
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid, potent, and persistent covalent chemical probes to deconvolute PI3Kα signaling†
Lukas
Bissegger‡
 a,
Theodora A.
Constantin‡
a,
Erhan
Keles‡
a,
Luka
Raguž
a,
Isobel
Barlow-Busch
c,
Clara
Orbegozo
a,
Thorsten
Schaefer
a,
Valentina
Borlandelli
a,
Thomas
Bohnacker
a,
Rohitha
Sriramaratnam
a,
Alexander
Schäfer
b,
Matthias
Gstaiger
b,
John E.
Burke
cd,
Chiara
Borsari§‡
a and
Matthias P.
Wymann‡
*a
a,
Theodora A.
Constantin‡
a,
Erhan
Keles‡
a,
Luka
Raguž
a,
Isobel
Barlow-Busch
c,
Clara
Orbegozo
a,
Thorsten
Schaefer
a,
Valentina
Borlandelli
a,
Thomas
Bohnacker
a,
Rohitha
Sriramaratnam
a,
Alexander
Schäfer
b,
Matthias
Gstaiger
b,
John E.
Burke
cd,
Chiara
Borsari§‡
a and
Matthias P.
Wymann‡
*a
aDepartment of Biomedicine, University of Basel, Mattenstrasse 28, 4058 Basel, Switzerland. E-mail: matthias.wymann@unibas.ch; Tel: +41 61 207 5046
bDepartment of Biology, Institute of Molecular Systems Biology, ETH Zurich, Otto-Stern-Weg 3, 8093 Zürich, Switzerland
cDepartment of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia V8W 2Y2, Canada
dDepartment of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia V6T 1Z3, Canada
First published on 12th November 2024
Abstract
Chemical probes have gained importance in the elucidation of signal transduction in biology. Insufficient selectivity and potency, lack of cellular activity and inappropriate use of chemical probes has major consequences on interpretation of biological results. The catalytic subunit of phosphoinositide 3-kinase α (PI3Kα) is one of the most frequently mutated genes in cancer, but fast-acting, high-quality probes to define PI3Kα's specific function to clearly separate it from other class I PI3K isoforms, are not available. Here, we present a series of novel covalent PI3Kα-targeting probes with optimized intracellular target access and kinetic parameters. On-target TR-FRET and off-target assays provided relevant kinetic parameters (kchem, kinact and Ki) to validate our chemical probes. Additional intracellular nanoBRET tracer displacement measurements showed rapid diffusion across the cell membrane and extremely fast target engagement, while investigations of signaling downstream of PI3Kα via protein kinase B (PKB/Akt) and forkhead box O (FOXO) revealed blunted pathway activity in cancer cell lines with constitutively activated PI3Kα lasting for several days. In contrast, persistent PI3Kα inhibition was rapidly bypassed by other class I PI3K isoforms in cells lacking functional phosphatase and tensin homolog (PTEN). Comparing the rapidly-diffusing, fast target-engaging chemical probe 9 to clinical reversible PI3Kα-selective inhibitors alpelisib, inavolisib and 9r, a reversible analogue of 9, revealed 9's superior potency to inhibit growth (up to 600-fold) associated with sustained suppression of PI3Kα signaling in breast cancer cell lines. Finally, using a simple washout protocol, the utility of the highly-selective covalent PI3Kα probe 9 was demonstrated by the quantification of the coupling of insulin, EGF and CXCL12 receptors to distinct PI3K isoforms for signal transduction in response to ligand-dependent activation. Collectively, these findings along with the novel covalent chemical probes against PI3Kα provide insights into isoform-specific functions in cancer cells and highlight opportunities to achieve improved selectivity and long-lasting efficacy.
