
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzocyclobutenone synthesis exploiting acylsilanes as photofunctional directing groups†
Rowan L.
Pilkington
 ,
Hannah J.
Ross
,
Liselle
Atkin
and
Daniel L.
Priebbenow
*
,
Hannah J.
Ross
,
Liselle
Atkin
and
Daniel L.
Priebbenow
*
Medicinal Chemistry Theme, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. E-mail: daniel.priebbenow@monash.edu
First published on 25th October 2024
Abstract
The visible-light irradiation of acylsilane tethered vinyl ketones promotes an intramolecular Stetter-type reaction via siloxycarbene intermediates. To exploit this unique mode of reactivity, we herein describe the innovative use of acylsilanes as photofunctional directing groups. First, an acylsilane directed ruthenium catalysed C–H olefination reaction was developed to generate benzoylsilanes bearing vinyl ketone functionality. Then, visible-light irradiation initiated the 1,4-conjugate addition of transient siloxycarbene intermediates with pendent vinyl ketones to afford unique benzocyclobutenone scaffolds primed for further synthetic elaboration.
Introduction
The use of acyl carbanion equivalents as Umpolung synthons is a well-established strategy in chemical synthesis.1 Common methods employed to access acyl carbanion equivalents include: (i) the deprotonation of 1,3-dithianes;2 (ii) the deprotonation of cyanohydrins;3 and (iii) the reaction of aldehydes with N-heterocyclic carbene (NHC) organocatalysts to generate Breslow intermediates.4 Acyl carbanion equivalents readily participate in transformations including 1,2-carbonyl addition (e.g. the benzoin reaction),5 1,4-conjugate addition (the Stetter reaction),6 and nucleophilic attack of electrophiles such as alkyl halides (e.g. the Corey–Seebach reaction).7Acylsilanes8 are versatile reagents that can be engaged as radical precursors, carbenoid precursors or electrophiles.9 Acylsilanes can also be considered precursors to acyl carbanion equivalents. To this end, acylsilanes—when activated photochemically via direct irradiation or triplet energy transfer photocatalysis—undergo 1,2-Brook rearrangement to generate siloxycarbene intermediates that represent acyl carbanion equivalents.10 In addition to X–H insertion (including insertion into heterocyclic C–H bonds) and [2 + 1]-cycloadditions, siloxycarbenes also participate in benzoin-type reactions with fluorinated ketones, aldehydes, and esters (Fig. 1).10c,11 However, the ability of visible-light induced siloxycarbenes to participate in 1,4-conjugate addition processes (i.e. Stetter-type reactions) is unexplored.12
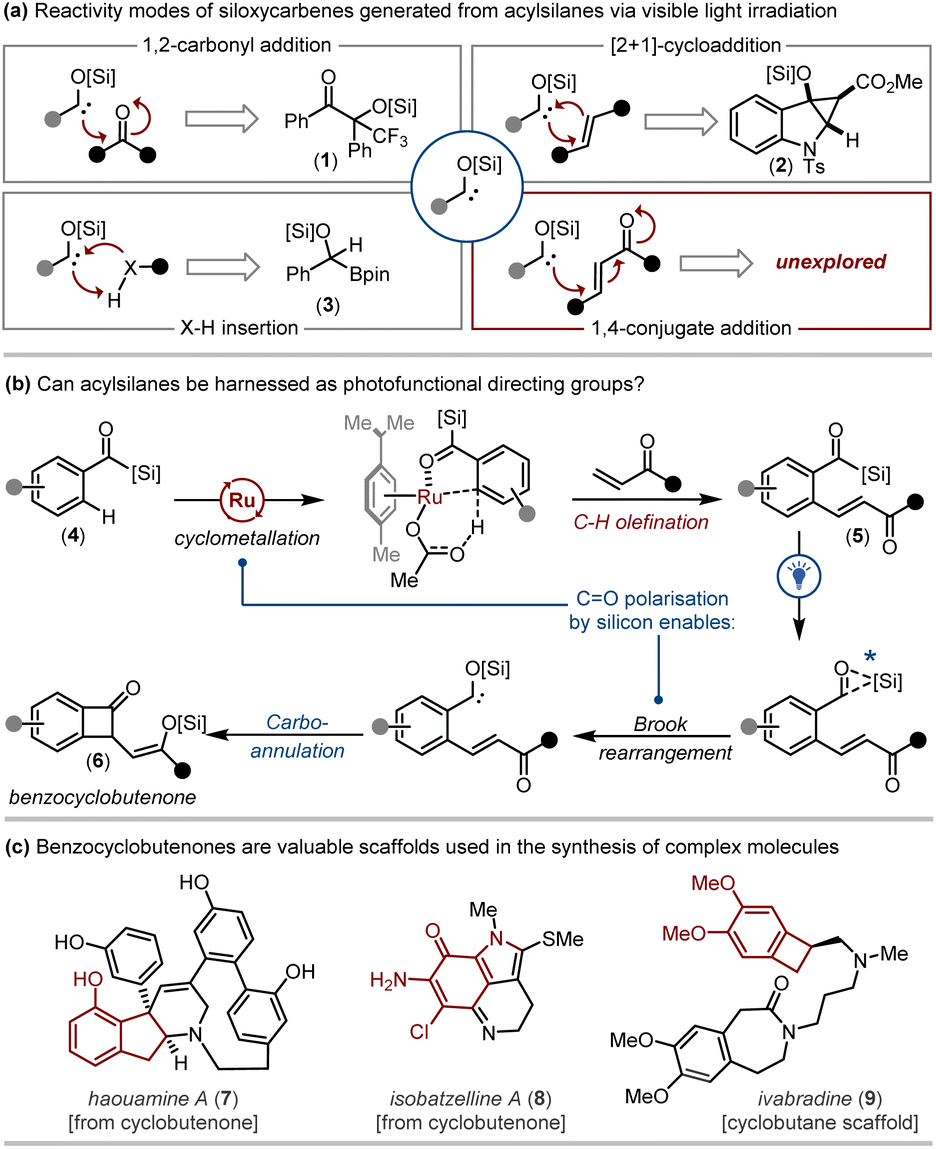 | ||
| Fig. 1 A protocol harnessing acylsilanes first as directing groups then as acyl anion precursors was designed to explore the photochemical Stetter-type carboannulation reaction to access benzocyclobutenone scaffolds. | ||
As part of our ongoing research into acylsilanes, we sought a representative framework that would enable us to investigate this elusive mode of carbene reactivity. Unfortunately, harnessing the unique reactivity of visible-light induced siloxycarbenes in chemical synthesis has been somewhat hampered by the lack of robust strategies to access complex scaffolds bearing the acylsilane linchpin.13
The directed functionalisation of inert C–H bonds by transition metal catalysis has emerged as an invaluable means to rapidly generate molecular complexity.14 To date, a range of directing groups have been employed that are derived from strongly-chelating pyridyl, amido, or imino templates or weakly-coordinating carbonyl derivatives.15 Within this context, acylsilanes have emerged as effective weakly-coordinating directing groups for the site selective C–H functionalisation of benzoylsilanes employing half-sandwich Cp*Co(III), Cp*Ir(III) and Cp*Rh(III) catalysts.16
The concomitant drawback of many effective directing groups employed in C–H functionalisation is their requisite removal or interconversion post-functionalisation where factors such as chemoselectivity, atom- and step-economy require careful consideration.17 To address this inefficiency, recent studies have focused on developing non-directed C–H functionalisation protocols, or those that employ native, traceless, or transient directing groups.18
Inspired by these efforts, we envisaged that the acylsilane motif could be exploited as a ‘photofunctional’ directing group. Here, acylsilanes could be initially engaged as directing groups to achieve the selective functionalisation of proximal C–H bonds, and then as acyl carbanion equivalents to drive secondary transformations via the photochemical conversion of the acylsilane to a reactive siloxycarbene intermediate.
