
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of dialkyl ketones from deoxygenative cross-coupling of carboxylic acids and alcohols†
Bo
Yang
 *ab and
Ri-Yuan
Tang
*ab
*ab and
Ri-Yuan
Tang
*ab
aKey Laboratory for Biobased Materials and Energy of Ministry of Education, College of Materials and Energy, South China Agricultural University, Guangzhou 510642, P. R. China. E-mail: boyang@scau.edu.cn
bState Key Laboratory of Green Pesticide, South China Agricultural University, Guangzhou 510642, P. R. China. E-mail: rytang@scau.edu.cn
First published on 8th October 2024
Abstract
Carboxylic acids and alcohols are widely commercially available, structurally diverse, benchtop stable, and ubiquitous in both natural products and pharmaceutical agents, making them ideal coupling partners for organic synthesis. Though various transformations have been developed by enabling the activation and subsequent cross-coupling of carboxylic acids and alcohols in separate contexts, the direct coupling of these two structural motifs to build value-added molecules is rare. Herein, we developed a direct deoxygenative cross-coupling between carboxylic acids and alcohols for dialkyl ketone synthesis via photoredox/nickel dual catalysis. This protocol provides a powerful platform to construct a wide range of structurally diverse ketone scaffolds with broad substrate scope, good functional group tolerance, step-economy and mild reaction conditions, using simple and readily available substrates. Moreover, the large-scale synthesis and late-stage functionalization of biological molecules also demonstrate the potential practicality.
Due to the prevalence of ketones in natural products and bioactive drugs1 and their central role as versatile reactants in synthetic chemistry,2 the development of powerful methods for ketone synthesis is highly desirable. In this context, a vast number of methods have been developed to construct ketones. Typically, ketone synthesis most often relies upon the addition of an organometallic reagent to an aldehyde followed by oxidation3 or more recently, the use of carboxylic acid derivatives to couple with various nucleophiles (Fig. 1a).4 While significant contributions have been made to this field, these methods typically necessitate a prefunctionalization step and often require nonabundant starting materials, such as air- and moisture-sensitive alkyl organometallics,4e,k–l and organoboron and organosilicon reagents,4c–e which are not step-economical and might lead to issues with functional group tolerance and waste generation, thereby limiting the reaction scope and practicality. To address this problem, we sought to develop a robust platform to deliver ketones utilizing easily accessible and commercially available starting materials under mild conditions.
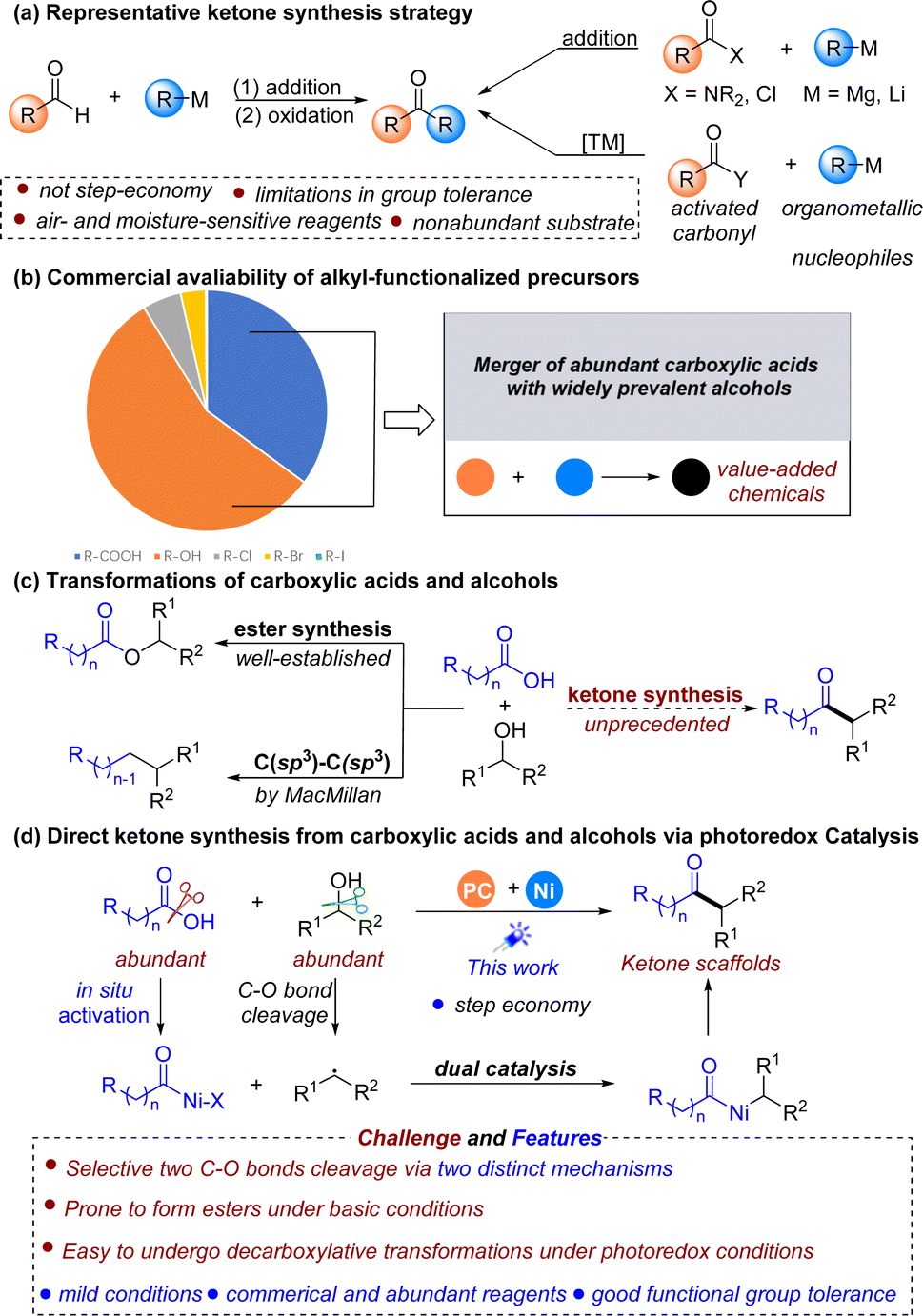 | ||
| Fig. 1 Background of ketone synthesis and cross-coupling of carboxylic acids and alcohols. | ||
