
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The ubiquitous P(o-tol)3 ligand promotes formation of catalytically-active higher order palladacyclic clusters†
David R.
Husbands
 a,
Theo
Tanner
a,
Adrian C.
Whitwood
a,
Neil S.
Hodnett
b,
Katherine M. P.
Wheelhouse
b and
Ian J. S.
Fairlamb
*a
a,
Theo
Tanner
a,
Adrian C.
Whitwood
a,
Neil S.
Hodnett
b,
Katherine M. P.
Wheelhouse
b and
Ian J. S.
Fairlamb
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk
bMedicine Development & Supply, GSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
First published on 23rd October 2024
Abstract
The Herrmann–Beller catalyst, Pd[(C^P)(μ2-OAc)]2, is readily formed by reaction of the cyclic trimer of ‘Pd(OAc)2’ with P(o-tol)3. In the presence of hydroxide, Pd(C^P)(μ2-OAc)]2 converts to [Pd(C^P)(μ2-OH)]2. Here, we report how this activated Pd precatalyst species, and related species, serve as a conduit for formation of higher order Pdn clusters containing multiple cyclopalladated P(o-tol)3 ligands. The catalytic competency of a Pd4-palladacyclic cluster is demonstrated in an arylated Heck cross-coupling, which is comparable to the base-activated form of Herrmann's catalyst, namely [Pd(C^P)(μ2-OH)]2. The findings show that ‘simple’ ubiquitous phosphine ligands can promote higher order Pd speciation, moving beyond well-known phosphine-ligated Pd1 and Pd2 complexes. The findings challenge the status quo in the field, in that phosphine ligands can ligate higher order Pdn species which are catalytically competent species in cross-coupling reactions.
Introduction
Tri-ortho-tolyl phosphine, P(o-tol)3, is a bench-stable phosphine ligand which is widely applied in some of the most challenging Pd-catalyzed cross-coupling reactions. P(o-tol)3 has an unusually large ligand cone angle (194°)1 and facile ability to undergo cyclometallation, especially at PdII.2 Reaction screening campaigns often show that P(o-tol)3 effectively competes with specialist ‘designer’ electron-rich, sterically bulky phosphines.3 Of the many interesting applications of the P(o-tol)3 ligand, perhaps one of the most curious and interesting findings was deuteration of its ortho-methyl substituents influences branched/linear isomer product ratios in the Suzuki–Miyaura cross-couplings (SMCCs) of non-activated Csp3-boronic acids with aryl halides, an outcome that is not readily explained with simple Pd0(P(tol)3)n catalyst models.4 Indeed, the related ‘simpler’ ligand, PPh3, can form multiple catalyst species, including higher order Pd3 clusters and nanoparticles.5 Furthermore, neutral PPh3 can be converted to anionic PPh2 ligands in PdII pre-catalyst activation processes.6 Out of curiosity emerges the question whether P(o-tol)3 can engage in similar ligand promiscuity to PPh3, influencing downstream Pd speciation and potentially different reaction outcomes.Herrmann–Beller palladacycle [Pd(C^P)(μ2-OAc)]21 is particularly effective for high temperature arylative Heck reactions,7 allowing for ultra-low Pd-catalyst loadings (TONs up to 1 × 106).8 Application of the Pd(OAc)2/P(o-tol)3 pre-catalyst system is also effective (forming 1in situ) for an eclectic array of transformations (Fig. 1).9 Various mechanistic proposals8,10 have been put forward for these pre-catalyst systems, from higher order Pd nanoparticles involving low-order Pd leaching, through to the operation of higher oxidation state PdIV species.
 | ||
| Fig. 1 Highlighting the previous work: the historical findings summarized for 1 and recent implication of hydroxo-bridged species 2 in SMCC reactions. This work, revealing the hidden complexity and formation of higher order palladacycles involving P(o-tolyl)3. | ||
Despite the mechanistic conundrums11 involving the study of PdII pre-catalyst systems, it is evident that there is an inherent complexity meriting deeper investigation. Indeed, in our recent work we have found that a stable [Pd(C^P)(μ2-OH)]22 palladacycle is readily formed from the reaction of 1 with hydroxide base, giving a highly active Pd catalyst system for SMCC reactions under mild conditions.12 The facile formation and stability of 2 supports the water-assistive activation of 1 required for arylative Heck reactions.13 Our previous findings complement findings on SMCC reaction mechanisms, where intermediate ‘PdII–OH’ species have been implicated in productive catalysis.14
Here, we report how 2 and related derivatives can form higher order Pdn clusters (n = 4, 6 and 8), under different reaction conditions, and show that Pd4-cluster 3 is a competent pre-catalyst for Heck reactions. It is clear from our studies that the P(o-tol)3 ligand readily stabilizes higher order PdII palladacyclic cluster species, while undergoing P–C bond cleavage under specific conditions. Furthermore, palladacycles derived from P(o-tol)3 are able to react with cross-coupling reagents.