Introduction
Small-molecule chemical probes are excellent tools to interrogate specific enzyme functions, and to dissect convoluted signaling pathways.1,2 Additionally, high-quality chemical probes are essential to validate target tractability, on-target efficacy and safety in early stage drug development.3 Despite their vital importance, many commonly used chemical probes do not meet the fundamental quality criteria regarding biochemical (<100 nM) and cellular potency (<1 μM), target selectivity (>30 fold to sequence related proteins), and a proof of target engagement.4–6 The use of probes with inferior quality often leads to corrupted data and false conclusions,7 highlighting the necessity to develop advanced chemical probes which meet stringent quality criteria.Investigating class I phosphoinositide 3-kinase (PI3K) signaling using small molecule chemical probes has been a challenging endeavor, also because class I PI3Ks include four isoforms (class IA: PI3Kα, PI3Kβ, PI3Kδ; class IB: PI3Kγ) with highly conserved ATP binding sites.8–10 In response to growth factor receptor engagement, these PI3Ks catalyze the phosphorylation of phosphatidylinositol(4,5)-bisphosphate (PIP2) to generate phosphatidylinositol(3,4,5)-trisphosphate (PIP3)11–13 and regulate cell metabolism, growth, proliferation and survival.14,15 Here signaling is convoluted, as the ubiquitously expressed PI3Kα and PI3Kβ have both PI3K distinct or redundant, context- and receptor-dependent cellular functions. For example, PI3Kα is activated by tyrosine kinase receptors (RTKs) and operates downstream of GTP-loaded Ras, while PI3Kβ can also be activated in response to G protein-coupled receptor (GPCRs) activation.16–18
In addition, membrane receptor activation can cause concurrent signaling through PI3K and mitogen-activated protein kinase (MAPK) pathways, leading to inter-pathway cross-signaling – depending on ligand, cell type and genetic context.19 To date, attempts to deconvolute receptor–PI3K isoform-coupling yielded incomplete or conflicting data.17,20–22
Activating mutations of catalytic subunit of PI3Kα (p110α, encoded by the PIK3CA locus) are often located in the helical (hotspot E542K/E545K) or kinase domain (hotspot H1047R),23–25 and are frequent in cancer.26 Despite its well-known oncogenic role, the current understanding of PI3Kα isoform-specific contributions in cellular signaling events and disease mechanisms remains poor. This delayed the clinical development of reversible small molecule PI3K inhibitors, which still lack specificity at effective concentrations, causing a high incidence of adverse reactions. Moreover, reversible inhibitors only provide transient target inhibition, leading to rapid rebound signaling and loss of drug action.27–33 Increased isoform specificity and duration of action is therefore necessary to widen the therapeutic window of PI3Kα inhibition and improve potency.
Covalent inhibitors, designed from reversible scaffolds via deliberate addition of weakly reactive ‘warheads’, have been successfully used to target oncogenic proteins, including BTK,34–36 EGFR/HER2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37–40 and KRAS.41,42 The covalent bond formation with non-conserved amino acids provides exceptional selectivity and potency, while simplifying pharmacokinetics, as the target remains inactive until its re-synthesis. Despite distinct advantages over reversible ATP-competitive inhibitors, covalent irreversible inhibitors and probes of PI3Kα have not been thoroughly investigated. We previously developed a method for Covalent Proximity Scanning (CoPS) to target remote, solvent-exposed cysteines.43 We applied CoPS to target the non-conserved Cys862 of PI3Kα located ∼11 Å away from the reversible binding site and, using our preclinical candidate PQR514,44 generated covalent inhibitor tool compounds with low intrinsic reactivity and excellent target engagement in vitro. Although these compounds were already accepted as high-quality covalent small molecule chemical probes,45 their cellular potency remained moderate due to low cellular permeability. Here, we report the strategic improvement of covalent small molecule chemical probes to achieve optimal potency and cellular permeability. We describe modifications in the linker between the reversible binding core module and the warhead which ultimately lead to optimized Cys862 targeting. Additionally, with the aid of the novel, highly cell-permeable chemical probe 9, we show for the first time the biological impact of isolated PI3Kα isoform inhibition in cancer cells. In particular, we demonstrate that a covalent inhibition of PI3Kα not only provides exceptional selectivity after washout, but also markedly increases potency and circumvents pathway reactivation in sensitive cell lines. These major improvements provide for prolonged duration of efficacy in comparison to reversible ATP-competitive PI3Kα inhibitors.
