
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-resonance emitters with room-temperature phosphorescence in amorphous state and excited by visible light†
Baoyun
Du
ab,
Yuliang
Wu
ab,
Xingdong
Wang
a,
Hongkun
Tian
 a,
Shiyang
Shao
*ac and
Lixiang
Wang
*ab
a,
Shiyang
Shao
*ac and
Lixiang
Wang
*ab
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: ssyang@ciac.ac.cn; lixiang@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, P. R. China
cSchool of Materials Sciences and Engineering, Hainan University, Haikou 570228, P. R. China
First published on 24th October 2024
Abstract
Unlike boron, nitrogen-containing multi-resonance emitters with thermally activated delayed fluorescence, here we report boron, sulfur (B, S)-based multi-resonance emitters with room-temperature phosphorescence (RTP) by inserting thiophene into a 5,9-dithia-13b-boranaphtho[3,2,1-de]anthracene skeleton that simultaneously realizes large singlet–triplet energy splitting and strong spin–orbital coupling, leading to efficient room-temperature phosphorescence in an amorphous state. Unlike most RTP emitters with ultraviolet excitation, the multi-resonance RTP emitters exhibit strong phosphorescence under daily-use blue/white LED lamps owing to their intense absorption in the visible-light region (400–486 nm). Meanwhile, such RTP behavior can be tuned by the number and fusing pattern of the thiophene moieties, with the emitters containing thiophene linked to boron atoms via α-positions exhibiting bathochromatically shifted emissions and longer phosphorescence lifetimes (47.7–119.4 ms) than those with β-position linkages. Given these features, amorphous RTP films with different emission colors and lifetimes are fabricated by dispersing the emitters in a poly(methyl methacrylate) matrix, and their applications in multi-color anti-counterfeiting are presented. These findings thus open a way to develop multi-resonance emitters as a new family of pure organic RTP materials that can work in an amorphous state and under visible-light excitation.
Introduction
Room-temperature phosphorescence (RTP) emitters have attracted increasing attention from researchers owing to their ability to utilize triplet excitons and long excited-state lifetimes, showing great potential in light-emitting diodes,1 anti-counterfeiting,2–4 biological imaging5,6 and sensors.7 So far, most RTP emitters are inorganics or organometallic complexes containing noble metals (Ir, Os, or Pt).8 In comparison, pure organic RTP emitters could be promising alternatives due to their abundant resources, bio-compatibility and structural flexibility.9 However, it is challenging to realize efficient RTP in pure organic molecules due to the non-radiative deactivation of triplet states. To address this issue, approaches such as crystallization10–13 and host–guest assembly systems14 have been developed to reduce molecular motion and suppress non-radiative decay. Nevertheless, such a crystalline state and host–guest assembly could increase the difficulty of material processing and limit many practical applications requiring amorphous emissive films.15–21 Moreover, most reported RTP materials are excited by high-energy ultraviolet (UV) light sources, which could cause phototoxicity to humans and are not readily available in daily usage scenarios.22 Compared to UV light, visible light is safer to health and can be easily accessible in daily life. In this regard, it is attractive to develop novel organic RTP emitters in an amorphous state that can be excited by visible-light sources.23–31Multi-resonance (MR) emitters based on heteroatom-doped polycyclic aromatic hydrocarbons (PAHs) have emerged as attractive luminescent materials for organic light-emitting diodes due to their high luminescent efficiencies and narrowband emissions.32–48 Owing to the opposite resonance effects of electron-rich and electron-deficient atoms in a PAH skeleton, the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) are localized alternately on adjacent atoms, forming non-bonding molecular orbitals that can intrinsically restrict vibrational motion under excitation.33 Meanwhile, rotational motion between molecular segments can also be suppressed by the rigid and fused polycyclic structure of MR emitters without the need for an extra-rigid environment. In this regard, MR emitters could be promising motifs for developing organic RTP emitters in an amorphous state because non-radiative decay of the triplet state can be inhibited by restricted molecular motion. However, the MR emitters ((B, N)-doped PAHs, as shown in Fig. 1a), reported so far are mainly thermally activated delayed fluorescence (TADF) chromophores, with their emission in essence coming from the radiative transition of singlet states.49 RTP coming from the direct radiative transition of triplet states has barely been observed for MR emitters as yet. This situation raises the question of whether and how MR emitters can actually be developed as RTP materials using molecular design.
 | ||
| Fig. 1 Molecular design for multi-resonance emitters with room-temperature phosphorescence. (a) Jablonski diagram for emission mechanism of previously reported multi-resonance emitters with TADF and RTP emitters in this work (Ex: excitation, PF: prompt fluorescence, DF: delayed fluorescence, Phos: phosphorescence). (b) Molecular structures for the designed emitters. (c) Excitation wavelength–phosphorescence mapping for α-DThBSS-Cz in doped film (1 wt% in PMMA). (d) Schematic diagram for α-DThBSS-Cz film with RTP emission excited by a blue LED lamp. | ||
Unlike MR-TADF emitters with (B, N)-doped PAH skeletons, here we demonstrate a strategy toward multi-resonance emitters with RTP by inserting thiophene into a boron, sulfur (B, S)-doped multi-resonance PAH skeleton (5,9-dithia-13b-boranaphtho[3,2,1-de]anthracene, BSS), which exhibits efficient phosphorescence in amorphous films under visible-light excitation (Fig. 1). (B, S)-Doped PAHs with multi-resonance character are selected as skeletons because their non-bonding molecular orbital features can intrinsically restrict vibrational relaxation to suppress the non-radiative transition of triplet states. Meanwhile, thiophene units inserted in the BSS skeleton can lower the triplet state and enlarge singlet–triplet energy splitting (ΔEST), leading to a slow reverse intersystem crossing (RISC) process from the T1 to the S1 state. Moreover, the heavy-atom effect of the embedded sulfur atoms can enhance spin–orbital coupling (SOC) and accelerate the T1 → S0 transition, providing an efficient RTP with a lifetime of >0.1 s and a quantum efficiency of 32% even in an amorphous state. Remarkably, unlike most RTP systems with absorption bands in the UV range, these multi-resonance RTP emitters can be excited by visible light owing to the small Stokes shift (<25 nm), making UV light excitation sources dispensable. By tuning the number and fusing pattern of the thiophene moieties for the emitters, amorphous RTP films with different emission colors and lifetimes are fabricated by dispersing them in a poly(methyl methacrylate) (PMMA) matrix, and their applications in multi-color anti-counterfeiting are presented. These findings not only provide new insights into the emission behavior of multi-resonance emitters but also open up a way to develop multi-resonance emitters as a new family of pure organic RTP materials that can work in an amorphous state and under visible-light excitation.