Results and discussion
While the [Pd(C^P)(μ2-OH)]22 pre-catalyst is an air and water stable complex,12 under dehydrating or limited water conditions, the complex readily loses H2O to form a new species, which was determined to be Pd4(μ4-O)(μ2-OH)2 cluster 3 by single crystal X-ray diffraction (XRD) analysis (Fig. 2). In solution, 2 exists as a mixture of trans and cis isomers (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio), but only as the trans-isomer in the solid-state. The act of removing solvent from a pure sample of 2 in solution is enough to form small quantities of 3 (observed as a color change from a cream to a yellow solid). Due to steric limitations, only the minor cis isomer of 2 can dimerize (inferred by the structure of 3). One of the unusual structural features of 3 is a four-coordinate tetrahedral oxygen (μ4-O) atom connecting four separate Pd atoms. There are two Pd–O bonds that are shorter (2.070(2) and 2.070(2) Å) and two Pd–O bonds that are marginally longer (2.089(2) and 2.088(2) Å). The former represents the anionic component to the μ4-O-bond arrangement, whereas the latter are dative in nature. The Pd–OH bond lengths (2.202(2) Å) are significantly longer than the Pd–O bonds (2.070(2) Å). The Pd–OH bond distances are also longer than seen in 2 {for 2, Pd–OH 2.115(2)}, so there appears to be an element of ‘relaxation’ in the structure of 3 compared with 2.
:1 ratio), but only as the trans-isomer in the solid-state. The act of removing solvent from a pure sample of 2 in solution is enough to form small quantities of 3 (observed as a color change from a cream to a yellow solid). Due to steric limitations, only the minor cis isomer of 2 can dimerize (inferred by the structure of 3). One of the unusual structural features of 3 is a four-coordinate tetrahedral oxygen (μ4-O) atom connecting four separate Pd atoms. There are two Pd–O bonds that are shorter (2.070(2) and 2.070(2) Å) and two Pd–O bonds that are marginally longer (2.089(2) and 2.088(2) Å). The former represents the anionic component to the μ4-O-bond arrangement, whereas the latter are dative in nature. The Pd–OH bond lengths (2.202(2) Å) are significantly longer than the Pd–O bonds (2.070(2) Å). The Pd–OH bond distances are also longer than seen in 2 {for 2, Pd–OH 2.115(2)}, so there appears to be an element of ‘relaxation’ in the structure of 3 compared with 2.
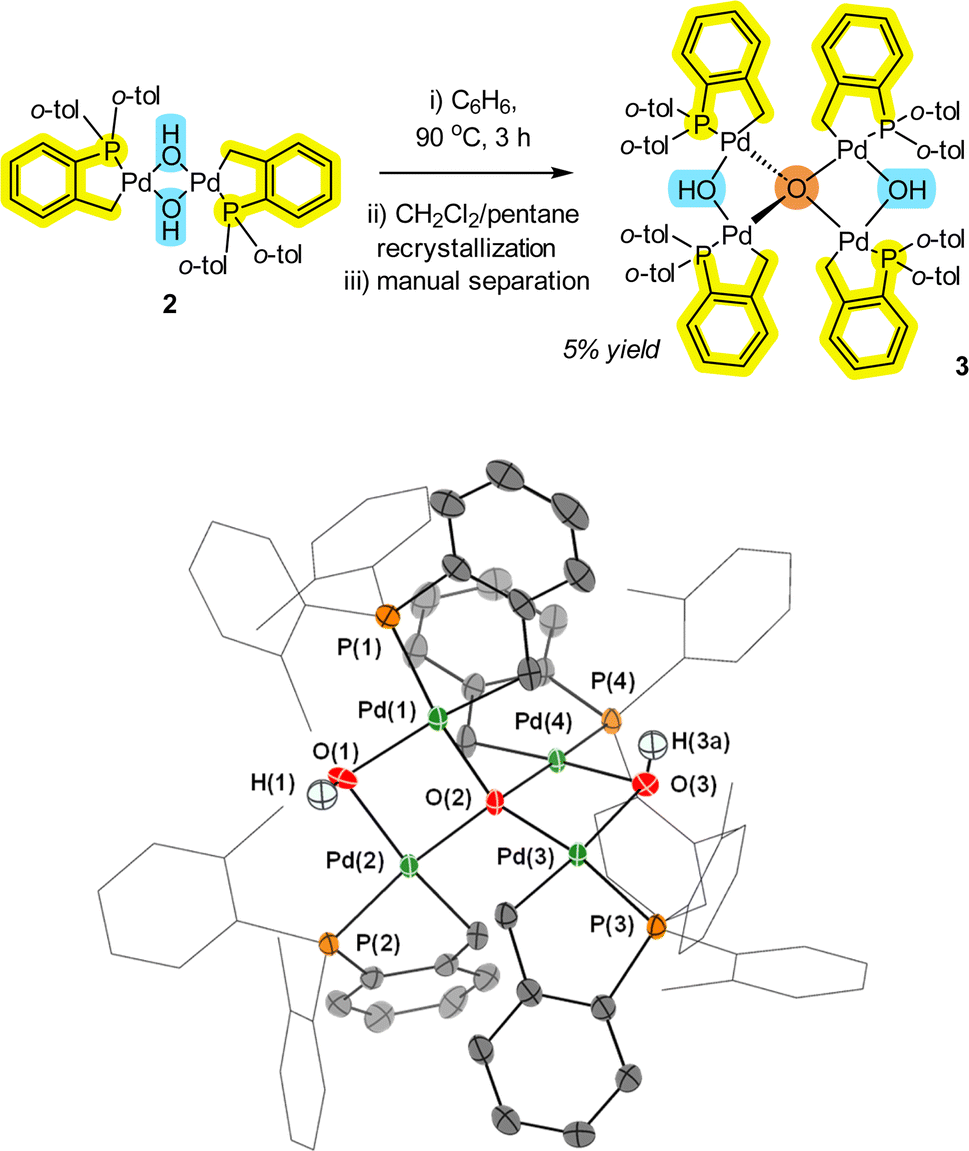 | ||
| Fig. 2 Formation of Pd4 cluster 3 from 2. The single crystal XRD structure of 3 is shown with selected ellipsoids (set to 50%) for clarity (all H-atoms with the exception of the bridging-hydroxo ligands have been hidden from view). Selected interatomic lengths/Å: Pd1–C1 = 2.032(4); Pd1–P1 = 2.1841(9); Pd1–O1 = 2.202(2); Pd1–O2 = 2.070(2); Pd1–Pd2 = 3.1785(4). Selected interatomic angles/°: P1–Pd1–C1 = 84.52(12); P1–Pd1–O1 = 102.74(18); Pd1–O1–Pd2 = 92.89(10); Pd1–O2–Pd2 = 99.66(10); Pd3–O2–Pd4 = 97.35(9)>. | ||