37–40 and KRAS.41,42 The covalent bond formation with non-conserved amino acids provides exceptional selectivity and potency, while simplifying pharmacokinetics, as the target remains inactive until its re-synthesis. Despite distinct advantages over reversible ATP-competitive inhibitors, covalent irreversible inhibitors and probes of PI3Kα have not been thoroughly investigated. We previously developed a method for Covalent Proximity Scanning (CoPS) to target remote, solvent-exposed cysteines.43 We applied CoPS to target the non-conserved Cys862 of PI3Kα located ∼11 Å away from the reversible binding site and, using our preclinical candidate PQR514,44 generated covalent inhibitor tool compounds with low intrinsic reactivity and excellent target engagement in vitro. Although these compounds were already accepted as high-quality covalent small molecule chemical probes,45 their cellular potency remained moderate due to low cellular permeability. Here, we report the strategic improvement of covalent small molecule chemical probes to achieve optimal potency and cellular permeability. We describe modifications in the linker between the reversible binding core module and the warhead which ultimately lead to optimized Cys862 targeting. Additionally, with the aid of the novel, highly cell-permeable chemical probe 9, we show for the first time the biological impact of isolated PI3Kα isoform inhibition in cancer cells. In particular, we demonstrate that a covalent inhibition of PI3Kα not only provides exceptional selectivity after washout, but also markedly increases potency and circumvents pathway reactivation in sensitive cell lines. These major improvements provide for prolonged duration of efficacy in comparison to reversible ATP-competitive PI3Kα inhibitors.
Results and discussion
Targeted optimization of PI3Kα-specific inhibiting covalent chemical probes
Compounds 1 and 2 (Fig. 1a) established using CoPS43 had an excellent in vitro, but only a moderate cellular on-target activity (IC50s for phosphorylation of Ser473 of PKB/Akt in SKOV3 cells were 86 for 1 and 82 nM for 2). We hypothesized that this originated from the highly polar nature of both molecules (LogD1: 1.12; 2: 0.97) and that introduction of more lipophilic moieties should increase their cellular permeability, leading to improved intracellular potency. To this end, we first analyzed interactions of compounds 1 and 2 (Fig. S1†) with p110α to identify exchangeable structural elements that lower hydrophilicity while retaining activity using the respective co-crystal structures (PDB-ID: 7R9V and 7R9Y). While the ATP binding site-occupying core formed well-established hydrogen bonds with residues Val851 and Asp810 for 1 and 2, additional linker-specific protein interactions were noted (Fig. S1†). Hydrogen bonds essential for isoform selectivity formed between the proximal, piperazine-adjacent carbonyl groups and residue Gln85946–49 in the case of both compounds and, additionally, between the distal, piperidine-adjacent carbonyl group of 1 and residue Thr856 (Fig. S1†). To explore the importance of each carbonyl group individually while simultaneously aiming to reduce hydrophilicity, we synthesized 3 and 4, which lack one of the two carbonyl groups (Fig. 1a). In either case, LogD values increased and TPSA decreased with 3 being the more hydrophobic compound (Table 1). While compound 4 showed comparable cellular activity (IC50 pPKB: 69 nM) as 1 and 2, compound 3 was more potent (IC50 pPKB: 38 nM, Fig. 1d). We continued by replacing the tertiary amine in compound 3 with an aromatic ring which led to compounds 5 and 6. Although this change resulted in higher LogD values (5: 3.32; 6: 2.66), the cellular activities remained comparable to 3, presumably caused by the rigidity of the linkers, thus preventing an optimal orientation of the warhead towards Cys862. Another negative feature of 5 and 6 was a drop in kinetic solubility (363 μM for 3vs.7 and 46 μM for 5 and 6, respectively), caused by the replacement of a very polar tertiary amine by a highly hydrophobic and rigid phenyl ring.
