
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of gem-difluorination on the conformation and properties of a model macrocyclic system†
T. J.
Cogswell‡
 *a,
R. J.
Lewis
b,
C.
Sköld
c,
A.
Nordqvist
a,
M.
Ahlqvist
d and
L.
Knerr
*a
*a,
R. J.
Lewis
b,
C.
Sköld
c,
A.
Nordqvist
a,
M.
Ahlqvist
d and
L.
Knerr
*a
aMedicinal Chemistry, Early Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden. E-mail: thomas.cogswell@acerta-pharma.com; laurent.knerr@astrazeneca.com
bMedicinal Chemistry, Early Respiratory and Immunology, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
cDrug Design and Discovery, Department of Medicinal Chemistry, BMC, Uppsala University, P.O. Box 574, SE751 23 Uppsala, Sweden
dDMPK, Early Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
First published on 6th November 2024
Abstract
Conformational control of drug candidates to engineer improved potency and ADME properties is an ongoing area of research. Macrocyclic rings tend to offer a greater degree of rigidity than non-cyclised small molecules, and, as a result they are perfect platforms to instil conformational controls. In this study, the difluoroalkoxyphenyl moiety is examined as a tool to alter the conformation of macrocycles. A fluorinated and non-fluorinated macrocyclic matched pair is compared in terms of conformation preferences and related ADME properties. The synthesised macrocycles are found to give similar major conformations exhibiting a trans amide in the macrocyclic backbone. However, for the fluorinated macrocycle, the major trans amide conformation is in equilibrium with a cis amide minor conformation, seen by 1H NMR in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of trans/cis. The conformational fits for the minor fluorinated isomer demonstrate the out of plane preference of the difluoroalkoxy system encouraging the amide within the macrocycle backbone to adopt a cis conformation. The fluorinated macrocycle was less metabolically stable compared to the non-fluorinated, postulated to be a result of the interconversion of trans amide to the cis amide, which potentially could be more readily metabolised.
:1 ratio of trans/cis. The conformational fits for the minor fluorinated isomer demonstrate the out of plane preference of the difluoroalkoxy system encouraging the amide within the macrocycle backbone to adopt a cis conformation. The fluorinated macrocycle was less metabolically stable compared to the non-fluorinated, postulated to be a result of the interconversion of trans amide to the cis amide, which potentially could be more readily metabolised.
Introduction
In the past decade, there has been an intense renewed interest in ‘beyond the rule of five’ modalities in drug discovery.1 As the complexity and diversity of protein targets and drug mechanisms of action increase, medicinal chemists need to access a broader range of modalities.2 Macrocycles can offer unique advantages over traditional small molecules with the cyclic structure conveying an innate preorganization on the molecular conformation.3 This cyclic nature results in a lowered entropic penalty on target binding, that can lead to higher target affinity and selectivity, especially important when targeting extended binding pockets and macromolecular interactions.4 Nature has made full use of this phenomenon, with 20% of all known natural products being macrocyclic.5 However, even with the numerous reports documenting the benefits of macrocyclization on the potency of drug candidates,6 a lack of understanding on how to best modulate their physicochemical properties and cell permeabilities to produce robust clinical candidates remains.7 As a result, macrocycles remain an under-exploited modality in drug discovery.3To capitalize on the preorganisation of macrocyclic structures, there is a significant interest into ways to bias and alter the conformation of macrocycles to optimize their target binding interactions through a rational design approach.8 Common approaches include the incorporation of stereocenters,9 the addition of alkyl groups to backbone atoms,10 and the installation of both saturated and unsaturated heterocycles into the macrocyclic backbone.11 One interesting approach takes advantage of the stereoelectronic interactions that polarized C–F bonds have on neighbouring groups, where orbital alignments favour stabilized conformations.12 Incorporation of fluorine atoms has previously been identified as a means to change conformational preference in many functional systems such as organocatalysis,13 pharmaceuticals,14 and liquid crystals.15