As the synthesis of 3 involves refluxing 2 at 90 °C for 3 h, it is of particular note that the stability of this palladacycle is maintained at elevated temperatures. Interestingly, Pd4 cluster 3 is similar to a structure formed involving reactions of Pd3(OAc)6 as reported by Bedford et al.15 namely [{Pd3(OAc)5}2(μ4-O)]. Under water-limiting conditions (note: not water-free) this demonstrates that aggregation of the PdII pre-catalyst species is possible for both 1 and ubiquitous [Pd3(OAc)6] vide infra. Similar higher order Pdn clusters have been implicated (by kinetic studies) in arylative Pd-catalyzed cyanation reactions mediated by [Pd(OAc)2(HNR2)2] pre-catalysts.16 Our findings therefore make a connection between non-phosphine and phosphine-ligated Pdn clusters.
In a separate reaction we found that [Pd(C^P)(μ2-Cl)]24 (see ESI† for synthesis), when reacted with AgC6F5 at room temperature, formed a small number of bright yellow single crystals (compared to red crystals for target compound 6).
Interestingly, pre-heating with AgC6F5 at 100 °C under vacuum to remove residual EtCN from the synthesis, and changing the reaction solvent from DCM to THF results in the exclusive formation of the transmetallation product 6 and its monomer.12 Single crystal XRD analysis of the yellow crystals showed that a related Pd4 cluster 5 was formed, which is the chloro-analogue of 3 (Fig. 3). The interesting implication here is that palladacycle compounds containing four Pd atoms can readily form, templated by a (μ4-O)-bonding arrangement (the O is likely from adventitious oxygen in the reaction vessel), such as seen earlier for non-phosphine ligated Pd3 cluster [{Pd3(OAc)5}2(μ4-O)].15
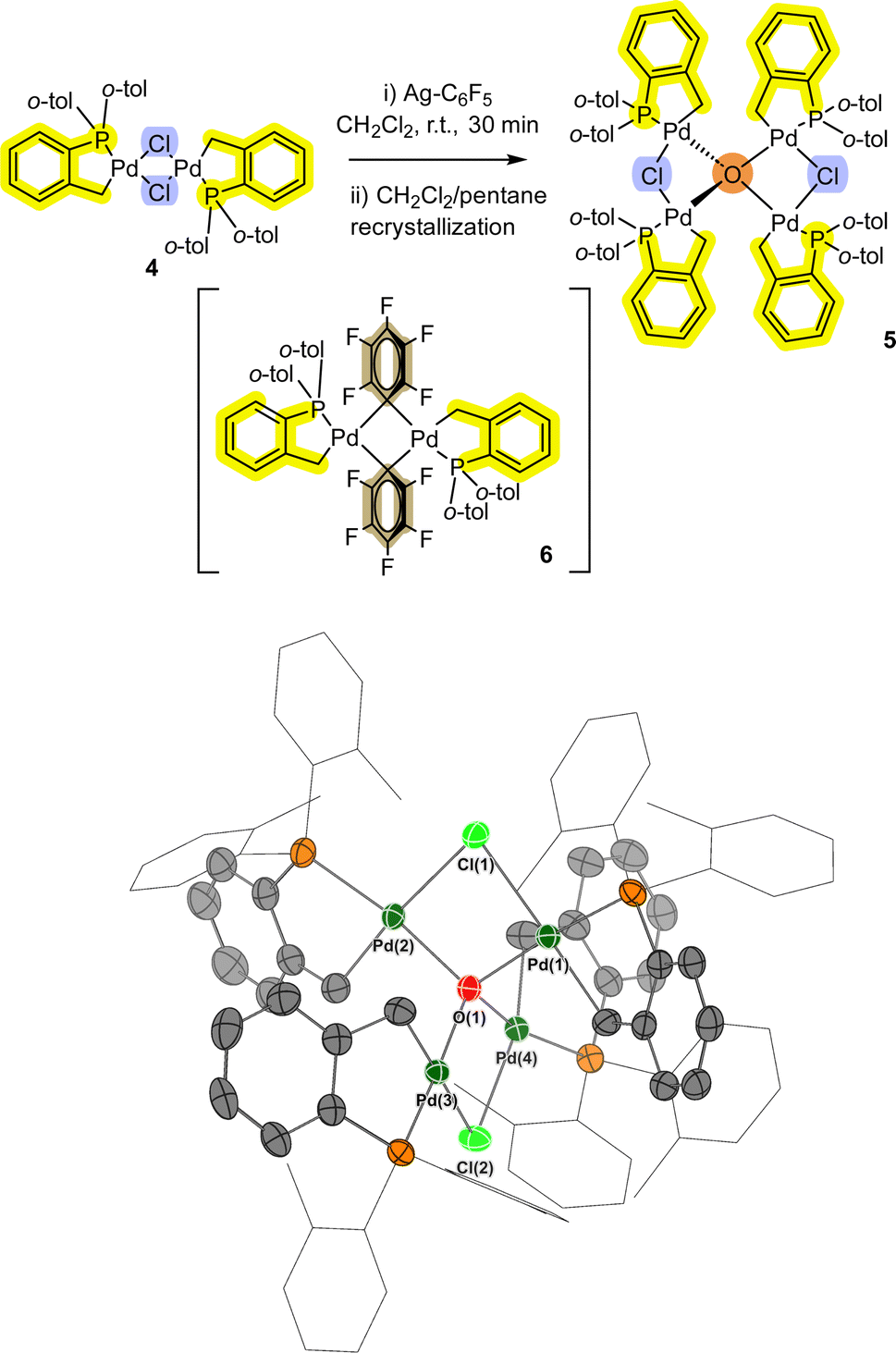 | ||
| Fig. 3 Formation of Pd4 cluster 5 from palladacycle 4. The single crystal XRD structure of 5 is shown with selected ellipsoids (set to 50%) for clarity (all H-atoms, with the exception of the bridging-hydroxo ligands, have been hidden from view). Selected interatomic lengths/Å: Pd1–Cl1 = 2.4737(8), Pd1–O1 = 2.101(2), Pd1–P1 = 2.2093(9), Pd1–C1 = 2.052(3). Selected interatomic angles/°: Pd1–Cl1–Pd2 = 84.35(3), Pd3–Cl2–Pd4 = 84.67(3), Pd1–O1–Pd4 = 103.46(9), Pd2–O1–Pd3 = 113.65(10). | ||
Returning to Pd4 cluster 3, on one occasion under specific reaction conditions we obtained single crystals of a new species which was determined by XRD analysis to be Pd6 cluster 7 (Fig. 4). In this case, Pd[(C^P)(μ2-OH)]22 was refluxed in benzene, filtered, crystallized from THF/water at 5 °C, which was exposed to air via a bleed needle for a period of 2 months. Structural features of note are: (1) a planar Pd3 motif with capping OH groups; (2) a bridging P(o-tol)2 palladacycle which has had one o-tol group cleaved onto a neighboring Pd atom; (3) the appearance of an ionic O2PMe2 group which implies that two o-tol methyl substituents have been cleaved from a P(o-tol)3 ligand.