Results and discussion
Molecular design and synthesis
To design multi-resonance RTP emitters, two kinds of (B, S)-doped PAHs are constructed by inserting thiophene moieties into a 5,9-dithia-13b-boranaphtho[3,2,1-de]anthracene (BSS) skeleton, with the thiophene moieties linked to boron atoms via either α-positions (α-ThBSS, α-DThBSS and α-DThBSS-Cz) or β-positions (β-ThBSS, β-DThBSS and β-DThBSS-Cz) (Fig. 1b). The BSS skeleton is selected due to its multi-resonance structure that can restrict molecular vibration and rotation relaxation under excitation.50 Meanwhile, thiophene moieties are inserted into BSS to extend the conjugation of the PAH skeleton to lower the triplet state and enlarge ΔEST, aiming to inhibit consumption of triplet states through RISC. Moreover, the heavy-atom effect of the embedded S atoms can enhance SOC, which not only accelerates the ISC process to populate the triplet states but also favors the T1 → S0 transition to produce phosphorescence.51–53 Unlike α-ThBSS and β-ThBSS with one fused thiophene ring, α-DThBSS and β-DThBSS have two thiophene moieties fused in BSS skeletons, providing a motif to investigate the effect of thiophene number on emission properties. Finally, 3,6-bis(tert-butyl)carbazole (Cz) moieties are introduced into the periphery of the BSS skeleton (α-DThBSS-Cz and β-DThBSS-Cz), which are expected to tune the emission color and luminescent efficiency of the resultant emitters.Synthetic procedures for the emitters are shown in Scheme 1. α-ThBSS and β-ThBSS with one thiophene moiety are synthesized using 3-bromobenzo[b]thiophene (1) or 2-bromobenzo[b]thiophene (5) as starting materials. These materials undergo sulfhydrylation, followed by two-step nucleophilic aromatic substitution with 2,5-dibromo-1,3-difluorobenzene and sodium thiophenolate to form bromide precursors 4 and 8, containing two different arylsulfide groups at the ortho-positions of 2-bromine atoms. The bromide precursors are then lithiated and undergo intramolecular electrophilic borylation to afford the key bromide polycyclic intermediates α-ThBSS-Br and β-ThBSS-Br, which are debrominated under Pd(dppf)Cl2 and NaBH4 to produce α-ThBSS and β-ThBSS in yields of 85% and 80%, respectively. The symmetrical emitters containing two thiophene moieties (α-DThBSS and β-DThBSS) are synthesized through a similar protocol, except that two equivalent benzo[b]thiophene-thiols (2 and 6) are reacted with 2,5-dibromo-1,3-difluorobenzene to provide bromide precursors 9 and 10 and then bromide polycyclic intermediates α-DThBSS-Br and β-DThBSS-Br. The emitters containing carbazole moieties (α-DThBSS-Cz and β-DThBSS-Cz) are obtained by Buchwald–Hartwig C–N coupling from α-DThBSS-Br and β-DThBSS-Br with 3,6-di-tert-butylcarbazole using Pd2dba3 as catalyst and t-BuONa as base. The key intermediates and final compounds were characterized using 1H, 13C NMR, elemental analysis and high-resolution mass spectra (see ESI†).
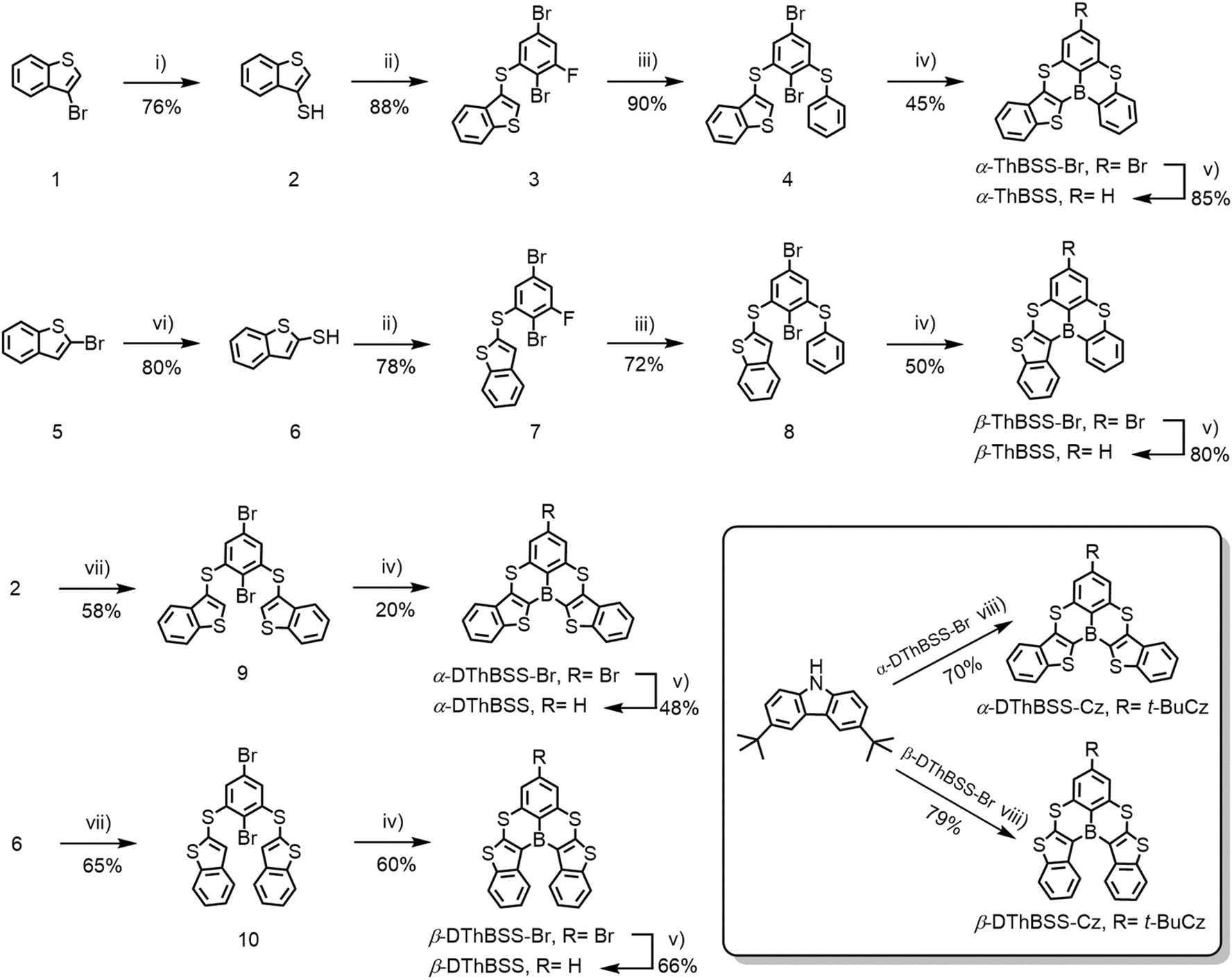 | ||
| Scheme 1 Synthetic routes for multi-resonance emitters with room-temperature phosphorescence. Reagents and conditions: (i) n-BuLi, S8, Et2O, −78 °C, 3 h. (ii) 2,5-Dibromo-1,3-difluorobenzene, K2CO3, NMP, 60 °C, 24 h. (iii) PhSNa, NMP, 60 °C, 24 h. (iv) n-BuLi, BBr3, i-Pr2NEt, m-xylene, 120 °C, 24 h. (v) Pd(dppf)Cl2, TMEDA, NaBH4, THF, 25 °C, 12 h. (vi) CH3SNa, DMF, reflux, 12 h. (vii) 2,5-Dibromo-1,3-difluorobenzene, K2CO3, NMP, 60 °C, 48 h. (viii) 3,6-Bis(tert-butyl)carbazole, Pd2(dba)3, t-Bu3P·HBF4, t-BuONa, toluene, 100 °C, 12 h. | ||
Single-crystal structures of the emitters are given in Fig. 2. The emitters exhibit trigonal geometries for PAH skeletons, with B–C bond lengths in the range of 1.536–1.554 Å and S–C bond lengths of 1.717–1.769 Å. It is found that the molecular geometries of the emitters are highly dependent on the linking patterns for the fused thiophene moieties. For α-ThBSS, α-DThBSS and α-DThBSS-Cz, where the α-position of thiophene was bonded to the central B atom, the skeletons show an almost planar configuration with a small dihedral angle (Φ) between the aromatic rings linked to B and S atoms. For example, the dihedral angle between two terminal phenyl rings (D and F rings) for α-ThBSS is only 12.76°, while those for α-DThBSS and α-DThBSS-Cz (D and D′ rings) are even smaller (Φ = 2.20°–2.83°). In contrast, for β-ThBSS, β-DThBSS and β-DThBSS-Cz with the β-position of thiophene bonded to the central B atom, the skeletons show a twisted configuration with large dihedral angles between the aromatic rings (47.72°, 50.87° and 43.11° between D and D′/F rings for β-ThBSS, β-DThBSS and β-DThBSS-Cz, respectively). Such a twisted configuration could be related to steric hindrance between hydrogen atoms on the benzene rings.35,54 Notably, the different molecular geometries for the emitters can result in different stacking patterns in the crystalline state. As shown in Fig. 2, α-ThBSS and α-DThBSS with a (quasi-)planar configuration exhibit close layer-by-layer stacking between adjacent molecules with π–π interaction distances of 3.554–3.639 Å. In contrast, in β-ThBSS and β-DThBSS with twisted structures, the emitters are packed much more loosely, with the π–π stacking distance between peripheral benzene rings in the range of 6.605–7.165 Å, indicating that the intermolecular π–π interaction is weakened in these emitters.