In a seminal publication in 2014, Hunter et al. reported the use of the fluorine gauche effect to demonstrate, for the first time, that stereoselective fluorination could alter the conformation of a peptidic macrocycle.16 Each synthesised isomer of the vicinal fluorinated Unguisin A occupied a unique low energy conformation which differed from the parent non-fluorinated compound. In 2016, O'Hagan and co-workers found that when installing a CF2-group into different positions of the aliphatic backbone of a family of musk lactones, the macrocyclic ring was constrained into a rectangular conformation with the CF2-group always situated at a corner site.17
In a non-macrocyclic system, Durley and co-workers reported an intriguing fluorination effect whilst developing a library of cholesteryl ester transfer protein (CETP) inhibitors.18 When compared to the alkoxyphenyl analogue 1, the incorporation of a fluorinated side chain in 2 led to an 8-fold increase in potency which was attributed to a more optimal configuration adopted within the enzyme active site (Fig. 1A). After studying the conformation through ab initio calculations, they confirmed that the non-fluorinated alkyl chain of 1 aligned with the plane of the aromatic ring whereas the fluorinated chain occupied an orthogonal conformation.19 This rotation of the OCF2 bond allowed the lone pairs of the oxygen to donate into the low lying 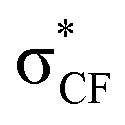 antibonding orbitals leading to a stabilization of the orthogonal conformation (Fig. 1B). Since this first example, O'Hagan reported that the same conformational effect also occurs when a sulfur linker replaces the oxygen.20
antibonding orbitals leading to a stabilization of the orthogonal conformation (Fig. 1B). Since this first example, O'Hagan reported that the same conformational effect also occurs when a sulfur linker replaces the oxygen.20
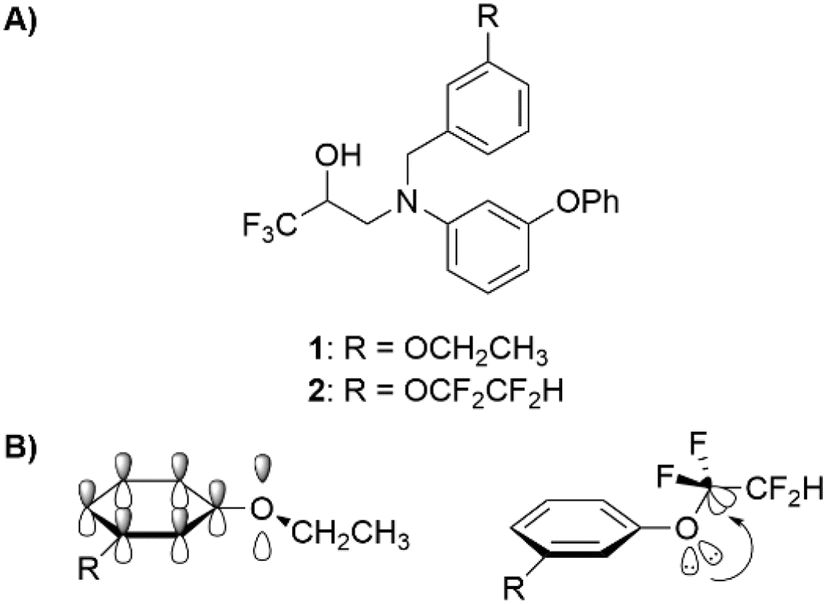 | ||
| Fig. 1 (A) The structures of CETP inhibitors reported by Durley and co-workers.19 The non-fluorinated analogue 1 has a IC50 of 1.6 μM while the fluorinated analogue 2 has an IC50 of 0.2 μM. The values were determined in a buffer transfer assay. (B) Hyperconjugative π–σ* C–F interaction rationale for the reduced in-plane conformational preference of a fluoroalkoxyaryl group.19 | ||
A survey of available crystallographic data for difluoromethoxyaryl-containing structures supported by density functional theory (DFT) calculations confirmed that the orthogonal arrangement of two endo fluorines was one of the two major conformations adopted by this module.21 An endo–exo arrangement was also predicted to have a similar energy in the gas phase leading to postulations that, due to this low barrier to rotation, this module has the potential to access different conformations when subjected to different micro-environments.22
Intrigued by the reported impact of the gem-difluorination of alkoxyphenyl group in acyclic systems, we decided to expand these studies by incorporating this fluorinated module into a macrocyclic core. While there is some exemplification of this chemistry in recent patent literature,23 there is still very little data reported on the impact of that design on the macrocycle properties. Therefore, we aimed to expand the toolbox of small, fluorinated units available to increase the conformational diversity and adaptability of macrocyclic scaffolds. Conformational changes through fluorination has been shown to alter physicochemical properties such as polarity, permeability, and solubility, therefore, an in-depth study into these relationships was also conducted.24
Results and discussion
Synthesis
To study this fluorination effect in macrocycles a model system was selected, based on the BACE-1 inhibitor 3. This was envisaged to provide entry into a simplified and medicinal chemistry relevant macrocycle ring system (Fig. 2).25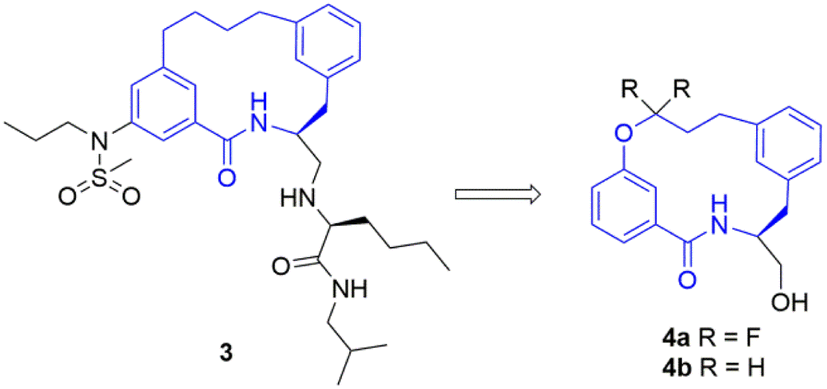 | ||
| Fig. 2 The structure of a BACE-1 inhibitor 3 developed by researchers at Merck for the treatment of Alzheimer's disease (left).25 The target model macrocyclic structures 4a and 4b to be studied herein (right). | ||
The fluorinated and non-fluorinated macrocycles 4a and 4b were prepared using the synthetic route reported in Scheme 1. The 1,1-difluorinated allylated phenol starting material 5a was synthesised in a previous publication from our laboratories utilizing methodology developed by Ichikawa et al.26,27 The starting materials (5a and 6) were combined through a Heck reaction followed by hydrogenation providing 8a in good yield. Ester hydrolysis and Boc deprotection gave the precyclisation precursor, that upon macrolactamisation yielded the desired macrocyclic scaffold 4a. Using the same synthetic route, the non-fluorinated match pair macrocycle 4b was also prepared.
