
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA potent and selective anti-glutathione peroxidase 4 nanobody as a ferroptosis inducer†
Xinyu
Li
ab,
Yaru
Li
c,
Aowei
Xie
 d,
Fenglin
Chen
ab,
Jing
Wang
ab,
Jianfeng
Zhou
ab,
Ximing
Xu
abe,
Zhenlin
Xu
*c,
Yong
Wang
*ab and
Xue
Qiu
*ab
d,
Fenglin
Chen
ab,
Jing
Wang
ab,
Jianfeng
Zhou
ab,
Ximing
Xu
abe,
Zhenlin
Xu
*c,
Yong
Wang
*ab and
Xue
Qiu
*ab
aKey Laboratory of Marine Drug, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China. E-mail: wangyong8866@ouc.edu.cn; qiuxue@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
cGuangdong Provincial Key Laboratory of Food Quality and Safety, College of Food Science, South China Agricultural University, Guangzhou, 510642, China. E-mail: xzlin@scau.edu.cn
dSchool of Food Science and Engineering, Ocean University of China, Qingdao 266003, China
eMarine Biomedical Research Institute of Qingdao, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266071, Shandong, P. R. China
First published on 29th October 2024
Abstract
Glutathione peroxidase 4 (GPX4) plays a crucial role in the ferroptosis pathway, emerging as a potential drug target in the treatment of refractory tumors. Unfortunately, the development of GPX4-targeted treatment has been very limited due to the poor selectivity and drug-like properties of current GPX4 inhibitors. Here, we report a proof-of-concept study of potent anti-GPX4 nanobodies, successfully identified through immunizing Bactrian camels and constructing a phage library. Utilizing a cell-penetrating peptide fusion strategy, these nanobodies with high affinities to GPX4 efficiently internalized in cells and formed the basis for further applications. In particular, 12E significantly inhibited cellular GPX4 and consequently induced remarkable ferroptosis in cancer cells. Furthermore, 12E could impair zebrafish dorsal organizer formation in vivo, as evidenced by a phenotype comparable to that observed in zebrafish with the gpx4b gene knocked out. The new GPX4-inhibiting nanobody described here exhibits superior proteome-wide selectivity and a vastly improved safety profile compared to existing GPX4 inhibitors. These incredible features of 12E, as an anti-GPX4 nanobody, may not only contribute to ferroptosis-related anticancer treatment but also establish a new paradigm for nanobodies in drug development for traditionally undruggable targets.
Introduction
Ferroptosis, a novel form of cell death defined in recent years, is characterized by iron-dependent cell death resulting from unrestricted lipid peroxidation (LPO) and subsequent membrane damage.1,2 A growing number of studies demonstrate that ferroptosis plays an important role in pathological processes, including tumors, neurodegenerative diseases, and tissue ischemia-reperfusion injuries.3–8 Glutathione peroxidase 4 (GPX4) is at the intersection of cellular iron metabolic homeostasis and redox homeostasis, playing a key role in ferroptosis.9,10 A wide range of cancers undergoing drug treatments is associated with a high mesenchymal therapy-resistant cell state, which has high dependency on GPX4 for survival. Studies have shown that using GPX4 inhibitors can effectively regulate the ROS levels and induce ferroptosis in drug-treated cancer-resistant strains, making GPX4 an important drug target for addressing tumor drug resistance.3,4Compared to well-developed drug targets like kinases and G protein-coupled receptors, GPX4 shows a relatively flat structure around its active site, lacking a conventional drug-binding pocket, making it a challenging target for drug development.11–13 Therefore, the development of GPX4 inhibitors with high selectivity has been challenging.14,15 Owing to the strong nucleophilicity of selenocysteine 46 in the active site of GPX4, medicinal chemists have attempted to design small molecular covalent inhibitors with electrophilic groups targeting GPX4. Most reported covalent inhibitors of GPX4, like RSL3 and ML162, exert their activities through chloroacetamide warheads; however, the weak selectivity and low bioavailability of these compounds significantly limit their in vivo applications.14–18 Although researchers have continuously sought other electrophilic reagents targeting GPX4, evaluations indicate that these reagents struggle to efficiently inhibit GPX4 when their electrophilic moieties are less active than chloroacetamide.19 Macromolecular drugs are emerging as robust therapeutic agents, treating various diseases with improved efficacy and safety profiles. Regarding GPX4 inhibition, only attempts using peptides have been reported, but these have shown relatively low binding affinity and inactive cellular anti-GPX4 activities.12 These results underline the extreme challenge in developing GPX4 inhibitors with high selectivity and potent cellular effects.
Nanobodies (NBs) have consistently garnered attention in drug development since their discovery.20,21 Compared with conventional antibodies, NBs have larger CDR1 and CDR3 regions, with the extended CDR3 loop offering greater flexibility to bind to antigenic cavities that are inaccessible to conventional antibodies, thus ensuring better affinity for antigens.22–24 Additionally, NBs possess favorable biochemical characteristics compared to full-size IgGs: (i) they maintain remarkable stability under harsh conditions, including extreme temperatures, the presence of proteases, high pressure and low pH, making them suitable for in vivo administration;25–28 (ii) post-administration, NBs diffuse rapidly throughout the body and have excellent tissue permeability;29 (iii) besides intravenous injection, NBs can be administered orally and intraperitoneally.30 Owing to their high stability, excellent tissue penetration, low immunogenicity and high selectivity, NBs have been developed for the treatment of a wide range of diseases, including cancers,31–33 neurodegenerative diseases,34 cardiovascular diseases,35 bacterial infections,36 rheumatoid arthritis and viral infections.37 For example, caplacizumab was the first approved NB drug, with its outstanding therapeutic effect against thrombotic thrombocytopenic purpura (TTP) accelerating the development of NBs as therapeutic inhibitors.38
Given the above, the various advantages of NBs may provide a significant opportunity to overcome the current challenge of GPX4 inhibitors. Herein, we present a proof-of-concept study of a NB targeting GPX4 as a ferroptosis inducer. We developed a potent NB with remarkable GPX4 inhibition and superior selectivity, ultimately inducing strong ferroptosis in cancer cells aided by cell-penetrating peptides (CPPs). The new GPX4-targeting NB, 12E, exhibited exceptional proteome-wide selectivity and a vastly improved safety profile compared to existing GPX4 small molecular inhibitors, as demonstrated in both cellular examinations and in vivo zebrafish evaluations. The unprecedented behaviors of 12E as an anti-GPX4 NB may contribute not only to ferroptosis-related anticancer treatments, but also accentuate the full potential of NBs as a new strategy for drug development against traditionally non-druggable targets.
Results and discussion
Construction of a NB library and screening of GPX4-specific NBs
Wild-type GPX4 (GPX4WT), a selenocysteine-containing peroxidase with low expression in most cells, has a complex purification process prone to activity loss, making it difficult to extract sufficient GPX4 from natural sources. A mutant GPX4U46C was firstly resolved by Scheerer et al. in 2007,11 and after that, they expressed and purified a GPX4 mutant AllCys(-)GPX4 by mutating all of the cysteines into alanine or serine, and they could finally resolve the crystal structure of the protein containing selenocysteine.13 It was demonstrated that the structure of GPX4 was highly conserved, and there were only minor differences between the structures of the two mutants (PDB entries 2OBI, 6ELW).11,13 A GPX4 mutant AllCys(-)GPX4U46C, with all cysteine residues mutated to non-cysteine residues and the 46-site selenocysteine replaced by a cysteine, was designed and generated (Fig. S1A†), and the circular dichroism (CD) spectra (Fig. S2†) of AllCys(-)GPX4U46C and a commercially available recombinant GPX4WT protein showed that the two proteins exhibit very similar structural features. AllCys(-)GPX4U46C was thus chosen as an immunogenic prototype for the subsequent experiments.The preparation and screening of anti-GPX4 NBs are illustrated in Fig. 1A. Initially, healthy Bactrian camels were carefully selected and immunized with AllCys(-)GPX4U46C. After six rounds of immunization, the serum inhibition rate against AllCys(-)GPX4U46C in these camels exceeded 85%. Subsequently, the total RNA was extracted from isolated blood lymphocytes (Fig. S3†) for cDNA synthesis. After two rounds of nested PCR, NB sequences were amplified successfully and cloned into pComb3X vectors.39,40 The recombinant vectors were then transformed in E. coli TG1 by electroporation. Following phage infection with TG1, a phage library with a concentration of 1012 cfu mL−1 was obtained for panning. Eventually, a total of ten NBs were obtained by four rounds of biopanning. By comparing their amino acid sequences, seven unique NBs with distinct CDR3 regions were successfully identified (Fig. 1B). These NBs were subsequently expressed in E. coli, with four (named 5C, 4C, 5F, and 12E) being successfully produced. The remaining three posed challenges in purification due to their low solubilities (Fig. S1B†). Consequently, NBs 5C, 4C, 5F, and 12E were chosen for the further investigation of anti-GPX4 activity.
