DOI:
10.1039/D4SC05422A
(Edge Article)
Chem. Sci., 2024,
15, 19322-19327
Installation of superacidic carbon acid moieties into polymer materials via post-polymerization modification†
Received
13th August 2024
, Accepted 21st October 2024
First published on 22nd October 2024
Abstract
In the fields of polymer and material chemistries, strong acid units have mainly included sulfonic acids, which has limited the extension of related material chemistries. Here, a unique carbon acid functionality, namely the bis[(trifluoromethyl)sulfonyl]methyl group, was integrated with polymers via a simple postpolymerization modification with the outstandingly electrophilic 1,1-bis[(trifluoromethyl)sulfonyl]ethylene. The proposed synthesis protocol was verified as an efficient process even for solid-state reactions. The synthesis afforded an organic material with a surface decorated with bis[(trifluoromethyl)sulfonyl]methyl units. The fabricated membranes featuring surface bis[(trifluoromethyl)sulfonyl]methyl units functioned as efficient organocatalysts with high catalytic activity for the Mukaiyama aldol reaction. This study provides a simple method for installing superacidic carbon acid moieties onto the surfaces of materials without tedious chemical treatments.
Introduction
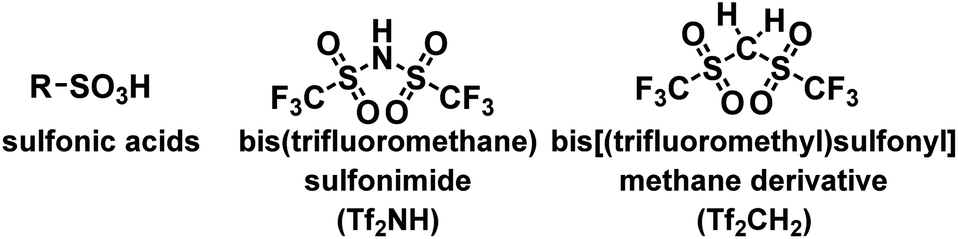
Polymers with strong acid functionalities have been used in a wide range of academic and industrial applications as polyelectrolytes, immobilized acid catalysts, ion-exchange membranes, proton-conducting membranes, etc.1–5 Among such polymers, those containing sulfonic acids are undoubtedly the most important because of the established commercialization of a wide range of monomers and polymers containing sulfonic acid units and their acidity due to their sulfonic acid functionalities.6,7 Despite the high availability of polymers with sulfonic acid units, their physicochemical properties are strongly influenced by the chemical and physical nature of the sulfonic acid moieties. For example, the acidities of sulfonic acids are identical, and their highly ionic nature leads to a decrease in their lipophilicity. Thus, the expansion of strong acid moieties in polymeric materials should advance materials and polymer science.
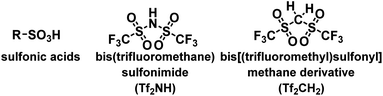
In this regard, achievements in superacid chemistry could facilitate the replacement of sulfonic acids.8 Considering the acidity of sulfonic acids, bis(trifluoromethane)sulfonimide (Tf2NH, triflylimide) is the most suitable alternative to sulfonic acids.9 As shown in Scheme 1, the sulfonic acid bears a releasable hydrogen atom on the oxygen atom. However, Tf2NH features a strong acidic proton at the nitrogen atom, chemically and physically distinguishing it from its sulfonic acid counterparts. Thus, Tf2NH and related nitrogen acids have been introduced into polymer science and used in various applications, offering a new class of polymers with strong acid moieties and their derivatives.10 Among polymers with strong acid moieties, bis[(trifluoromethyl)sulfonyl]methane (Tf2CH2; Tf = CF3SO2) is attractive because the tetravalency of its central carbon atom enables the synthesis of several derivatives through the replacement of one proton on the carbon atom. Such derivatives exhibit strong acidities comparable to those of organic sulfonic acids and sulfuric acid because the presence of two adjacent (trifluoromethyl)sulfonyl groups effectively enhances the acidity of the remaining C–H moiety. Owing to the strong acidities of Tf2CH-based carbon acids, their conjugate bases are highly stable “carbon” anions. Overall, Tf2CH-based carbon acid derivatives hold promise in polymer chemistry, and their incorporation into polymer materials could enrich the chemistry of polymer acids.