Carboxylic acids and alcohols are widely commercially available, structurally diverse, benchtop stable, relatively nontoxic, and ubiquitous in both natural products and pharmaceutical agents,5 making them ideal coupling partners for organic synthesis (Fig. 1b). In recent years, a variety of transformations have been developed by enabling the activation and subsequent cross-coupling of carboxylic acids and alcohols via metallaphotoredox catalysis in separate contexts.6 The direct coupling of these two prevalent structural motifs to build value-added molecules is significantly rare but highly of interest. Conventionally, alcohols and carboxylic acids are most commonly coupled to form esters,7 and fragment cross-coupling of these two structural motifs has been explored to a lesser extent. Recently, the efficient direct coupling of carboxylic acids and alcohols to forge new C(sp3)–C(sp3) bonds has been developed via an N-heterocyclic carbene (NHC)-promoted deoxygenation process by the MacMillan group (Fig. 1c, left).8 Despite this great achievement, developing new types of cross-coupling reactions between these two molecules has remained an appealing yet elusive goal. Considering the importance of ketone scaffolds, we wondered if diverse ketones could be accessed from the direct coupling of abundant carboxylic acids and alcohols, where acids serve as acyl electrophiles, and alcohols serve as nucleophiles (Fig. 1c, right). On this subject, Hong developed a photoinduced method for synthesizing ketones from alcohols and carboxylic acid derivatives through NHC catalysis under mild reaction conditions.6g This approach worked well for benzoic acid, but was not effective for the alkanoic acid substrates. As a consequence, developing an efficient and new catalytic methodology to convert carboxylic acids and alcohols into dialkyl ketone scaffolds is still highly of interest and would complement Hong's strategy.
However, direct coupling of these two structural motifs forming ketones in a desired manner is not as easy as might be expected due to the potential competing cleavage of two C–O bonds in these two molecules. The main challenge for realizing this transformation was how to selectively achieve C–O bond cleavage in both alcohols and carboxylic acids via two distinct mechanisms. In recent years, the combination of photoredox and nickel catalysis has emerged as a powerful tool in chemical bond construction,9 which might provide an alternative protocol for the ketone preparations from alcohols10,11 and acids. In such a reaction, a transition-metal catalytic unit could engage sequentially with the acyl electrophiles formed in situ from carboxylic acids12 and radicals generated from alcohols through oxidative addition13 and radical capture.12a,14 Then the resulting diorganonickel adduct undergoes reductive elimination to afford the desired ketone scaffolds (Fig. 1d). Nevertheless, to achieve this goal, other potential competing reactions, such as the esterification reaction6l and decarboxylative transformation,15,6e,h,i which are commonly encountered under basic and photoredox conditions, are also a big problem and need to be avoided.