More specifically, we strategised a protocol involving the acylsilane directed ortho C–H olefination of benzoylsilanes 4 employing vinyl ketones to access olefinated acylsilanes 5. Subsequently, visible light irradiation might generate benzocyclobutenones 6via a photochemical 1,4-conjugate addition process (Fig. 1). The anticipated benzocyclobutenone scaffolds are valued chemotypes that can undergo transformations including ring expansion19 to access complex natural products including haouamine A (7)20 and isobatzelline A (8).21 The benzocyclobutane core is also central to therapeutic agents such as ivabradine (9, Fig. 1).22 The realisation of our strategy to explore the photochemical 1,4-conjugate addition of siloxycarbenes while concurrently exploiting acylsilanes as photofunctional directing groups is described herein.
Results and discussion
We first focused on the acylsilane directed C–H olefination, where optimal conditions for the ortho-alkenylation of benzoylsilanes 4 with vinyl ketones to afford 5 were sought. Initial attempts revealed that in the presence of [Ru(p-cymene)Cl2]2 (5 mol%), AgBF4 (10 mol%), and Cu(OAc)2 (2.0 equiv.), 4-(tert-butyl)benzoyl(trimethyl)silane (4a) underwent reaction with methyl vinyl ketone (2.0 equiv.) at 60 °C for 5 h to afford 5a in 36% yield (Fig. 2a, entry 1). Further investigation revealed that silver hexafluoroantimonate was the optimal additive (48% yield, Fig. 2a, entry 2), and chlorinated solvents were critical for reaction success (Fig. 2a).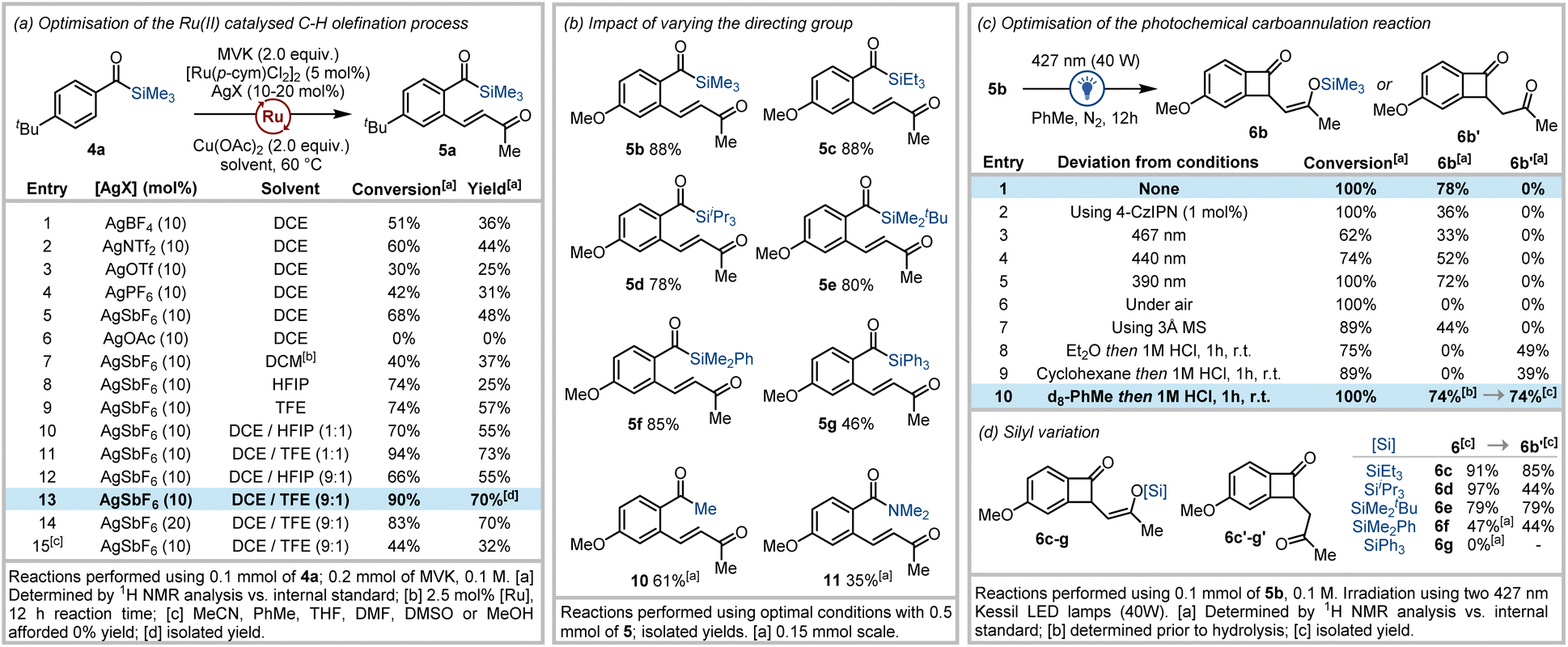 | ||
| Fig. 2 (a) Optimisation of the (p-cymene)Ru(II) catalysed olefination of benzoyl silanes with methyl vinyl ketone (MVK). (b) Studies into the impact of varying the directing group on the outcome of the olefination process. (c) Optimisation of the visible-light induced carboannulation reaction. (d) Studies into the impact of varying the silyl group on benzocyclobutenone formation and subsequent hydrolysis. | ||
During previous studies, we observed that neutral or electron deficient benzoylsilanes reacted poorly in acylsilane directed C–H functionalisation reactions to afford reduced yields relative to electron rich benzoylsilanes. In an attempt to overcome this reduced reactivity, we recently explored structural modifications to the cyclopentadienyl ligand within Cp*Rh(III) catalysts.16b Fluorinated alcoholic solvents are also known to increase reaction rates and yields in C–H functionalisation processes employing weakly coordinating directing groups.23 These effects are attributed to the low nucleophilicity yet strong hydrogen bond donor ability of these solvents which improves solubility, stabilises cationic intermediates, and activates weakly-coordinating directing groups to enhance electrophilic metalation during C–H functionalisation. Accordingly, the use of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 2,2,2-trifluoroethanol (TFE) was explored in an attempt to improve the acylsilane directed C–H olefination process.