 |
| | Scheme 1 Organic acids with acidities comparable to those of sulfonic acids. | |
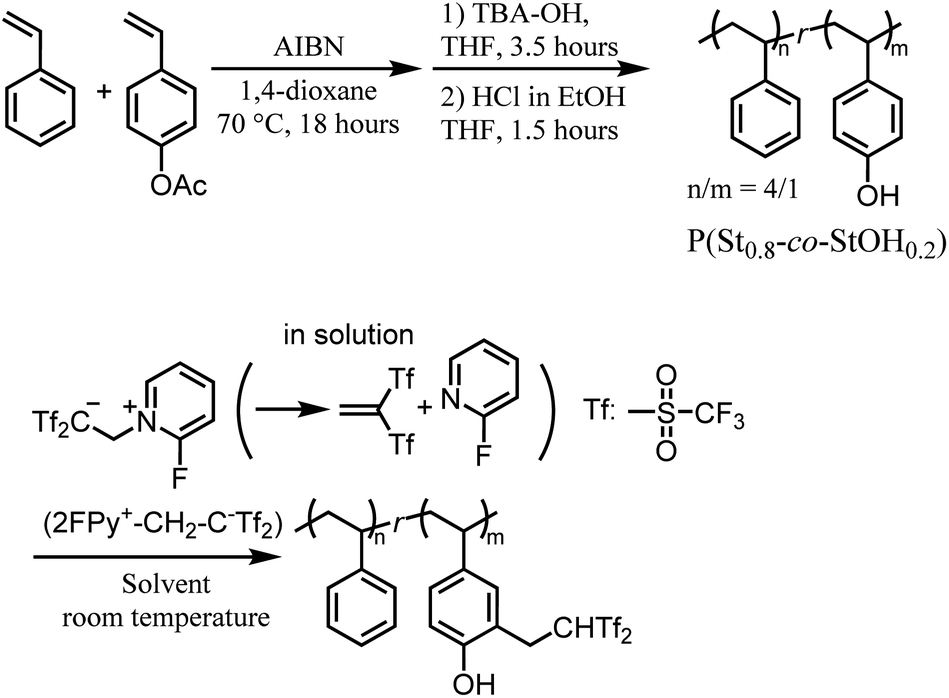
Despite the structural advantages of Tf2CH2 derivatives, their synthesis has proved extremely difficult. The lack of a feasible synthesis protocol has prevented intensive research into their material applications.11 Recently, an innovative synthetic pathway was discovered for the chemical decoration of small organic molecules with the Tf2CH functionality. Specifically, 2-(2-fluoropyridin-1-ium-1-yl)-1,1-bis(trifluoromethylsulfonyl)ethan-1-ide (2FPy+-CH2-C−Tf2), a zwitterionic compound bearing a stabilized carbanion moiety, was developed as an easy-to-handle and effective reagent for the in situ generation of a highly electron-deficient 1,1-bis[(trifluoromethyl)sulfonyl]ethylene (Tf2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) (Scheme 2).12 The Tf2CCH2 generated in a solution rapidly reacted with coexisting nucleophiles to yield Tf2CH-decorated products (Scheme 2). However, the utility of 2FPy+-CH2-C−Tf2 is limited in the chemical transformations of small molecules, and its applications in polymer and material sciences have not been demonstrated.13–16
CH2) (Scheme 2).12 The Tf2CCH2 generated in a solution rapidly reacted with coexisting nucleophiles to yield Tf2CH-decorated products (Scheme 2). However, the utility of 2FPy+-CH2-C−Tf2 is limited in the chemical transformations of small molecules, and its applications in polymer and material sciences have not been demonstrated.13–16
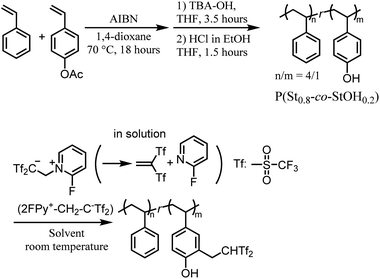 |
| | Scheme 2 Schematic overview of the synthesis of polymers bearing Tf2CH2 derivatives via postpolymerization reactions of polymeric phenol derivatives with 2FPy+-CH2-C−Tf2. | |
Thus, this study investigated the applicability of 2FPy+-CH2-C−Tf2 in polymer and material sciences. To achieve this, postpolymerization modifications17 based on the reactivity of 2FPy+-CH2-C−Tf2 were performed to generate polymers with Tf2CH2 derivatives (Scheme 2). In addition, the utility of 2FPy+-CH2-C−Tf2 in surface reactions was verified through radiation-induced graft polymerization (RIGP).18–21 Finally, the obtained organic materials with surfaces decorated with Tf2CH2 derivatives were preliminarily evaluated as immobilized organocatalysts.22,23
Results and discussion
Postpolymerization modification reactions with 2FPy+-CH2-C−Tf2 in solution
As previously mentioned, this study aimed to verify the chemical utility of 2FPy+-CH2-C−Tf2 in line with polymer synthesis. Although the released Tf2CCH2 has been reported to exhibit high reactivity with various nucleophiles, we selected polymeric phenol derivatives since the phenol moiety is ubiquitous in polymer and associated material chemistry. Thus, we designed postpolymerization reactions of polymeric phenol derivatives with 2FPy+-CH2-C−Tf2. To prevent problems arising from potential hydrogen-bonding network formation within phenol OH groups and changes in polymer solubility during postpolymerization conversions, the phenol unit incorporation was adjusted to 20 mol% in the copolymers. Thus, P(St0.8-co-StOH0.2) was used as a starting polymer prepared via free radical copolymerization and the subsequent acetyl deprotection of styrene (St) and 4-acetoxystyrene (StOAc) (Scheme 2). The postpolymerization modifications of P(St0.8-co-StOH0.2) were performed using 2FPy+-CH2-C−Tf2 (Scheme 2).