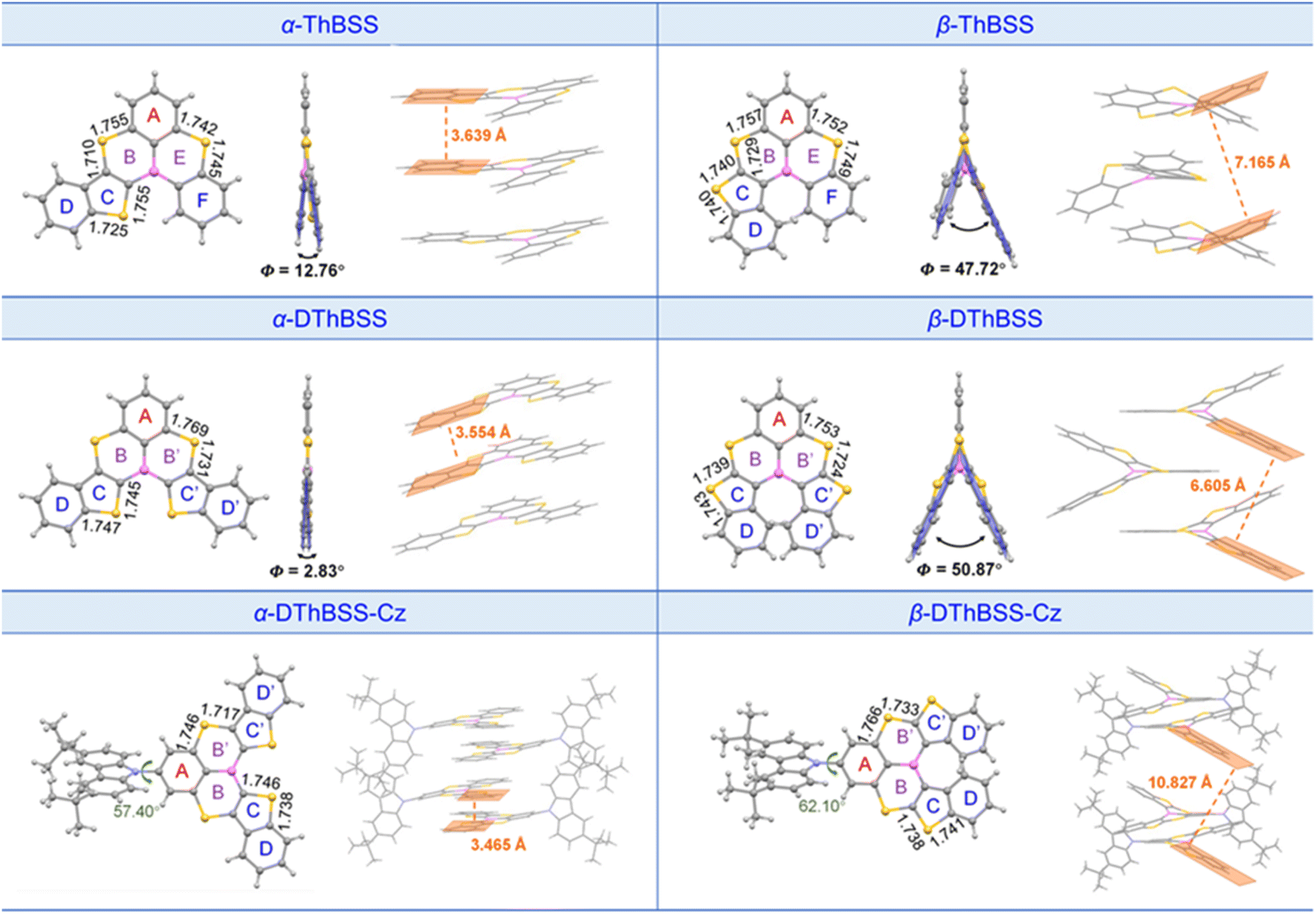 | ||
| Fig. 2 Single-crystal structures and intermolecular packing patterns for the emitters. | ||
Multiple resonance effect
To gain insight into the photophysical properties of the emitters, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations (M062X/def2SVP level) were carried out to evaluate their distributions of frontier molecular orbitals (FMOs) and natural transition orbitals (NTOs). Meanwhile, spin-component scaling second-order approximate coupled-cluster (SCS-CC2) calculations were also conducted to estimate the excited-state energy levels (SCS-CC2/cc-pVDZ).55 As shown in Fig. 3, the highest occupied NTOs (HONTOs) of the S1 state for both α-isomers (α-ThBSS and α-DThBSS) and β-isomers (β-ThBSS and β-DThBSS) are mainly distributed on bridging S atoms and their ortho-/para-positions of phenyl rings, as well as S atoms and α-/β-positions of thiophene moieties, whereas the lowest unoccupied NTOs (LUNTOs) are located predominantly on the B atom and its ortho-/para-positions of surrounding phenyl and thiophene rings, corresponding to atomically separated NTO distributions for typical multi-resonance emitters.56 For α-DThBSS-Cz and β-DThBSS-Cz with carbazole units, the HONTOs and LUNTOs are mainly distributed on the polycyclic skeleton rather than on carbazole moieties, indicating that the multi-resonance character can be retained in these emitters. Similar to NTO distributions, the frontier molecular orbitals also exhibit a typical multi-resonance character with alternately distributed HOMOs and LUMOs (Fig. S16, ESI†). Compared to β-isomers with HOMO levels of −6.77 to −6.79 eV and LUMO levels of −1.14 to −1.24 eV, the corresponding α-isomer counterparts exhibit shallower HOMO levels (−6.51 to −6.60 eV) and deeper LUMO levels (−1.44 to −1.60 eV) owing to the planar geometry and extended electron delocalization, giving narrower energy gaps (Eg) of 4.98–5.15 eV than those of the β-isomers (5.53–5.65 eV). From α-ThBSS with one thiophene to α-DThBSS with two thiophene units, the HOMO level is raised by 0.08 eV, and the LUMO level is lowered by 0.09 eV. In contrast, from β-ThBSS to β-DThBSS, the HOMO is slightly lowered by 0.01 eV and the LUMO is raised by 0.10 eV. It is noted that the emitters exhibit weak structural relaxation under excitation with small reorganization energies (λS, < 0.13 eV) and a small change in average bond-length (0.003–0.011 Å) between the S0 and S1 states regardless of the fusing pattern of thiophene (Fig. S7–S14†). These results indicate the small change in molecular configuration between ground and excited states, leading to small Stokes shifts between the absorption and emission spectra. The S1 energy level for α-ThBSS is 3.12 eV, lower than that of β-ThBSS (S1 = 3.30 eV), in line with the more planar configuration for α-ThBSS. Upon increasing the thiophene number from α-ThBSS to α-DThBSS, the S1 state is lowered by 0.03 eV due to extended conjugation. However, from β-ThBSS to β-DThBSS, the S1 energy level is slightly raised from 3.30 eV to 3.39 eV, attributed to the more distorted PAH skeleton for the latter. For α-DThBSS-Cz and β-DThBSS-Cz, the S1 energy levels are higher than those of their parent emitters (α-DThBSS and β-DThBSS), indicating that their emission colors could be blue-shifted by introducing peripheral carbazole substituents into a multi-resonance skeleton.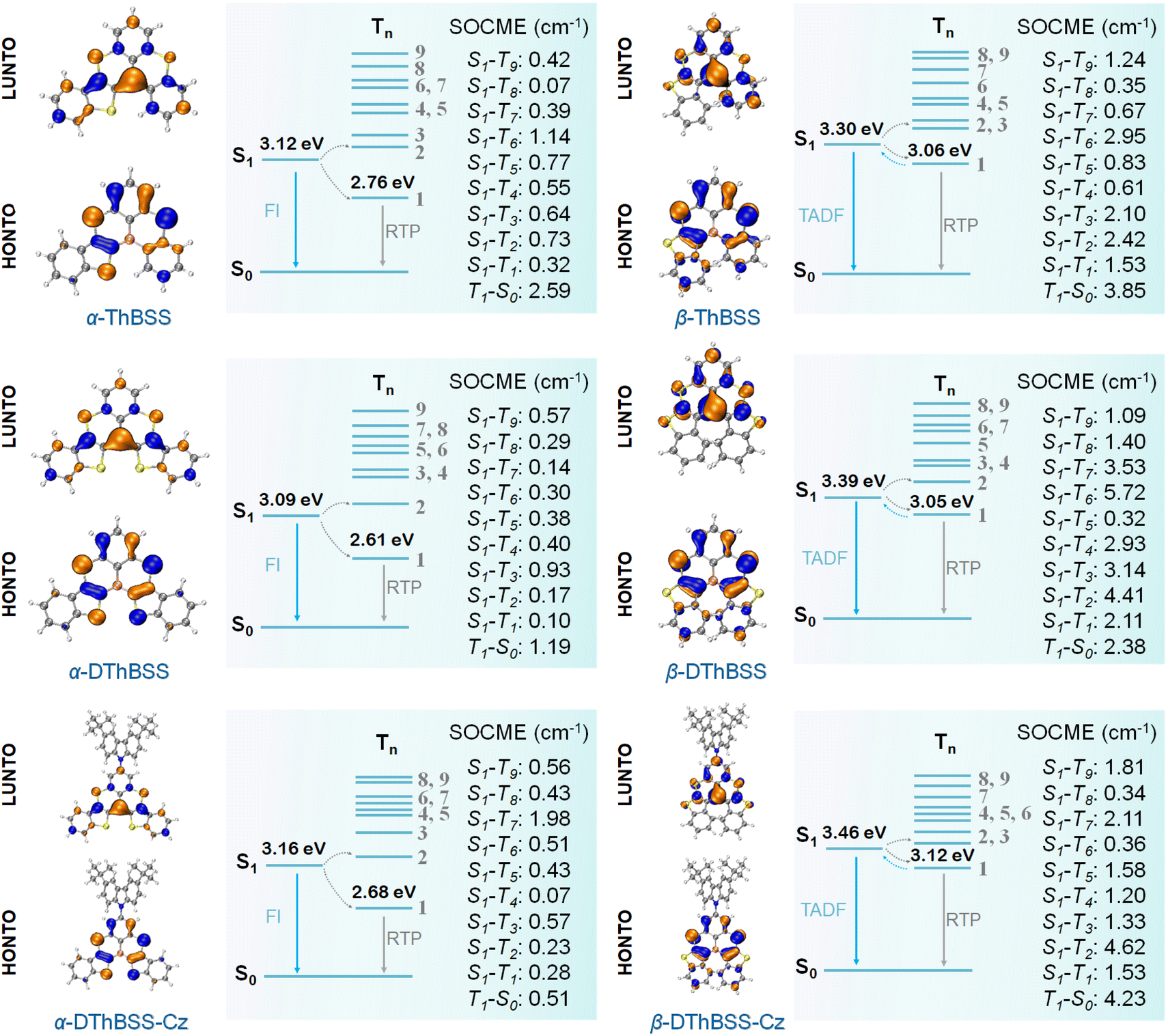 | ||