Investigations into the use of fluorinated alcoholic solvents led to increased yields of 5 when TFE was employed exclusively as solvent (57%, Fig. 2a, entry 9), whereas reduced yields of 5 were obtained using only HFIP (25%, Fig. 2a, entry 8). Subsequent attempts using reduced amounts of TFE and HFIP in combination with 1,2-DCE (in either a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 9:1 ratio) revealed that only 10% TFE as a co-solvent was required to deliver enhanced yields (70%, Fig. 2a). Additional studies highlighting the beneficial effect of TFE on the acylsilane directed C–H olefination process are outlined in the ESI.†24
:1 or 9:1 ratio) revealed that only 10% TFE as a co-solvent was required to deliver enhanced yields (70%, Fig. 2a). Additional studies highlighting the beneficial effect of TFE on the acylsilane directed C–H olefination process are outlined in the ESI.†24
With high-yielding conditions in hand for the C–H olefination, we next explored the impact on both reaction rates and yield when varying the silyl group within 4-methoxy benzoylsilanes.24 Kinetic studies revealed that reaction rates were not influenced when the steric bulk around the silicon atom was increased, which is consistent with metal coordination occurring via the carbonyl oxygen.24 This was reflected in the fact that high yields of 5a–f (70–88%) were obtained within 2–4 hours for reactions involving trimethylsilyl (TMS), triethylsilyl (TES), triisopropylsilyl (TIPS), tert-butyldimethylsilyl (TBS) and dimethylphenylsilyl (DMPS) derived benzoylsilanes with methyl vinyl ketone (Fig. 2b). Reduced yields were obtained in the case of the reaction of the triphenylsilyl (TPS) analogue (5g, 46%), primarily due to subsequent decomposition of the product. Arenes containing other weakly-coordinating carbonyl-based directing groups (including acetophenone and N,N-dimethylbenzamide) were also subjected to the optimised reaction conditions to afford previously unreported methyl vinyl ketone tethered arenes 10 and 11 in good yields (Fig. 2b).
Next, the photochemical carboannulation reaction was investigated. Gratifyingly, visible-light irradiation of the ortho-olefinated benzoylsilane 5b using blue LEDs (427 nm) delivered clean conversion to the (Z)-benzocyclobutenone-1,4-silyl enol ether 6b after 12 hours (Fig. 2c). The photochemical cyclisation reaction was further optimised to afford either the silyl enol ether 6b, or corresponding benzocyclobuta-1,4-dione 6b′ after hydrolysis.24 The carboannulation process performed best in aprotic non-polar solvents, where toluene resulted in the most efficient formation of desired product (78%). Employing longer wavelengths (440 nm or 467 nm) slowed conversion of the acylsilane, whilst the reaction was retarded in the presence of molecular sieves or photosensitisers such as 4-CzIPN.25 The use of an inert atmosphere also proved critical for obtaining high yields. After the photochemical irradiation step, hydrolysis of the silyl enol ether was routinely accomplished via addition of aqueous hydrochloric acid to the reaction mixture with stirring for 1 hour to generate the benzocyclobuta-1,4-dione 6b′ in 74% isolated yield.
Structural variations within the silyl group that might impact both initial siloxycarbene formation and the 1,4-conjugate addition process was explored (Fig. 2d). The structure of the silyl group appeared to have little impact on the reaction process with excellent yields obtained for formation of the benzocyclobutenone silyl enol ether for TMS, TES, TIPS and TBDMS analogues. Reduced yields were observed in the case of the DMPS variant, which was not isolated due to instability, whilst the TPS analogue proved unreactive. While acylsilanes containing the TMS group were predominantly used for the remainder of our studies as the resultant silyl enol ether underwent expedient hydrolysis, the TES and TBDMS enol ethers were also readily hydrolysed to afford high yields of the 1,4-dione 6b′. These silyl enol ethers were expectedly more stable than the trimethylsilyl enol ether and could be isolated via column chromatography on neutralised silica gel. However, the TIPS silyl enol ether adduct 6d resisted complete hydrolysis under standard conditions affording a reduced yield for 6b′ (Fig. 2d). The notable enhanced stability of 6d is advantageous where retention of the silyl enol ether functional group is desired for downstream transformations.