 | ||
| Fig. 1 (A) Flowchart of anti-GPX4 NB screening (created with BioRender). (B) Amino acid sequences of NBs with different CDR3 domains. | ||
Investigation of the affinity and binding site of NBs to GPX4
Considering the convenience and sensitivity of Förster resonance energy transfer (FRET) assays based on quantum dot donors to other fluorophore acceptors,41,42 we first developed a straightforward QD-to-dye FRET assay to determine the GPX4 affinities of NBs. If not mentioned specifically, the recombinant AllCys(-)GPX4U46C protein was used for the subsequent cell-free experiments. NBs with poly-histidine tags were randomly labeled with Cy5 (NB-Cy5, Fig. S4A and Table S1† for absorption spectra and conjugation ratios) and attached directly to the Zn-rich surface of QD605 via metal-affinity mediated self-assembly, generating an energy transfer from QD605 to Cy5. Potential binding of NBs with GPX4 would lead to their displacements from the QD605 surface, reducing the FRET signal from QD605 to Cy5 (Fig. 1A), and recovering the fluorescence (FL) intensity of QD605. The emission spectrum of the FRET donor QD605 overlaps well with the absorption spectrum of the FRET acceptor Cy5, which provides a Förster distance of 5.6 nm (Fig. 2A). The metal affinity-mediated self-assembly strategy allows for highly efficient quantitative labeling without the need for separation and purification. This reaction is rapid and allows for direct labeling before using, offering convenience and avoiding denaturation issues associated with long-term storage. | ||
| Fig. 2 (A) QD-to-dye FRET assay to determine the GPX4 affinities of NBs. Top: absorption (dotted) and FL emission (solid) spectra of QD605 (green; 545–680 nm; resolution: 1 nm; excitation: 450 ± 5 nm; concentration: 1 μM) and Cy5-NHS (red; 610–785 nm; resolution: 1 nm; excitation: 540 ± 5 nm; concentration: 1 μM). Bottom: saturation binding curve of GPX4 and NBs from QD-to-Cy5 FRET assay. Kd values were fitted using the ‘One site-total’ model in Prism 8.0. Data are presented as the mean ± SD from three independent experiments. (B) Kinetic binding curves of GPX4 and NBs determined by SPR. The arrow indicates that the concentrations of NBs increased from 1.95 nM to 500 nM. (C) FRET measurement of the formation of the NB-GPX4-RSL3 ternary complex. Top-left: schematic representation of the FRET assay created with BioRender. Top-right: absorption (dotted) and emission (solid) spectra of Cy3-NHS (green) and Cy5-NHS (red). Optical band-pass filters (gray) were used for Cy3 (550/10 nm) and Cy5 (665/8 nm) detection in two detection channels. Bottom: FRET responses of GPX4 to different combinations of RSL3-Cy5 (left)/ML162-Cy5 (right) and four NBs. The assays included constant concentrations of NB-Cy3 and RSL3-Cy5 and varying concentrations of GPX4. FRET data are presented as the mean ± SD from three independent experiments. | ||
As shown in Fig. S5 and 2A,† the FL intensities of QD605 increased with increasing GPX4 concentrations for all NBs. This indicated that NBs-Cy5 were displaced from the QD605 surface, and the curves began to stabilize at a GPX4 concentration of 200 nM, beyond which no further displacement of NBs-Cy5 occurred. The Kd values reflecting the capacities of GPX4 to displace four of the NBs from the QD surface were calculated to be 143.1 nM, 74.98 nM, 106.6 nM, and 207.6 nM for 4C, 5C, 5F, and 12E, respectively (Fig. 2A). The surface plasmon resonance (SPR) technique was employed to validate the affinities of these NBs to GPX4 and the associations of the four NBs with GPX4 shown in SPR assays were extremely strong and fast (Fig. 2B); in contrast, the conjugations of ML162 and RSL3 with GPX4 were relatively slow (Fig. S6A and B†), as reflected in Table S4† of their association rate constants (ka) and dissociation rate constants (kd). The maximum responses of SPR angles with different concentrations of NBs are plotted in Fig. S6C,† and 4C, 5C, 5F and 12E provided as low as 5.8 nM, 4.3 nM, 4.1 nM, and 11.2 nM binding affinities (Kd) with GPX4, respectively. These parallel results from FRET and SPR measurements demonstrated that the screened NBs have exceptional affinities to GPX4.
Encouraged by the impressive GPX4 affinities of the screened NBs, we developed a new FRET assay to investigate whether the NBs could bind to the active site of GPX4 (Fig. 2C). RSL3 and ML162, known to bind to the active site of GPX4, were used as competitive inhibitors.15 We first synthesized RSL3-Cy5 as a FRET acceptor by conjugating RSL3 with Cy5 using a click-chemistry approach. Four NBs 4C, 5C, 5F, and 12E, which had been validated to bind to GPX4, were labeled with Cy3-NHS, and the NB-Cy3 conjugates were used as FRET donors (the absorption spectra and conjugation ratios of NBs-Cy3 are shown in Fig. S4B and Table S2†). The emission spectrum of the FRET donor Cy3 overlaps well with the absorption spectrum of the FRET acceptor Cy5, which provides a Förster distance of 5.1 nm (Fig. 2C). When NB-Cy3 and RSL3-Cy5 bound to different sites of GPX4, they formed a ternary complex of Cy3-NBs-GPX4-RSL3-Cy5, where FRET occurred. Conversely, when NB-Cy3 and RSL3-Cy5 bound to the same site on GPX4 or partially shared a binding site, fewer ternary complexes were formed, resulting in lower FRET signals. As shown in Fig. 2C, 4C and 5F yielded relatively high FRET signals, indicating that they bound to different GPX4 sites compared with RSL3. Conversely, only minor FRET acceptor sensitizations were observed for 12E and 5C, suggesting that they may bind to a similar site as RSL3 and occupy the active pocket of GPX4. A similar assay using ML162 as a competitive inhibitor was also performed, leading to the consensus that 12E and 5C can bind to the active site of GPX4, indicating their significant potential to block the activity of GPX4.
Although these NBs may bind to different sites of GPX4, both the NB-on-QD displacement assay (QD-to-Cy5 FRET) and the NB-GPX4-RSL3/ML162 binding assay (Cy3-to-Cy5 FRET) mentioned above evidenced their exceptional affinities to GPX4, indicating their direct application in rapid, homogeneous and sensitive quantification of GPX4 in solution. As shown in Fig. S7,† without any optimization, 4C-Cy3 and 12E-Cy3 can detect as low as 0.5 μg of GPX4 in western blot (WB) measurement, verifying the applicability of the fluorescently labeled NBs for selective staining of GPX4. On the other hand, they represent very promising tools for GPX4-related disease treatment to overcome the current challenge of GPX4 inhibitors. For instance, 4C and 5F can be used as ligands for the protein of interest (POI) to degrade GPX4 via the nanobody-based protein degradation strategy,43,44 whereas 12E and 5C may enable the deactivation of GPX4 directly by occupying the active site of GPX4.