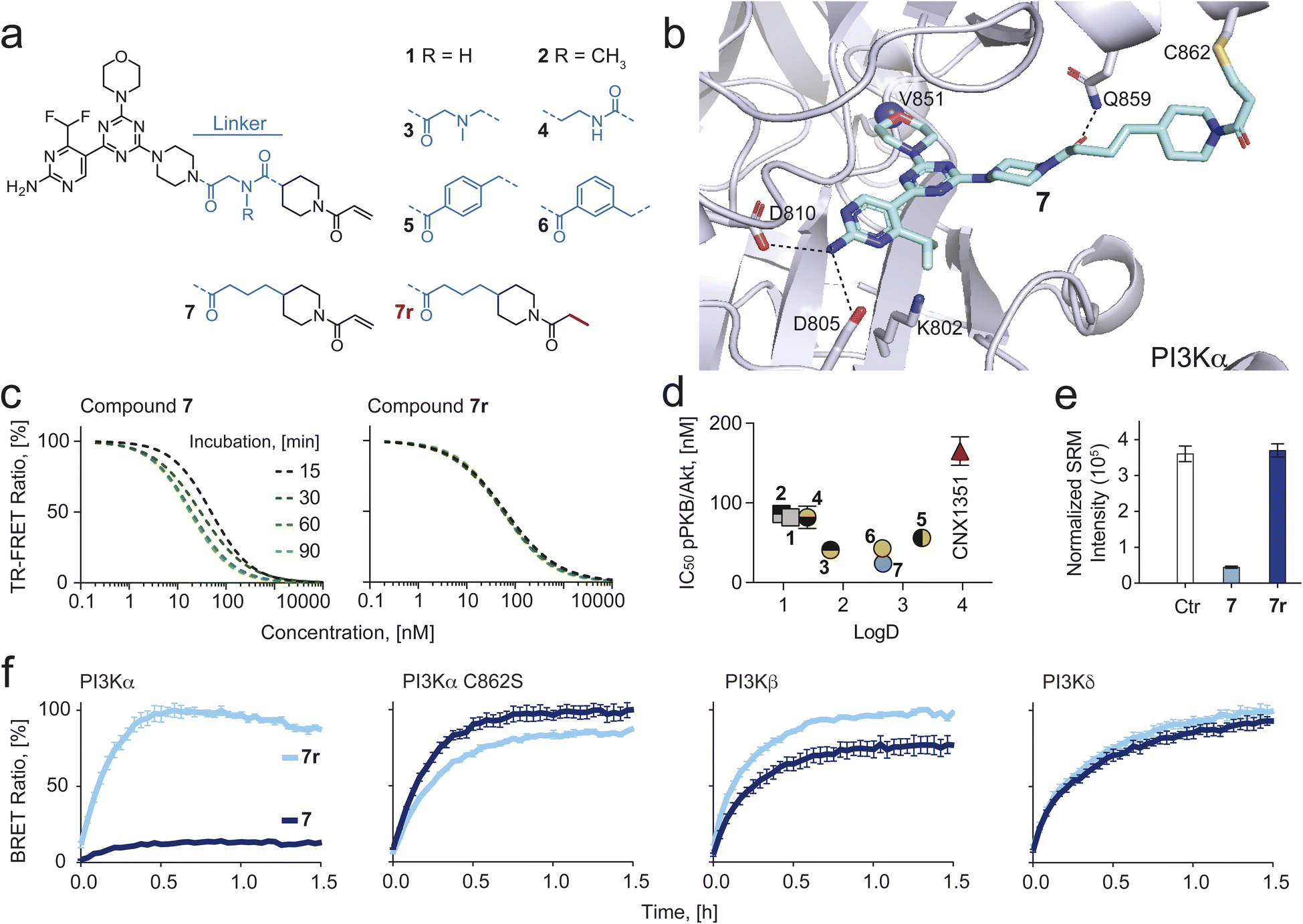 | ||
| Fig. 1 (a) Chemical structure of eight PI3Kα-selective inhibiting chemical probes. (b) Modelling of compound 7 bound covalently to Cys862 of p110α (PDB ID: 7R9V was used as a starting point). H-bonds are depicted as dashed black lines. (c) Time-dependent IC50 shift derived from time-resolved fluorescence resonance energy transfer (TR-FRET) ratios (Fig. S2† for all compounds), comparing compound 7 and its reversible analogue 7r. (d) IC50 values for PKB phosphorylation (pPKB, Ser473) measured in SKOV3 cells by in-cell western (ICW) plotted against logD values (measured by Bienta Enamine Biology Services). Data shown are mean ± SD from at least n = 3 independent experiments. Error bars are not shown when smaller than symbols. (e) LC-SRM quantification of covalent Cys862-modification by 7 and no covalent modification by 7r. (f) Bioluminescence resonance energy transfer (BRET) target occupancy assay in live HEK293 cells. The cells were transiently transfected with constructs encoding a Nanoluciferase (Nanoluc) fused to different PI3K isoforms (PI3Kα, PI3Kβ and PI3Kδ) or a Cys862 to Ser mutated PI3Kα. 24 h after transfection, cells were incubated with 3 μM of 7 or 7r for 2 h. The probes were washed out (twice for 10 min with Opti-MEM) and a cell-permeable Py-BODIPY moiety bearing a fluorescent energy transfer probe (BRET tracer) was added to the cells (0.2 μM final concentration). Recovery of the BRET signal, resulting from displacement of the inhibitor from the ATP-binding pocket, was monitored for 1.5 h. Prolonged target occupancy after probe washout indicates covalent bond formation. Data shown are mean ± SEM (n = 3). | ||
| Compound | TPSAa [Å2] | LogDb |
Solubilityc [μM] | k chem × 104 [M−1 s−1] | k inact × 104 [s−1] | K i [nM] | k inact/Ki × 105 [nM−1 s−1] | pPKB/Aktd,f [nM] |