| Fig. 3 Natural transition orbital distributions (HONTO and LUNTO) of S1 states, and spin–orbit coupling matrix element (SOCME) for the emitters calculated using the TD-DFT method at M062X/def2SVP level. | ||
To investigate the absorption and emission properties of the emitters, the UV-vis absorption and steady-state photoluminescence (PL) spectra are measured. As shown in Fig. 4, the absorption spectra of α-ThBSS and α-DThBSS contain two sets of absorption bands at 300–375 nm and 400–486 nm in toluene, which are assigned to the π–π* transitions of polycyclic skeletons and intramolecular charge-transfer (ICT) transitions, respectively. For α-DThBSS-Cz, an additional absorption band at 375–390 nm is observed, which could be attributed to the π–π* absorption of the carbazole unit. Similar absorption bands are observed for β-isomers (β-ThBSS, β-DThBSS and β-DThBSS-Cz), with those at 300–360 nm and 385–450 nm assigned to the π–π* transitions of polycyclic skeletons and ICT transitions, respectively, while that at ∼380 nm for β-DThBSS-Cz is attributed to the carbazole unit. For the PL spectra, three α-isomers exhibit intense and sharp emissions at 473–481 nm in toluene, despite shoulder emission bands being observed at 496–511 nm, giving small FWHM values of 26–28 nm typical for multi-resonance emitters. However, for β-isomers, the PL spectra show emission bands mainly at 437–451 nm with relatively weak shoulder emissions, corresponding to FWHM values of 26–27 nm (Table S3†). Compared to α-ThBSS and α-DThBSS showing emission maxima of 473–481 nm, β-ThBSS and β-DThBSS exhibit blue-shifted emission peaks at 451 and 438 nm, respectively, consistent with their higher S1 states predicted by TD-DFT calculations. By introducing carbazole groups in para-positions to boron atoms for α-DThBSS-Cz and β-DThBSS-Cz, the emitters exhibit a hypochromatic shift in emission by 1–6 nm without broadening the FWHMs. Notably, the Stokes shifts for the emitters are only 16–24 nm (Table S3†), meaning that the absorption bands are close to the emission bands, which is favorable for the emitters to be excited by visible light. According to onsets of fluorescence and phosphorescence spectra at 77 K, ΔEST values are determined to be 0.42, 0.49 and 0.48 eV for α-ThBSS, α-DThBSS and α-DThBSS-Cz, and 0.26, 0.33 and 0.34 eV for β-ThBSS, β-DThBSS and β-DThBSS-Cz, respectively. Such ΔESTs are much larger than that of the parent emitter BSS (0.15 eV (ref. 50)) because the fused thiophene lowers the T1 state more drastically than the S1 state. For example, from BSS to α-ThBSS and α-DThBSS, the S1 energy level decreases from 2.84 to 2.76 and 2.75 eV (reduced by 0.09 eV in total), while the T1 energy level decreases from 2.69 to 2.34 and 2.26 eV (reduced by 0.43 eV in total), respectively.