Validation of the binding site of NBs by biomolecular modeling and GPX4 mutation
To gain structural insight into the interaction of GPX4 with NBs, the binding modes of GPX4 with 12E, 5C, and 4C were predicted using the state-of-the-art AlphaFold via ColabFold.45,46 Complexes with a pLDDT39 score,47 which measures the quality of the local model for each residue, of over 90 were selected from the predicted structures to determine the optimal combination mode. The analysis was conducted using the molecular graphics program PyMol.48 As shown in Fig. 3A, the three NBs 12E, 5C and 4C interacted with GPX4 primarily through polar interactions, forming stable binding interfaces but with different binding patterns and sites. 12E and 5C bound to the active site of GPX4, while 4C bound to a different site away from the active site. These dockings were coincident with the FRET measurements, which showed that only 12E and 5C would compete for the binding site of RSL3 or ML162.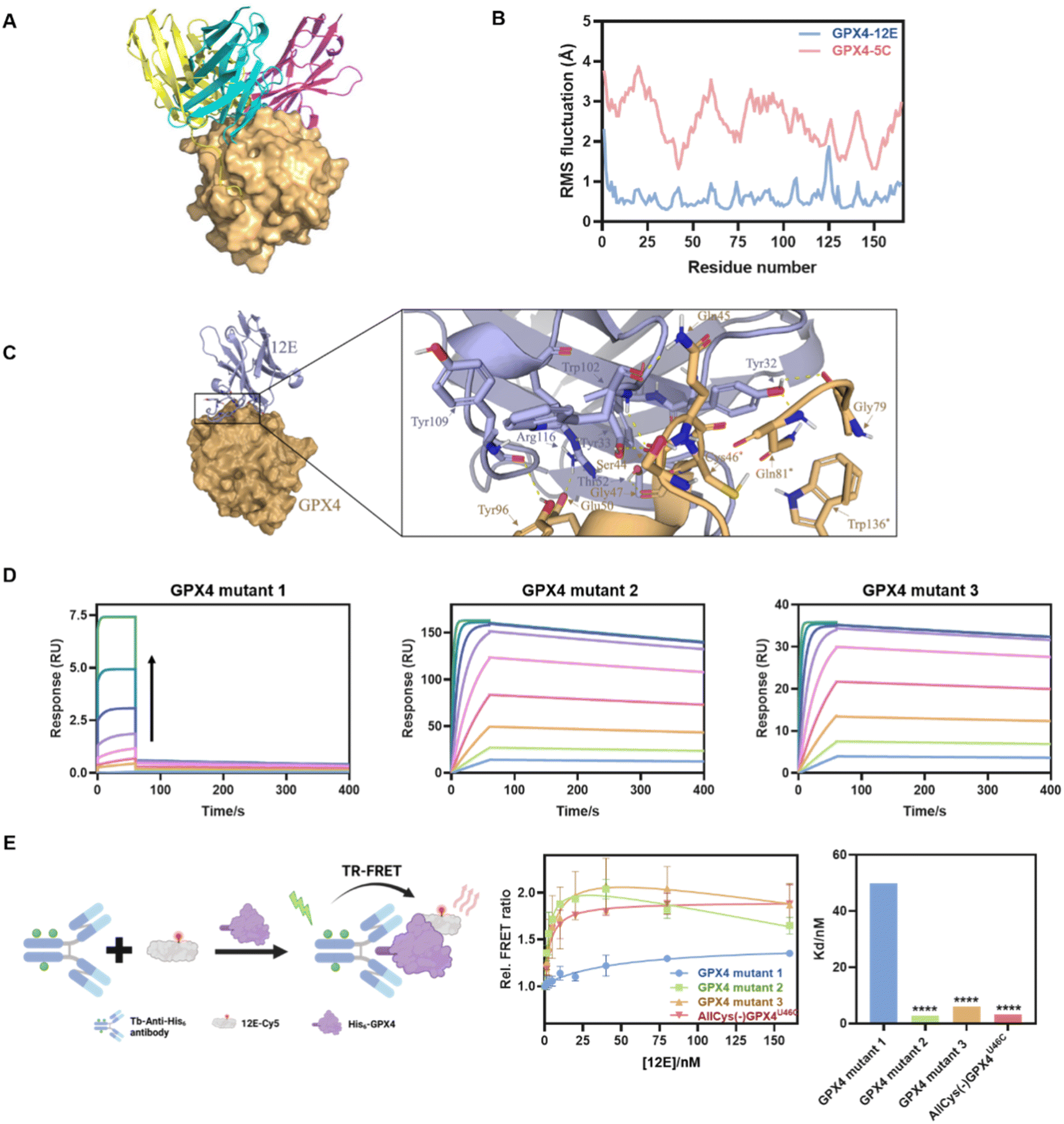 | ||
| Fig. 3 (A) Molecular modeling of GPX4 and NBs 12E (blue), 4C (purple) and 5C (yellow). (B) RMSF of GPX4 within GPX4-12E or GPX4-5C complexes. (C) Molecular surface and zoomed-in views of interactions of the GPX4-12E complex. (D) Kinetic binding curves of GPX4 mutants (mutants 1, 2 and 3) and 12E determined by SPR. The arrow indicates that the concentrations of NBs increased from 1.95 nM to 500 nM. AllCys(-)GPX4U46C,S44A, Q45A, K48A, E50A, AllCys(-)GPX4U46C, I129A, L130A, K135A, W136A and AllCys(-)GPX4U46C, P155A, M156A, E157A were named GPX4 mutants 1, 2, and 3, respectively. (E) TR-FRET measurement for the affinity tests of GPX4 mutants and 12E. Left: schematic representation of the TR-FRET measurement. Middle: affinity binding curves of GPX4 mutants and 12E via TR-FRET. The results were repeated two times and presented as mean ± SD (n = 2). Right: the Kd values of GPX4 mutants and 12E via TR-FRET. Kd was obtained by fitting the relative FRET ratio with the ‘One site-total binding’ model in Prism 8.0. ****p < 0.0001 vs. the vehicle control. | ||
Although 12E and 5C bound to similar sites, the results of molecular dynamics simulation in 100 ns indicated that 12E exhibited better structural complementarity and the GPX4-12E complex was more stable than the GPX4-5C complex (Fig. 3B and S8†). From the residue-based RMSD (root-mean-square deviation) graph, it can be seen that the binding between NBs and GPX4 has gradually equilibrated over the course of the simulation. The RMSF (root-mean-square fluctuation) revealed that the alpha-C in each amino-acid residue of GPX4 protein in the GPX4-12E complex exhibited lower flexibility compared to that of the GPX4-5C complex. Specifically, the residues of Trp102 of 12E bound firmly and simultaneously to both Ser44 and Gln45 side chains of GPX4. The located interactions surrounding Trp102 of 12E involve Tyr109, Arg166 of 12E and Tyr96, Glu50 of GPX4, suggesting a high degree of shape complementarity (Fig. 3C). Tyr32 of 12E bound to Gln81 and Gly79 of GPX4, while the neighboring Tyr33 bound to Ser44, constituting the binding interface of 12E with GPX4. The modeling results clearly showed that 12E could cover the flat pocket surrounded by the active and catalytic triad of GPX4, consisting of Cys46, Gln81 and Trp136. Both FRET measurement and molecular modeling suggested that 12E and 5C could bind to the active site of GPX4. However, 12E presents significant potential to inhibit GPX4 activity as it may form a more stable complex with GPX4 compared to 5C. In contrast, 4C most probably bound to a non-active site of GPX4 and likely presented no substantial effect on GPX4 function.
Considering the stable binding interface of 12E and GPX4 through polar interactions with the amino acid cluster centered on C46 indicated by molecular modeling, several GPX4 mutants were further prepared to confirm the binding site of 12E. The amino acid cluster centered on C46 and two other amino acid clusters centered on I129 and M156, which were spatially far or close to the active site, were also chosen randomly as comparison. We therefore constructed three GPX4 mutants, AllCys(-)GPX4U46C, S44A, Q45A, K48A, E50A, AllCys(-)GPX4U46C, I129A, L130A, K135A, W136A and AllCys(-)GPX4U46C, P155A, M156A, E157A, named GPX4 mutants 1, 2, and 3, respectively (Fig. S1A†), and applied SPR and FRET to test the affinities of GPX4 mutants with 12E. The results of binding-dissociation kinetics and affinities in SPR measurements (Fig. 3D and S6D†) showed that GPX4 mutant 1 almost lost its ability to bind to 12E, while GPX4 mutants 2 and 3 did not show significant changes in their affinities to 12E. A time-resolved FRET (TR-FRET) assay using a lanthanide Tb complex as a FRET donor and Cy5 as a FRET acceptor was also developed to investigate the affinities of different GPX4 mutants to 12E (Fig. 3E).49,50 The binding of 12E-Cy5 to different GPX4 mutants depended on its affinities to them, and this binding can be successfully determined by time-resolved Tb-to-Cy5 FRET with the help of the His-tag antibody conjugated with the Tb complex (His-tag antibody-Tb, Fig. S4C and Table S3† show absorption spectra and conjugation ratios). As shown in Fig. 3E, very low FRET signals were observed for GPX4 mutant 1 compared to the other mutants, which means that GPX4 mutant 1 lost most of its affinity to 12E. Taking all the results together, we could conclude that 12E bound to GPX4 mainly through the amino acids around the active site of GPX4, which aligned well with the MD stimulation.