In this work, we developed a photoredox-catalyzed synthetic protocol for diverse dialkyl ketone synthesis from naturally abundant carboxylic acids and alcohols under mild conditions with good functional group compatibility, and broad substrate scope. This protocol features no protection and deprotection steps. Given the structural diversity of carboxylic acids and alcohols, the success of this protocol could potentially enhance the synthesis of complex ketones. More significantly, ketones can be directly constructed from two abundant starting materials, thus expanding the existing ketone synthetic routes.
To start our investigation, the synthetic method for ketones was explored with the commercially available carboxylic acid 1 and alcohol 2 as the model substrate (Table 1). Based on previously reported elegant carboxylic acid activations in ketone synthesis,6h,j,12a,14 Boc2O was chosen as the activating reagent to generate mixed anhydride in situ from carboxylic acids. After extensive reaction condition screening (see the ESI† for details), we were pleased to find that the corresponding ketone 3 was obtained in 73% isolated yield using N4 as the alcohol-activating agent and Cs2CO3/K2CO3 and pyridine as bases in the presence of a catalytic amount of NiBr2·DME and L1 under visible light irradiation in DMA/1,4-dioxane using Ir(ppy)2(dtbbpy)PF6 as the photocatalyst (entry 1). The thiazole-based NHC reagent N1 and other simple triazole-based NHC molecules N2 and N3 are ineffective in this transformation (entry 2). These initial optimization studies revealed the importance of NHC types for the reaction efficiency. A slightly lower yield was obtained when tBuOMe was used instead of 1,4-dioxane (entry 3). Other solvents, such as benzotrifluoride, tetrahydrofuran and acetonitrile were also tested and the results indicated that 1,4-dioxane was more suitable for this transformation (entry 4). Screening of a range of ligands revealed that acylation product 3 could also be generated, albeit in diminished yields (entry 5). A slight increase in the yield was observed on reducing the amount of NiBr2·DME and L1 (entry 6). Light irradiation was essential for this transformation as it did not progress under dark conditions (entry 7). Further control experiments showed that NHC and the photocatalyst were indispensable in this transformation (entries 8–9). It was worth noting that the side products due to decarboxylation and esterification could be detected.
|
|
||
|---|---|---|
| Entry | Variation from optimized conditions | Yieldb (%) |
| a Reaction conditions: 1 (0.3 mmol), 2 (0.42 mmol), 1 mol% Ir(ppy)2(dtbbpy)PF6, 10 mol% NiBr2·DME, 15 mol% L1, Cs2CO3 (0.45 mmol), K2CO3 (1.3 equiv.), DMA (3 mL), N4 (1.5 mmol), pyridine (1.4 equiv.), 1,4-dioxane (3 mL), Boc2O (1.3 equiv.), 450–455 nm LEDs. b Yields of 3 were determined by 1H NMR spectroscopy with mesitylene as an internal standard and the isolated yield is shown in parentheses. r.t., room temperature; NHC, N-heterocyclic carbene; DME, 1,2-dimethoxyethane; DMA, N,N-dimethylacetamide. | ||
| 1 | None | 75 (73) |
| 2 | N1, N2, and N3 instead of N4 | 0, trace, 0 |
| 3 | BuOMe instead of 1,4-dioxane | 60 |
| 4 | PhCF, THF, and MeCN instead of 1,4-dioxane | 54, 13, <10 |
| 5 | L2, L3, L4, and L5 instead of L1 | 37, 32, 30, trace |
| 6 | 5 mol% NiBr2·DME, 7.5 mol% L1 | 78 (75) |
| 7 | No light irradiation | 0 |
| 8 | No NHC | 0 |
| 9 | No PC | 0 |
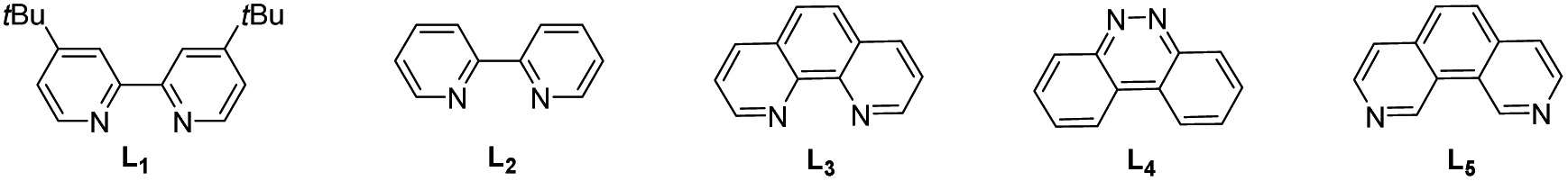
|
||
With the optimized reaction conditions in hand, we then investigated the scope of carboxylic acids and alcohols (Fig. 2). We first probed the ability of various aliphatic acids for cross-coupling in our system (3–22). Substituted phenyl propionic acid and butyric acid derivatives yielded desired products in moderate to good yields (3–10). A range of aliphatic acids, including linear and cyclic acids, were amenable substrates, providing the cross-coupling products in good to excellent yields (11–18). Notably, carboxylic acids with additional functionalities were also compatible with this protocol. For example, various functional groups, such as alkyl chloride, ester, protected amine and ketone remain intact to furnish the corresponding cross-coupling