|---|---|---|---|---|---|---|---|---|
|
a TPSA calculated using Marvin/JChem v21.
b LogD as measured by Bienta Enamine Biology Services (for cLogP values see extended Table S1).
c Kinetic solubility measured at pH 7.4 in PBS (protocol see ESI).
d Calculated from n ≥ 3 independent measurements; SD values are reported in Table S1.
e Approximation due to incomplete baseline separation between compound 4 and its adduct with βME.
f Phosphorylation of PKB/Akt on Ser473 was determined in SKOV3 cells by in-cell western assays after 1 h of compound exposure.
|
||||||||
| 1 | 175.9 | 1.12 | 333 | 3.7 | 9.27 | 2.23 | 41.6 | 86 |
| 2 | 167.1 | 0.97 | 383 | 3.5 | 11.8 | 5.26 | 22.5 | 82 |
| 3 | 150.0 | 1.79 | 363 | 2.7 | 1.57 | 29.5 | 0.53 | 38 |
| 4 | 158.8 | 1.40 | 393 | 5.2e | 1.13 | 13.7 | 0.82 | 69 |
| 5 | 146.8 | 3.32 | 7 | 3.2 | 0.85 | 8.4 | 1.03 | 54 |
| 6 | 146.8 | 2.66 | 46 | 2.9 | 2.14 | 11.6 | 1.85 | 41 |
| 7 | 146.8 | 2.67 | 243 | 2.5 | 3.09 | 5.00 | 6.24 | 22 |
| 7r | 146.8 | n.d. | n.d. | — | — | 6.26 | — | 40 |
| 8 | 156.0 | 1.70 | 381 | 2.9 | 10.0 | 4.15 | 24.4 | 19 |
| 9 | 156.0 | 2.43 | 357 | 3.0 | 48.2 | 8.68 | 56.1 | 19 |
| 9r | 156.0 | n.d. | n.d. | — | — | 2.9 | — | 48 |
| CNX1351 | 99.2 | 3.96 | 10 | 2.2 | 7.79 | 43.3 | 1.82 | 172 |
| Ibrutinib | 107.6 | 4.08 | 10 | 10 | — | — | — | — |
| BYL719 | 100.2 | 3.16 | 49 | — | — | 0.83 | — | 162 |
| GDC-0077 | 109.5 | 1.10 | 357 | — | — | 0.50 | — | 10 |
We speculated that a more flexible hydrophobic linker would improve target engagement and increase kinetic solubility. Thus, we designed, modelled (Fig. 1b) and synthesized compound 7, which had the cationic nitrogen from compound 3 replaced by a methylene. Indeed, 7 showed an improved cellular potency (pPKB IC50 of 22 nM, Fig. 1d and Table 1; full datasets and SD in Table S1†), while retaining good solubility (243 μM) and an optimal LogD (2.67).