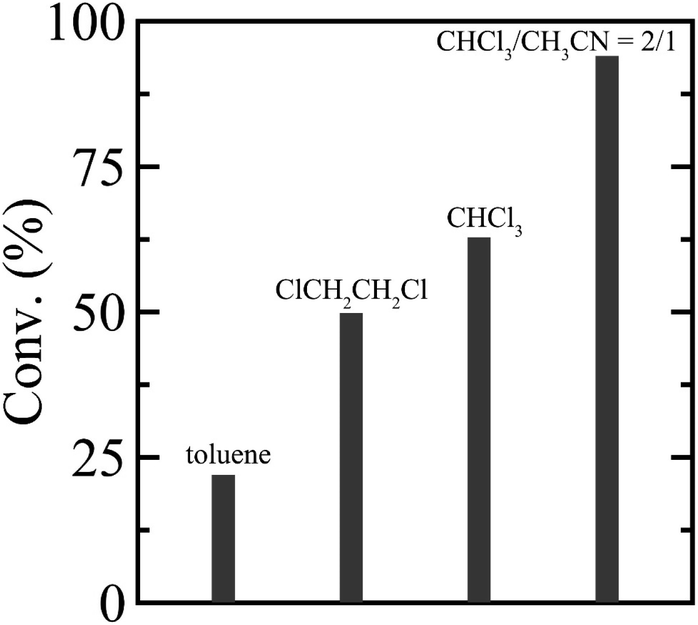
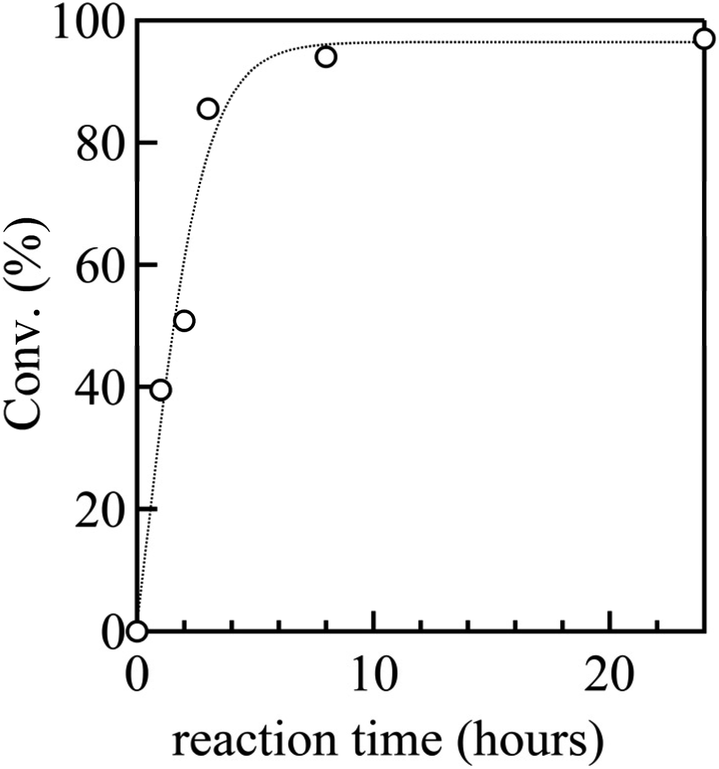
To establish a versatile reaction platform, the postpolymerization reactions of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 were optimized. The postpolymerization reactions of P(St0.8-co-StOH0.2) were conducted for 8 h at room temperature ([PhOH]0/[2FPy+-CH2-C−Tf2]0 = 1/1.5). The reaction solvent was varied because the solubility and solvation of 2FPy+-CH2-C−Tf2 should be decisive for the kinetic release of Tf2CCH2 molecules (Fig. 1). Owing to the polymer solubility and chemical stability of 2FPy+-CH2-C−Tf2, only halogenated solvents, toluene and acetonitrile, were used. Thus, the postpolymerization reaction correlated well with the polarity of the solvents. Using apolar toluene, the reaction achieved only 22.0% conversion, which was lower than that achieved using polar halogenated solvents (49.8% in 1,2-dichloroethane and 62.8% in chloroform). In line with the polarity effect, the addition of aprotic polar acetonitrile had a drastic effect on the reaction. Specifically, the postpolymerization reaction achieved a practically quantitative (94.0%) conversion in a mixed solvent system of chloroform and acetonitrile (v/v = 2/1). This was consistent with the chemical nature of the salt compound, 2FPy+-CH2-C−Tf2, where the solubility and solvation of the inner salt were assisted by the polar acetonitrile. After optimizing the reaction, a kinetic investigation was conducted for the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 in chloroform and acetonitrile (v/v = 2/1) (Fig. 2). As shown in Fig. 2, the postpolymerization modification reaction practically achieved a quantitative conversion at 8 h, which was fairly rapid reaction kinetics for postpolymerization modification reactions.