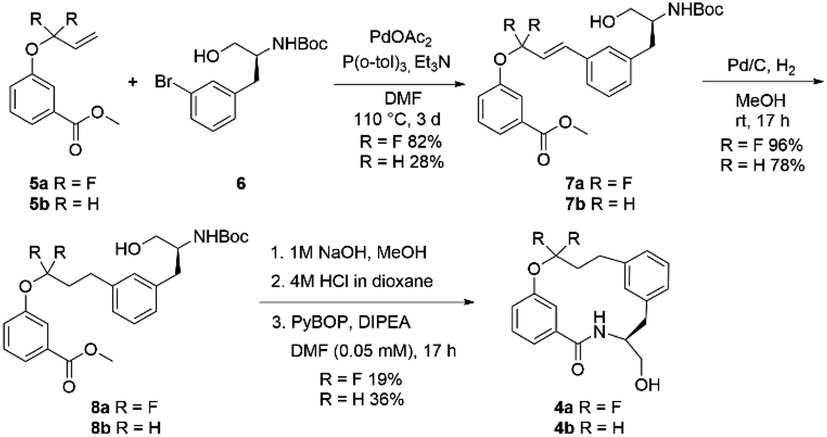 | ||
| Scheme 1 The synthesis of the model fluorinated macrocycle and non-fluorinated match pair. | ||
For macrocycle 4a, two unexpected features of the proton NMR spectrum were immediately apparent (Fig. 3). Firstly, two conformations were visible in an approximate ratio of 4:1 with slow exchange on the NMR timescale. Secondly, the minor conformation exhibited a highly shielded aromatic proton at 4.59 ppm. This signal was 2.2 ppm more shielded than the equivalent proton in the major conformation. Most other protons exhibited differences in chemical shift of 0.2 to 0.9 ppm between the conformations. This conformational signature suggests a significant change in conformation between major and minor forms.
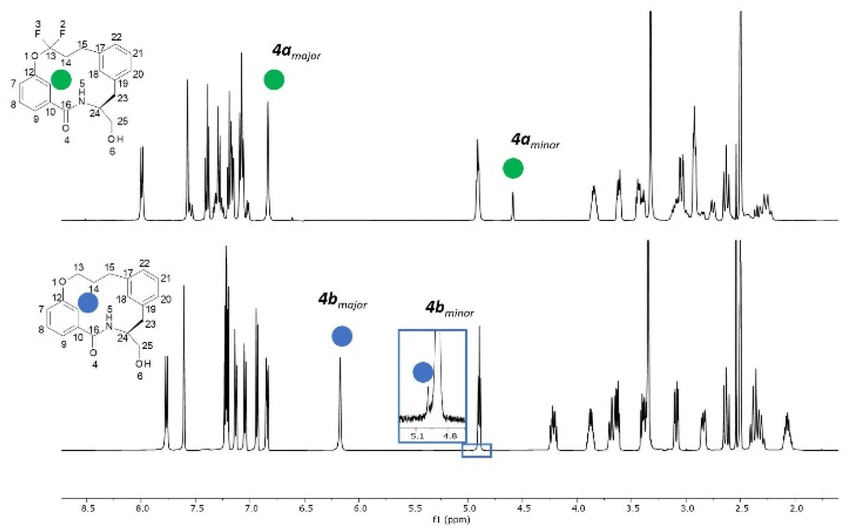 | ||
| Fig. 3 The stacked 1H NMR spectra in DMSO-d6 of the non-fluorinated macrocycle 4b (top) and fluorinated macrocycle 4a (bottom). | ||
In contrast, the proton spectrum for non-fluorinated macrocycle 4b did not clearly exhibit slowly exchanging conformations, though a ROESY NMR experiment did reveal the presence of a second conformation (visible as exchange cross peaks from certain protons – see ESI†). The ratio between the major and minor forms was 200:1, a significant difference to that observed in 4a.
Conformational analysis
To investigate the influence of the difluoromethoxyaryl system on the conformation of the macrocyclic backbone of 4a and 4b, calculated conformations were fit to experimental NMR data using the Mspin program.28 The results of the MSpin program was confirmed by an independent fit of the major conformations using a least squares approach using QM optimized conformations.Conformational searches for both 4a and 4b were performed that contained examples of both cis and trans amides. After QM optimization, data was fit in the MSpin program using proton–proton NOE distances, coupling constants and calculated proton chemical shifts. The use of chemical shifts was especially important for the minor conformation as only a few proton–proton NOE distances could be accurately determined. Proton-fluorine NOEs were used qualitatively to distinguish between the different conformational fits found for 4a.
For the major conformation of 4a (4amajor), two conformations were found, approximately equally distributed, accounting for about 90% of the population (Fig. 4). The two conformations differ from each other primarily in the relative orientations of the two aromatic rings.