Confirmation of covalent targeting of Cys862 in PI3Kα
We synthesized a reversible analog of 7, dubbed 7r, which lacks the Michael acceptor (Fig. 1a, red) and therefore is not able to form a covalent bond with Cys862 of PI3Kα. TR-FRET experiments using 7 and 7r showed a time-dependent IC50 shift for 7, implying covalent engagement of PI3Kα, but not for the reversible analogue 7r (Fig. 1c). Further, we confirmed covalent modification of Cys862 by bottom-up LC-MS/MS based proteomics. The existence of covalent adducts between the tryptic peptide containing Cys862 (NSHTIMQIQCK) was investigated following incubation of recombinant PI3Kα and BSA with 7 or 7r. As a covalent modification induces a characteristic mass shift of the peptide, and peptide precursor MS1 spectra were recorded with high mass accuracy (10 ppm), XIC traces for the modified forms of the peptides could be extracted. XIC peaks for the peptide covalently modified with 7 were identified in the samples treated with 7, but not 7r or DMSO (Fig. S3†). The presence of covalently modified peptide NSHTIMQIQCK was demonstrated by peptide-spectrum matching (Fig. 1e). MS2 spectra were searched against 20308 human protein entries from the Uniprot database, and high confidence (>0.9) matches were considered (see proteomics data in ESI†). Among these, the only compound-modified peptide was NSHTIMQIQCK, modified with 7 but not with 7r (Table S2†). Importantly, all other covered cysteines (28/32 cysteines in p110α, 5/6 cysteines in p85α, 33/35 cysteines in BSA) were not covalently modified, supporting exclusive targeting of Cys862 in p110α by 7.Next, a bioluminescence resonance energy transfer (BRET) compound displacement assay was used to confirm covalent binding of 7 to PI3Kα in live cells. A stable Nanoluciferase (Nanoluc)50,51 was fused to different class IA PI3K isoforms and expressed in HEK293 cells. The light emitted from the Nanoluc-PI3K fusion proteins is transferred to a cell-permeable fluorescent tracer (energy transfer probe 3 in ref. 52) when bound to the ATP binding site of PI3K, causing light emission at a longer wavelength. In HEK293 cells expressing wild-type (WT) PI3Kα, tracer addition following exposure to 7 and subsequent washout did not increase BRET signal over time (Fig. 1f). In contrast, mutation of Cys862 to Ser (C862S) in PI3Kα allowed tracer binding after compound washout, demonstrating selective and irreversible bond formation between 7 and Cys862 in PI3Kα in living cells. Importantly, the reversible analogue 7r was readily washed out from WT and C862S PI3Kα and neither compound formed covalent bonds with PI3Kβ and PI3Kδ (Fig. 1f).
Inhibition of PI3K signaling in cancer cell lines
Isoform-selective reversible inhibitors BYL719 (alpelisib, PI3Kα-selective),46 TGX221 (PI3Kβ-selective),53 and CAL101 (idelalisib, PI3Kδ-selective)54 were investigated alongside 7 in two PI3Kα-mutated cell breast cancer cell lines (H1047R in T47D, E545K in MCF7), and two PTEN-deficient cancer cell lines (prostate cancer PC3 and melanoma A2058). In T47D and MCF7, 7 showed, respectively, a 5- and 8-fold better cellular potency (lower pPKB IC50) as compared to BYL719, whereas TGX221 and CAL101 did not prevent PKB phosphorylation (Fig. 2a). These data indicate that mutated PI3Kα dominates PI3K output in T47D and MCF7 cell lines and suggest that inhibition of PI3Kβ and PI3Kδ by the pan-PI3K binding module of 7 does not contribute to the prominent potency of 7. In contrast, PTEN-deficient cell lines showed moderate responses to all reversible inhibitors, while 7 most potently inhibited phosphorylation of PKB, likely due its reversible pan-PI3K inhibition achieved by the its ATP-binding site core module (Fig. 2a). | ||
| Fig. 2 (a) PI3Kα or PTEN-mutated cancer cell lines were incubated for 1 h with a range of concentrations (3 μM to 0.8 nM) of isoform-selective PI3K inhibitors (7, covalent PI3Kα-selective; 7r, reversible analogue of 7; BYL719, reversible PI3Kα-selective, TGX221, reversible PI3Kβ-selective; CAL101, reversible PI3Kδ-selective). Phosphorylation of PKB (pPKB, Ser473) was determined by in-cell western (ICW) assay. Data shown are mean ± SEM from n = 3 independent experiments. (b) Cells were incubated for 2 h with 3 μM of inhibitor, followed by drug washout (twice for 10 min with growth medium supplemented with 10% FCS). Subsequently, cells were incubated at 37 °C and 5% CO2 for the indicated times before fixation. Phosphorylation PKB (pPKB, Ser473) was determined in at least n = 3 independent experiments by ICW assay. Data shown are mean ± SEM from n = 4 independent