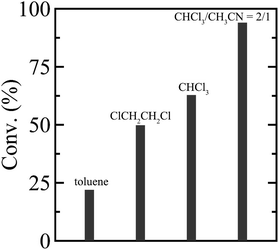 |
| | Fig. 1 Solvent effect on the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2. | |
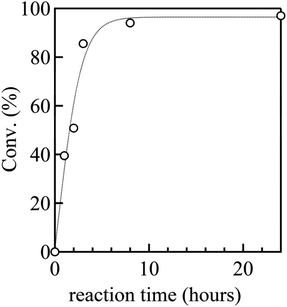 |
| | Fig. 2 Kinetic plots of the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 (the dashed line is an eye guide). | |
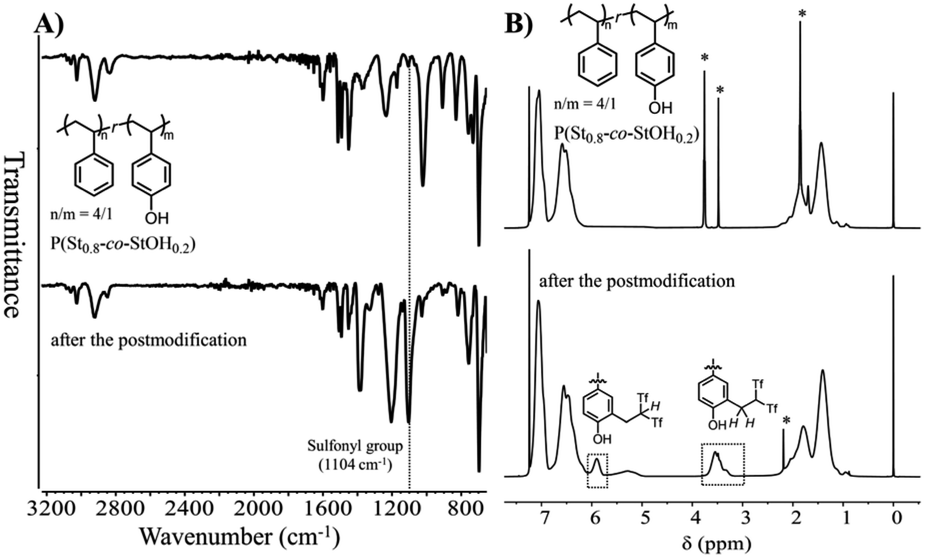
Owing to the feasible conversion of the phenol units in P(St0.8-co-StOH0.2), the obtained polymers were chemically and structurally evaluated prior to and after the reaction with 2FPy+-CH2-C−Tf2 in chloroform and acetonitrile (v/v = 2/1). The evaluations were performed via Fourier transform infrared (FT-IR), proton nuclear magnetic resonance (1H NMR), and size-exclusion chromatography (SEC) measurements. Fig. 3 shows the FT-IR and 1H NMR spectra of the polymers prior to and after the postpolymerization modification of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2. The FT-IR measurements revealed a strong peak at 1104 cm−1 owing to sulfonyl groups in the spectrum of the polymer after the reaction with 2FPy+-CH2-C−Tf2, supporting the installation of the Tf2CH unit into the polymers. Similarly, the 1H NMR measurements reinforced the installation of the Tf2CH unit. In the spectrum of the polymer after the reaction with 2FPy+-CH2-C−Tf2, peaks were observed at 3.6 and 5.9 ppm, corresponding to Tf2CH-CH2-methylene and methine protons, respectively. These peaks were consistent with the small-molecular-weight model compound reported by some of the authors.12 The 1H NMR and FT-IR measurements showed that 2FPy+-CH2-C−Tf2 smoothly reacted with P(St0.8-co-StOH0.2).
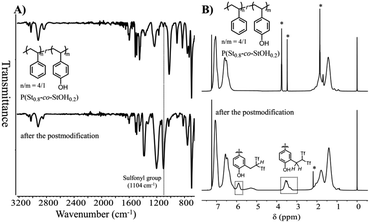 |
| | Fig. 3 Attenuated total reflectance-mode Fourier transform infrared spectra (A) and 1H nuclear magnetic resonance spectra in CDCl3 (B) of polymers prior to (upper) and after (lower) the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 (* refers to the residual solvents). | |
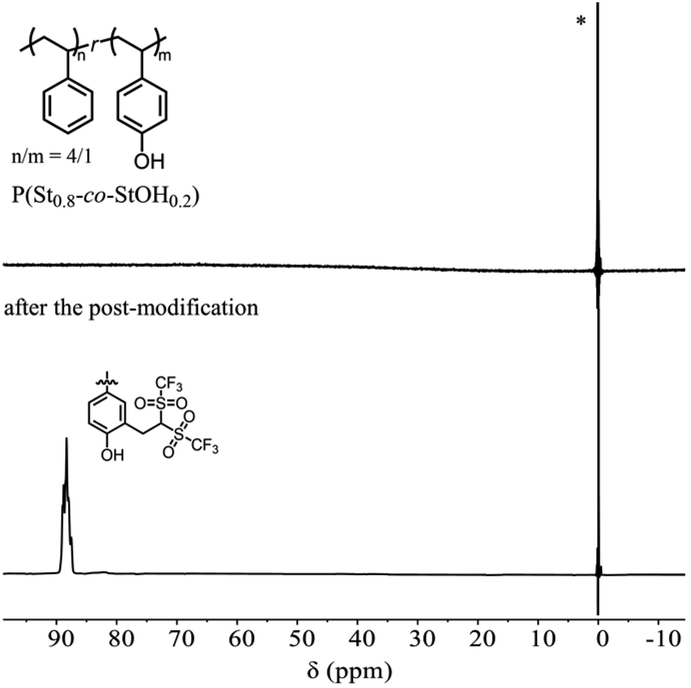
To directly confirm the attachment of the Tf2CH unit onto the polymer structures, P(St0.8-co-StOH0.2) was subjected to 19F NMR measurements prior to and after the postpolymerization modification with 2FPy+-CH2-C−Tf2 (Fig. 4). Prior to the postpolymerization modification, the 19F NMR spectra of P(St0.8-co-StOH0.2) did not exhibit any peaks due to fluorine atoms, whereas a broad distinct peak was observed at 88.3 ppm after the postpolymerization modification with 2FPy+-CH2-C−Tf2. Furthermore, the 19F NMR measurements supported the modification of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2.