products, potentially allowing for the subsequent orthogonal functionalization (15–19). In particular, carboxylic acids with synthetic handles, such as halide (10 and 15), were readily incorporated into the accessible ketone scaffolds, which highlights the potential applications for the incorporation of these scaffolds into more complex targets. Products derived from alkenyl acids were also tolerated, as demonstrated by β,β-dimethylacrylic acid (20) and lineoic acid (21). These results show the great potential for structural modification and resource utilization of naturally existing carboxylic acids. Heterocycle-containing carboxylic acid reacted smoothly in this system, affording the deoxygenated cross-coupling product in moderate yield (22). Of particular note is that substituted phenyl acetic acid and hindered carboxylic acids (23), such as N-Boc proline (24) and 2-phenyl propionic acid (25) were not suitable for this transformation, yielding no product. Additionally, experiments with various benzoic acid derivatives were also performed under the standard conditions. Unfortunately, these substrates were not compatible with our system (26–27). This phenomenon could be attributed to the diminished reactivity in carboxylic acid activation.
 | ||
| Fig. 2 Substrate scope for ketone synthesis. Standard conditions: carboxylic acid (0.3 mmol), alcohol (0.42 mmol), 1 mol% Ir(ppy)2(dtbbpy)PF6, 10 mol% NiBr2·DME, 15 mol% L1, Cs2CO3 (0.45 mmol), K2CO3 (1.3 equiv.), DMA (3 mL), N4 (1.5 mmol), pyridine (1.4 equiv.), 1,4-dioxane (3 mL), Boc2O (1.3 equiv.), 450–455 nm LEDs. Isolated yield. | ||
Having established that this transformation tolerates various carboxylic acids, we turned our attention towards evaluating the scope of alcohol components. Consistent with our expectation, we were pleased to find that a wide variety of primary alcohols were successfully applied in this protocol, furnishing the desired ketones in moderate yields (28–32). The instability of the corresponding alkyl radicals originating from alcohols could be responsible for the relatively lower yield (29–32). Of particular note is that the developed protocol was also tolerant of the alcohol containing protected amine, as demonstrated by 31, which was isolated in 40% yield. Notably, the alkene-retained product (32) was obtained in 39% yield, while intramolecular radical cyclization was not observed. This result suggests the faster capture of the alkyl radical than 5-endo-trig cyclizations under the specific reaction conditions. Secondary alcohols, especially cyclic alcohols, ranging from four- to seven-membered rings, were found to be viable coupling partners, successfully delivering the corresponding products in 40–61% yields (33–38). It is noteworthy that sterically encumbered polycyclic alcohols, such as 2-adamantanol, were employed without an appreciable decrease in the reaction efficiency (37). A relatively increased yield was obtained when benzyl alcohols were used, which is in line with the stability of the corresponding radical intermediates from alcohols (38–41). With these positive results in hand, we finally tested the feasibility of tertiary alcohols, such as 1-methylcyclohexanol and tert-butanol, and the experimental results indicated that no corresponding cross-coupling product could be observed (42–43).
Given the exceptionally mild and simple conditions, we sought to demonstrate the utility of this operationally convenient method in the late-stage functionalization of complex molecules. As shown in Fig. 3, the oxaprozin analogue 44 could be generated efficiently with our strategy in 55% yield. Lithocholic acid analogues could also underwent smoothly, providing the deoxygenative ketone in 60% and 62% yield, respectively (45 and 46). With stearic acid as an acyl donor, the transformation proceeded efficiently, yielding 47 in 75% yield. A naturally occurring steroid was also successfully employed and afforded the corresponding product 48 in moderate yield. Bronchodilator proxyphylline showed good reactivity to deliver the product 49 in 21% yield. These results show great potential for the structural modification of an array of complex biological molecules, especially in medicinal chemistry.