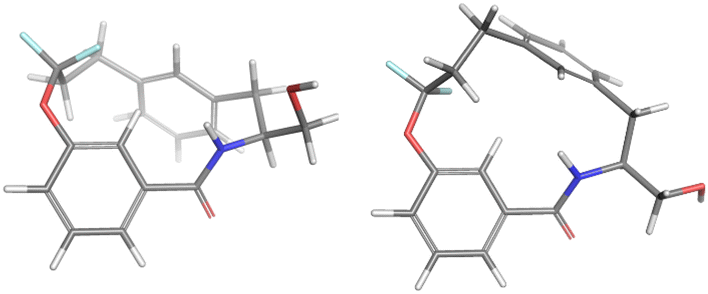 | ||
| Fig. 4 Conformations 4amajor_#1 and 4amajor_#2 from Mspin making up 90% of the population of 4amajor. | ||
For the minor conformation of 4a (4aminor), the same procedure was followed to yield a fit composed of two conformers 4aminor_#1 at 65% and 4amajor_#2 at 30% (Fig. 5). Interestingly, the conformers both bear the cis amide in place of the trans, demonstrating a significantly different conformation compared to 4amajor. The highly shielded proton seen in the NMR spectrum (Fig. 3), was found to make an edge/face interaction with the second aromatic ring accounting for the unusual shielding.29
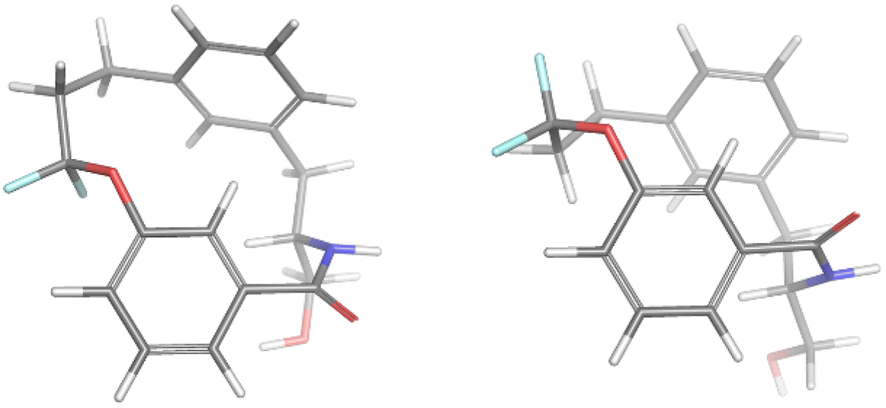 | ||
| Fig. 5 The conformational fits from Mspin for 4aminor. Two cis amides 4aminor_#1 (left) and 4aminor_#2 at 30% (right). | ||
A similar approach was followed to find two clusters of similar conformations for the non-fluorinated macrocycle 4b. The two clusters are shown in Fig. 6. On the left are two conformations differing only in the OH bond orientation accounting for 55% of the population and on the right, three conformations accounting for 25% of the population which differ in OH orientation and the amide bond portion of the macrocycle.
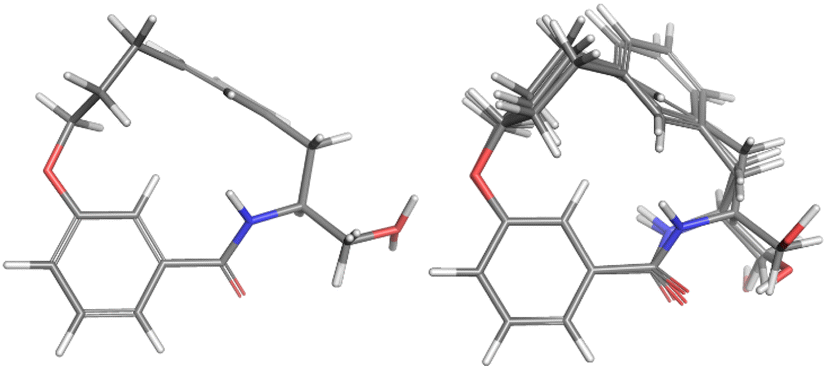 | ||
| Fig. 6 The conformational fits from Mspin for 4b. The overlay consisting of conformations 4b_#1 and 4b_#2 (left). The overlay of conformations 4b_#3, 4b_#4 and 4b_#5 accounting for 25% of the population (right). | ||
The results from Mspin were compared with conformations selected by comparing the sum of the absolute difference between the experimentally derived 1H–1H NOE distances for 4a and 4b respectively (See ESI†) and the corresponding atom distance of a calculated conformational ensemble after QM optimization.
A combination of two conformations was proposed for 4amajor since both fluorine substituents displayed a strong 1H–19F NOE to H11. These two conformations agreed with the solutions found by Mspin (Fig. 4). For 4b an overall best fit was found for a conformation comprising a trans amide bond and the ether substituent in the plane of the phenyl ring, this also best reflected one of the suggested solutions from Mspin (Fig. 6).