 | ||
| Fig. 4 Formation of Pd6 cluster 7 from palladacycle 2. The single crystal XRD structure of 7 is shown with selected ellipsoids (set to 50%) for clarity (all H-atoms with the exception of the bridging-hydroxo ligands have been hidden from view). Selected bond lengths (Å) and angles (°); Pd1–P1 = 2.201(2); Pd1–O1 = 2.141(6); Pd1–Pd2 = 3.0422(9); Pd2–P3 = 2.218(2); Pd3–P4 = 2.238(2); O2–P2 = 1.631(6); P2–C22 = 1.878(9); Pd1–Pd2–Pd3 = 59.30(2); O1–Pd1–O2 = 70.5(2); Pd1–O2–P2 = 129.7(3); C22–P2–C23 = 109.7(4). | ||
Alternative possibilities for the O2PMe2 anion were fully considered. Our computational calculations (using DFT methods) suggest that the O2PMe2 anion is the most likely (compared with other plausible anions to O2SMe2, O2PO2 and O2P{(OH)2}) (see ESI†). Clearly the formation of Pd6 cluster 7 has involved at least two P–C bond activations, with some palladacyclic motifs remaining intact, an observation that is testament to the stabilization conferred by cyclopalladation. The formation of “Pd(o-tol)” corner motifs is particularly noteworthy. It is interesting to note that Sunada et al. reported a nuclearity expansion in Pdn clusters triggered by a migrating phenyl group from cyclooligosilanes, a process that is reminiscent of that observed in the formation of 7.17
Along with Pd4 clusters 3 and 5, Pd6 cluster 7 demonstrates that under water-limiting conditions that higher order PdII clusters can be formed, which is relevant to catalytic cross-coupling reaction conditions (vide infra).
Next, in a study involving reaction of 2 by aryl boronic acids,12 further complexity involving higher order Pdn species possessing palladacyclic motifs was revealed. Reaction of 2 with arylboronic acid 9 (2 equiv.), in the presence of excess boric acid (4 equiv.) and octafluoronaphthalene (1 equiv.) showed the formation of a broad lump between δ 30–40 ppm by 31P NMR spectroscopic analysis, accounting for 93% of the observed 31P signals (see ESI†). This indicates that other Pd species are likely formed. Upon addition of 4-fluorobromobenzene, no reaction was observed, implying these species contain oxidised PdII centres vide infra (noting that Pd0 would undergo an oxidative addition). As such the crude reaction mixture was set-up for crystallisation (i.e. using THF/pentane, slow vapor diffusion over 17 months). Single crystal XRD analysis revealed the formation of a remarkable Pd8 cluster 8 (Fig. 5). While there is disorder in the structure, a suitable model was constructed (see ESI,† Section 6). Firstly, Pd8 cluster 8 possesses a distinctive bridging μ4-F atom. Pd8 cluster 8 also possesses bridging oxygens from two stabilizing-boronate anions. Therefore, the Pd8 cluster core motif in 8 carries an overall +2 charge. Two of the central Pd atoms are not cyclopalladated, leaving 6 palladacyclic motifs within this unique Pd8-cluster.