CPP-appended 12E as a bio-macromolecular inhibitor inducing ferroptosis in OS-RC-2 cells
To explore the cellular effects of NBs, cell-penetrating peptides (CPPs) were utilized to facilitate the entry of NBs into cells. CPPs originated from the human immunodeficiency virus (HIV) transcriptional transactivator (TAT) protein and the Drosophila tentacle homologous structural domain.51,52 Numerous CPPs have been investigated in preclinical and clinical research as powerful tools for delivering various cargoes in infection, inflammation, and cancer treatments.53–55 Especially, Hackenberger et al. developed several simple and highly effective CPP-based strategies for the intracellular delivery of functional cargoes such as proteins and nanobodies against different endogenous targets.56–59 For instance, they demonstrated that co-incubation of fusion-expressed proteins with an arginine-rich (R10) CPP and a thio-nitro-benzoic-acid-activated R10 peptide, TNB-R10, can significantly increase the intracellular delivery of nanobodies and full-size IgG.60 The same strategy was applied herein, where R10 was fused to the C-terminus of NBs 12E and 4C to facilitate their entry into cells, with an SGSGSG linker inserted between the C-terminus of the NBs and the CPP to maintain CPP flexibility (Fig. 4A). After obtaining the fusion-expressed 12E-R10 and 4C-R10 proteins (Fig. S1C†), we labeled them with Cy5 (the conjugation ratios of NBs-R10-Cy5 are shown in Table S5†) to visualize their ability to enter cells with the assistance of TNB-R10. Confocal laser scanning microscopy (CLSM) images of renal cell carcinoma OS-RC-2 cells (Fig. 4A) and neuroblastoma SH-SY5Y cells (Fig. S9†) revealed that, with the aid of TNB-R10, both 12E-R10 and 4C-R10 successfully entered cells with similar delivery efficiencies (the averaged cytosolic fluorescence intensities were quantified) in complete cell medium and were primarily distributed in the cytosol (no significant localization was observed inside the nucleus) after one hour of incubation. As for the control measurement, where cells were incubated with NB-R10-Cy5 without the help of TNB-R10, very weak fluorescence was only detected in OS-RC-2 cells (Fig. 4A). The result highlighted the necessity and efficiency of applying CPP-appended NBs and also introducing TNB-R10 for NB-based drug administration for intracellular targets.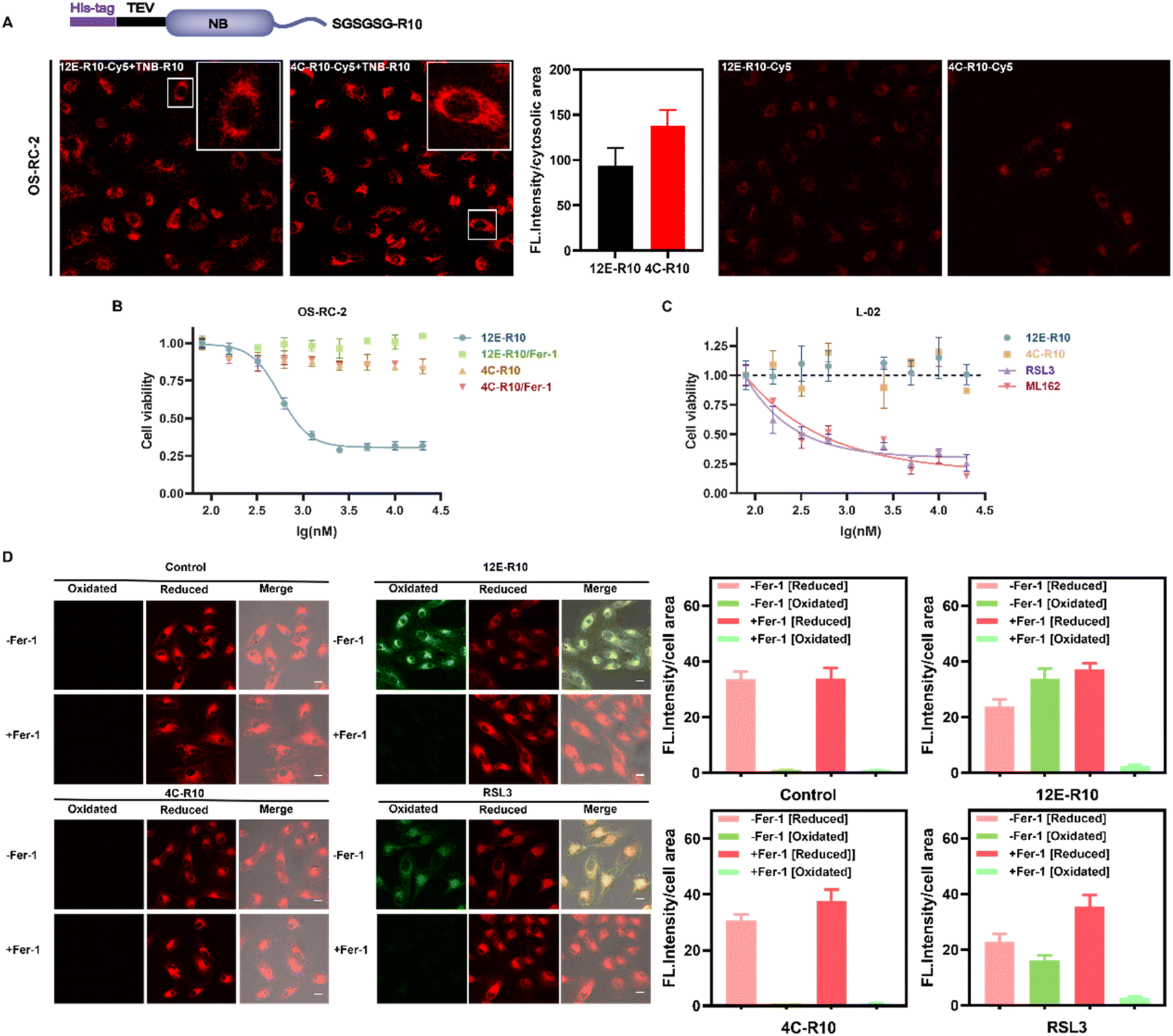 | ||
| Fig. 4 (A) Intracellular delivery efficiency of the NB-R10 fusion protein with TBN-R10 additives. Top: schematic design of NB-R10 fusion protein. Bottom: CLSM images of OS-RC-2 cells incubated with 5 μM 12E-R10-Cy5 or 4C-R10-Cy5 in the presence or absence of 10 μM TNB-R10 for 1 h at 37 °C. Excitation: 581 nm; emission: 580–650 nm; scale bar: 20 μm. Individual cytosolic fluorescence intensity of the CLSM images in the presence of TNB-R10 was processed with Image J software. (B) Viabilities of OS-RC-2 cells incubated with 12E-R10/4C-R10 and 10 μM TNB-R10 in the presence or absence of Ferrostatin-1 (−Fer-1, ferroptosis inhibitor) for 12 h at 37 °C. (C) Viabilities of L-02 cells incubated with 4C-R10/12E-R10 and 10 μM TNB-R10 additives, or small molecular inhibitors RSL3 and ML162 at 37 °C for 72 h. The results were repeated three times and presented as mean ± SD (n = 3). (D) Lipid peroxidation induced by 12E-R10, 4C-R10 and RSL3 based on C11 BODIPY581/591 cell imaging. OS-RC-2 cells were first incubated with 5 μM 12E-R10/4C-R10 (in the presence of 10 μM TNB-R10) and RSL3 (1 μM) at 37 °C for 2 h, and then incubated with 1 μM C11 BODIPY581/591 for 30 min for imaging; scale bar: 20 μm. Cells were also pre-incubated with (+Fer-1) or without Fer-1 (−Fer-1). Individual cell fluorescence intensity in different channels of the CLSM images (processed with Image J software) was indicated on the right of the images. | ||
Given the outstanding GPX4 affinities of the screened NBs and their high cell-entry efficiency with the aid of CPPs, we explored the potential of NBs to induce ferroptosis in ferroptosis-sensitive OS-RC-2 cells. Ferrostatin-1 (Fer-1) is regarded as a ferroptosis-specific inhibitor that can scavenge the initiating alkoxyl radicals produced by ferrous iron from lipid hydroperoxides, thereby reversing the occurrence of ferroptosis in cells.61 Thus, Fer-1 was co-incubated with the NBs to determine the ability of 12E-R10 and 4C-R10 to induce ferroptosis. As shown in Fig. 4B, 12E-R10 exhibited strong antiproliferative activity with an IC50 of 0.56 μM and the addition of Fer-1 completely reversed the cytotoxicity. This data demonstrated the potency of 12E-R10 in inducing ferroptosis in cells, likely due to its ability to target the active site of GPX4. In contrast, 4C-R10 was almost non-cytotoxic in the presence or absence of Fer-1, even at a high concentration of 20 μM (Fig. 4B), indicating that 4C-R10 lacked the ability to induce ferroptosis in OS-RC-2 cells. These results corresponded well to the aforementioned illustration that 4C-R10 was bound to the non-active site of GPX4.