experiments. (c–h) Concentration and time dependent nuclear translocation of FOXO1 KTR probe in response to 7 or BYL719. Cytosol/nuclear ratio of FOXO1 KTR probe was determined with batch analysis using Cell Profiler software (for details see Methods). (c and f) Concentration-dependent IC50 determination in (c) T47D and (f) A2058 cells incubated with compounds for 1 h. (d and g) Time-dependent FOXO1 KTR probe translocation into the nucleus following treatment with 3 μM of 7, BYL719 or DMSO in (d) T47D and (g) A0258 cells. (e and h) Cells were incubated with 7, BYL179 or DMSO (3 μM) for 1 h, followed by washout (twice for 10 min with growth medium supplemented with 10% FCS). (i) Representative images of (left) T47D and (right) A2058 cells incubated with 7 or BYL719 (3 μM) for 1 h (prior to washout) and 6 h after inhibitor washout. | ||
To confirm these findings, we carried out washout studies using 7, which provide an unambiguous discrimination of PI3Kα dependencies in these cell lines. Because 7 binds reversibly to all class I PI3K isoforms, but covalently only to PI3Kα, washout results in pure PI3Kα inhibition. In all cell lines, removal of reversible inhibitors BYL719 and 7r after 2 h incubation reactivated PI3K/PBK signaling within the first hour (Fig. 2b). On the other hand, incubation with 7 followed by washout resulted in efficacious, long-term (>18 h) inhibition of the pathway in T47D and MCF7 cells, confirming PI3Kα as the main driver of PI3K/PKB signaling and demonstrating prolonged engagement of PI3Kα. In PTEN-deficient cell lines, PI3K/PKB signaling returned to ∼50% after washout of 7, suggesting significant contribution from other class IA PI3K isoforms.
Dynamic tracking of PI3K signaling in living cells
To study the dynamics of downstream pathway signaling through PKB, we utilized cell lines stably transfected with a kinase translocation reporter (KTR) probe based on a modified PKB substrate (Forkhead box protein O1, FOXO1) fused to a fluorescent reporter.55 This probe translocates from the nucleus to the cytoplasm in response to phosphorylation by activated PKB,56,57 which can be quantified in real-time using high-content microscopy. We first assessed the cytosol/nuclear ratio of FOXO1-KTR in live cells as a function of increasing concentrations of covalent compound 7 or BYL719. In PI3Kα-mutant T47D cells, 1 h incubation with 7 resulted in complete nuclear import of the FOXO1-KTR (IC90 = 50 nM), whereas BYL719 was not able to induce full nuclear translocation at the concentrations used here, suggestive of residual PKB activity (Fig. 2c). In PTEN-deficient A2058 cells, nuclear translocation of FOXO1-KTR required higher concentrations of 7 (IC90 = 1657 nM), whereas BYL719 was completely ineffective, as expected (Fig. 2f). We next investigated time-dependent PKB signaling dynamics upon treatment with 7 and BYL719 using live cell imaging. In T47D cells, treatment with 7 or BYL719 (3 μM) resulted in rapid (<20 min) import of FOXO1-KTR into the nucleus (Fig. 2d). Subsequent washout resulted in a fast recovery of cytosolic signal in cells treated with BYL719, whereas persistent nuclear FOXO1-KTR localization (>10 h) was achieved in cells treated with 7 (Fig. 2e and i). In A2058 cells, only exposure to compound 7 (3 μM) induced nuclear FOXO1 KTR localization, and this occurred at a slower rate (>40 min) than in T47D cells (Fig. 2g). Subsequent washout in A2058 cells led to complete cytosolic FOXO1 KTR translocation (Fig. 2h and i). These data demonstrate that only covalent inhibition of PI3Kα by compound 7 can completely block signaling through PKB in sensitive PI3Kα-mutant cell lines, and that residual upstream PI3K activity is sufficient to fully activate downstream pathway signaling.Fine-tuning linker–protein interaction
Despite 7 being an excellent covalent PI3Kα small molecule chemical probe, we hypothesized that further optimization of linker–protein surface interactions would result in compounds with superior target engagement and improved cellular potency. To electrostatically complement the positively charged protein surface, we synthesized 8, bearing an ether moiety within the linker, and 9, bearing an additional (S)-methyl moiety on the morpholine ring to increase lipophilicity (Fig. 3a). Indeed, TR-FRET experiments with 8 and 9 showed, a 4- and 9-fold improvement in the kinact/Ki ratio, respectively, as compared to 7 (Fig. 3b and Table 1), while LogD values were 1.70 for 8 and 2.43 for 9 (Table 1). While the kinact/Ki values of 8 and 9 were in the same range as for 1 and 2, cellular potency was significantly improved (Table 1).