 | ||
| Fig. 5 Formation of Pd8 cluster 8 from palladacycle 2 (the Pd atoms have been formatted in bold for clarity). The single crystal XRD structure of 8 is shown with selected ellipsoids (set to 50%) for clarity (all H-atoms have been hidden from view). Selected interatomic lengths/Å: P1–Pd1 = 2.1856(10); Pd1–C7 = 2.024(5); Pd1–F1A = 2.02(3); Pd2A–F1A = 2.10(2); Pd3A–F1A = 2.06(3); Pd4–F1A = 2.04(3); Pd4–O1 = 1.991(3); Pd4–O4 = 1.980(3). Selected interatomic angles/°: O4–Pd4–O1 = 95.56(13); Pd1–F1A–Pd2A = 98.7(10); Pd1–F1A–Pd4 = 97.7(13); Pd2A–F1A–Pd3A = 131.2(14); Pd1–F1A–Pd3A = 120.6(13); P1–Pd1–C7 = 83.77(14). | ||
Given the changes seen in both 31P and 19F NMR spectral data we postulate the involvement of intermediates (Int-I and Int-II),12 in the transformation leading to the formation of 8. It is pertinent to mention the formation of organic by-/side-products, fluorobenzene (formed by protodeborylation) and 4,4′-difluoro-biphenyl (formed by homocoupling of the 4′-fluorophenyl boronic acid). The appearance of fluoride ion in 8 suggests an abstraction of fluoride from either octofluoronaphthalene, p-F–C6H4–B(OH)2 or other fluoride-containing by- or side-product.
Catalysis
We chose the Heck reaction of an aryl halide 9 with terminal alkene 10 (forming 11, as a single E-isomer) to benchmark the catalytic performance of Pd4 cluster 3 relative to known12 dinuclear complex 2 (Fig. 6). This class of catalytic reaction is underpinned by a significant body of mechanistic information involving palladacyclic pre-catalysts, making it the ideal candidate in which to assess the catalytic competency of 3.13 The catalytic arylative Heck reaction was further amenable to reaction monitoring using in situ IR (Mettler-Toledo RIR15 with a flexible AgX diamond probe), which allows the solution changes in the IR stretching bands of the different reaction components (see Fig. 6 caption details) to be monitored in real-time (note: the second derivative was used to process the raw spectral data, in-keeping with the kinetic analyses conducted on Suzuki–Miyaura cross-couplings as part of an extended but related study examining the catalytic behaviour of 2).12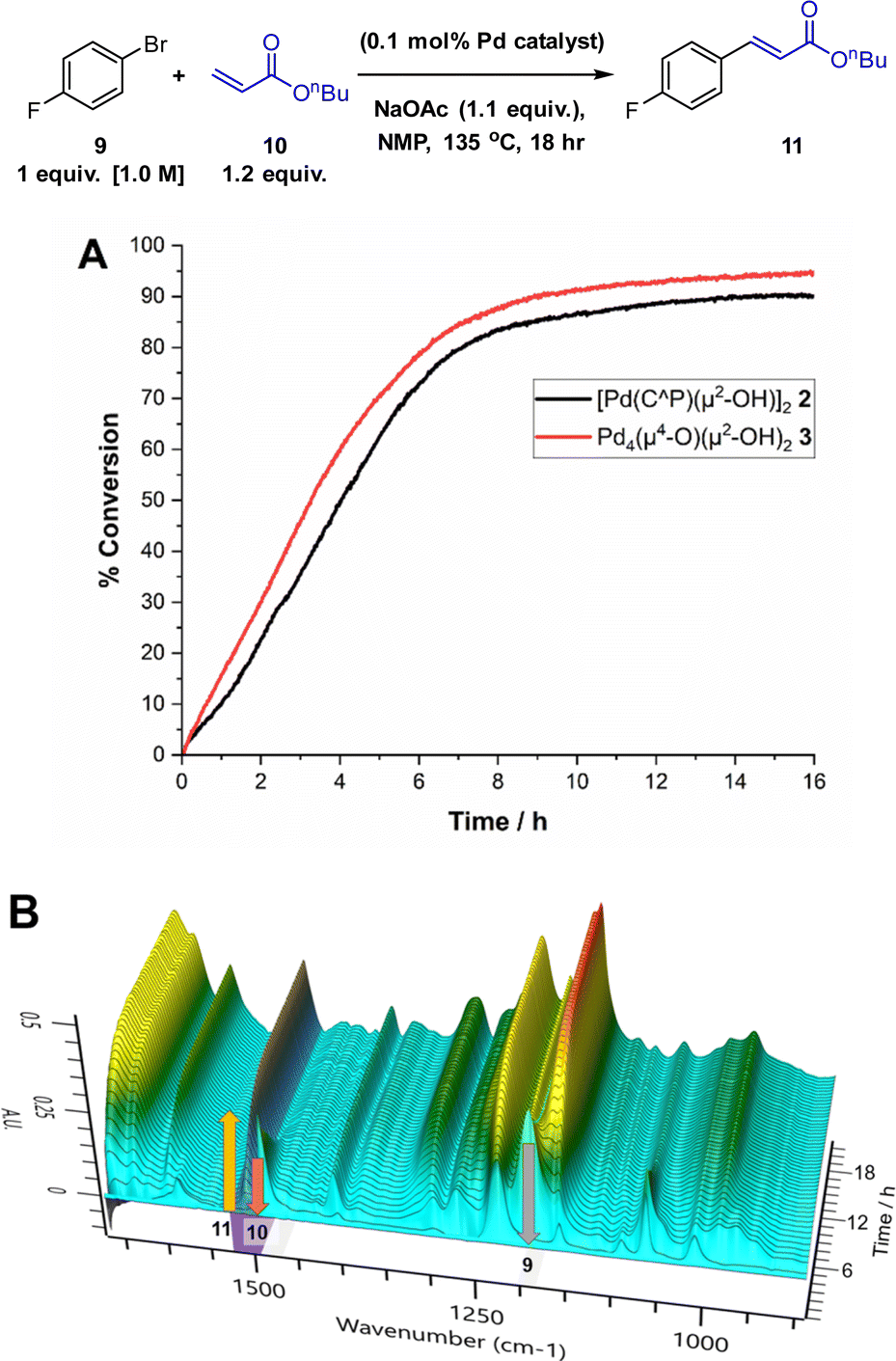 | ||
| Fig. 6 Model Heck arylation reaction to test Pd4 cluster 3 against intermediate dinuclear Pd species 2. (A) Kinetic profiles showing the formation of n-butyl 4-fluorocinnamate 11 (product appearance at 1509 cm−1). (B) Temporal IR spectral changes underpinning the kinetic changes shown in A. Formation of 11 at 1509 cm−1 and loss of 1-bromo-4-fluorobenzene 10 at 1484 cm−1 and n-butyl acrylate 9 at 1190 cm−1 (reaction mixture compared with reference standards in NMP solvent). | ||