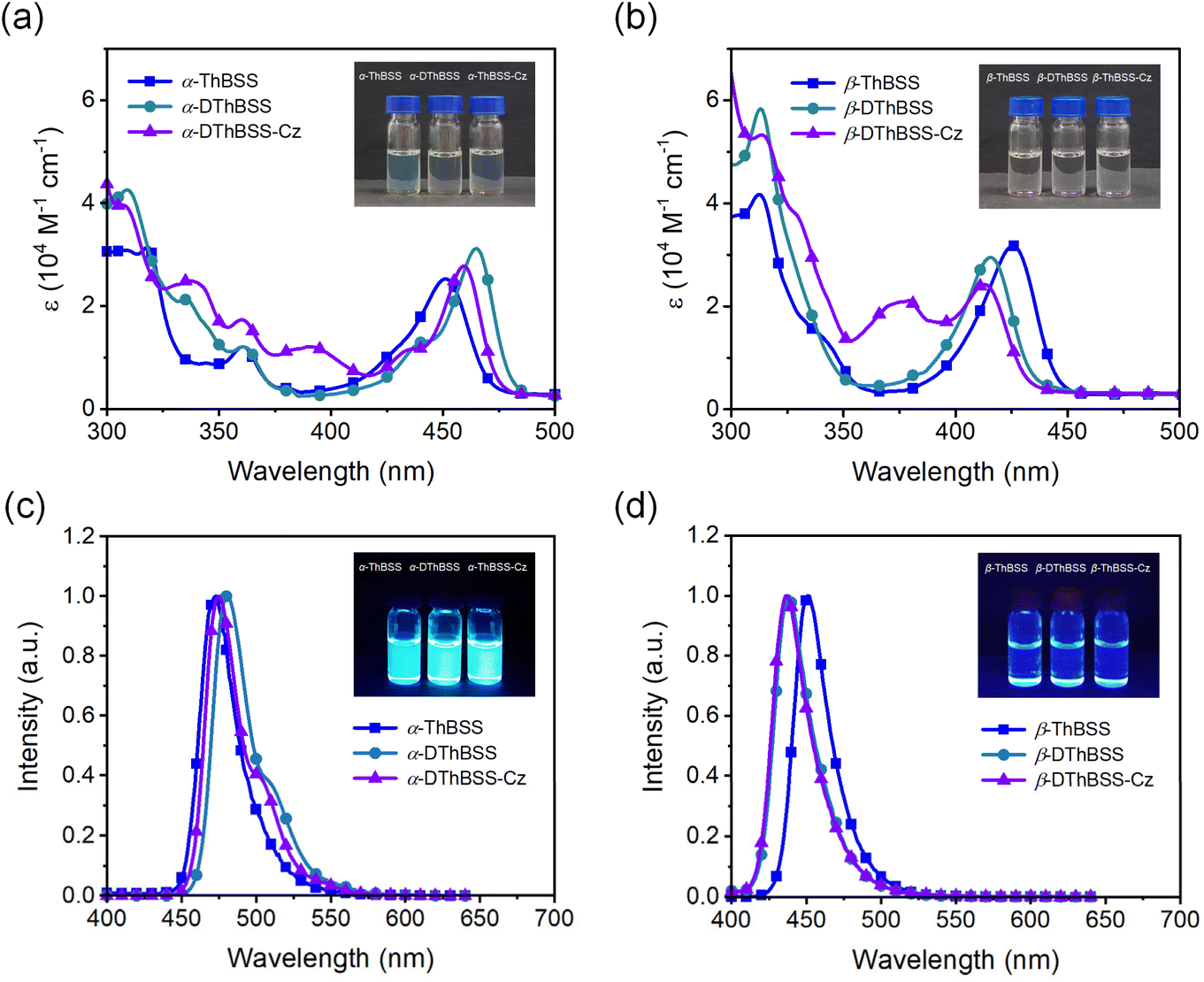 | ||
| Fig. 4 Photophysical properties for the emitters in toluene solution: (a) and (b) absorption spectra (10−5 mol L−1); (c) and (d) steady-state PL spectra (10−5 mol L−1). | ||
Room-temperature phosphorescence behavior
Unlike solution-state PL spectra with only blue emission bands, PL spectra of the emitters in the solid state (1 wt% in PMMA) exhibit dual emission bands with one blue emission and one green/yellow emission (Fig. 5). For α-ThBSS, α-DThBSS and α-DThBSS-Cz, the blue emissions exhibit single-exponential decay profiles that have nanosecond-scale lifetimes (3.4–5.5 ns, Table 1) and are insensitive to temperature (150–300 K, Fig. 5d and S22†), indicating that they are fluorescence emissions. In contrast, for β-ThBSS, β-DThBSS and β-DThBSS-Cz, the blue emissions exhibit double-exponential decay characteristics containing prompt (lifetime: 1.3–3.8 ns) and delayed component (lifetime: 1.4–10.4 ms). The intensity ratio of the latter gradually intensifies as the temperature rises, indicative of their TADF characters. Remarkably, the green/yellow emissions for both kinds of emitters display long lifetimes with the PL decay curves extending to 0.1–1 s at 300 K. With increasing temperature, the PL intensity decays faster, corresponding to phosphorescent emission behavior. This assignment is consistent with time-resolved emission spectra (TRES) where the long-wavelength phosphorescence emissions gradually evolve to be the main emissions at a time scale of >100 ns (Fig. 5b and c). Taking α-DThBSS-Cz as examples, fluorescence at 473 nm is the dominant emission at a time scale of <60 ns but becomes weaker at a time of >140 ns. In comparison, phosphorescence at 577 nm gradually evolves into the main emission as time increases to 920 ns. The phosphorescence lifetimes for the emitters are found to be very dependent on the linkage pattern of thiophene moieties. For instance, α-DThBSS containing two thiophenes linked to the B atom through the α-position exhibits a phosphorescence lifetime of 113.4 ms, which is 15 folds longer than that of β-DThBSS with β-position linkage (7.5 ms). It is noted that the phosphorescence spectra of the emitters exhibit broad emission bands, which may be due to enhanced conjugation by incorporation of thiophene units. Actually, we notice that T1 states for the emitters exhibit larger reorganization energies (λS +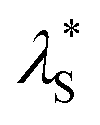 = 0.275 to 0.544 eV) than those of S1 states (λS +
= 0.275 to 0.544 eV) than those of S1 states (λS + 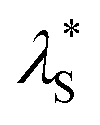 = 0.174 to 0.234 eV, Fig. S14†), indicating greater structural relaxation for T1 states. Unlike many RTP emitters requiring hydrogen bonds or other intermolecular interactions provided via crystalline or host–guest assembly systems, the phosphorescence of MR emitters is observed in doped films with an amorphous state (supported by the absence of diffraction signals observed for the doped films via X-ray diffraction, Fig. S24†) and does not rely on a specific matrix. Additionally, besides PMMA, other materials such as polystyrene (PS) and 1,3-bis(carbazol-9-yl)benzene (mCP) can be used as a matrix for the emitters to exhibit RTP emission (Fig. S26 and S27†). Moreover, phosphorescence for the emitters can be readily excited by visible light (Fig. S28†). Taking α-DThBSS-Cz as an example, the phosphorescence can be observed with an excitation wavelength ranging from 400 to 460 nm (Fig. 5g), unlike many organic RTP emitters needing UV light excitation sources.57–59 The phosphorescence quantum yields (ΦPh) for α-ThBSS, α-DThBSS, and α-DThBSS-Cz are 26%, 14% and 32%, respectively, while those for β-ThBSS, β-DThBSS and β-DThBSS-Cz are 9%, 19% and 22%, respectively.
= 0.174 to 0.234 eV, Fig. S14†), indicating greater structural relaxation for T1 states. Unlike many RTP emitters requiring hydrogen bonds or other intermolecular interactions provided via crystalline or host–guest assembly systems, the phosphorescence of MR emitters is observed in doped films with an amorphous state (supported by the absence of diffraction signals observed for the doped films via X-ray diffraction, Fig. S24†) and does not rely on a specific matrix. Additionally, besides PMMA, other materials such as polystyrene (PS) and 1,3-bis(carbazol-9-yl)benzene (mCP) can be used as a matrix for the emitters to exhibit RTP emission (Fig. S26 and S27†). Moreover, phosphorescence for the emitters can be readily excited by visible light (Fig. S28†). Taking α-DThBSS-Cz as an example, the phosphorescence can be observed with an excitation wavelength ranging from 400 to 460 nm (Fig. 5g), unlike many organic RTP emitters needing UV light excitation sources.57–59 The phosphorescence quantum yields (ΦPh) for α-ThBSS, α-DThBSS, and α-DThBSS-Cz are 26%, 14% and 32%, respectively, while those for β-ThBSS, β-DThBSS and β-DThBSS-Cz are 9%, 19% and 22%, respectively.