The fluorinated analogue 4a is expected to prefer that the O–CF2 bond is out of plane of the bonded aromatic ring. However, this bond is constrained by being part of a macrocyclic ring, thus moving this bond out of plane likely introduces higher energy conformational elements elsewhere in the macrocycle. These conformational compromises can be seen born out in conformations 4amajor_#1, 4amajor_#2, 4aminor_#1 and 4aminor_#2. Conformation 4amajor_#2 is similar to the 4b conformations in that the O–CF2 bond is almost co-planar with the aromatic ring and the ideal chain geometry is only slightly distorted (maximum deviation 18°). In conformation 4amajor_#1 however, the O–CF2 bond is 32° out of plane from the aromatic ring but the ideal chain geometry is more distorted (maximum deviation 37°). When the amide is in a cis geometry (conformations 4aminor_#1 and 4aminor_#2), the O–CF2 bond can move further out of plane (60° and 46°) and the ideal chain geometry is less distorted (maximum deviation 23°). This comes at the expense of some greater deviation from planarity of the amide bond (36° and 27°). 4b adopts a conventional conformation with a trans amide bond and has torsion angles of bonded sp3 atoms (syn, anti, gauche) deviating by no more than 12° from an ideal chain (+60°, 180°, −60°). The macrocyclic O–CH2 bond is within 6° of co-planarity of the aromatic ring except in conformation 4b_#5 when it is 15° out of plane. The amide bond is distorted from planarity by 12–17°. Full details of the torsions observed can be seen in Table S3.†
Using the ratios of trans to cis conformer for 4a and 4b, it is possible to estimate an energy penalty of at least 10 kJ mol−1 for positioning the fluorine atoms approximately in plane with the aromatic ring compared to the proton atoms. This assumes only that the remote fluorine proton exchange has no direct effect on the energetics of the amide bond.
Physicochemical and ADME properties
To explore further the effects of fluorination on the macrocycle, in vitro physicochemical and ADME profiling was performed, and the two systems compared head-to-head (Table 1). The fluorination effect on measured lipophilicity (logD7.4) was minimal, which is consistent with literature reports.3 The intrinsic permeability between the two macrocycles was also comparable, with 4a and 4b, having Papp values of 96 and 74, respectively, in the Caco-2 in vitro assay. Interestingly, a 10-fold difference in solubility between the two systems was identified, which was not correlated with any significant measured lipophilicity or polarity (as measured by ePSA)30 differences.
| Fluorinated macrocycle 4a | Non-fluorinated macrocycle 4b | |
|---|---|---|
| logD7.4 |
2.4 | 2.4 |
| ePSA (Å2) | 70 | 69 |
| Human hepatocytes metabolic stability t1/2 (min) | <2.3 | 10 |
| Human liver microsomes metabolic stability t1/2 (min) | <2.3 | 31 |
| Chemical stability | 11 | >72 |
| Aq. pH1 t1/2 (h) | ||
| Chemical stability | >72 | >72 |
| Aq. pH7.4 t1/2 (h) | ||
| Chemical stability | >72 | >72 |
| Aq. pH10 t1/2 (h) | ||
| Solubility (μM) | 21 | 243 |
| Intrinsic permeability | 96 | 74 |
| Caco AB pH 6.5 (inhibition) Papp (1 × 10−6 cm s−1) |
A difference in the metabolic stability was observed between the two systems. In both human hepatocytes and human liver microsomes, the half-life (t1/2) of fluorinated 4a was below a measurable time point whereas the non-fluorinated 4b had a t1/2 of 10 and 31 minutes respectively.
At first glance, this result seems to be counter intuitive as fluorination has been widely reported to improve the metabolic stability of drug candidates.31 To look in more detail, a metabolic stability study was undertaken to identify the metabolites in both the fluorinated and non-fluorinated examples upon incubation in human hepatocytes (Schemes 2 and 3). The amide of 4a was found to be rapidly hydrolysed to the open compound as a major metabolite. Whereas 4b showed little amide hydrolysis and instead was predominantly dealkylated at the phenolic site leading to the phenol derivative as the main metabolite. As expected, the fluorinated macrocycle was completely protected from this dealkylation metabolic pathway. We postulate that the presence of a significant cis amide population due to the conformational impact of the gem-difluorinated system is a major driver in the promotion of metabolism through amide cleavage. This represented a dramatic switch of metabolism pathway upon fluorination and whilst the magnitude is surprising in this case similar effect has been previously reported in the literature.32
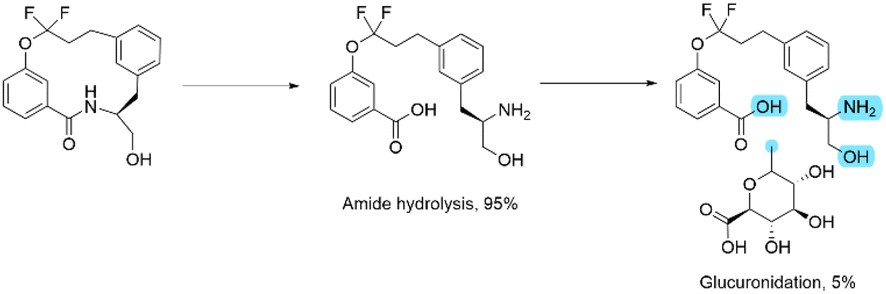 | ||
| Scheme 2 The metabolism profile of 4a upon incubation with human hepatocytes. The percentage values are semi-quantitative, based on MS-response. Where the exact position of glucuronidation could not be determined, the potential sites are labelled in blue. | ||
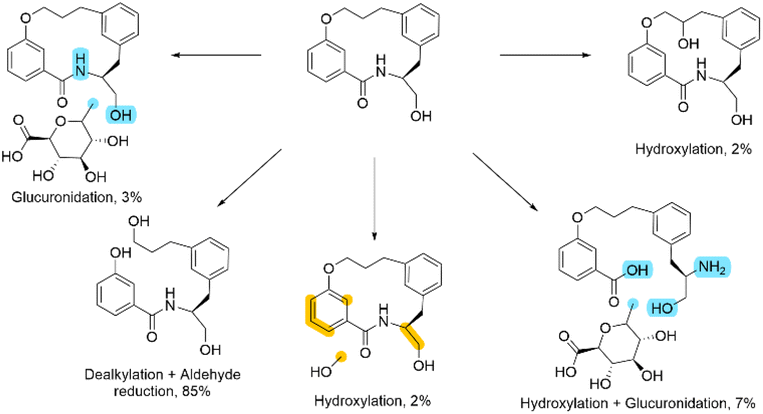 | ||
| Scheme 3 The metabolism profile of 4b upon incubation with human hepatocytes. The percentage values are semi-quantitative, based on MS-signal. Where the exact positions could not be determined, the potential sites of glucuronidation are labelled in blue and the potential sites of oxidation labelled in orange. | ||