It is noteworthy that the delivery efficiency and cytotoxicity of 12E-R10 and 4C-R10 on the human normal liver cell line L-02 were tested, and interestingly, with the successful cytosolic delivery of 12E-R10 and 4C-R10 (Fig. S9†), neither of them exhibited significant toxicity to these healthy cells at any of the tested concentrations (Fig. 4C). To determine whether the differential cytotoxicity of 12E-R10 in L-02 and OS-RC-2 cells was due to GPX4 inhibition, we used WB to assess the expression levels of GPX4 in both cell lines. The results indicated that the level of GPX4 in L-02 cells was lower than that in OS-RC-2 cells (Fig. S10†). We could conclude that the altered toxicities of 12E-R10 in OS-RC-2 and L-02 cells (Fig. 4B and C) were caused by the different GPX4 levels and 12E-R10 exhibited high toxicity to OS-RC-2 cells because of its highly specific inhibition on GPX4 activity. In contrast, small molecular GPX4 inhibitors RSL3 and ML162 were reported to exhibit cytotoxicity in human normal cells due to their off-target effect as covalent inhibitors, and indeed, they demonstrated strong cytotoxicity in L-02 cells (Fig. 4C). These results established the unique safety of GPX4-targeting NBs on non-cancerous cells.
Unrestricted lipid peroxidation is a hallmark of ferroptosis and can be evaluated in cells by multiple approaches. To verify the ferroptosis inducing activity of NBs, we used C11BODIPY581/591 as a fluorescent indicator to monitor ROS levels in OS-RC-2 cells. As shown in Fig. 4D, the presence of both 12E-R10 and RSL3 caused C11BODIPY581/591 to emit green fluorescence due to the blue shift in the emission wavelength upon oxidation, while 4C-R10 did not induce oxidation of C11BODIPY581/591. Furthermore, the accumulation of lipid peroxides triggered by 12E-R10 can be reversed by the addition of Fer-1, emphasizing its remarkable ferroptosis selectivity. Malondialdehyde (MDA), an intermediate metabolite of the lipid peroxidation reaction, is commonly used to reflect lipid peroxide levels. As shown in Fig. 5A, the intracellular MDA amount increased significantly after treatment of OS-RC-2 cells with 12E-R10 compared to the negligible effect of 4C-R10 on MDA levels, further confirming the facilitation of lipid peroxidation within cells by 12E-R10. The mRNA levels of prostaglandin-endoperoxide synthase 2 (PTGS2) were recognized as an important ferroptosis indicator, as ferroptosis could cause an expression upregulation of PTGS2. In our case, as shown in Fig. 5B, PTGS2 transcription in OS-RC-2 cells treated with 12E-R10 increased 2.7-fold compared to the control, illustrating the ferroptosis induced by 12E-R10. Furthermore, the release of cytosolic enzymes such as lactate dehydrogenase (LDH) is used extensively to identify necrotic cells. The concentration of LDH in OS-RC-2 cells was measured after being treated with 12E-R10, and the results showed that 12E-R10 does not induce LDH release, indicating that it does not induce cell necrosis (Fig. 5C). Together, the above data signify the remarkable ability and selectivity of 12E-R10 to induce cellular ferroptosis.
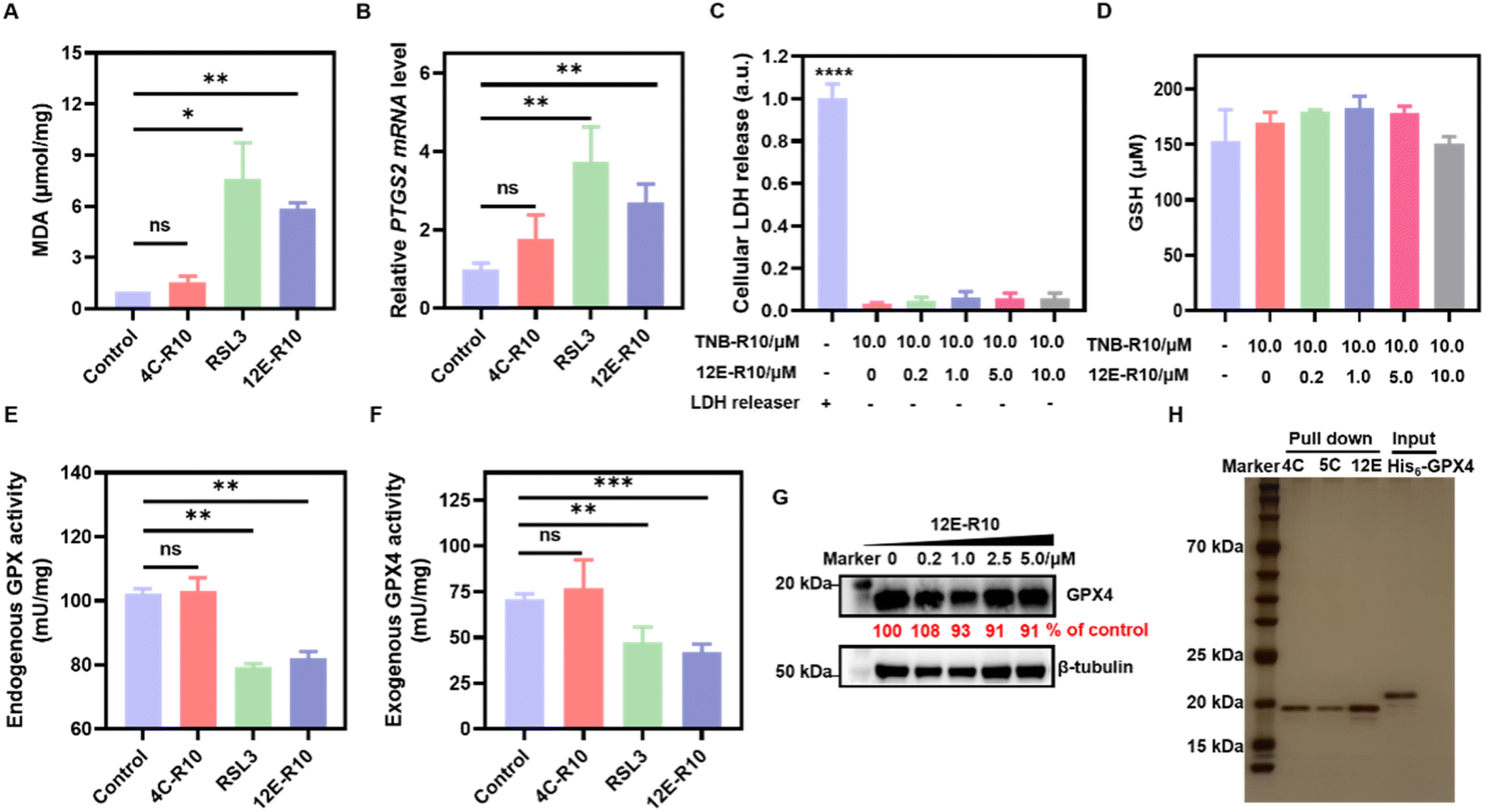 | ||
| Fig. 5 Bio-evaluations of ferroptosis related parameters. (A) MDA assay. (B) PTGS2 mRNA detection. (C) LDH release. (D) GSH concentrations. (E) Endogenous GPX activity. (F) Exogenous GPX4WT activity. (G) WB analysis of GPX4 degradation. (H) Proteome profiles of 4C-R10, 5C-R10 and 12E-R10 assessed by SDS-PAGE (silver staining). Lane 1: marker; Lanes 2–4: 4C-R10, 5C-R10 and 12E-R10 pull-down of OS-RC-2 cell lysates; Lane 5: recombinant GPX4 protein sample. OS-RC-2 cells were incubated with 5 μM 12E-R10/4C-R10 in the presence of 10 μM TNB-R10 or 0.8 μM RSL3 for 4 h (A, B, and E). (A–F) Results were repeated three times and presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs. the vehicle control. | ||
GPX4 inhibition and proteome profiling of 12E-R10
We further confirmed the remarkable ferroptosis-inducing potency of 12E-R10 evoking through GPX4 inhibition rather than GSH depletion or GPX4 degradation. The intracellular levels of GSH and GPX4 in OS-RC-2 cells treated with 12E-R10 were determined and the results showed that the GSH concentrations did not show obvious variation (Fig. 5D), and WB indicated that no significant degradation of GPX4 was observed under the tested concentrations (Fig. 5G). Moreover, the direct effects of 12E-R10 and 4C-R10 on the function of GPX4 were investigated. We first evaluated their impacts on intact GPX4 activity by measuring NADPH consumption in cell lysates. As shown in Fig. 5E, NADPH consumption in OS-RC-2 cells significantly decreased after treatment with 12E-R10, while 4C-R10 showed no effect. Subsequently, the activity of a recently available recombinant GPX4WT protein was tested directly in cell-free solution in the presence of 12E-R10, 4C-R10 or RSL3. The activity of GPX4WT measured with a commercial glutathione peroxidase kit (DTNB) showed that under identical conditions, 12E-R10 and RSL3 decreased the activity of GPX4 by 40% and 33%, respectively, while 4C-R10 did not show any detectable GPX4 inhibition (Fig. 5F). These data were consistent with the ferroptosis inducing behaviors of NBs in cells. Overall, these systematic enzymatic and cellular experiments validated that 12E-R10 induced superior ferroptosis in OS-RC-2 cells primarily by specially targeting and inhibiting the activity of GPX4.One of the major challenges in developing GPX4 small molecular inhibitors for clinical therapeutics is their high chemical reactivity, which results in relatively low proteome-wide selectivity.14 In light of the significant GPX4 inhibition and the strong ferroptosis-inducing ability of 12E-R10, it is interesting to examine the proteome profile of 12E-R10 in cells. Consequently, a cellular proteome affinity profiling experiment was conducted to verify the cellular specificity of 12E-R10 and 4C-R10. As shown in Fig. 5H and S11,† both 12E-R10 and 4C-R10 demonstrated negligible affinities for other cellular proteins and exhibited markedly high selectivity for GPX4. This proteome affinity profiling was vastly superior to that of RSL3 and ML162, which engaged in numerous covalent interactions with multiple cellular proteins.14 The distinct cellular selectivity of NBs promises to overcome a key limitation of small molecular GPX4 inhibitors, making them a favorable option for further development.