With a high-yielding workflow established for benzocyclobutenone synthesis exploiting acylsilanes as photofunctional directing groups, we next investigated the diversity of benzoylsilanes and vinyl ketones applicable to this process. Initially, a range of benzoylsilanes were subjected to the optimised olefination reaction conditions. A variety of substituents on the aryl ring of the benzoylsilane were tolerated in the C–H olefination reaction with methyl vinyl ketone where benzoylsilanes containing electron donating groups afforded the product in generally higher yields than the corresponding benzoylsilanes containing electron withdrawing groups (Scheme 1). Substrates containing (thio)ether (5k), alkenyl (5p), ester (5q) and halide (5h, 5l, 5o) substituents provided the corresponding products in satisfactory yields as did the furan- and thiophene-derived heterocyclic acylsilanes (5r, 5s).
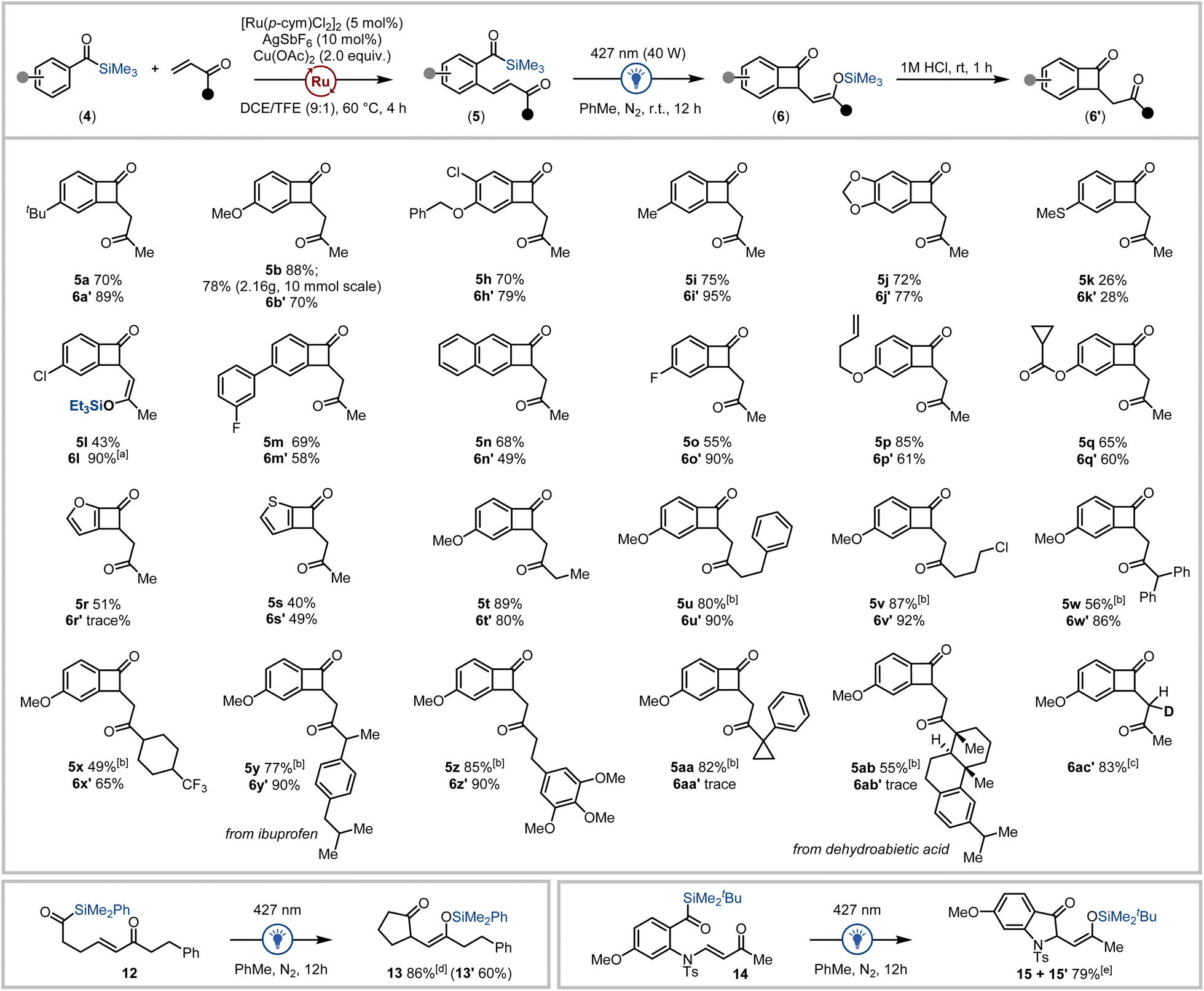 | ||
| Scheme 1 Reaction scope detailing the outcome of the Ru(II) C–H olefination of benzoylsilanes with vinyl ketones (unless otherwise noted, isolated yields reported and reactions were conducted at 0.50 mmol scale using 2.0 equivalents of vinyl ketone) followed by photochemical carboannulation (unless otherwise noted, isolated yields reported and reactions conducted at 0.10 mmol scale). [a] Isolated as the triethylsilyl enol ether; [b] 1.5 equivalents of vinyl ketone used; [c] hydrolysis step conducted using DCl/D2O. [d] not subjected to hydrolysis step, yield determined by 1H NMR analysis of reaction mixture, then product isolated as the 1,4-dione 13′ (60%) with hydrolysis during silica gel chromatography; [e] not subjected to hydrolysis step, combined isolated yields of silyl enol ether (15, 42%) and 1,4-dione (15′, 37%) obtained following partial hydrolysis during silica gel chromatography. | ||
Variation in the vinyl ketone reagent was also explored, where a series of linear, carbocyclic, and saturated heterocyclic vinyl ketones all performed well to afford diverse substrates in high yield, despite less of the olefin being used (1.5 equiv.) for the more complex vinyl ketones (5u–5ab, Scheme 1). The acylsilane and vinyl ketone derivatives that did not produce synthetically useful amounts of the desired olefinated products are outlined in the ESI.†24 Of note, the use of acroleins instead of vinyl ketones as coupling partners in acylsilane directed C–H functionalisation predominantly affords silyl indene products under both (p-cym)Ru(II) and Cp*Rh(III) catalysis.16d,g
The generality of the photochemical carboannulation reaction was then explored using the benzoylsilanes prepared via C–H olefination. Irradiation of the olefinated benzoylsilanes under visible light followed by hydrolysis of the intermediate silyl enol ether enabled the facile preparation of a series of benzocyclobutenone-1,4-dione scaffolds 6′ (Scheme 1). Yields for this process were typically high in the case of the methyl vinyl ketone derivatives (60–95%), generating a range of benzocyclobutenone analogues (6a′–6s′). Notably, the silyl enol ether of 4-chlorobenzoylsilane analogue 6l containing the triethylsilyl enol ether was readily isolable following chromatography. The thiomethylphenyl (6k′, 28%), naphthyl (6n′, 49%) and thiophene acylsilanes (6s′, 49%) afforded moderate yields in the two-step process, although irradiation of the furanoyl silane derivative afforded only trace amounts of the cyclised product 6r′. Benzoyl silanes containing more highly functionalised vinyl ketone tethers also proved amenable to the photochemical carboannulation reaction affording a series of benzocyclobutenones in 65–90% yield (including the benzoylsilane prepared from the ibuprofen-derived vinyl ketone 6y′).