To understand the chemical stability profiles of both macrocycles, we complemented the metabolization study with hydrolytic stability studies at pH 1, 7.4 and 10. We could show that, except for the lesser stability of fluorinated macrocycle in acidic conditions (half-life 11 hours at pH 1), both scaffolds were similarly stable with half-lives above 3 days in all conditions tested. Based on pKa measurements for two benzoic acids possessing the fluorinated and non-fluorinated allyloxy groups (see ESI†) we could also verify that the inductive electron withdrawing effect of the fluorinated analogue was modest thus strengthening the evidence for impact on chemical stability of this fluorinated module being limited.
Additionally, we could show that the fluorinated analogue peculiar metabolic stability profile was specific to the macrocyclic scaffold by studying a matched pair of related linear analogues (see ESI†). Interestingly this demonstrated that while the linear non-fluorinated analogue showed comparable stability to its macrocyclic counterpart, the stability of the linear fluorinated analogue was considerably higher than the fluorinated macrocycle based on their half-lives upon incubation with human microsomes or human hepatocytes. The improved stability for the linear fluorinated analogue could not be attributed to a change in lipophilicity as both fluorinated analogues had a logD7.4 of 2.4.
Conclusions
To study the conformational effects that gem-difluorination has on a macrocyclic system, model fluorinated and non-fluorinated match pair 4a and 4b were synthesized. The measured NMR data of 4a and 4b were input into MSpin software to identify likely conformational fits. The fluorinated macrocycle was found to have two conformations on the NMR timescale in a ratio of 4:1 whereas the non-fluorinated analogue favours a single conformation with a 200:1 ratio. The conformational fits for 4amajor and 4b showed the alkoxy phenyl chain to be in a similar geometry and a trans amide present on the other side of the macrocyclic scaffold. However, the fit for 4aminor showed an altered geometry of the fluorinated chain and, seemingly to allow this to occur within the macrocycle, an edge/face interaction between the two aromatic rings and the cis amide were present. Using the difference in ratio between the cis:trans conformers of 4a and 4b, we were able to estimate the strength of the fluorination effect to be at least 10 kJ mol−1.
The physicochemical and ADME properties for 4a and 4b were examined, revealing a difference in metabolic stability in both human hepatocytes and human liver microsomes between the two macrocycles. Metabolite identification studies revealed that for 4a, amide cleavage was the predominant metabolic pathway, whereas this was a minor pathway for 4b.
This is the first time this gem-difluorinated alkoxyphenyl conformational effect has been demonstrated experimentally in a macrocyclic system and, interestingly, it was found to have a profound effect on the properties of the compound. The fluorination pattern studied here represents another potential conformational design tool to modulate the conformer population of macrocyclic scaffolds and optimize their interaction with biological targets. Future work to install this group into a diverse array of macrocyclic rings could shed further light on how this effect could be harnessed to tune the conformations of macrocycles. The inclusion of this motif into an active medicinal chemistry regime where target-binding and target-selectivity data are generated represents an important future goal for this area of research.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
TC: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing. RJL: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. CS: conceptualization, writing – review & editing. AN: molecular modelling calculations and fitting conformations to calculated NOE distances, writing – review & editing. MA: data curation, investigation, visualization, writing – review & editing. LK: conceptualization, investigation, project administration, supervision, writing – original draft, writing – review & editing.Conflicts of interest
RJL, AN, MA and LK are employed by AstraZeneca and RJL, AN, MA and LK own shares in the company. TC is currently employed by Acerta Pharma, a member of the AstraZeneca group. The postdoctoral position of TC, at the time the work was undertaken, was part funded by AstraZeneca.Acknowledgements
This work was supported by the Initial Training Network, FLUOR21(TC), funded by the FP7 Marie Curie Actions of the European Commission (FP7-PEOPLE-2013-ITN-607787). The authors would like to acknowledge the support of the ADME group of AstraZeneca Gothenburg. CS acknowledges support from KLOSS Akademi Ut (INNOV 2015/29) and Vinnova (grant no. 2016-02110).Notes and references
- (a) M.-Q. Zhang and B. Wilkinson, Curr. Opin. Biotechnol., 2007, 18(6), 478–488 CrossRef CAS PubMed; (b) B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21(9), 1115–1142 CrossRef CAS PubMed; (c) G. B. Santos, A. Ganesn and F. S. Emery, ChemMedChem, 2016, 2245–2251 CrossRef CAS PubMed; (d) D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, J. Med. Chem., 2018, 61(7), 2636–2651 CrossRef CAS PubMed.
