis of Disilabenzene-Bridged Covalent Organic Frameworks, Nat. Chem., 2023, 15(1), 136–142 CrossRef CAS.
- T. Niu, C. Hua and M. Zhou, On-Surface Synthesis toward Two-Dimensional Polymers, J. Phys. Chem. Lett., 2022, 13(34), 8062–8077 CrossRef CAS PubMed.
- G. Zhan, Z.-F. Cai, K. Strutyński, L. Yu, N. Herrmann, M. Martínez-Abadía, M. Melle-Franco, A. Mateo-Alonso and S. D. Feyter, Observing Polymerization in 2D Dynamic Covalent Polymers, Nature, 2022, 603(7903), 835–840 CrossRef CAS PubMed.
- B. T. Koo, R. F. Heden and P. Clancy, Nucleation and Growth of 2D Covalent Organic Frameworks: Polymerization and Crystallization of COF Monomers, Phys. Chem. Chem. Phys., 2017, 19(15), 9745–9754 RSC.
- V. Nguyen and M. Grünwald, Microscopic Origins of Poor Crystallinity in the Synthesis of Covalent Organic Framework COF-5, J. Am. Chem. Soc., 2018, 140(9), 3306–3311 CrossRef CAS.
- W. Hao, C. Sui, G. Cheng, J. Li, L. Miao, G. Zhao, Y. Sang, J. Li, C. Zhao, Y. Zhou, Z. Zang, Y. Zhao, X. He and C. Wang, Dynamic Insights into the Growth Mechanisms of 2D Covalent Organic Frameworks on Graphene Surfaces, ACS Nano, 2024, 18(15), 10485–10494 CrossRef CAS PubMed.
- R. Iftimie, P. Minary and M. E. Tuckerman, Ab Initio Molecular Dynamics: Concepts, Recent Developments, and Future Trends, Proc. Natl. Acad. Sci. U.S.A., 2005, 102(19), 6654–6659 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik and H. M. Aktulga, The ReaxFF Reactive Force-Field: Development, Applications and Future Directions, npj Comput. Mater., 2016, 2(1), 1–14 CrossRef.
- A. C. Van Duin, S. Dasgupta, F. Lorant and W. A. R. F. F. Goddard, A Reactive Force Field for Hydrocarbons, J. Phys. Chem. A, 2001, 105(41), 9396–9409 CrossRef CAS.
- H. Li, A. D. Chavez, H. Li, H. Li, W. R. Dichtel and J.-L. Bredas, Nucleation and Growth of Covalent Organic Frameworks from Solution: The Example of COF-5, J. Am. Chem. Soc., 2017, 139(45), 16310–16318 CrossRef CAS.
- H. Li, A. M. Evans, W. R. Dichtel and J.-L. Bredas, Quantitative Description of the Lateral Growth of Two-Dimensional Covalent Organic Frameworks Reveals Self-Templation Effects, ACS Mater. Lett., 2021, 3(4), 398–405 CrossRef CAS.
- J. Tian, K. A. Treaster, L. Xiong, Z. Wang, A. M. Evans and H. Li, Taming Two-Dimensional Polymerization by a Machine-Learning Discovered Crystallization Model, Angew. Chem., Int. Ed., 2024, e202408937 CAS.
- A. Chatterjee and D. G. Vlachos, An Overview of Spatial Microscopic and Accelerated Kinetic Monte Carlo Methods, J. Comput.-Aided Mater. Des., 2007, 14, 253–308 CrossRef.
- B. Puchala, M. L. Falk and K. Garikipati, An Energy Basin Finding Algorithm for Kinetic Monte Carlo Acceleration, J. Chem. Phys., 2010, 132(13), 134104 CrossRef.
- J. R. Gissinger, B. D. Jensen and K. E. Wise, REACTER: A Heuristic Method for Reactive Molecular Dynamics, Macromolecules, 2020, 53(22), 9953–9961 CrossRef CAS.