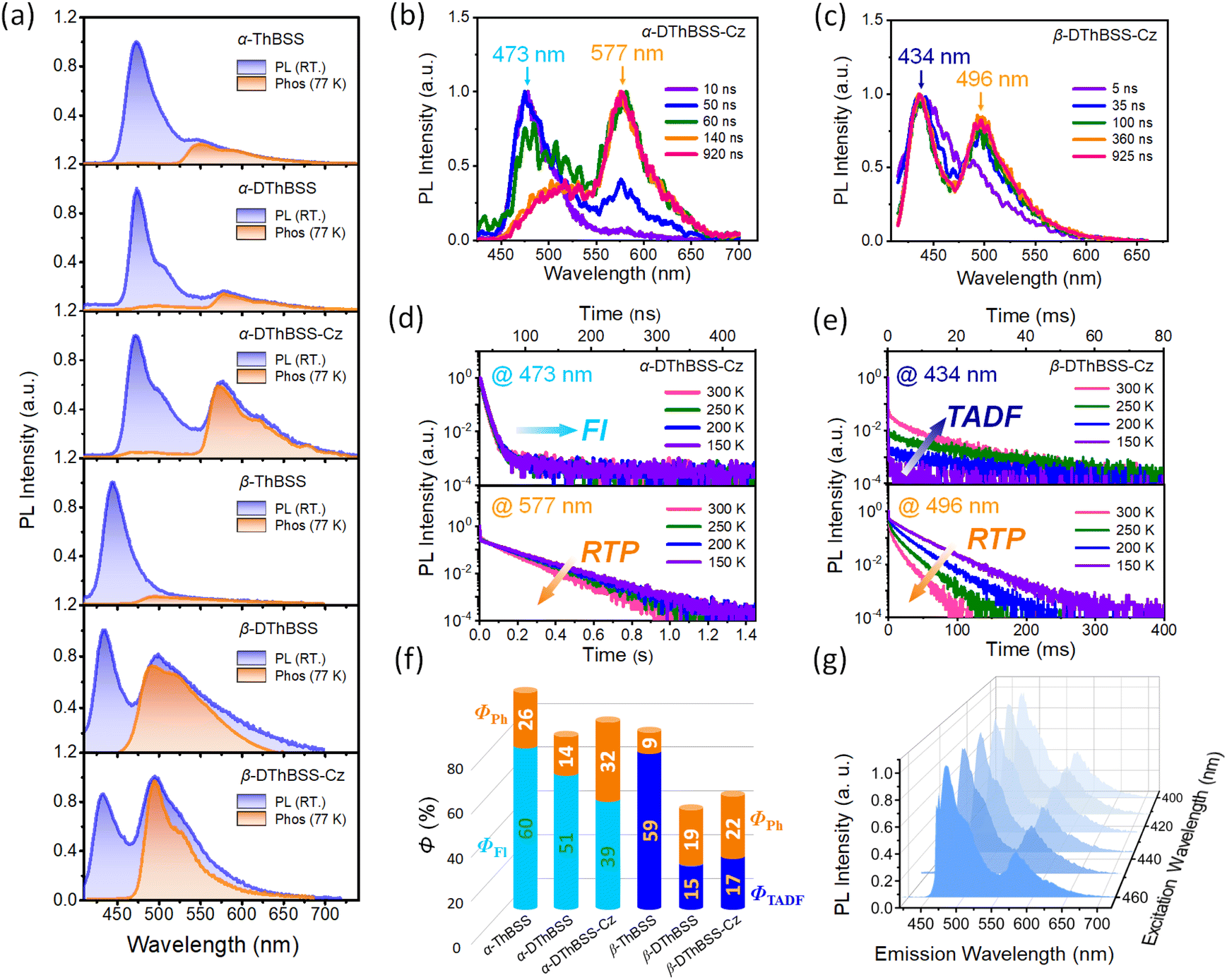 | ||
| Fig. 5 Room-temperature phosphorescence characteristics for the emitters in the doped films (1 wt% in PMMA). (a) Steady-state PL and phosphorescence spectra. (b) and (c) Time-resolved PL spectra for α-DThBSS-Cz and β-DThBSS-Cz. (d) and (e) Temperature-dependent PL decay characteristics. (f) Fluorescence (ΦFl), TADF (ΦTADF) and phosphorescence (ΦPh) quantum yields. (g) Steady-state PL spectra with excitation wavelength varying from 400 to 460 nm for α-DThBSS-Cz. | ||
| Compounds | λ em [nm] | τ F [ns] | τ p/τdc [ns ms−1] | τ Ph [ms] | Φ PL [%] | Φ Ph [%] | S1/T1g [eV] | ΔESTh [eV] |
|---|---|---|---|---|---|---|---|---|
| a Emission peak wavelength. b Lifetimes for fluorescence emission. c Lifetimes of prompt (τp) and delayed fluorescence (τd) for TADF emission. d Lifetimes for phosphorescence emission. e Total PL quantum yield (ΦPL) with experimental errors of ±2%. f Phosphorescence quantum yield (ΦPh). g Energy levels for lowest singlet (S1) and triplet (T1) state. h Singlet–triplet energy gap. | ||||||||
| α-ThBSS | 474, 545 | 5.1 | — | 47.7 | 86 | 26 | 2.76/2.34 | 0.42 |
| α-DThBSS | 475, 580 | 5.5 | — | 113.4 | 65 | 14 | 2.75/2.26 | 0.49 |
| α-DThBSS-Cz | 473, 577 | 3.4 | — | 119.4 | 71 | 32 | 2.76/2.28 | 0.48 |
| β-ThBSS | 445, 493 | — | 1.3/1.4 | 6.8 | 68 | 9 | 2.94/2.68 | 0.26 |
| β-DThBSS | 436, 498 | — | 3.8/3.3 | 7.5 | 34 | 19 | 2.98/2.65 | 0.33 |
| β-DThBSS-Cz | 434, 496 | — | 1.5/10.4 | 12.5 | 39 | 22 | 3.00/2.66 | 0.34 |
To explore the origin of RTP for the emitters, we have calculated their SOC matrix elements (SOCME) between singlet and triplet states. It is found that SOCME between S1 and Tn states (〈S1|ĤSOC|Tn〉 (n = 1–9)) are quite high (up to 4.62 cm−1, Fig. 3) compared to typical (B, N)-based MR-TADF emitters (for instance, DABNA-1 (ref. 32) with a maximum SOCME of ∼0.52 cm−1, Fig. S15†), resulting in fast ISC processes (kISC: 0.25 to 7.33 × 108 s−1, Table S4†) that can rapidly populate triplet (Tn) states. According to Kasha's rule, the formed triplet (Tn) states can readily decay to the T1 state, which are then either up-converted to the S1 state through RISC to generate TADF emission or deactivated to the S0 state to give phosphorescence via radiative transition. Owing to the large ΔESTs (0.42–0.49 eV) for α-ThBSS, α-DThBSS and α-DThBSS-Cz, no TADF emissions are observed for these emitters, indicating that the RISC process is not favored in this case. Therefore, triplet excitons mostly decay to the ground state to form phosphorescence, which is favored due to the high SOCME for T1 → S0 transitions (〈T1|ĤSOC|S0〉, 0.51–2.59 cm−1) that are 3.6–18.5 folds that of DABNA-1 (〈T1|ĤSOC|S0〉, 0.14 cm−1, Fig. S15†). In contrast, for the β-isomers with thiophene linked to the B atom via the β-position, the moderate ΔESTs (0.26–0.34 eV) allow some T1 excitons to be converted to the S1 states through RISC (kRISC = 0.44 to 5.17 × 103 s−1), rendering simultaneous TADF and phosphorescence emissions. Notably, the parent BSS emitter with small ΔEST (0.15 eV) exhibits only TADF emission with no phosphorescence observed,50 supporting the importance of the inserted thiophene moieties and enlarged ΔESTs in generating RTP emission. Finally, the nonradiative transition rates for the T1 states of the emitters are quite slow (kpnr = 5.7 to 131.8 s−1, Table S4†), indicating that dissipation of triplet states can be effectively suppressed by the (B, S)-doped PAH multi-resonance skeleton, which also favors RTP emission.