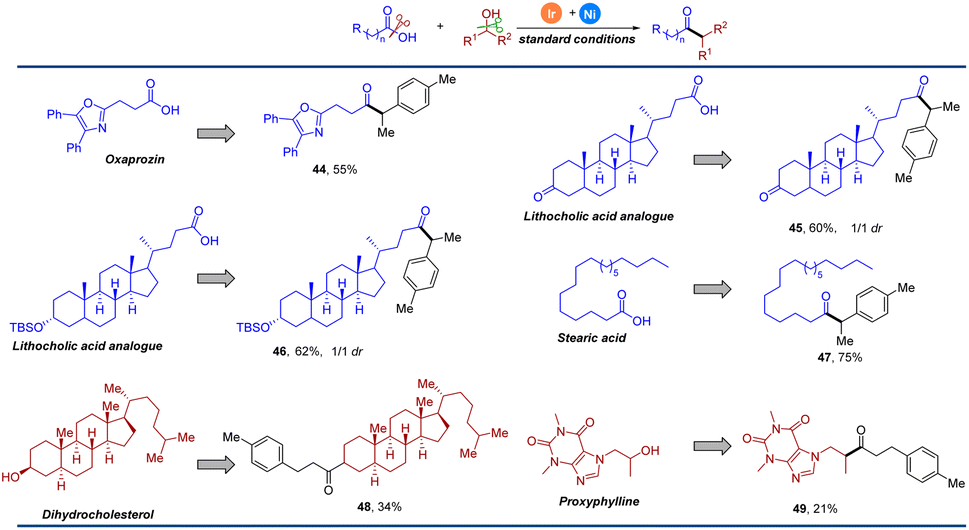 | ||
| Fig. 3 Late-stage functionalization. Standard conditions: carboxylic acid (0.3 mmol), alcohol (0.42 mmol), 1 mol% Ir(ppy)2(dtbbpy)PF6, 10 mol% NiBr2·DME, 15 mol% L1, Cs2CO3 (0.45 mmol), K2CO3 (1.3 equiv.), DMA (3 mL), N4 (1.5 mmol), pyridine (1.4 equiv.), 1,4-dioxane (3 mL), Boc2O (1.3 equiv.), 450–455 nm LEDs. Isolated yield. | ||
To further showcase the synthetic utility of this developed strategy, a large-scale experiment was conducted, providing the desired ketone 3 in 71% yield (Fig. 4). There was almost no change in the chemical yield, suggesting that large-scale chemical production might be possible.
 | ||
| Fig. 4 Large-scale synthesis. | ||
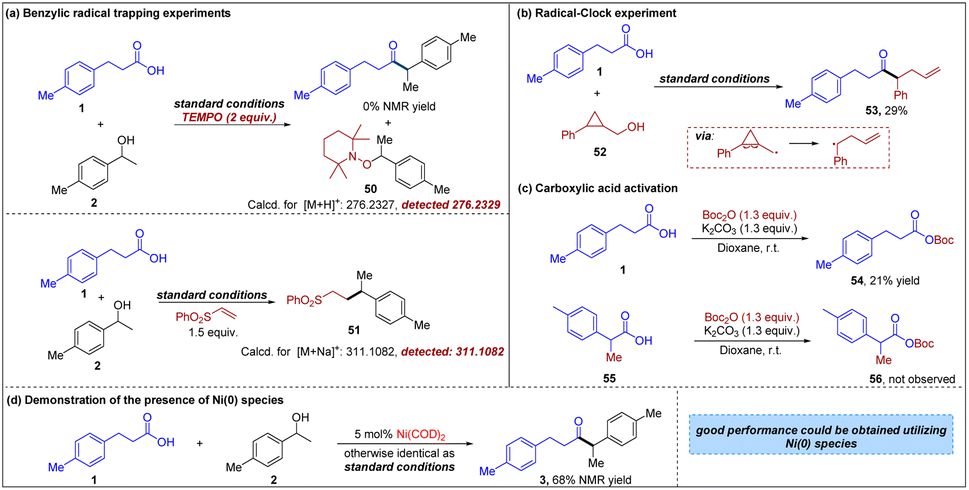
To gain further insight into the reaction mechanism, a series of mechanistic studies were performed (Fig. 5). In the presence of radical trap TEMPO, the reaction was completely shut down (Fig. 5a), indicating that a radical intermediate might be involved in this transformation. More importantly, a benzyl-trapped product, 2,2,6,6-tetramethylpiperidin-1-yl benzoate 50 was observed via high resolution mass spectrometry, further supporting that the reaction proceeds through a radical deoxygenative pathway and the intermediacy of a benzylic radical. Furthermore, the generation of a benzylic radical from 2 in the reaction also could be demonstrated by the observation of 51 when phenyl vinyl sulfone was added to the system. Additionally, to further elucidate the possible reaction pathway, a radical clock experiment was performed with cyclopropanemethanol 52 and the observation of ring-opening product 53 suggested the involvement of a radical intermediate (Fig. 5b). In our hypothesis, carboxylic acid is activated by Boc2O, leading to the corresponding acyl-Ni oxidative insertion complex. The control experiments are consistent with this hypothesis. First, when primary carboxylic acid 1 is treated with Boc2O and K2CO3, moderate conversion to the expected mixed anhydride is observed within 3 h (Fig. 5c). In the parallel experiment using secondary carboxylic acid 55, there is no observable formation of the mixed anhydride 56 at the same time point, resulting in the formation of the acyl–Ni complex being difficult. These results are in line with the limitations of carboxylic acids shown in Fig. 2. It is noteworthy that the desired ketone 3 could be detected in 68% NMR yield when Ni(COD)2 was used in place of NiBr2·DME (Fig. 5d), indicating the presence of Ni(0) species.