Intriguingly, benzoylsilanes bearing tertiary substituted vinyl ketones (including the benzoylsilane prepared from dehydroabietic acid-derived vinyl ketone 5ab) failed to undergo photochemical cyclisation. This can be rationalized as the effect of steric encumbrance on either conjugate addition or more likely the silyl transfer process by the proximal tertiary carbon in substrates 5aa and 5ab. Finally, a deuterated analogue 6ac′ was accessible via hydrolysis of the silyl enol ether using DCl/D2O. Additional benzoylsilane derivatives that were successfully olefinated yet did not appreciably undergo photocyclisation are outlined in the ESI.†24
To further explore the photochemical carboannulation reaction, we prepared alkyl acylsilane 12 bearing a pendent vinyl ketone moiety. Irradiation using blue light (427 nm) promoted cyclisation to the corresponding cyclic ketone-1,4-silyl enol ether (Scheme 1, bottom left). Cyclopentanone 13 was generated in 86% yield via irradiation of 12, ultimately affording the corresponding 1,4-dione hydrolysis product (13′) in 60% yield after column chromatography. It was also discovered that 2-sulfonamido benzoylsilane 14 containing a tethered vinyl ketone underwent efficient carboannulation to afford the corresponding indoline 15/15′ under visible-light irradiation (Scheme 1, bottom right).
To provide mechanistic insights into the photochemical carboannulation reaction, the potential reaction pathways were explored using density functional theory (DFT) analysis (Fig. 3a). The photoinitiated 1,2-Brook rearrangement of acylsilanes is well established.8a,b After initial photonic excitation of the ground state benzoylsilane (5), the vertically excited acylsilane singlet state (S*) undergoes intersystem crossing (ISC) to generate the triplet excited acylsilane (T*), from which 1,2-silyl transfer proceeds to generate the triplet siloxycarbene (INT1T).25b Conversion of the triplet carbene to the more stable singlet siloxycarbene (INT1S) is favourable (ΔG = −14.9 kcal mol−1), due to stabilisation of the singlet carbene via hyperconjugative contributions from the oxygen lone pair to the adjacent carbene vacant 2p orbital.10b
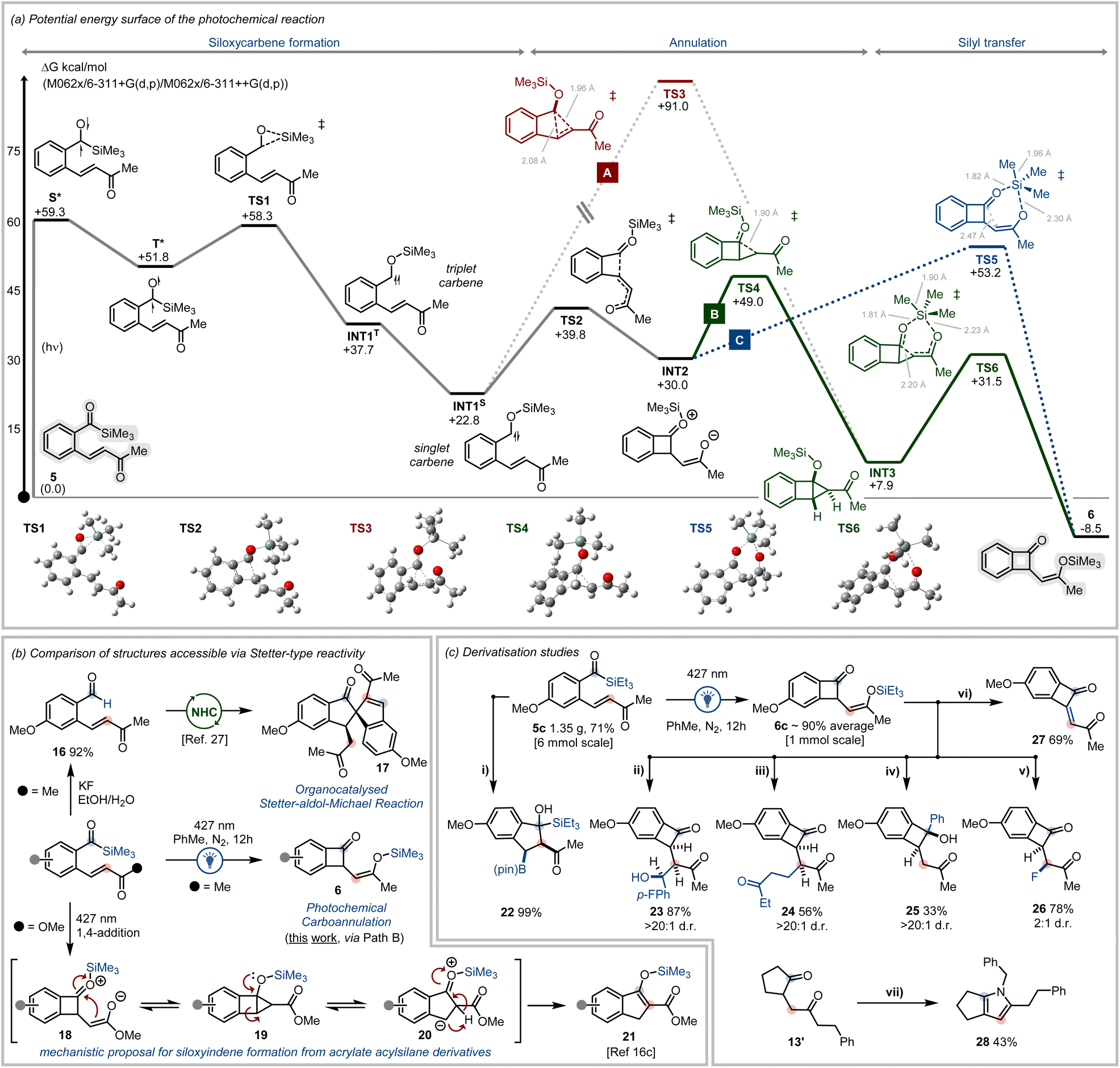 | ||
| Fig. 3 (a) DFT analysis of the potential energy surface for the carboannulation process via a siloxycarbene intermediate, the optimised geometries and stationary point bond lengths of key transition states are illustrated. (b) Comparison of the products generated via the NHC-catalysed Stetter reaction, the photochemical irradiation of vinyl ketone tethered acylsilanes and the photochemical irradiation of acrylate tethered acylsilanes; desilylation conditions: KF, EtOH/H2O, 60 °C, 16 h; (c) derivatisation of substrates prepared during investigations into acylsilanes as photofunctional directing groups: (i) CuI, LiOtBu, B2(pin)2, THF, r.t., 12 h; (ii) 4-fluorobenzaldehyde, BF3·OEt2, THF, −78 °C to r.t., 12 h; (iii) ethyl vinyl ketone, BF3·OEt2, THF, −78 °C to r.t., 12 h; (iv) PhMgBr, THF, −78 °C to r.t., 1 h; (v) Selectfluor®, MeCN, 0 °C to r.t., 12 h; (vi) NCS, DCM, 0 °C to r.t., 12 h; (vii) BnNH2, MeOH, AcOH, 70 °C, 12 h. | ||
From the singlet siloxycarbene, multiple reaction pathways were considered to approach the benzocyclobutenone product 6. We recently reported that photochemically generated siloxycarbenes undergo intramolecular cyclopropanation with tethered alkenes bearing electron withdrawing groups.10a It was thus considered that cyclopropane-fused structures could be implicit in the present system to give benzocyclobutenones. The fused cyclopropane INT3 was calculated to be relatively stable with respect to acylsilane 5 (ΔGrel = +7.9 kcal mol−1) and, although a stationary point for a concerted [2 + 1]-cycloaddition viaTS3 was located, the energy barrier associated with this transition state appears prohibitively high (Fig. 3a, Path A) and is unlikely to be traversable.