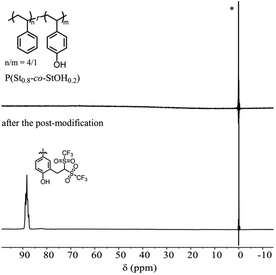 |
| | Fig. 4 The 19F nuclear magnetic resonance spectra in CDCl3 of the polymers prior to (upper) and after (lower) the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 (* refers to the internal reference, C6F6). | |
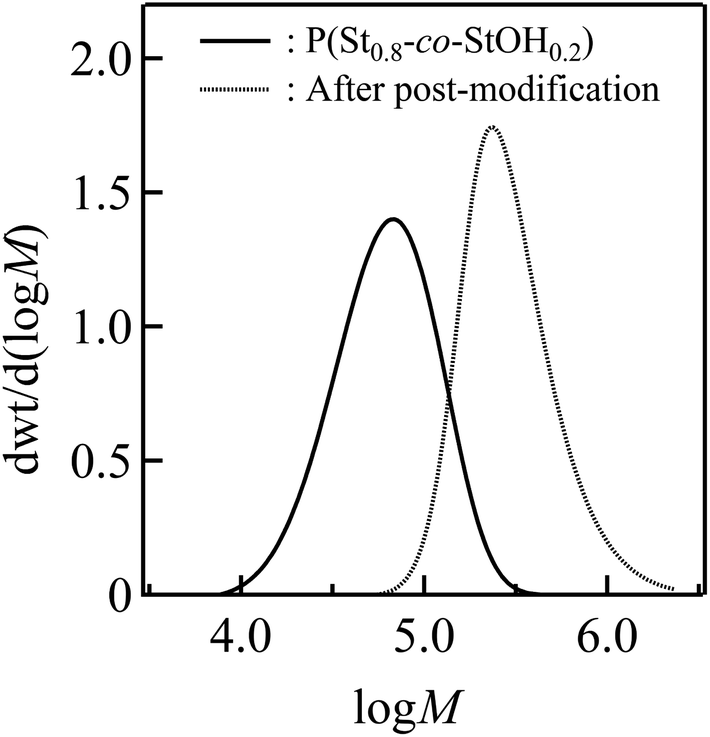
Finally, SEC in dimethylformamide (DMF) containing LiCl was employed to characterize P(St0.8-co-StOH0.2) prior to and after the postpolymerization process (Fig. 5). Fig. 5 shows a remarkable change in the SEC results of the polymers triggered by the postpolymerization process. The apparent number-average molecular weight (Mn) increased from 49.9 kg mol−1 for the starting P(St0.8-co-StOH0.2) to 248 kg mol−1 for the modified product. This corresponded with the structural change in the polymers. The modified product featured structurally bulky bis[(trifluoromethyl)sulfonyl]methane units in the pendant structures, which probably caused the increase in the apparent molecular weights. In addition, SEC-determined molecular weights are highly affected by the hydrodynamic volumes in the solution state. Thus, polymers bearing Tf2CH units should be ionized by the coexisting LiCl salts to afford polymers with Tf2CLi units during SEC measurements because Tf2CH units are known to exhibit high acidity comparable to that of sulfuric acids. Naturally, we expected that polymers bearing Tf2CLi units would have larger hydrodynamic volumes than those bearing Tf2CH units because of their ionic nature, leading to a large increase in the apparent molecular weights. The experimental data strongly supported the postpolymerization modification of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2, and the modified product was designated as a polymer bearing Tf2CH units, denoted as P(St0.8-co-StOH-CH2CTf2H0.2). For the first time, as far as we know, 2FPy+-CH2-C−Tf2 was revealed to be compatible with polymer synthesis and produced polymers bearing Tf2CH units without tedious chemical transformation.
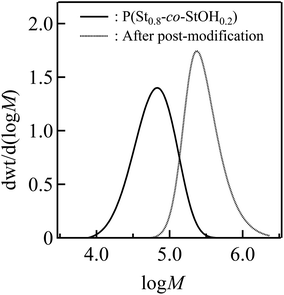 |
| | Fig. 5 Size-exclusion chromatography measurements of the polymers prior to (solid) and after (dashed) the postpolymerization reaction of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2. The measurement condition was 0.35 mL min−1 in dimethylformamide (DMF) containing 0.01 mol L−1 LiCl at 40 °C. | |
Fabrication of membranes bearing strong carbon acids via surface postpolymerization modification with 2FPy+-CH2-C−Tf2
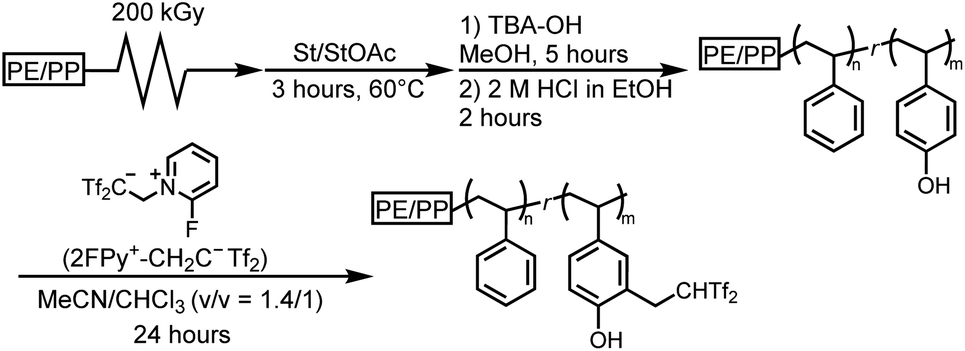
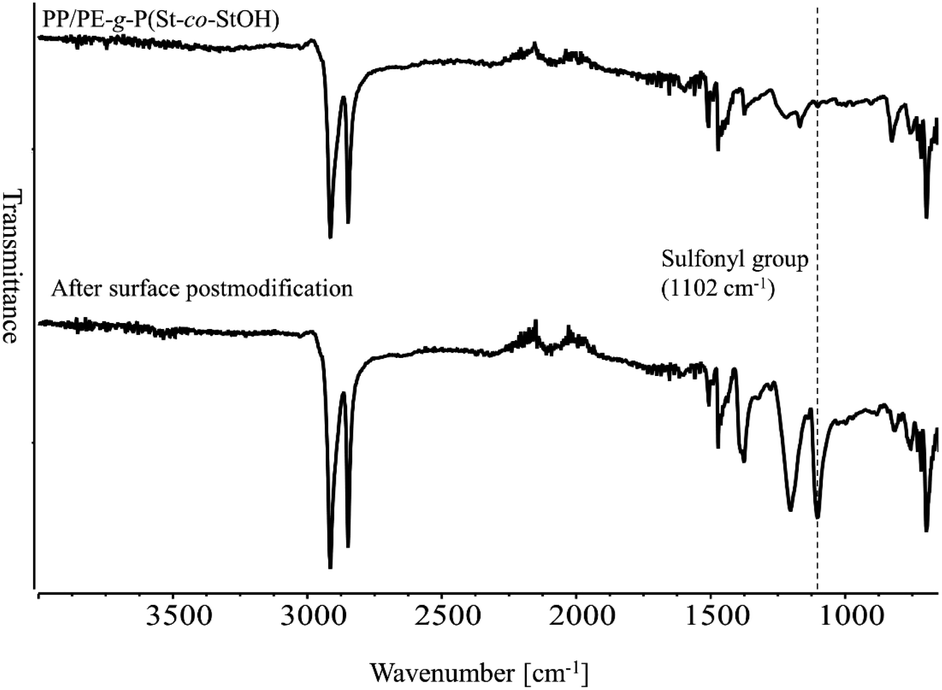