Nanobody 12E impairs zebrafish dorsal organizer formation
Previously, we demonstrated that knocking out the gpx4b gene in zebrafish led to a dorsalized phenotype in the maternal mutant, characterized by a reduction in the ventral caudal fin at 26 hours post-fertilization (hpf).62 Gpx4b showed a high degree of sequence identity with its human homolog GPX4 (Fig. S12†). Therefore, we utilized the wild-type zebrafish embryos to evaluate the dorsalizing effect of the NBs we generated. Equivalent doses of 12E, 4C, ML162, and RSL3 (7 μM) were injected into 1- to 2-cell zebrafish embryos, respectively (Fig. 6A). Ventral caudal fins were significantly reduced in most (49 out of 60, 82%) zebrafish embryos at 26 hpf following treatment with 12E (Fig. 6B). The consistent phenotype with the gpx4-null mutants confirmed the effective targeting of GPX4 in zebrafish by 12E. Yet 4C presented similar results to PBS-injected zebrafish embryos, showing no neutralizing effect on the GPX4 function. Regarding small molecular GPX4 inhibitors, the administration of ML162 led to a similar phenotype to that of 12E, but with a relatively lower occurrence (28 out of 60, 47%). This result indicated that the in vivo potency of targeting GPX4 of 12E was clearly stronger than that of ML162, when giving the same dose without an additional cell penetrating process. The injection of RSL3 into zebrafish embryos significantly impacted their normal developmental trajectory, exhibiting the hypoplasia of the cephalo–caudal axis and even leading to mortality in some embryos (Fig. 6B). However, 12E, 4C, and ML162 did not exhibit such effects on the development of zebrafish embryos. These findings indicated that 12E specifically targeted GPX4 in zebrafish, and as expected from the characteristics of NBs, its toxicity was greatly reduced compared to that of RSL3. The significant in vivo GPX4 reduction and the favorable safety profile in zebrafish embryos underscore the considerable potential for GPX4-related mechanism research and drug development. | ||
| Fig. 6 Zebrafish dorsal organizer formation impaired by NB 12E. (A) Schematic representation of the observation for early development of zebrafish. (B) Brightfield images of the wild-type (WT) embryos at the indicated stages after the injection of 7 μM RSL3/12E/4C and PBS (the injection volume is 1 nL). The red arrow indicates the reduced ventral tail fin. The frequency of embryos with the indicated patterns is shown in the bottom right corner of each panel. Lateral views with anterior to the left. Scale bar = 500 μm. | ||
Conclusions
In this manuscript, GPX4-targeting NBs have been developed as optimal candidates to overcome the challenges posed by small molecular inhibitors in targeting GPX4 for therapy-resistant cancer. Based on meticulous design and the applications of multiple biophysical techniques such as FRET and SPR, a few potent NBs with high affinities to GPX4 have been successfully identified. In particular, biomolecular simulation and protein mutation studies revealed that 12E can occupy the active and catalytic triad of GPX4, leading to its superior proteome-wide selectivity and remarkable inhibition on GPX4, ultimately triggering significant ferroptosis in cancer cells through the aid of CPPs. The ability of 12E to induce ferroptosis has been further evidenced by multiple approaches, including lipid accumulation experiments and ferroptosis biomarker investigation, showing negligible damage to human normal cells in contrast to the serious cytotoxicity of currently used small molecular GPX4 inhibitors. More promisingly, the subsequent in vivo evaluation of 12E highlighted its specific diminishing effect on GPX4, comparable to knocking out the gpx4b gene in zebrafish, but without obvious toxicity issues affecting their normal development. Thus, as the first cell-permeant biomacromolecule targeting GPX4, this work positions 12E as a superior alternative to currently known GPX4 inhibitors as a ferroptosis inducer. This may also pave the way for NBs as a new strategy in drug development against traditionally non-druggable targets.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. Q., Y. W., Z. X. designed and supervised the study. X. L., Y. L., F. C., J. W. performed the experiments and analyzed the data. A. X., X. X. did the stimulations. J. Z. guided the zebrafish study. X. Q., Y. W., X. L. wrote the manuscript with input from all co-authors.Conflicts of interest
X. L., Y. L., Z. X., Y. W., and X. Q. are inventors of patent applications related to this work.Acknowledgements
This work has been supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2022QNLM0003-2), the National Natural Science Foundation of China (No. 82104120), the Taishan Scholar Youth Expert Programs in Shandong Province (tsqn202211064 and tsqn202103035), the Excellent Young Scholars of Shandong Provincial Natural Science Foundation (ZR2023YQ013 and 2022HWYQ069), and the Fundamental Research Funds for the Central Universities, Ocean University of China (No. 202262015). All experimental protocols were approved by and conducted in accordance with the Ethical Committee of Experimental Animal Care, Ocean University of China.References
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley and W. S. Yang, et al., Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell, 2012, 149(5), 1060–1072 CrossRef PubMed.
- B. R. Stockwell, Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications, Cell, 2022, 185(14), 2401–2421 CrossRef PubMed.
- M. J. Hangauer, V. S. Viswanathan, M. J. Ryan, D. Bole, J. K. Eaton, A. Matov, J. Galeas, H. D. Dhruv, M. E. Berens and S. L. Schreiber, et al., Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition, Nature, 2017, 551(7679), 247–250 CrossRef PubMed.
- V. S. Viswanathan, M. J. Ryan, H. D. Dhruv, S. Gill, O. M. Eichhoff, B. Seashore-Ludlow, S. D. Kaffenberger, J. K. Eaton, K. Shimada and A. J. Aguirre, et al., Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway, Nature, 2017, 547(7664), 453–457 CrossRef PubMed.
- R. Shah, M. S. Shchepinov and D. A. Pratt, Resolving the role of lipoxygenases in the initiation and execution of Ferroptosis, ACS Cent. Sci., 2018, 4(3), 387–396 CrossRef PubMed.