Applications
In view of the RTP feature for the emitters, their preliminary application in anti-counterfeiting is demonstrated using α-ThBSS, α-DThBSS and α-DThBSS-Cz as model samples considering their long phosphorescence lifetimes. As shown in Fig. 6, butterfly-shaped patterns are fabricated by coating the emitter inks (1 wt% in PMMA, 10 mg mL−1) on wood-free paper through a hollowed-out mask. The patterns are barely seen under daylight, but are observed as blue (α-ThBSS and α-DThBSS) or white (α-DThBSS-Cz) images under 365 nm UV irradiation owing to simultaneous fluorescence and phosphorescence emission. After ceasing excitation, the colors of the patterns change to olivine or yellow because phosphorescence dominates the emission. Interestingly, besides UV light sources, the patterns can also be excited by visible light with excitation wavelength extending to ∼480 nm, as presented in an excitation–photoluminescence mapping diagram (Fig. 6c), making visible light such as blue or white light suitable as an excitation source. For example, by exciting the α-DThBSS-Cz-doped films under commercial blue (450 nm, 0.5 W) or white LED lamps (400–700 nm, 0.8 W) and then ceasing excitation, a yellow afterglow can be observed by the naked eyes, similar to the observation under 365 nm excitation (Fig. 6d). Such properties for the emitters can make them convenient for use in anti-counterfeiting applications. As shown in Fig. 6e, numeric codes of “23918” are written on wood-free paper with α-DThBSS-Cz ink (1 wt% in PMMA, 10 mg mL−1). After drying, the numbers are nearly invisible under daylight. Upon exposure under blue LED illumination, the numeric codes are still invisible due to a strong blue-light background. However, after turning off the lamp, the encrypted numbers “23918” can be observed.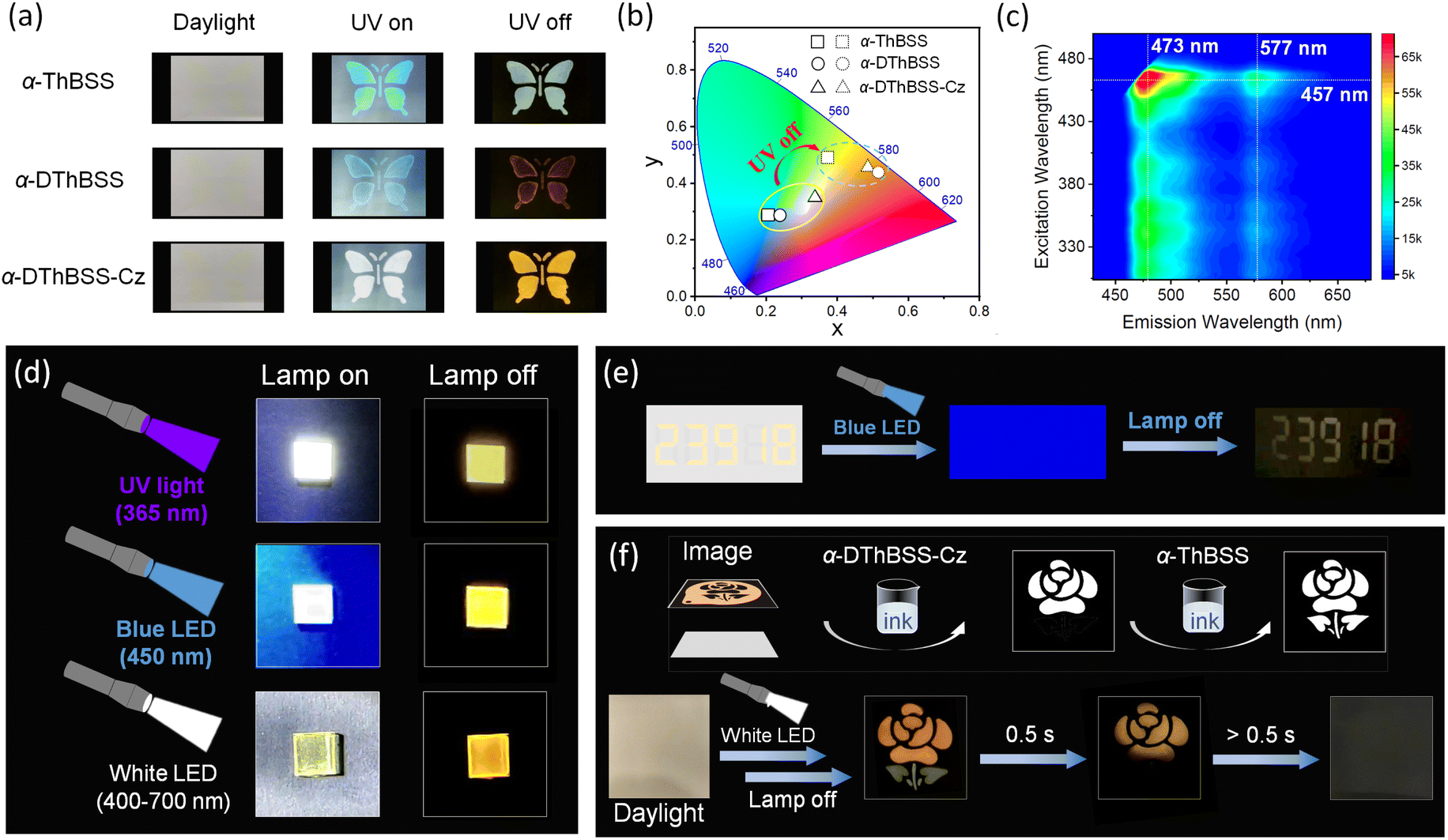 | ||
| Fig. 6 Anti-counterfeiting applications. (a) Photographs for butterfly-shaped patterns fabricated by coating the emitter inks (1 wt% in PMMA, 10 mg mL−1 in chlorobenzene) on wood-free paper. (b) CIE coordinates for the patterns under and after excitation. (c) Excitation wavelength–fluorescence mapping for α-DThBSS-Cz-doped film. (d) Images for α-DThBSS-Cz-doped films under different excitation sources. (e) Photographs for numeric codes written with α-DThBSS-Cz ink on wood-free paper under and after illumination by a blue LED lamp. (f) Photographs for multi-color anti-counterfeiting with α-ThBSS and α-DThBSS-Cz ink under a white LED excitation source. | ||
It is noteworthy that the tunable emission colors and phosphorescence lifetimes for MR emitters enable further multi-color anti-counterfeiting applications. As shown in Fig. 6f, by successively printing the α-DThBSS-Cz ink through a flower-shaped mask and α-ThBSS ink through a leaf-shaped mask on the same paper, a “flower + leaf” pattern can be formed. After drying, a nearly blank paper with no patterns can be observed under daylight. After exciting the paper using a white LED lamp and then ceasing excitation, a two-color picture with a yellow-emitting flower from α-DThBSS-Cz and an olivine-emitting leaf from α-ThBSS can be observed first. Subsequently, the olivine leaf disappears, but the yellow flower remains observable owing to the longer phosphorescent lifetime of α-DThBSS-Cz than α-ThBSS. As time goes on further (>0.5 s), the yellow flower also disappears. Such multi-color afterglows from MR emitters under visible-light excitation not only provide an approach toward high-level anti-counterfeiting applications but also expand the usage scenarios by making UV sources dispensable.
Conclusions
In summary, unlike reported multi-resonance TADF materials, we propose a strategy toward multi-resonance emitters with RTP by inserting thiophene into a (B, S)-doped multi-resonance PAH (5,9-dithia-13b-boranaphtho[3,2,1-de]anthracene, BSS) skeleton, exhibiting efficient phosphorescence emission in an amorphous state under visible-light excitation. The emitters reveal fast intersystem crossing and favored T1 → S0 transition because of enhanced SOC by the heavy-atom effect of S atoms, but have a slow RISC process owing to the lowered T1 state and enlarged singlet–triplet energy splitting. Moreover, the multi-resonance (B, S)-doped PAHs act as promising skeletons to suppress the non-radiative transition of triplet states, giving phosphorescence with a lifetime of >0.1 s and a quantum yield up to 32% in amorphous films. Unlike many organic RTP emitters with absorption bands in the UV range, MR emitters can be excited by visible light, making UV light excitation sources dispensable. Employing the emitters, amorphous RTP films are fabricated by dispersing them in a PMMA matrix, and their applications in multi-color anti-counterfeiting are presented. These findings provide new insights into modulating emission behaviors for multi-resonance emitters and shed light on their potential as a new family of pure organic RTP materials that can work in an amorphous state and under visible-light excitation.Data availability
All data supporting this study are available in the ESI.†Author contributions
B. D. synthesized the emitters, carried out theoretical calculations and performed experiments on steady-state photophysical properties and anti-counterfeiting application. Y. W., X. W. and S. W. contribute to transient-state photophysical measurements. H. T. carried out single-crystal analysis. S. S. and L. W. conceived the idea, designed the experiments and wrote the manuscript. All the authors contributed to the discussion of results and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (52073282, 52122309, 52261135541, and 91833306), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0520102), the Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm2024024), the Innovational Fund for Scientific and Technological Personnel of Hainan Province (KJRC2023C09), and the Startup Scientific Research Foundation from Hainan University (KYQD(ZR)22174). The authors also acknowledge the staff of the BL17B beamline of National Facility for Protein Science in Shanghai (NFPS) at the Shanghai Synchrotron Radiation Facility for assistance in crystal structure data collection and the Network and Computing Center, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences for help with theoretical calculations.Notes and references
- Z. Chen, M. Li, Q. Gu, X. Peng, W. Qiu, W. Xie, D. Liu, Y. Jiao, K. Liu, J. Zhou and S. J. Su, Adv. Sci., 2023, 2207003 CrossRef CAS PubMed.