Alternatively, it was considered that the reaction proceeds via the intramolecular 1,4-conjugate addition of the siloxycarbene INT1S to the tethered vinyl ketone (Fig. 3a) to form zwitterionic INT2viaTS2. A 1,6-silicon transfer viaTS5 (ΔGINT2-TS5 = 23.2 kcal mol−1) might then occur to irreversibly trap the silyl enol ether and afford 6, a pathway (Fig. 3a, Path C) which is overall highly exergonic (ΔGINT2-PROD = −38.4 kcal mol−1).
It was also considered that a cyclopropane intermediate (INT3) might be generated via intramolecular addition of the enolate onto the oxonium ion in INT2. A reasonable transition state for ring closure to INT3 was located (viaTS4), with conversion to 6 possible via a concerted ring-opening/silicon transfer process via a 6-membered transition state (TS6, Fig. 3a, Path B). This transition state possesses a relatively low energy stationary point compared to TS5 due to the entropic effects of molecular pre-organisation in INT3, however both pathways share a very similar activation energy (ΔG ∼ 23 kcal mol−1).
Experimentally, this photocyclisation process requires significantly longer reaction times (up to 24 h) than that needed for related intramolecular transformations (e.g. 5–10 min).10a,16c,26 This suggests a high degree of reversibility within the reaction mechanism involving re-formation of the starting material from INT2 until silyl transfer can proceed to trap the final product via Path B or C. Ultimately Path B (Fig. 3a) was computed to be the kinetically favoured pathway to afford benzocyclobutenone 6via the concerted silyl-transfer/ring-opening of a putative fused cyclopropane adduct INT3.
Of note, the benzocyclobutenone adducts generated via the photochemical carboannulation reaction are unique and differ from the products generated from closely related systems (Fig. 3b). For example, under NHC-catalysis, vinyl ketone tethered benzaldehydes (e.g.16) undergo a domino Stetter–aldol–Michael reaction to afford spirocyclic bis-indanes (17).27 In this example, an intermolecular Stetter reaction from the Breslow intermediate occurs with a secondary substrate rather than the expected intramolecular Stetter reaction with the tethered vinyl ketone. Furthermore, photochemical irradiation of acrylate tethered benzoylsilanes affords siloxyindene adducts (21) rather than the benzocyclobutenone scaffolds generated from vinyl ketone tethered benzoylsilanes.16c Mechanistically, the photochemical carboannulation of acrylate tethered benzoylsilanes was originally proposed to proceed via a 6π-electrocyclisation followed by a 1,5-H shift to afford 21. However, based on the outcomes of this study, we now consider that siloxyindene formation may also proceed via 1,4-conjugate addition followed by rearrangement and H-shift (Fig. 3b).28
To demonstrate additional synthetic utility, the derivatisation of various substrates generated throughout our study was next investigated. Firstly, hydrolytic desilylation of 5b was accomplished to provide benzaldehyde 16 in high yield (Fig. 3b). Intriguingly, the copper catalysed conjugate borylation of the ortho-olefinated benzoylsilane 6c was accompanied by subsequent aldol reaction with the acylsilane group to afford a densely functionalised boryl indane 22 (Fig. 3c). Silyl enol ethers are versatile carbon-based nucleophiles, and as such, the ability of the benzocyclobutenone adducts 6 to be expeditiously diversified was explored. Triethylsilyl enol ether 6b was repeatedly isolated on larger scales and in high yield (average yield 90%) following irradiation. This compound could be engaged as a nucleophile in the Mukaiyama aldol addition with aldehydes to afford 23 in 87% yield, and underwent Michael addition reaction with ethyl vinyl ketone to give 24 in 56% yield (Fig. 3c). The silyl enol ether could also function as a protecting group enabling the selective derivatisation of the cyclobutanone ketone via reaction with phenylmagnesium bromide to afford 25 (Fig. 3c). Reaction of 6c with Selectfluor® afforded the mono-fluorinated adduct 26 while reaction with N-chlorosuccinimide afforded the des-hydro product 27 proposedly by a spontaneous halogenation/elimination sequence (Fig. 3c).