- (a) S. Surade and T. L. Blundell, Chem. Biol., 2012, 19(1), 42–50 CrossRef CAS PubMed; (b) E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56(35), 10294–10323 CrossRef CAS PubMed.
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discov., 2008, 7, 608–624 CrossRef CAS PubMed; (b) V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS; (c) A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- (a) J. Mallinson and I. Collins, Future Med. Chem., 2012, 4(11), 1409–1438 CrossRef CAS PubMed; (b) A. Russo, C. Aiello, P. Grieco and D. Marasco, Curr. Med. Chem., 2016, 23(8), 748–762 CrossRef CAS PubMed.
- A. T. Frank, S. N. Farina, N. Sawwan, O. R. Wauchope, M. Qi, E. M. Brzostowska, W. Chan, F. W. Grasso, P. Haberfield and A. Greer, Mol. Diversity, 2007, 11, 115–118 CrossRef CAS PubMed.
- For selected examples see: (a) N. Proisy, S. Y. Sharp, K. Boxall, S. Connelly, S. M. Roe, C. Prodromou, A. M. Z. Slawin, L. H. Pearl, P. Workman and C. J. Moody, Chem. Biol., 2006, 13(11), 1203–1215 CrossRef CAS PubMed; (b) Z.-F. Tao, L. Wang, K. D. Stewart, Z. Chen, W. Gu, M.-H. Bui, P. Merta, H. Zhang, P. Kovar, E. Johnson, C. Park, R. Judge, S. Rosenburg, T. Sowin and N.-H. Lin, J. Med. Chem., 2007, 50(7), 1514–1527 CrossRef CAS PubMed; (c) U. Lücking, G. Siemeister, M. Schäfer, H. Briem, M. Krüger, P. Lienau and R. Jautelat, ChemMedChem, 2007, 2(1), 63–77 CrossRef PubMed; (d) J. E. DeLorbe, J. H. Clements, B. B. Whiddon and S. F. Martin, ACS Med. Chem. Lett., 2010, 1(8), 448–452 CrossRef CAS PubMed; (e) D. I. Oumana, N. K. Yilmaz, K. L. Prachanronarong, C. A. A. Ali and C. A. Schiffer, ACS Chem. Biol., 2016, 11(4), 900–909 CrossRef PubMed.
- (a) F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57(2), 278–295 CrossRef CAS PubMed; (b) D. S. Nielson, H. N. Hoang, R.-J. Lohman, T. A. Hill, A. J. Lucke, D. J. Craik, D. J. Edmonds, D. A. Griffith, C. J. Rotter, R. B. Ruggeri, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 12059–12063 CrossRef PubMed; (c) A. T. Bockus, K. W. Lexa, C. R. Pye, A. S. Kalgutkar, J. W. Gardner, K. C. R. Hund, W. H. Hewtt, J. A. Schwochert, E. Glassey, D. A. Price, A. M. Mathiowetz, S. Liras, M. P. Jacobson and R. S. Lokey, J. Med. Chem., 2015, 58(11), 4581–4589 CrossRef CAS PubMed; (d) B. Over, P. Matsson, C. Tyrchan, P. Artursson, B. C. Doak, M. A. Foley, C. Hilgendorf, S. E. Johnston, M. D. Lee IV, R. J. Lewis, P. McCarren, G. Muncipinto, U. Norinder, M. W. D. Perry, J. R. Duvall and J. Kihlberg, Nat. Chem. Biol., 2016, 12, 1065–1074 CrossRef CAS PubMed.
- (a) D. P. Fairlie, G. Abbenante and D. R. March, Curr. Med. Chem., 1995, 2, 654–686 CrossRef CAS; (b) J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488–499 CrossRef CAS; (c) M. Porel, D. N. Thornlow, N. N. Phan and C. A. Alabi, Nat. Chem., 2016, 8, 590–596 CrossRef CAS PubMed; (d) S. D. Appavoo, S. Huh, D. B. Diaz and A. K. Yudin, Chem. Rev., 2019, 119, 9724–9752 CrossRef CAS PubMed.
- For selected examples see: (a) M. P. Glenn, M. J. Kelso, J. D. A. Tyndall and D. P. Fairlie, J. Am. Chem. Soc., 2003, 125, 640–641 CrossRef CAS PubMed; (b) A. Sharma, S. Sharma, R. P. Tripathi and R. S. Ampapathi, J. Org. Chem., 2012, 77, 2001–2007 CrossRef CAS PubMed.
- For selected examples see: (a) H. Luesch, W. Y. Yoshida, R. E. Moore and V. J. Paul, Bioorg. Med. Chem., 2002, 10, 1973–1978 CrossRef CAS PubMed; (b) T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Coillins, J. J. Cui, J. G. Deal, Y.-L. Deng and D. Dihn, et al. , J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed; (c) J. Elleraas, J. Ewanicki, T. W. Johnson, N. W. Sach, M. R. Collins and P. F. Richardson, Angew. Chem., 2016, 128, 3654–3659 CrossRef.