- X. X. Fang, H. Wang, D. Han, E. J. Xie, X. Yang, J. Y. Wei, S. S. Gu, F. Gao, N. L. Zhu and X. J. Yin, et al., Ferroptosis as a target for protection against cardiomyopathy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(7), 2672–2680 CrossRef PubMed.
- W. Y. Sun, V. A. Tyurin, K. Mikulska-Ruminska, I. H. Shrivastava, T. S. Anthonymuthu, Y. J. Zhai, M. H. Pan, H. B. Gong, D. H. Lu and J. Sun, et al., Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal, Nat. Chem. Biol., 2021, 17(4), 465–476 CrossRef CAS PubMed.
- Y. Lv, C. Liang, Q. Sun, J. Zhu, H. Xu, X. Li, Y.-Y. Li, Q. Wang, H. Yuan and B. Chu, et al., Structural insights into FSP1 catalysis and ferroptosis inhibition, Nat. Commun., 2023, 14(1), 5933 CrossRef CAS.
- I. Ingold, C. Berndt, S. Schmitt, S. Doll, G. Poschmann, K. Buday, A. Roveri, X. X. Peng, F. P. Freitas and T. Seibt, et al., Selenium utilization by GPX4 is required to prevent hydroperoxide-induced Ferroptosis, Cell, 2018, 172(3), 409–422 CrossRef CAS PubMed.
- G. C. Forcina and S. J. Dixon, GPX4 at the crossroads of lipid homeostasis and Ferroptosis, Proteomics, 2019, 19(18), 1800311 CrossRef.
- P. Scheerer, A. Borchert, N. Krauss, H. Wessner, C. Gerth, W. Hohne and H. Kuhn, Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4), Biochemistry, 2007, 46(31), 9041–9049 CrossRef CAS PubMed.
- K. Sakamoto, S. Sogabe, Y. Kamada, S. Matsumoto, A. Kadotani, J. Sakamoto and A. Tani, Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis, Biochem. Biophys. Res. Commun., 2017, 482(2), 195–201 CrossRef CAS PubMed.
- A. Borchert, J. Kalms, S. R. Roth, M. Rademacher, A. Schmidt, H. G. Holzhutter, H. Kuhn and P. Scheerer, Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2018, 1863(9), 1095–1107 CrossRef CAS PubMed.
- J. K. Eaton, L. Furst, R. A. Ruberto, D. Moosmayer, A. Hilpmann, M. J. Ryan, K. Zimmermann, L. K. L. Cai, M. Niehues and V. Badock, et al., Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles, Nat. Chem. Biol., 2020, 16(5), 497–506 CrossRef CAS PubMed.
- H. R. Liu, F. Forouhar, A. N. J. Lin, Q. Wang, V. Polychronidou, R. K. Soni, X. Xia and B. R. Stockwell, Small-molecule allosteric inhibitors of GPX4, Cell Chem. Biol., 2022, 29(12), 1680–1693 CrossRef CAS PubMed.
- D. Allimuthu and D. J. Adams, 2-Chloropropionamide as a low-reactivity electrophile for irreversible small-molecule probe identification, ACS Chem. Biol., 2017, 12(8), 2124–2131 CrossRef CAS PubMed.
- J. K. Eaton, R. A. Ruberto, A. Kramm, V. S. Viswanathan and S. L. Schreiber, Diacylfuroxans are masked nitrile oxides that inhibit GPX4 covalently, J. Am. Chem. Soc., 2019, 141(51), 20407–20415 CrossRef CAS PubMed.
- W. Li, J. Yu, J. Wang, X. J. Fan, X. M. Xu, H. Wang, Y. Xiong, X. Y. Li, X. M. Zhang and Q. E. Zhang, et al., How does ferrocene correlate with ferroptosis? Multiple approaches to explore ferrocene-appended GPX4 inhibitors as anticancer agents, Chem. Sci., 2024, 15(27), 10477–10490 RSC.
- J. K. Eaton, L. Furst, L. L. Cai, V. S. Viswanathan and S. L. Schreiber, Structure-activity relationships of GPX4 inhibitor warheads, Bioorg. Med. Chem. Lett., 2020, 30(23), 127538 CrossRef PubMed.
- C. Hamerscasterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hamers, E. B. Songa, N. Bendahman and R. Hamers, Naturally occurring antibodies devoid of light chains, Nature, 1993, 363(6428), 446–448 CrossRef.
- S. Muyldermans, A guide to: generation and design of nanobodies, FEBS J., 2021, 288(7), 2084–2102 CrossRef PubMed.
- M. Arbabi-Ghahroudi, Camelid single-domain antibodies: promises and challenges as lifesaving treatments, Int. J. Mol. Sci., 2022, 23(9), 5009 CrossRef.
- C. Castrignano, F. Di Scipio, F. Franco, B. Mognetti and G. N. Berta, Reviving a classic antigen with a cutting-edge approach: nanobodies for HER2+ breast cancer, Pharmaceuticals, 2023, 16(6), 794 CrossRef PubMed.
- H. P. Tang, Y. Gao and J. Y. Han, Application progress of the single domain antibody in medicine, Int. J. Mol. Sci., 2023, 24(4), 4176 CrossRef PubMed.
- M. Lauwereys, M. A. Ghahroudi, A. Desmyter, J. Kinne, W. Holzer, E. De Genst, L. Wyns and S. Muyldermans, Potent enzyme inhibitors derived from dromedary heavy-chain antibodies, FEBS J., 1998, 17(13), 3512–3520 Search PubMed.
- J. M. J. Perez, J. G. Renisio, J. J. Prompers, C. J. van Platerink, C. Cambillau, H. Darbon and L. G. J. Frenken, Thermal unfolding of a llama antibody fragment: a two-state reversible process, Biochemistry, 2001, 40(1), 74–83 CrossRef.
- M. Dumoulin, K. Conrath, A. Van Meirhaeghe, F. Meersman, K. Heremans, L. G. J. Frenken, S. Muyldermans, L. Wyns and A. Matagne, Single-domain antibody fragments with high conformational stability, Protein Sci., 2002, 11(3), 500–515 CrossRef.
- G. Hussack, T. Hirama, W. Ding, R. MacKenzie and J. Tanha, Engineered single-domain antibodies with high protease resistance and thermal stability, PLoS One, 2011, 6(11), e28218 CrossRef PubMed.
- F. Van Bockstaele, J. B. Holz and H. Revets, The development of nanobodies for therapeutic applications, Curr. Opin. Invest. Drugs, 2009, 10(11), 1212–1224 Search PubMed.
- B. K. Jin, S. Odongo, M. Radwanska and S. Magez, Nanobodies: a review of generation, diagnostics and therapeutics, Int. J. Mol. Sci., 2023, 24(6), 5994 CrossRef PubMed.
- S. Y. Sun, Z. Q. Ding, X. M. Yang, X. Y. Zhao, M. L. Zhao, L. Gao, Q. Chen, S. X. Xie, A. Q. Liu and S. H. Yin, et al., Nanobody: a small antibody with big implications for tumor therapeutic strategy, Int. J. Nanomed., 2021, 16, 2337–2356 CrossRef PubMed.
- J. C. Maza, D. M. García-Almedina, L. E. Boike, N. X. Hamlish, D. K. Nomura and M. B. Francis, Tyrosinase-mediated synthesis of nanobody-cell conjugates, ACS Cent. Sci., 2022, 8(7), 955–962 CrossRef PubMed.
- A. Maali, M. Gholizadeh, S. Feghhi-Najafabadi, A. Noei, S. S. Seyed-Motahari, S. Mansoori and Z. Sharifzadeh, Nanobodies in cell-mediated immunotherapy: on the road to fight cancer, Front. Immunol., 2023, 14, 1012841 CrossRef.
- Z. Asefy, S. Hoseinnejhad and Z. Ceferov, Nanoparticles approaches in neurodegenerative diseases diagnosis and treatment, Neurol. Sci., 2021, 42(7), 2653–2660 CrossRef.
- L. A. Bocancia-Mateescu, D. Stan, A. C. Mirica, M. G. Ghita, D. Stan and L. L. Ruta, Nanobodies as diagnostic and therapeutic tools for cardiovascular diseases (CVDs), Pharmaceuticals, 2023, 16(6), 863 CrossRef CAS.