Using 2 as the precatalyst, the reaction 9 + 10 → 11 reaches 80% conversion (to product 11) after 8 h (comparable to 1H NMR spectroscopic analysis), providing a good window to compare the catalytic performance of Pd4 cluster 3. Under anhydrous reaction conditions, single crystals of 3 exhibited catalytic activity commensurate with 2, as shown by the similar rates of product formation and shape of the product (11) evolution curves. In the presence of water (a necessary additive, likely generating 2in situ12) the Herrmann–Beller catalyst 1 exhibits similar catalytic activity to 2 and 3 (see ESI†). The results taken together allow us to make a link between 1, 2 and 3 for the Heck reaction 9 + 10 → 11 (note ∼2% homocoupled product, 4,4′-difluoro-biphenyl, was also observed by NMR spectroscopic analysis).
The overall outcome from these comparative catalytic reactions support the assertion that under water-limiting conditions (<50 ppm H2O at 0.1 mol% Pd catalyst loading), both Pd2 complex 2 and Pd4 cluster 3 are stepping-stones to higher order Pdn species. PdNP catalysed arylative Heck reactions are well known.8,13d Thus, the catalytic competency of 3 provides evidence that P(o-tol)3 based Pd pre-catalysts can bridge the gap to formation of PdNPs via this route involving higher order Pdn cluster species.
Further discussion regarding Pd4 cluster formation
It is pertinent to reflect on the formation of the higher order Pd4 cluster 3 and how it connects to Pdn species of lower nuclearity (Fig. 7). It is evident that both solvent and water need to be considered in equilibrium events described in Fig. 7. The equilibrium balance will be dependent on the solvent type and concentration of water. Palladacyclic dinuclear complexes, such as 2, are in equilibrium with mononuclear Pd1 complexes.18 The equilibrium position will be expected to change when employing polar aprotic solvents and where there are heterocyclic systems present (possessing strong 2-electron nitrogen donor atoms, like pyridine,18 for example).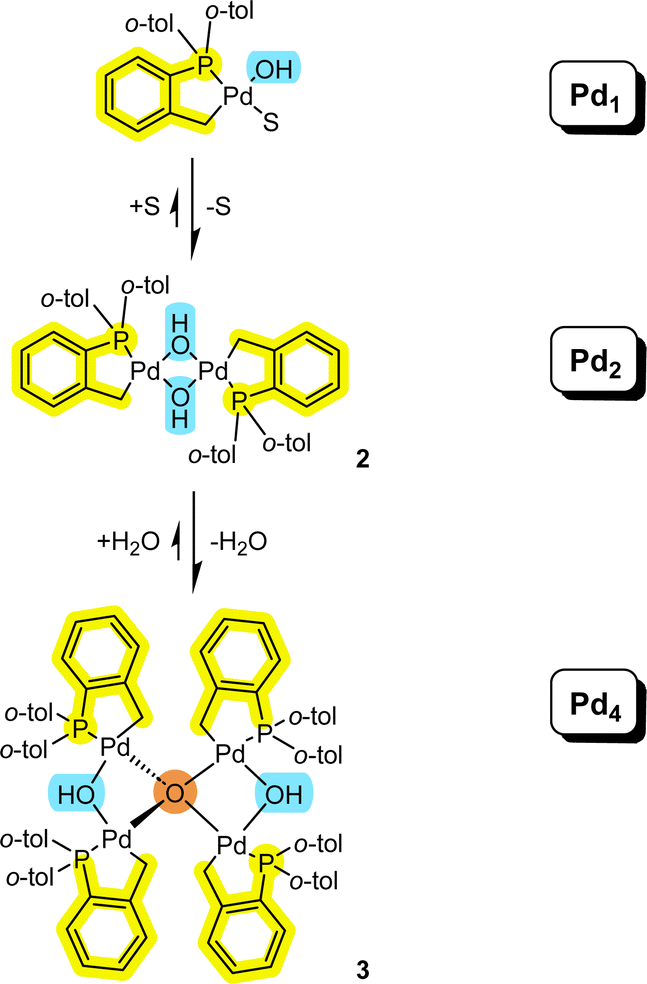 | ||
| Fig. 7 Connecting mononuclear palladacyclic species with the higher order Pd4 type cluster 3 (S = solvent). | ||
The formation of Pdn clusters 7 and 8 is quite remarkable – they are ‘a needle in a haystack’ type result. That said, their formation demonstrates that the P(o-tol)3 ligand can be chemically-fragmented, templating and stabilising these higher order Pdn species. It is an important serendipitous result.