Having demonstrated the utility of 2FPy+-CH2-C−Tf2 in synthetic polymer chemistry, we attempted to fabricate membrane materials whose surfaces were decorated with Tf2CH units. To achieve this, we combined RIGP and surface postpolymerization modification. This is because RIGP enables surface functionalization for most organic materials without compromising their material properties whereas plasma treatment has been also integrated with material chemistry.24 To be specific, scalability and robustness of the RIGP was emphasized despite that plasma chemistry is also powerful from the viewpoint of easy handling. Thus, polyethylene-coated polypropylene nonwoven fabric (PE/PP) was selected as a base material and subjected to RIGP and subsequent acetyl deprotection of St and StOAc (Scheme 3). The PE/PP membrane was irradiated with electron beams at a dose of 200 kGy and subjected to RIGP, using St and StOAc as a monomer and a solvent, respectively, for 3 h at 60 °C ([St]0/[StOAc]0 = 1/1). This afforded PE/PP-g-P(St-co-StOAc) with a weight gain (termed the grafting degree) of 233%. PE/PP-g-P(St-co-StOAc) was subjected to simple acetyl deprotection, which afforded a PE/PP functionalized with P(St-co-StOH), denoted as PE/PP-g-P(St-co-StOH). Similar to the postpolymerization modification reaction with 2FPy+-CH2-C−Tf2 in solution, PE/PP-g-P(St-co-StOH) was reacted with 2FPy+-CH2-C−Tf2 in the mixed solvent of CHCl3 and CH3CN. To obtain insights into the surface functionalization, PE/PP-g-P(St-co-StOH) was subjected to FT-IR measurements prior to and after the postmodification with 2FPy+-CH2-C−Tf2 (Fig. 6). Similar to the solution phase reaction, the FT-IR measurements revealed a sharp peak caused by sulfonyl groups at 1102 cm−1 in the spectrum of the membrane that was reacted with 2FPy+-CH2-C−Tf2, supporting the surface anchoring of the Tf2CH units.
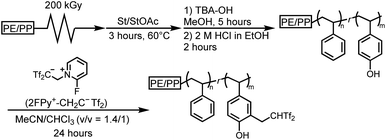 |
| | Scheme 3 Schematic overview of the synthesis of polyethylene-coated polypropylene nonwoven fabric bearing surface Tf2CH2 derivatives via surface postpolymerization reactions with 2FPy+-CH2-C−Tf2. | |
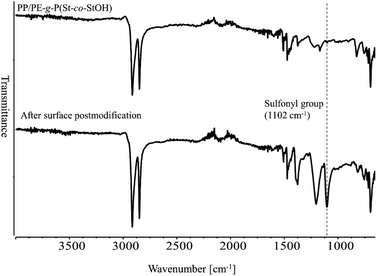 |
| | Fig. 6 Attenuated total reflectance-mode Fourier transform infrared spectra of membranes prior to (upper) and after (lower) the surface reaction of PE/PP-g-P(St-co-StOH) with 2FPy+-CH2-C−Tf2. | |
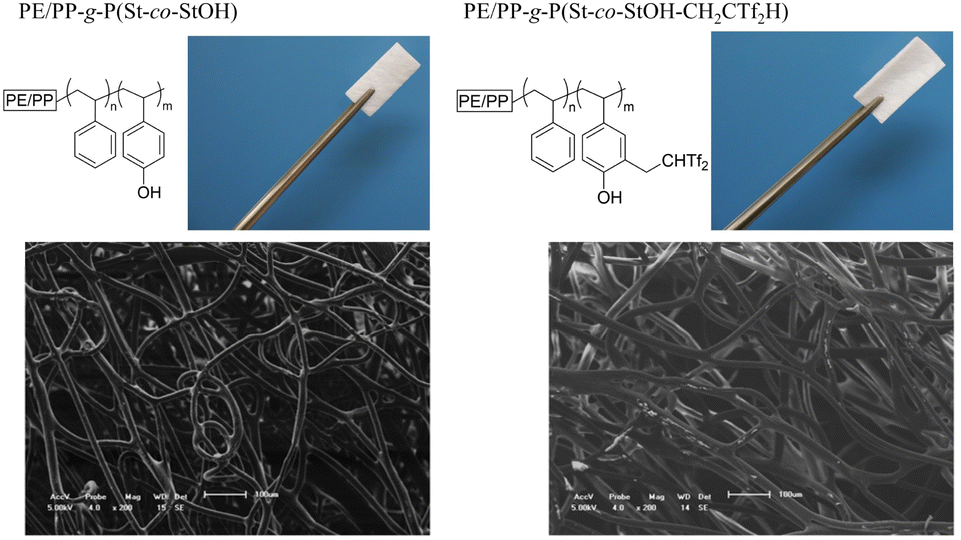