- Q. Qin, H. Liu, W. B. He, Y. C. Guo, J. X. Zhang, J. J. She, F. Zheng, S. C. Zhang, S. Muyldermans and Y. R. Wen, Single domain antibody application in bacterial infection diagnosis and neutralization, Front. Immunol., 2022, 13, 1014377 CrossRef.
- S. Ahangarzadeh, Z. Payandeh, R. Arezumand, K. Shahzamani, F. Yarian and A. Alibakhshi, An update on antiviral antibody-based biopharmaceuticals, Int. Immunopharmacol., 2020, 86, 106760 CrossRef.
- M. Scully, S. R. Cataland, F. Peyvandi, P. Coppo, P. Knöbl, J. A. K. Hovinga, A. Metjian, J. de la Rubia, K. Pavenski and F. Callewaert, et al., Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura, N. Engl. J. Med., 2019, 380(4), 335–346 CrossRef PubMed.
- Y. Q. Zhang, Z. L. Xu, F. Wang, J. Cai, J. X. Dong, J. R. Zhang, R. Si, C. L. Wang, Y. Wang and Y. D. Shen, et al., Isolation of Bactrian camel single domain antibody for parathion and development of one-step dc-FEIA method using VHH-alkaline phosphatase fusion protein, Anal. Chem., 2018, 90(21), 12886–12892 CrossRef.
- F. Wang, Z. F. Li, Y. Y. Yang, D. B. Wan, N. Vasylieva, Y. Q. Zhang, J. Cai, H. Wang, Y. D. Shen and Z. L. Xu, et al., Chemiluminescent enzyme immunoassay and bioluminescent enzyme immunoassay for tenuazonic acid mycotoxin by exploitation of nanobody and nanobody-nanoluciferase fusion, Anal. Chem., 2020, 92(17), 11935–11942 CrossRef.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, FRET as a biomolecular research tool-understanding its potential while avoiding pitfalls, Nat. Methods, 2019, 16(9), 815–829 CrossRef.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Energy transfer with semiconductor quantum dot bioconjugates: a versatile platform for biosensing, energy harvesting, and other developing applications, Chem. Rev., 2017, 117(2), 536–711 CrossRef PubMed.
- F. F. Shen, G. Zheng, M. Setegne, K. Tenglin, M. Izadi, H. Xie, L. T. Zhai, S. H. Orkin and L. M. K. Dassama, A cell-permeant nanobody-based degrader that induces fetal hemoglobin, ACS Cent. Sci., 2022, 8(12), 1695–1703 CrossRef PubMed.
- H. P. He, C. J. Zhou and X. Chen, ATNC: versatile nanobody chimeras for autophagic degradation of intracellular unligandable and undruggable proteins, J. Am. Chem. Soc., 2023, 145(45), 24785–24795 Search PubMed.
- R. Evans, M. O'Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Zídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. M. Jumper, and D. Hassabis, Protein complex prediction with AlphaFold-Multimer, bioRxiv, 2021, preprint, DOI: 10.1101/2021.10.04.463034.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19(6), 679–682 CrossRef PubMed.
- P. Bryant, G. Pozzati and A. Elofsson, Improved prediction of protein-protein interactions using AlphaFold2, Nat. Commun., 2022, 13(1), 1265 CrossRef PubMed.
- W. L. Delano, The PyMOL Molecular Graphics System, 2002 Search PubMed.
- Y. Li, M. Q. Qian, Y. P. Liu and X. Qiu, APPROACH: Sensitive Detection of Exosomal Biomarkers by Aptamer-Mediated Proximity Ligation Assay and Time-Resolved Förster Resonance Energy Transfer, Biosensors-Basel, 2024, 14(5), 15 Search PubMed.
- N. C. Payne, A. S. Kalyakina, K. Singh, M. A. Tye and R. Mazitschek, Bright and stable luminescent probes for target engagement profiling in live cells, Nat. Chem. Biol., 2021, 17(11), 1168–1177 CrossRef.
- A. H. Joliot, A. Triller, M. Volovitch, C. Pernelle and A. Prochiantz, Alpha-2,8-polysialic acid is the neuronal surface-receptor of antennapedia homeobox peptide, New Biol., 1991, 3(11), 1121–1134 Search PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, The 3RD helix of the antennapedia homeodomain translocates translocates through biological-membranes, J. Biol. Chem., 1994, 269(14), 10444–10450 CrossRef.
- F. H. Wang, Y. Wang, X. Zhang, W. J. Zhang, S. R. Guo and F. Jin, Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery, J. Controlled Release, 2014, 174, 126–136 CrossRef PubMed.
- G. Guidotti, L. Brambilla and D. Rossi, Cell-penetrating peptides: from basic research to clinics, Trends Pharmacol. Sci., 2017, 38(4), 406–424 CrossRef.
- A. Borrelli, A. L. Tornesello, M. L. Tornesello and F. M. Buonaguro, Cell penetrating peptides as molecular carriers for anti-cancer agents, Molecules, 2018, 23(2), 295 CrossRef PubMed.
- H. D. Herce, D. Schumacher, A. F. L. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M. A. Kasper, S. Reinke, E. Krause and H. Leonhardt, et al., Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells, Nat. Chem., 2017, 9(8), 762–771 CrossRef PubMed.
- A. F. L. Schneider, L. S. Benz, M. Lehmann and C. P. R. Hackenberger, Cell-permeable nanobodies allow dual-color super-resolution microscopy in untransfected living cells, Angew. Chem., Int. Ed., 2021, 60(40), 22075–22080 CrossRef CAS.
- J. V. V. Arafiles, J. Franke, L. Franz, J. Gómez-González, K. Kemnitz-Hassanin and C. P. R. Hackenberger, Cell-surface-retained peptide additives for the cytosolic delivery of functional proteins, J. Am. Chem. Soc., 2023, 145(45), 24535–24548 CAS.
- L. Franz, T. Rubil, A. Balázs, M. Overtus, K. Kemnitz-Hassanin, C. Govaerts, M. A. Mall, and C. P. R. Hackenberger, A cell-permeable nanobody to restore F508del cystic fibrosis transmembrane conductance regulator activity, bioRxiv, 2024, preprint, DOI: 10.1101/2024.04.26.591242.
- A. F. L. Schneider, M. Kithil, M. C. Cardoso, M. Lehmann and C. P. R. Hackenberger, Cellular uptake of large biomolecules enabled by cell-surface-reactive cell-penetrating peptide additives, Nat. Chem., 2021, 13(6), 530–539 CrossRef CAS.
- G. Miotto, M. Rossetto, M. L. Di Paolo, L. Orian, R. Venerando, A. Roveri, A. M. Vuckovic, V. B. Travain, M. Zaccarin and L. Zennaro, et al., Insight into the mechanism of ferroptosis inhibition by ferrostatin-1, Redox Biol., 2020, 28, 101328 CrossRef CAS.
- X. Z. Rong, Y. M. Zhou, Y. Z. Liu, B. B. Zhao, B. Wang, C. X. Wang, X. X. Gong, P. P. Tang, L. Lu and Y. Li, et al., Glutathione peroxidase 4 inhibits Wnt/β-catenin signaling and regulates dorsal organizer formation in zebrafish embryos, Development, 2017, 144(9), 1687–1697 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed materials and methods; SDS-PAGE and WB of NBs, GPX4 mutants and NBs-R10 after protein expression and purification; circular dichroism spectra of AllCys(-)GPX4U46C and recombinant GPX4WT protein; agarose gel plot of RNA samples extracted from blood lymphocytes; the absorption spectra and conjugation ratios of NB-Cy5, NB-R10, NB-Cy3 and His-tag-antibody-Tb conjugates; fluorescence spectra of NB-on-QD displacement assays (QD-to-Cy5 FRET); saturation binding curves and kinetic binding curves via SPR; association rate constant (ka) and dissociation rate constant (kd) of GPX4 with NB or RSL3/ML162; 4C-Cy3 and 12E-Cy3 for selective staining of GPX4 in WB; RMSD of GPX4-12E or GPX4-5C complexes; CLSM images of SH-SY5Y and L-02 cells incubated with 12E-R10-Cy5 and 4C-R10-Cy5; WB analysis of GPX4 levels in OS-RC-2 and L-02 cells; proteome profiling of 4C-R10, 12E-R10 and 5C-R10 on OS-RC-2 cells by WB; amino acid sequence alignment of human, mouse, and zebrafish GPX4/Gpx4 (PDF). See DOI: https://doi.org/10.1039/d4sc05448b |
| This journal is © The Royal Society of Chemistry 2024 |