Conclusions
The ubiquitous P(o-tol)3 ligand is capable of templating and stabilizing higher order Pdn clusters (where n = 4, 6 and 8). Our research findings challenge the status quo – higher order palladacycles are readily accessible under different reaction conditions and are stable and catalytically relevant. Pd4 cluster 3 was found to exhibit catalytic activity similar to both 1 and 2. Generally, the P(o-tol)3 ligand has unique electronic and steric properties, which has been widely applied in Pd-catalysed cross-coupling reactions since their discovery in the 1960s/1970s.1 It is common to see in the literature simple models used to describe ‘Pd–P(o-tol)3’ catalyst species, including ligand parameterization methodologies.2 Our findings show that higher order species need to be factored into more complete models for catalytic reaction manifolds, particularly with reaction complexity19 in mind. Our findings further add to related observations made for the aggregation of Pdn species, containing the Trost Modular ligand, made by Lloyd-Jones et al. in asymmetric allylic alkylation reactions.20 Formation of higher order palladacyclic clusters also provides clues to pathways leading to the generation of PdNPs.Data availability
We have provided a comprehensive electronic version of ESI.† The raw spectroscopic data (e.g. NMR fid files) and kinetic data (.csv files) will be uploaded to the York Research Database (the open data repository for the University of York) on publication of the manuscript. We can be contacted by email for processed data files (e.g. MNova NMR files and Excel/Origin files), although we are unable to provide access to the licensed commercial software. The cif files for the single crystal XRD structures are available through the Cambridge Crystallographic Data Centre (CCDC).21Author contributions
N.·S.·H., K. M. P. W. and I. J. S. F. jointly conceived and designed the research project focusing on Herrmann's catalyst 1, securing iCASE funding from GSK and EPSRC (UKRI). D. R. H. and I. J. S. F. devised experiments, supported by input from N.·S. H., K. M. P. W. and D. R. H. conducted all of coordination chemistry and catalysis experiments described in the manuscript. I. J. S. F. and D. R. H. co-wrote the manuscript, with input provided by all authors. D. R. H. prepared the electronic ESI† document, with input from all authors. A. C. W. and T. T. were involved in the data collection and processing of single crystal X-ray diffraction data (A. C. W. further refined the XRD structural data following feedback by the expert X-ray crystallographic reviewer). The research results described in this manuscript are presented in the PhD thesis of D. R. H. (graduated 2024). N. S. H., K. M. P. W. and I. J. S. F. provided guidance during regular online/in-person meetings throughout D. R. H.‘s PhD studies. I. J. S. F. supervised the PhD studies of D. R. H. at the University of York.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge our technical specialists at the University of York: Drs Alex Heyam (now University of Leeds) and Heather Fish (for NMR) and Dr Karl Heaton (for MS). We are grateful to the Viking team at the University of York for the setting-up and management of our high-performance computing system for computational calculations using DFT methods. We acknowledge the following funders and supporters of this research: EPSRC for a full iCASE voucher award (19000077), supported by a GlaxoSmithKline Top-Up award (BIDS 3000034763); Royal Society for an Industry Fellowship (to I. J. S. F.); University of York for funding Mettler-Toledo ReactIR instrumentation (IC10 and RIR15); EPSRC grant (EP/P011217/1) for the purchase of a flexible ReactIR Di-probe (for the RIR15). Lastly, we are particularly grateful to the three reviewers of our manuscript for their constructive feedback. The input from the X-ray crystallographic expert was particularly informative, for which we are grateful for their time and insight.Notes and references
- (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704 CrossRef CAS PubMed; (b) J. Jover and J. Cirera, Dalton Trans., 2019, 48, 15036 RSC.
- (a) Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610 CrossRef CAS PubMed; (b) S. H. Newman-Stonebraker, S. R. Smith, E. Borowski, E. Peters, T. Gensch, H. C. Johnson, M. S. Sigman and A. G. Doyle, Science, 2021, 374, 301 CrossRef CAS PubMed.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 359, 429 CrossRef CAS PubMed.
- J. W. Lehmann, I. T. Crouch, D. J. Blair, M. Trobe, P. Wang, J. Li and M. D. Burke, Nat. Commun., 2019, 10, 1263 CrossRef PubMed.