1,4-Diketones are also established entry points to heterocyclic ring systems. Showcasing the utility of the photochemical carbonannulation protocols described herein, cyclopentanone 1,4-diketone 13 was readily converted to the corresponding 1,4,5,6-tetrahydrocyclopenta[b]pyrrole 28 after condensation with benzylamine (Fig. 3c).
Conclusions
In summary, a unique mode of reactivity for siloxycarbenes was discovered involving the visible light induced intramolecular Stetter-type reaction of acylsilanes bearing tethered vinyl ketones. To exploit this novel reactivity, acylsilanes were harnessed as photofunctional directing groups to achieve both the (i) [Ru(p-cymene)Cl2]2 catalysed C–H olefination of benzoylsilanes with vinyl ketones, and (ii) visible light induced carboannulation to afford benzocylobutenone-1,4-silyl enol ethers (and derivatives thereof). This reaction manifold served as a valuable platform to deliver further insights into the properties and reactivity of acylsilanes as both precursors for siloxycarbene intermediates (via visible light irradiation), and directing groups for C–H functionalisation, where the accelerating effect of trifluoroethanol proved critical for achieving high yields.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. L. P. conceived the project. R. L. P., H. J. R. and L. A. conducted the experiments. R. L. P. conducted the computational analysis. R. L. P., H. J. R., L. A. and D. L. P. analysed the data and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. P. acknowledges the Australian Research Council (ARC) for their generous funding support (DE200100949; DP240100102). R. L. P. acknowledges the Australian Government RTP for a PhD Scholarship, H. J. R. acknowledges Monash University's Faculty of Pharmacy and Pharmaceutical Sciences Foundation Board for the Cyril Tonkin PhD Scholarship.References
- D. Seebach, Angew. Chem., Int. Ed., 1979, 18, 239–258 CrossRef.
- (a) B.-T. GrÖBel and D. Seebach, Synthesis, 1977, 1977, 357–402 CrossRef; (b) A. B. Smith and C. M. Adams, Acc. Chem. Res., 2004, 37, 365–377 CrossRef CAS; (c) M. Haroon, A. F. Zahoor, S. Ahmad, A. Mansha, M. Irfan, A. Mushtaq, R. Akhtar, A. Irfan, K. Kotwica-Mojzych and M. Mojzych, in Molecules, vol. 28, 2023 Search PubMed; (d) M. Yus, C. Nájera and F. Foubelo, Tetrahedron, 2003, 59, 6147–6212 CrossRef CAS.
- (a) G. Stork and L. Maldonado, J. Am. Chem. Soc., 1971, 93, 5286–5287 CrossRef CAS; (b) G. Stork and L. Maldonado, J. Am. Chem. Soc., 1974, 96, 5272–5274 CrossRef CAS; (c) J. D. Albright, Tetrahedron, 1983, 39, 3207–3233 CrossRef CAS.
- (a) T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef; (b) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- (a) R. S. Menon, A. T. Biju and V. Nair, Beilstein J. Org. Chem., 2016, 12, 444–461 CrossRef CAS PubMed; (b) N. Gaggero and S. Pandini, Org. Biomol. Chem., 2017, 15, 6867–6887 RSC.
- (a) M. M. Heravi, V. Zadsirjan, K. Kafshdarzadeh and Z. Amiri, Asian J. Org. Chem., 2020, 9, 1999–2034 CrossRef CAS; (b) J. R. de Alaniz and T. Rovis, Synlett, 2009, 2009, 1189–1207 CrossRef PubMed.
- D. Seebach and E. J. Corey, J. Org. Chem., 1975, 40, 231–237 CrossRef CAS.
- For reviews on the topic see: (a) W. P. Hong, H. N. Lim and I. Shin, Org. Chem. Front., 2023, 10, 819–836 RSC; (b) D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS; (c) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC; (d) G. Zhou, Z. Guo and X. Shen, Angew. Chem., Int. Ed., 2023, 62, e202217189 CrossRef CAS.
- For selected recent examples see: (a) L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, ACS Catal., 2018, 8, 304–309 CrossRef CAS; (b) T. Inagaki, T. Ando, S. Sakurai, M. Yamanaka and M. Tobisu, Chem. Sci., 2023, 14, 2706–2712 RSC; (c) T. Inagaki, S. Sakurai, M. Yamanaka and M. Tobisu, Angew. Chem., Int. Ed., 2022, 61, e202202387 CrossRef CAS PubMed; (d) S. Sakurai, T. Inagaki, T. Kodama, M. Yamanaka and M. Tobisu, J. Am. Chem. Soc., 2022, 144, 1099–1105 CrossRef CAS PubMed; (e) J. Rong, R. Oost, A. Desmarchelier, A. J. Minnaard and S. R. Harutyunyan, Angew. Chem., Int. Ed., 2015, 54, 3038–3042 CrossRef CAS; (f) F.-G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364–8370 CrossRef CAS.
- For selected recent examples see: (a) A. Bunyamin, C. Hua, A. Polyzos and D. L. Priebbenow, Chem. Sci., 2022, 13, 3273–3280 RSC; (b) D. L. Priebbenow, J. Org. Chem., 2019, 84, 11813–11822 CrossRef CAS; (c) D. L. Priebbenow, R. L. Pilkington, K. N. Hearn and A. Polyzos, Org. Lett., 2021, 23, 2783–2789 CrossRef CAS; (d) Y. Zhang, G. Zhou, X. Gong, Z. Guo, X. Qi and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202202175 CrossRef CAS PubMed; (e) G. Zhou and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202115334 CrossRef CAS PubMed; (f) Z. Zhu, W. Zhang, Y. Zhang, S. Liu and X. Shen, CCS Chem., 2022, 5, 325–333 CrossRef; (g) C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS PubMed; (h) J. R. Vale, R. F. Gomes, C. A. M. Afonso and N. R. Candeias, J. Org. Chem., 2022, 87, 8910–8920 CrossRef CAS PubMed; (i) Z. Li, Z. Zhang, Z. Zhang, G. Zhou, Y. Zhang, S. Liu and X. Shen, Sci. China. Chem., 2024 DOI:10.1007/s11426-024-2203-5.