 | ||
| Fig. 5 Mechanistic studies. | ||
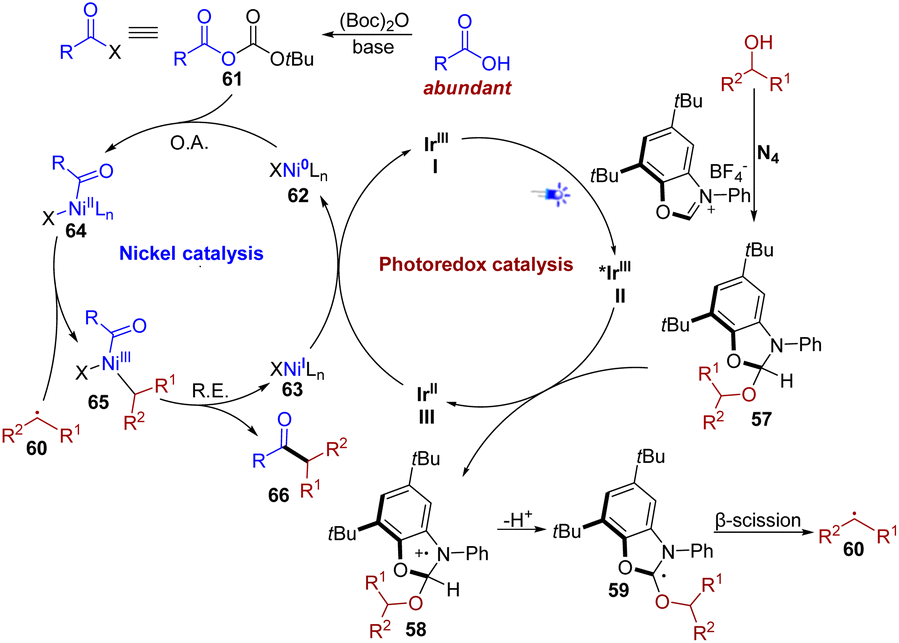
Based on the previously reported literature6d,f,16 and the aforementioned mechanistic studies, a plausible mechanism for this transformation is proposed in Fig. 6. The proposed mechanism starts with the condensation of alcohol and NHC (N4), providing activated alcohol 57. Upon irradiation with visible light, the photocatalyst Ir(ppy)2(dtbbpy)PF6I is known to access the highly oxidizing excited state species II (*IrIII) (Ered1/2 *IrIII/IrII = + 0.66 V vs. SCE),17 which could be reductively quenched by the activated alcohol 57, affording aminium radical cation 58. Then a deprotonation process occurred at the α-position of 58, yielding radical intermediate 59. Subsequent β-scission occurred, thus generating the key alkyl intermediate 60. The nickel catalytic cycle is initiated by the oxidative addition of the Ni(0) catalyst 62 to an in situ-activated carboxylic acid 61 formed by Boc2O under basic conditions, to afford Ni(II) species 64. Subsequently, efficient trapping of the alkyl radical 60 provides Ni(III) complex 65, which undergoes reductive elimination to yield the desired ketone 66 and Ni(I) complex 63. Finally, the single electron transfer between Ni(I) species 63 and reduced photocatalyst III (IrII) regenerates the ground-state photocatalyst I (IrIII) and the Ni(0) catalyst, completing both catalytic cycles.