- N. W. J. Scott, M. J. Ford, N. Jeddi, A. Eyles, L. Simon, A. C. Whitwood, T. Tanner, C. E. Willans and I. J. S. Fairlamb, J. Am. Chem. Soc., 2021, 143, 9682 CrossRef CAS PubMed.
- N. W. J. Scott, M. J. Ford, C. Schotes, R. R. Parker, A. C. Whitwood and I. J. S. Fairlamb, Chem. Sci., 2019, 10, 7898 RSC.
- (a) M. Beller, T. H. Riermeier, C. P. Reisinger and W. A. Herrmann, Tetrahedron Lett., 1997, 38, 2073 CrossRef CAS; (b) W. A. Herrmann, V. P. W. Böhm and C. P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS; (c) W. A. Herrmann, C. Brossmer, C.-P. Reisinger, T. H. Riermeier, K. Öfele and M. Beller, Chem.–Eur. J., 1997, 3, 1357 CrossRef CAS.
- A. H. M. Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef PubMed.
- P. Maity, V. V. R. Reddy, J. Mohan, S. Korapati, H. Narayana, N. Cherupally, S. Chandrasekaran, R. Ramachandran, C. Sfouggatakis, M. D. Eastgate, E. M. Simmons and R. Vaidya-nathan, Org. Process Res. Dev., 2018, 22, 888 CrossRef CAS.
- F. d'Orlyé and A. Jutand, Tetrahedron, 2005, 61, 9670 CrossRef.
- C. G. Baumann, S. De Ornellas, J. P. Reeds, T. E. Storr, T. J. Williams and I. J. S. Fairlamb, Tetrahedron, 2014, 70, 6174 CrossRef CAS.
- D. R. Husbands, T. Tanner, A. C. Whitwood, N. S. Hodnett, K. M. P. Wheelhouse and I. J. S. Fairlamb, ACS Catal., 2024, 14, 12769 CrossRef CAS PubMed.
- (a) T. Rosner, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 4621 CrossRef CAS PubMed; (b) T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848 CrossRef CAS PubMed; (c) C. Amatore, L. El Kaïm, L. Grimaud, A. Jutand, A. Meignié and G. Romanov, Eur. J. Org Chem., 2014, 4709 CrossRef CAS; (d) V. Sable, K. Maindan, A. R. Kapdi, P. S. Shejwalkar and K. Hara, ACS Omega, 2017, 2, 204 CrossRef CAS PubMed.
- (a) B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116 CrossRef CAS PubMed; (b) C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2012, 18, 6616 CrossRef CAS PubMed; (c) A. A. Thomas and S. E. Denmark, Science, 2016, 352, 329 CrossRef CAS PubMed; (d) A. A. Thomas, A. F. Zahrt, C. P. Delaney and S. E. Denmark, J. Am. Chem. Soc., 2018, 140, 4401 CrossRef CAS PubMed; (e) A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362 CrossRef CAS PubMed.
- R. B. Bedford, J. G. Bowen, R. B. Davidson, M. F. Haddow, A. E. Seymour-Julen, H. A. Sparkes and R. L. Webster, Angew. Chem., Int. Ed., 2015, 54, 6591 CrossRef CAS PubMed.
- J. T. W. Bray, M. J. Ford, P. B. Karadakov, A. C. Whitwood and I. J. S. Fairlamb, React. Chem. Eng., 2019, 4, 122 RSC.
- K. Shimamotoa and Y. Sunada, Chem. Commun., 2021, 57, 7649 RSC.
- S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656 RSC.
- G. E. Clarke, J. D. Firth, L. A. Ledingham, C. S. Horbaczewskyj, R. A. Bourne, J. T. W. Bray, P. L. Martin, J. B. Eastwood, R. Campbell, A. Pagett, D. J. MacQuarrie, J. M. Slattery, J. M. Lynam, A. C. Whitwood, J. Milani, S. Hart, J. Wilson and I. J. S. Fairlamb, Nat. Commun., 2024, 15, 3968 CrossRef CAS PubMed.
- (a) I. J. S. Fairlamb and G. C. Lloyd-Jones, Chem. Commun., 2000, 2447 RSC; (b) J. Eastoe, I. J. S. Fairlamb, J. M. Fernández-Hernández, E. Filali, J. C. Jeffery, G. C. Lloyd-Jones, A. Martorell, A. Meadowcroft, P.-O. Norrby, T. Riis-Johannessen, D. A. Sale and P. M. Tomlin, Faraday Discuss., 2010, 145, 27 RSC; (c) D. Agrawal, D. Schröder, D. A. Sale and G. C. Lloyd-Jones, Organometallics, 2010, 29, 3979 CrossRef CAS; (d) D. T. Racys, J. Eastoe, P.-O. Norrby, I. Grillo, S. E. Rogers and G. C. Lloyd-Jones, Chem. Sci., 2015, 6, 5793 RSC.
- CCDC numbers: Deposition numbers CCDC 2288572 (for 3), CCDC 228858 (for 5), CCDC 2288584 (for 7) and CCDC 2308805 (for 8) contain the supplementary crystallographic data for this paper..
Footnote |
| † Electronic supplementary information (ESI) available: All experimental details and compound characterization data and details of the catalysis studies. CCDC 2288572, 2288580, 2288584 and 2308805. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05346j |
| This journal is © The Royal Society of Chemistry 2024 |