Dissimilar to the solution phase reactions, surface functionalization could potentially impact the membrane materials. Thus, the material was characterized via optical microscopy, scanning electron microscopy (SEM), and energy-dispersive X-ray spectroscopy (EDX). First, optical microscopy and SEM were employed to investigate PE/PP-g-P(St-co-StOH) prior to and after the modification with 2FPy+-CH2-C−Tf2 (Fig. 7). Although the shape of the material could be affected by the installation of bulky and fluorinated segments, the shape remained intact, as confirmed during the optical microscopy. Furthermore, the surface functionalization with 2FPy+-CH2-C−Tf2 did not damage the fiber structures supported by the SEM results (Fig. 7). The SEM-EDX profiles and mapping revealed that elements corresponding to the Tf2CH units were detected and uniformly dispersed along the fiber structures (Fig. S1 and S2†). Overall, the experimental data showed that the surface reactions with 2FPy+-CH2-C−Tf2 smoothly proceeded to afford a PE/PP coated with surface Tf2CH units (PE/PP-g-P(St-co-StOH-CH2CTf2H)). Here, the utility of 2FPy+-CH2-C−Tf2 was demonstrated in the field of material chemistry for the first time; thus, chemists can install unique strong carbon acid moieties onto material surfaces while bypassing tedious chemical treatments. This is crucial for material chemists who do not wish to handle dangerous chemicals.
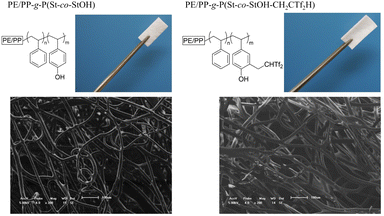 |
| | Fig. 7 Optical image (upper) and scanning electron microscopy (lower) images of membranes prior to and after the surface reaction of PE/PP-g-P(St-co-StOH) with 2FPy+-CH2-C−Tf2. | |
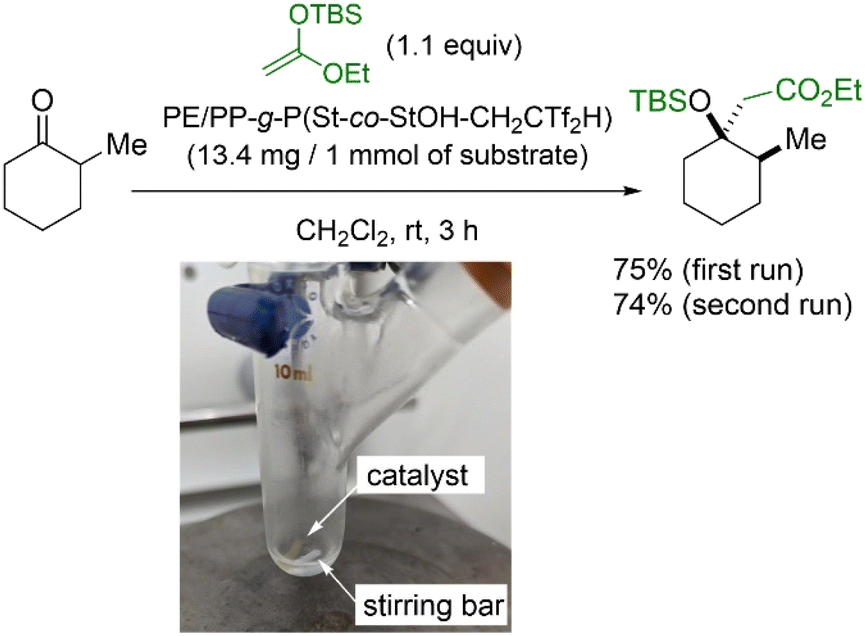
Finally, we demonstrated an intrinsic advantage of PE/PP-g-P(St-co-StOHCH2CTf2H) through a material application. In this context, PE/PP-g-P(St-co-StOHCH2CTf2H) was subjected to a preliminary assessment as an immobilized organocatalyst because the installed Tf2CH units are known to exhibit high acidity close to that of sulfuric acid and, thus, act as an efficient acidic organocatalyst. As a proof of concept, the Mukaiyama aldol reaction was designed (Fig. 8). Similar to previous studies, PE/PP-g-P(St-co-StOHCH2CTf2H) exhibited high catalytic activity during the Mukaiyama aldol reaction even in an immobilized state, and the aldol product was obtained in 75% yield. In addition, the polymer catalyst demonstrated reusability. Considering that the combination of RIGP and surface postpolymerization modification allows the functionalization of most organic materials regardless of their chemical and mechanical properties, we envision improving immobilized organocatalysts that are usually suffering from low catalytic activity.