 | ||
| Fig. 6 A plausible mechanism. | ||
Conclusions
In summary, we have developed a direct deoxygenative cross-coupling between carboxylic acids and alcohols for ketone synthesis via photoredox/nickel dual catalysis under mild conditions. This protocol provides a powerful platform to construct a wide range of structurally diverse ketone scaffolds with broad substrate scope, good functional group tolerance, step-economy and mild reaction conditions, using simple and readily available carboxylic acids and widely abundant alcohols as starting materials. Given the structural diversity of carboxylic acids and alcohols, the success of this metallaphotoredox-catalyzed deoxygenative cross-coupling protocol could potentially enhance the synthesis of complex ketones. In addition, this developed method will promote the resource utilization of naturally abundant acids and alcohols and enhance the preparation of ketone scaffolds. The exact roles of carboxylic acids and alcohols were demonstrated by mechanistic studies, as we hypothesized, that the carboxylic acids provide the acyl group and the alcohols afford the alkyl group. Asymmetric transformations of carboxylic acids and alcohols into ketones are underway in our laboratory and will be reported in due course.Data availability
The ESI† is available and includes experimental procedures for all reactions and characterization data for all products, including 1H and 13C spectra and HRMS data.Author contributions
Conceptualization and funding acquisition were done or provided by B. Y.; resources and supervision were done by B. Y.; project administration, data curation, investigation and formal analysis were done by B. Y. and R.-Y. T.; writing was done by B. Y. and revised by B. Y. and R.-Y. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22001079) and the Basic and Applied Basic Research Foundation of Guangdong Province (2019B151502052 and 2024B1515040004).Notes and references
- (a) R. McDaniel, A. Thamchaipenet, C. Gustafsson, H. Fu, M. Betlach, M. Betlach and G. Ashley, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1846–1851 CrossRef CAS PubMed; (b) P. K. Poutiainen, T. A. Venalainen, M. Perakyla, J. M. Matilainen, S. Vaisanen, P. Honkakoski, R. Laatikainen and J. T. Pulkkinen, Bioorg. Med. Chem., 2010, 18, 3437–3447 CrossRef CAS PubMed; (c) F. K. Lee, P. Krishnan, A. Muhamad, Y. Y. Low, T. S. Kam, K. N. Ting and K. H. Lim, Nat. Prod. Res., 2022, 36, 3972–3978 CrossRef CAS PubMed; (d) M. C. Cuquerella, V. Lhiaubet-Vallet, J. Cadet and M. A. Miranda, Acc. Chem. Res., 2012, 45, 1558–1570 CrossRef CAS PubMed.
- (a) D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC; (b) K. Ma, B. S. Martin, X. Yin and M. Dai, Nat. Prod. Rep., 2019, 36, 174 RSC; (c) N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed; (d) J. Kalia and T. R. Raines, Curr. Org. Chem., 2010, 14, 138–147 CrossRef CAS PubMed.
- N. J. Lawrence, J. Chem. Soc., Perkin Trans. 1, 1998, 1739–1750 RSC.
- (a) R. H. Vekariya and J. Aube, Org. Lett., 2016, 18, 3534–3537 CrossRef CAS PubMed; (b) G. Bram and M. Vilkas, Bull. Soc. Chim. Fr., 1964, 5, 945 Search PubMed; (c) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS; (d) J. Buchspies and M. Szostak, Catalysts, 2019, 9, 53 CrossRef; (e) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (f) J. Wang, M. E. Hoerrner, M. P. Watson and D. J. Weix, Angew. Chem., Int. Ed., 2020, 59, 13484 CrossRef CAS PubMed; (g) H. Yin, C. Zhao, H. You, K. Lin and H. Gong, Chem. Commun., 2012, 48, 7034–7036 RSC; (h) S. Ni, M. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS PubMed; (i) A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed; (j) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854 CrossRef CAS PubMed; (k) I. Aidhen and S. Balasubramaniam, Synthesis, 2008, 3707–3738 CrossRef; (l) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228–234 CrossRef CAS PubMed; (m) H. Huang, Q. S. Dai, H. J. Leng, Q. Z. Li, S. L. Yang, Y. M. Tao, X. Zhang, T. Qi and J. L. Li, Chem. Sci., 2022, 13, 2584–2590 RSC; (n) J. Masson-Makdissi, J. K. Vandavasi and S. G. Newman, Org. Lett., 2018, 20, 4094–4098 CrossRef CAS PubMed; (o) N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2016, 8, 75–79 CrossRef CAS PubMed; (p) J. Wang, B. P. Cary, P. D. Beyer, S. H. Gellman and D. J. Weix, Angew. Chem., Int. Ed., 2019, 58, 12081–12085 CrossRef CAS PubMed; (q) M. Zhang, J. Xie and C. Zhu, Nat. Commun., 2018, 9, 3517 CrossRef PubMed; (r) R. Ruzi, K. Liu, C. Zhu and J. Xie, Nat. Commun., 2020, 11, 3312 CrossRef CAS PubMed; (s) T. B. Boit, N. A. Weires, J. Kim and N. K. Garg, ACS Catal., 2018, 8, 1003–1008 CrossRef CAS PubMed.