- A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed.
- D. Wang, J. Gong, Y. Xiong, H. Wu, Z. Zhao, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2022, 33, 2208895 CrossRef.
- X. Yao, H. Ma, X. Wang, H. Wang, Q. Wang, X. Zou, Z. Song, W. Jia, Y. Li, Y. Mao, M. Singh, W. Ye, J. Liang, Y. Zhang, Z. Liu, Y. He, J. Li, Z. Zhou, Z. Zhao, Y. Zhang, G. Niu, C. Yin, S. Zhang, H. Shi, W. Huang and Z. An, Nat. Commun., 2022, 13, 4890 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS.
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102 CrossRef CAS PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Brase, Adv. Mater., 2021, 33, e2005630 CrossRef.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- S. Shen, G. V. Baryshnikov, Q. Xie, B. Wu, M. Lv, H. Sun, Z. Li, H. Agren, J. Chen and L. Zhu, Chem. Sci., 2023, 14, 970 RSC.
- Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Wurthner, C. M. Marian, L. Ji and T. B. Marder, Angew. Chem., Int. Ed., 2020, 59, 17137 CrossRef CAS.
- Z. Wu, F. Dinkelbach, F. Kerner, A. Friedrich, L. Ji, V. Stepanenko, F. Wurthner, C. M. Marian and T. B. Marder, Chem.–Eur. J., 2022, 28, e202200525 CrossRef CAS.
- D. Li, F. Lu, J. Wang, W. Hu, X. M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916 CrossRef CAS PubMed.
- J. Jovaisaite, S. Kirschner, S. Raisys, G. Kreiza, P. Baronas, S. Jursenas and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef CAS PubMed.
- Z. Ma, Z. Yang, L. Mu, L. Deng, L. Chen, B. Wang, X. Qiao, D. Hu, B. Yang, D. Ma, J. Peng and Y. Ma, Chem. Sci., 2021, 12, 14808 RSC.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206 CrossRef CAS PubMed.
- S. Kuila, S. Garain, S. Bandi and S. J. George, Adv. Funct. Mater., 2020, 30, 2003693 CrossRef CAS.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325 CrossRef CAS.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386 CrossRef CAS.
- M. Gmelch, T. Achenbach, A. Tomkeviciene and S. Reineke, Adv. Sci., 2021, 8, e2102104 CrossRef PubMed.
- Y. Zhang, L. Gao, X. Zheng, Z. Wang, C. Yang, H. Tang, L. Qu, Y. Li and Y. Zhao, Nat. Commun., 2021, 12, 2297 CrossRef CAS.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928 CrossRef CAS.
- K. Jiang, S. Hu, Y. Wang, Z. Li and H. Lin, Small, 2020, 16, e2001909 CrossRef.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, e1807887 CrossRef.
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W. W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 Search PubMed.
- Y. Fan, S. Liu, M. Wu, L. Xiao, Y. Fan, M. Han, K. Chang, Y. Zhang, X. Zhen, Q. Li and Z. Li, Adv. Mater., 2022, 34, e2201280 CrossRef PubMed.
- Y. Li, L. Jiang, W. Liu, S. Xu, T. Y. Li, F. Fries, O. Zeika, Y. Zou, C. Ramanan, S. Lenk, R. Scholz, D. Andrienko, X. Feng, K. Leo and S. Reineke, Adv. Mater., 2021, 33, e2101844 CrossRef PubMed.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS.
- S. Garain, S. Kuila, B. C. Garain, M. Kataria, A. Borah, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 12323 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678 CrossRef CAS.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311 CrossRef CAS PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468 CrossRef CAS PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier, A. Kohler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588 CrossRef CAS.
- J. Park, J. Lim, J. H. Lee, B. Jang, J. H. Han, S. S. Yoon and J. Y. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 45798 CrossRef CAS PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780 CrossRef CAS.
- K. R. Naveen, S. J. Hwang, H. Lee and J. H. Kwon, Adv. Electron. Mater., 2021, 8, 2101114 CrossRef.
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, K. Goushi, X. Tang, Y. Tsuchiya, H. Nakanotani and C. Adachi, Adv. Opt. Mater., 2022, 10, 2200682 CrossRef CAS.
- Y. C. Cheng, X. C. Fan, F. Huang, X. Xiong, J. Yu, K. Wang, C. S. Lee and X. H. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202212575 CrossRef CAS PubMed.
- Y. Zou, J. Hu, M. Yu, J. Miao, Z. Xie, Y. Qiu, X. Cao and C. Yang, Adv. Mater., 2022, 34, e2201442 CrossRef PubMed.
- X. F. Luo, S. Q. Song, H. X. Ni, H. Ma, D. Yang, D. Ma, Y. X. Zheng and J. L. Zuo, Angew. Chem., Int. Ed., 2022, 61, e202209984 CrossRef CAS PubMed.
- Y. K. Qu, D. Y. Zhou, F. C. Kong, Q. Zheng, X. Tang, Y. H. Zhu, C. C. Huang, Z. Q. Feng, J. Fan, C. Adachi, L. S. Liao and Z. Q. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201886 CrossRef CAS PubMed.
- X. Wang, L. Wang, G. Meng, X. Zeng, D. Zhang and L. Duan, Sci. Adv., 2023, 9, eadh1434 CrossRef CAS PubMed.
- Q. Wang, Y. Xu, T. Yang, J. Xue and Y. Wang, Adv. Mater., 2023, 35, e2205166 CrossRef PubMed.
- B. Lei, Z. Huang, S. Li, J. Liu, Z. Bin and J. You, Angew. Chem., Int. Ed., 2023, 62, e202218405 CrossRef CAS PubMed.
- X. Yao, Y. Li, H. Shi, Z. Yu, B. Wu, Z. Zhou, C. Zhou, X. Zheng, M. Tang, X. Wang, H. Ma, Z. Meng, W. Huang and Z. An, Nat. Commun., 2024, 15, 4520 CrossRef CAS PubMed.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 CrossRef CAS.
- F. Chen, L. Zhao, X. Wang, Q. Yang, W. Li, H. Tian, S. Shao, L. Wang, X. Jing and F. Wang, Sci. China: Chem., 2021, 64, 547 CrossRef CAS.
- M. Nagata, H. Min, E. Watanabe, H. Fukumoto, Y. Mizuhata, N. Tokitoh, T. Agou and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 20280 CrossRef CAS.
- T. Agou, K. Matsuo, R. Kawano, I. S. Park, T. Hosoya, H. Fukumoto, T. Kubota, Y. Mizuhata, N. Tokitoh and T. Yasuda, ACS Mater. Lett., 2019, 2, 28 CrossRef.
- T. Wang, Y. Zou, Z. Huang, N. Li, J. Miao and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202211172 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498 CrossRef CAS PubMed.
- A. Pershin, D. Hall, V. Lemaur, J. C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- K. R. Naveen, P. Palanisamy, M. Y. Chae and J. H. Kwon, Chem. Commun., 2023, 59, 3685 RSC.
- L. Ma, Y. Liu, H. Tian and X. Ma, JACS Au, 2023, 3, 1835 CrossRef CAS PubMed.
- S. Xiong, Y. Xiong, D. Wang, Y. Pan, K. Chen, Z. Zhao, D. Wang and B. Z. Tang, Adv. Mater., 2023, 35, e2301874 CrossRef PubMed.
- X. Zhang, C. Qian, Z. Ma, X. Fu, Z. Li, H. Jin, M. Chen, H. Jiang and Z. Ma, Adv. Sci., 2023, 10, e2206482 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization details and photophysical properties. CCDC 2239638–2239642 and 2239553. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05383d |
| This journal is © The Royal Society of Chemistry 2024 |