- For selected examples see: (a) S. Zaretsky, C. G. Scully, A. J. Lough and A. K. Yudin, Chem.–Eur. J., 2013, 19, 17668–17672 CrossRef CAS PubMed; (b) J. R. Frost, C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 CrossRef CAS PubMed.
- (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) L. Hunter, Beilstein J. Org. Chem., 2010, 38, DOI:10.3762/bjoc.6.38.
- L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860–11871 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61(14), 5822–5880 CrossRef CAS PubMed.
- M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- X.-G. Hu, D. S. Thomas, R. Griffith and L. Hunter, Angew. Chem., Int. Ed., 2014, 53, 6176–6179 CrossRef CAS PubMed.
- (a) M. J. Corr, R. A. Cormanich, C. N. von Hahmann, M. Bühl, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Org. Biomol. Chem., 2016, 14, 211–219 RSC; (b) R. Callejo, M. J. Corr, M. Yang, D. B. Cordes, A. M. Z. Slawin and D. O'Hagan, Chem.–Eur. J., 2016, 22(24), 8137–8151 CrossRef CAS PubMed.
- R. C. Durley, M. L. Grapperhaus, M. A. Massa, D. A. Mischke, B. L. Parnas, Y. M. Fobian, N. P. Rath, D. D. Honda, M. Zeng, D. T. Connolly, D. M. Heuvelman, B. J. Witherbee, K. C. Glenn, E. S. Krul, M. E. Smith and J. A. Sikorski, J. Med. Chem., 2000, 43(24), 4575–4578 CrossRef CAS PubMed.
- M. A. Massa, D. P. Spangler, R. C. Durley, B. S. Hickory, D. T. Connolly, B. J. Witherbee, M. E. Smith and J. A. Sikorski, Bioorg. Med. Chem. Lett., 2001, 11, 1625–1628 CrossRef CAS PubMed.
- R. Tomita, N. Al-Maharik, A. R. Garcia, M. Bühl and D. O'Hagan, Org. Biomol. Chem., 2018, 16, 1113–1117 RSC.
- (a) CSD Version 5.37 Search PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- Q. A. Huchet, N. Trapp, B. Kuhn, B. Wagner, H. Fischer, N. A. Kratochwil, E. M. Carreira and K. Müller, J. Fluorine Chem., 2017, 198, 34–46 CrossRef CAS.
- (a) G. R. Ott, K. Gibson, B. Lefker, P. Humphries, Orexin-2 receptor agonists, WO Pat., 167925, 2023 Search PubMed; (b) A. Dumoulin, P. M. Blom, A. Daugan, M. Laugeois, C. G. Housseman, A. Denis, Y. Lamotte, A. Le Tiran, K. Christensen, Leucine-Rich Repeat Kinase 2 inhbitors, WO Pat., 194976, 2022 Search PubMed.
- Q. A. Huchet, B. Kuhn, B. Wagner, N. A. Kratochwil, H. Fischer, M. Kansey, D. Zimmerli, E. M. Carreira and K. Müller, J. Med. Chem., 2015, 58(22), 9041–9060 CrossRef CAS.
- S. J. Stachel, C. A. Coburn, S. Sankaranarayanan, E. A. Price, B. L. Pietrak, Q. Huang, J. Lineberger, A. S. Espeseth, L. Jin, J. Ellis, M. K. Holloway, S. Munshi, T. Allison, D. Hazuda, A. J. Simon, S. L. Graham and J. P. Vacca, J. Med. Chem., 2006, 49(21), 6147–6150 CrossRef CAS PubMed.
- T. J. Cogswell, A. Dahlén and L. Knerr, Chem.–Eur. J., 2019, 25, 1184–1187 CrossRef CAS PubMed.
- T. Fujita, K. Sugiyama, S. Sanada, T. Ichitsuka and J. Ichikawa, Org. Lett., 2016, 18(2), 248–251 CrossRef CAS PubMed.
- Mnova – Mspin Search PubMed.
- A. Y. S. Balazs, R. J. Carbajo, N. L. Davies, Y. Dong, A. W. Hird, J. W. Johannes, M. L. Lamb, W. McCoull, P. Raubo, G. R. Robb, M. J. Packer and E. Chiarparin, J. Med. Chem., 2019, 62, 9418–9437 CrossRef CAS.
- G. H. Goetz, W. Farrell, M. Shalaeva, S. Sciabola, D. Anderson, J. Yan, L. Philippe and M. J. Shapiro, J. Med. Chem., 2014, 57, 2920–2929 CrossRef CAS PubMed.
- (a) P. Shah and A. D. Westwell, J. Enzym. Inhib. Med. Chem., 2007, 22(5), 527–540 CrossRef CAS PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) R. S. Obach, G. S. Walker and M. A. Brodney, Drug Metab. Dispos., 2016, 44, 634–646 CrossRef CAS PubMed; (b) Y. Pan, ACS Med. Chem. Lett., 2019, 10, 1016–1019 CrossRef CAS PubMed; (c) B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63(12), 6315–6386 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05424e |
| ‡ Current address: Acerta Pharma B.V., A Member of the AstraZeneca Group, Kloosterstraat 9, 5349 AB, Oss, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2024 |