- (a) P. Ertl and T. Schuhmann, J. Nat. Prod., 2019, 82, 1258–1263 CrossRef CAS PubMed; (b) T. Henkel, R. M. Brunne, H. Müller and F. Reichel, Angew. Chem., Int. Ed., 1999, 38, 643–647 CrossRef CAS; (c) P. Ertl, J. Cheminf., 2017, 9, 1 CrossRef PubMed.
- (a) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed; (b) J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522–6526 CrossRef CAS PubMed; (c) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed; (d) S. B. Beil, T. Q. Chen, N. E. Intermaggio, D. W. MacMillan and C. Acc, Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed; (e) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (f) Z. Dong and D. W. C. MacMillan, Metallaphotoredox-enabled deoxygenative arylation of alcohols, Nature, 2021, 598, 451–456 CrossRef CAS PubMed; (g) C.-Y. Tan, M. Kim and S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202306191 CrossRef CAS PubMed; (h) A. Whyte and T. P. Yoon, Angew. Chem., Int. Ed., 2022, 61, e202213739 CrossRef CAS PubMed; (i) X. Wu, J. Han, S. Xia, W. Li, C. Zhu and J. Xie, CCS Chem., 2022, 4, 2469–2480 CrossRef CAS; (j) X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed; (k) H.-M. Guo and X. Wu, Nat. Commun., 2021, 12, 5365 CrossRef CAS PubMed; (l) J.-B. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. D. Bel, G. M. Gallego, J. E. Spangler, J. Starr, S.-L. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398 CrossRef CAS PubMed.
- E. Fischer and A. Speier, Chem. Ber., 1895, 28, 3252–3258 CrossRef CAS.
- H. A. Sakai and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6185–6192 CrossRef CAS PubMed.
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS; (b) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (c) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (d) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016, 3, 522–526 RSC; (e) W.-J. Zhou, Y.-H. Zhang, Y.-Y. Gui, L. Sun and D.-G. Yu, Synthesis, 2018, 50, 3359–3378 CrossRef CAS; (f) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS.
- E. E. Stache, A. B. Ertel, R. Tomislav and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- (a) W.-D. Li, Y. Wu, S.-J. Li, Y.-Q. Jiang, Y.-L. Li, Y. Lan and J.-B. Xia, J. Am. Chem. Soc., 2022, 144, 8551–8559 CrossRef CAS PubMed; (b) L. Wang, Z. Li, Y. Zhou and J. Zhu, Org. Lett., 2024, 26, 2297–2302 CrossRef CAS PubMed.
- (a) L. J. Gooßen and K. Ghosh, Angew. Chem., Int. Ed., 2001, 40, 3458–3460 CrossRef; (b) Y. Wei, J. Lam and T. Diao, Chem. Sci., 2021, 12, 11414–11419 RSC.
- (a) R. Kakino, H. Narahashi, I. Shimizu and A. Yamamoto, Chem. Lett., 2001, 30, 1242–1243 CrossRef; (b) L. J. Gooben, N. Rodríguez and K. Gooben, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef PubMed.
- (a) N. Chalotra, S. Sultan and B. A. Shah, Asian J. Org. Chem., 2020, 9, 863–881 CrossRef CAS; (b) G. Bergonzini, C. Cassani and C.-J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed; (c) S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS PubMed; (d) J. Amani and G. A. Molander, Org. Lett., 2017, 19, 3612–3615 CrossRef CAS PubMed; (e) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS PubMed; (f) L. Huan, X. Shu, W. Zu, D. Zhong and H. Huo, Nat. Commun., 2021, 12, 3536 CrossRef CAS PubMed; (g) R. Kakino, H. Narahashi, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2002, 75, 1333–1345 CrossRef CAS.
- (a) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (b) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS PubMed.
- (a) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) X. Shu, D. Zhong, Q. Huang, L. Huan and H. Huo, Nat. Commun., 2023, 14, 125 CrossRef CAS PubMed; (d) Z. Li, C. Li, Y. Ding and H. Huo, Coord. Chem. Rev., 2022, 460, 214479 CrossRef CAS.
- H. A. Sakai, W. Liu, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds, and copies of 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4sc05420b |
| This journal is © The Royal Society of Chemistry 2024 |