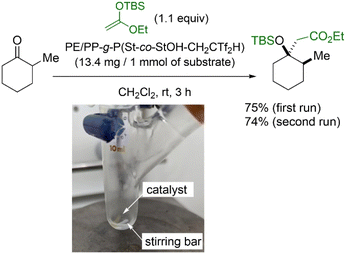 |
| | Fig. 8 Organocatalytic Mukaiyama aldol reaction of 2-methylcyclohexanone catalyzed by PE/PP-g-P(St-co-StOHCH2CTf2H). | |
Conclusions
This paper reports a synthetic utility of 2FPy+-CH2-C−Tf2 with respect to polymer and material chemistry. The postpolymerization modification of P(St0.8-co-StOH0.2) with 2FPy+-CH2-C−Tf2 proceeded practically quantitatively in a polar solvent system, and the 2FPy+-CH2-C−Tf2 only slightly exceeded the phenol units. Encouraged by the feasible post-modification reactions in solution phase, the utility of 2FPy+-CH2-C−Tf2 was further demonstrated during a solid-phase synthesis via a successful combination with RIGP. Thus, PE/PP was functionalized with surface strong carbon acid units of Tf2CH, which was preliminarily revealed to function as an efficient immobilized organocatalyst. Since a wide range of organic materials, including even fluorinated polymers, are compatible with RIGP technique in any form, the proposed protocol allows the fabrication of organic membranes featuring strong carbon acid moieties based on chemically intact materials, which should lead to new material applications such as ion-exchange membrane with taking advantage of unique acid of Tf2CH. Thus, we strongly believe that the study findings will promote the chemistry of polymeric materials with strong acid units.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
R. K. gratefully acknowledges the S-Membrane Project at Gunma University for the financial support.
Notes and references
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-i. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- R.-T. Liu, Z.-L. Xu, F.-M. Li, F.-Y. Chen, J.-Y. Yu, Y. Yan, Y. Chen and B. Y. Xia, Chem. Soc. Rev., 2023, 52, 5652–5683 RSC.
- K. Ayers, Curr. Opin. Electrochem., 2019, 18, 9–15 CrossRef CAS.
- M. Shabani, H. Younesi, M. Pontié, A. Rahimpour, M. Rahimnejad and A. A. Zinatizadeh, J. Cleaner Prod., 2020, 264, 121446 CrossRef CAS.
- Y. S. Kim, Adv. Sci., 2023, 10, 2303914 CrossRef CAS.
- J. Song, W. Zhao, L. Zhou, H. Meng, H. Wang, P. Guan, M. Li, Y. Zou, W. Feng, M. Zhang, L. Zhu, P. He, F. Liu and Y. Zhang, Adv. Sci., 2023, 10, 2303969 CrossRef CAS PubMed.
- H. Yanai and T. Taguchi, J. Fluorine Chem., 2015, 174, 108–119 CrossRef CAS.
- W. Zhao and J. Sun, Chem. Rev., 2018, 118, 10349–10392 CrossRef CAS.
- C. J. Chen, Z. L. Li, Q. Zhou, P. X. Han and G. L. Cui, eTransportation, 2024, 20, 100318 CrossRef.
- K. Ishihara, A. Hasegawa and H. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 4077–4079 CrossRef CAS.
- H. Yanai, Y. Takahashi, H. Fukaya, Y. Dobashi and T. Matsumoto, Chem. Commun., 2013, 49, 10091–10093 RSC.
- M. Toledano-Pinedo, T. Martinez del Campo, H. Yanai and P. Almendros, ACS Catal., 2022, 12, 11675–11681 CrossRef CAS.
- H. Yanai, S. Hoshikawa, Y. Moriiwa, A. Shoji, A. Yanagida and T. Matsumoto, Angew. Chem., Int. Ed., 2021, 60, 5168–5172 CrossRef CAS PubMed.
- H. Yanai, R. Takahashi, Y. Takahashi, A. Kotani, H. Hakamata and T. Matsumoto, Chem.–Eur. J., 2017, 23, 8203–8211 CrossRef CAS PubMed.
- H. Yanai, Chem. Rec., 2023, 23, e202300076 CrossRef CAS PubMed.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- M. Nasef, Prog. Polym. Sci., 2004, 29, 499–561 CrossRef CAS.
- N. Seko, M. Tamada and F. Yoshii, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 236, 21–29 CrossRef CAS.
- M. Tamada, N. Seko and F. Yoshii, Radiat. Phys. Chem., 2004, 71, 223–227 CrossRef CAS.
- R. Kakuchi, K. Matsubara, J. F. Madrid, B. J. D. Barba, M. Omichi, Y. Ueki and N. Seko, MRS Commun., 2022, 12, 552–564 CrossRef CAS.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed.
- K. Takasu, Synlett, 2009, 2009, 1905–1914 CrossRef.
- L. De Smet, G. Vancoillie, P. Minshall, K. Lava, I. Steyaert, E. Schoolaert, E. Van De Walle, P. Dubruel, K. De Clerck and R. Hoogenboom, Nat. Commun., 2018, 9, 1123 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The following files are available free of charge: experimental details and SEM-EDX spectra of the PE/PP-g-P(St-co-StOHCH2CTf2H) are included. See DOI: https://doi.org/10.1039/d4sc05422a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Takuma
Oguchi
a,
Minoru
Kuroiwa
a,
Yu
Hirashima
c,
Masaaki
Omichi
b,
Noriaki
Seko
*b and
Hikaru
Yanai
*a,
Takuma
Oguchi
a,
Minoru
Kuroiwa
a,
Yu
Hirashima
c,
Masaaki
Omichi
b,
Noriaki
Seko
*b and
Hikaru
Yanai