- W. Pisula, M. Kastler, C. Yang, V. Enkelmann and K. Müllen, Columnar Mesophase Formation of Cyclohexa-m-phenylene-Based Macrocycles, Chem.–Asian J., 2007, 2(1), 51–56 CrossRef CAS.
- J. P. Perdew and A. Zunger, Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23(10), 5048 CrossRef CAS.
- N. A. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, Organized Formation of 2D Extended Covalent Organic Frameworks at Surfaces, J. Am. Chem. Soc., 2008, 130(21), 6678–6679 CrossRef CAS PubMed.
- F. Haase and B. V. Lotsch, Solving the COF Trilemma: Towards Crystalline, Stable and Functional Covalent Organic Frameworks, Chem. Soc. Rev., 2020, 49(23), 8469–8500 RSC.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and Testing of a General Amber Force Field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model, J. Phys. Chem., 1993, 97(40), 10269–10280 CrossRef CAS.
- S. C. Lau and C. L. Bilodeau, Effect of Monovalent Cations on the Structure and Dynamics of Multimodal Chromatographic Surfaces, Langmuir, 2024, 40(13), 6694–6702 CrossRef CAS PubMed.
- X. Lyu, H. Wang, X. Liu, L. He, C. Do, S. Seifert, R. E. Winans, L. Cheng and T. Li, Solvation Structure of Methanol-in-Salt Electrolyte Revealed by Small-Angle X-Ray Scattering and Simulations, ACS Nano, 2024, 18(9), 7037–7045 CrossRef CAS PubMed.
- Z. Liu, M. Lin, C. Liu, X. Chen, Q. Chen, X. Li, X. Wu, Y. Wang, L. Wang and F. Yang, Development of (2-(Benzyloxy) Phenyl) Methanamine Derivatives as Potent and Selective Inhibitors of CARM1 for the Treatment of Melanoma, J. Med. Chem., 2024, 67(8), 6313–6326 CrossRef CAS.
- R. Gradisch, K. Schlögl, E. Lazzarin, M. Niello, J. Maier, F. P. Mayer, L. Alves da Silva, S. M. Skopec, R. D. Blakely and H. H. Sitte, Ligand Coupling Mechanism of the Human Serotonin Transporter Differentiates Substrates from Inhibitors, Nat. Commun., 2024, 15(1), 417 CrossRef CAS.
- S. Foiles, M. Baskes and M. S. Daw, Embedded-Atom-Method Functions for the Fcc Metals Cu, Ag, Au, Ni, Pd, Pt, and Their Alloys, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 7983 CrossRef CAS.
- G. M. Leuty, A. Abu-Nada and M. Tsige, Multilayer Adsorption of Methane and Chloromethane on the Molybdenum (100) Surface, J. Phys. Chem. C, 2012, 116(27), 14514–14525 CrossRef CAS.
- H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12–6 and 9–6 Lennard-Jones Potentials, J. Phys. Chem. C, 2008, 112(44), 17281–17290 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(11), 7413 CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100(13), 136406 CrossRef PubMed.
- M. Monteferrante, A. Tiribocchi, S. Succi, D. Pisignano and M. Lauricella, Capturing Free-Radical Polymerization by Synergetic Ab Initio Calculations and Topological Reactive Molecular Dynamics, Macromolecules, 2022, 55(5), 1474–1486 CrossRef CAS PubMed.
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A, 1992, 31(3), 1695–1697 CrossRef.
- L. Verlet, Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules, Phys. Rev., 1967, 159(1), 98 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16 Revision C. 01, 2016, Gaussian Inc., Wallingford CT, 2016, 1, 572 Search PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 1995, 117(1), 1–19 CrossRef CAS.
- T. Lu and F. M. Chen, A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- R. W. Hockney and J. W. Eastwood, Computer Simulation Using Particles, crc Press, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05168h |
| This journal is © The Royal Society of Chemistry 2024 |