- (a) K. Ishida, F. Tobita and H. Kusama, Chem.–Eur. J., 2018, 24, 543–546 CrossRef CAS PubMed; (b) L. Ma, Y. Yu, L. Xin, L. Zhu, J. Xia, P. Ou and X. Huang, Adv. Synth. Catal., 2021, 363, 2573–2577 CrossRef CAS.
- (a) This work was initially published as a preprint on ChemRxiv: R. L. Pilkington, H. J. Ross, L. Atkin, D. L. Priebbenow, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-9nt6c; during the finalisation of this manuscript, two studies describing the synthesis of related compounds via either cyclopropanation or 1,4-conjugate addition were published: see (b) S. S. Patel, S. Gupta and C. B. Tripathi, Chem.–Asian J., 2024, e202400240 CrossRef CAS PubMed; (c) Y. Liu, Z. Zhu, Y. Zhang, Y. Zhang, S. Liu and X. Shen, Org. Lett., 2024, 26, 5911–5916 CrossRef CAS PubMed.
- X. Wang, F. Liu, Y. Li, Z. Yan, Q. Qiang and Z.-Q. Rong, ChemCatChem, 2020, 12, 5022–5033 CrossRef CAS.
- (a) J. H. Docherty, T. M. Lister, G. McArthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed; (b) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- (a) K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. Da Silva Júnior and D. Maiti, Chem.–Eur. J., 2021, 27, 12453–12508 CrossRef CAS; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- For selected recent examples see: (a) L. Atkin and D. L. Priebbenow, Chem. Commun., 2022, 58, 12604–12607 RSC; (b) L. Atkin and D. L. Priebbenow, Angew. Chem., Int. Ed., 2023, 62, e202302175 CrossRef CAS; (c) P. Becker, D. L. Priebbenow, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 269–271 CrossRef CAS; (d) D. L. Priebbenow and C. Hua, Chem. Commun., 2021, 57, 7938–7941 RSC; (e) P. Becker, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2015, 54, 15493–15496 CrossRef CAS PubMed; (f) R. Vaggu, N. Thadem, M. Rajesh, R. Grée and S. Das, Org. Lett., 2023, 25, 2594–2599 CrossRef CAS PubMed; (g) T. Zhang, C. Zhang, X. Lu, C. Peng, Y. Zhang, Z. Zhu, G. Zhong and J. Zhang, Org. Biomol. Chem., 2024, 22, 466–471 RSC.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- (a) J. Das, W. Ali and D. Maiti, Trends Chem., 2023, 5, 551–560 CrossRef CAS; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (c) G. Rani, V. Luxami and K. Paul, Chem. Commun., 2020, 56, 12479–12521 RSC.
- (a) S. Ochi, Z. Zhang, Y. Xia and G. Dong, Angew. Chem., Int. Ed., 2022, 61, e202202703 CrossRef CAS; (b) H. Lu, T.-T. Zhao, J.-H. Bai, D. Ye, P.-F. Xu and H. Wei, Angew. Chem., Int. Ed., 2020, 59, 23537–23543 CrossRef CAS; (c) B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727–12731 CrossRef CAS PubMed; (d) R. Li, B. Li, H. Zhang, C.-W. Ju, Y. Qin, X.-S. Xue and D. Zhao, Nat. Chem., 2021, 13, 1006–1016 CrossRef CAS PubMed.
- K. H. Park, A. Rizzo and D. Y. K. Chen, Chem. Sci., 2020, 11, 8132–8137 RSC.
- Y. Yamashita, L. Poignant, J. Sakata and H. Tokuyama, Org. Lett., 2020, 22, 6239–6243 CrossRef CAS PubMed.
- (a) J. P. Vilaine, Pharmacol. Res., 2006, 53, 424–434 CrossRef CAS PubMed; (b) J. Peglion, J. Vian and C. Thollon, Can. J. Physiol. Pharmacol., 1994, 72, 95 Search PubMed; (c) C. Thollon, C. Cambarrat, J. Vian, J. F. Prost, J. L. Peglion and J. P. Vilaine, Br. J. Pharmacol., 1994, 112, 37–42 CrossRef CAS PubMed.
- (a) T. Bhattacharya, A. Ghosh and D. Maiti, Chem. Sci., 2021, 12, 3857–3870 RSC; (b) S. K. Sinha, T. Bhattacharya and D. Maiti, React. Chem. Eng., 2019, 4, 244–253 RSC; (c) J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394–400 RSC; (d) H. M. Begam, K. Pradhan, K. Varalaxmi and R. Jana, Chem. Commun., 2023, 59, 5595–5598 RSC.
- Refer to the ESI† for additional information.
- (a) K. Ishida, H. Yamazaki, C. Hagiwara, M. Abe and H. Kusama, Chem.–Eur. J., 2020, 26, 1249–1253 CrossRef CAS PubMed; (b) J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141, 16227–16231 CrossRef CAS PubMed.
- L. Atkin, H. J. Ross and D. L. Priebbenow, J. Org. Chem., 2023, 88, 14205–14209 CrossRef CAS PubMed.
- E. Sánchez-Larios, J. M. Holmes, C. L. Daschner and M. Gravel, Org. Lett., 2010, 12, 5772–5775 CrossRef PubMed.
- We recently obtained experimental evidence for structurally related systems that infers that siloxyindene formation might also proceed via initial 1,4-conjugate addition of the siloxycarbene to a tethered acrylate. Conversion of the cyclobutane adduct to the siloxyindene may involve a putative fused cyclopropane intermediate.
Footnote |
| † Electronic supplementary information (ESI) available: Computational and experimental details including characterisation data and NMR spectra. See DOI: https://doi.org/10.1039/d4sc05715e |
| This journal is © The Royal Society of Chemistry 2024 |