 | ||
| Fig. 3 (a) Chemical structure of three covalent PI3Kα-selective chemical probes and one reversible analogue (9r). (b) Comparison of the second order rate constant for 9 covalent chemical probes and CNX1351. Dissociation constants (Ki) and rate for covalent binding to PI3Kα constants (kinact) were used to characterize covalent binding efficiency. The kinetic parameters were calculated with global fitting from TR-FRET ratios (Fig. S5†) for numerical integration on a kinetic model using KinTek Global Kinetic Explorer modeling software. Values are shown as mean ± SD (n = 3). (c) Intrinsic reactivity (kchem) of 9 covalent chemical probes, CNX1351 and ibrutinib. For details and SD see Table S1.† (d) Modelling of on-target reactivity. Experimentally determined intrinsic reactivity, dissociation and rate constants (kchem, Ki and kinact) were used for each compound to model the target engagement with 10 nM PI3Kα and 100 nM compound over 40 minutes. (e) Modelling of off-target reactivity. Side reactions between compounds 1–9 (green line, intrinsic warhead reactivity below 10−3 M−1 s−1) and 7 mM GSH. Modelling of side reactions was carried out with KinTek Global Kinetic Explorer modeling software. (f) X-ray co-crystallographic structure of PI3Kα with 9 (green) (PDB-ID: 8TWY) bound covalently to Cys862 of p110α. H-bonds are depicted as dashed black lines (data collection and refinement statistics are reported in Table S3†). (g) Electron density maps of compound 9 (PDB-ID: 8TWY) compared with compounds 1 (PDB-ID: 7R9V) and 2 (PDB-ID: 7R9Y) covalently bound to p110α (h) lipophilic efficiency (LipE) analysis of covalent chemical probes and BYL719. IC50 values (pPKB, Ser473) measured in SKOV3 cells by in-cell western (ICW) plotted against logD values. Data shown are mean ± SD from at least n = 3 independent experiments (for calculation see Table S4†). (i) Covalent binding efficiency (kinact/Ki) plotted against LipE values. Data shown are mean ± SD from at least n = 3 independent experiments. (j) Diffusion coefficients of 6 covalent chemical probes, two reversible analogues (7r and 9r), CNX1351 and BYL719. Diffusion coefficients were obtained by fitting the NanoBRET data (Fig. 3l) with KinTek Global Kinetic Explorer software (see Methods for detailed calculations). Values are shown as mean ± SD (n = 3; for calculations see Table S5†). (k) Diffusion coefficients plotted against covalent binding efficiency (kinact/Ki) for 6 covalent chemical probes and CNX1351. Values are shown as mean ± SD (n = 3). Error bars are not shown when smaller than symbols. (l) Target engagement in live HEK293 cells using bioluminescence resonance energy transfer (BRET) assay. HEK293 cells transfected with N-terminal Nanoluc fused to PI3Kα were pre-incubated with Tracer K-3 (Promega) and NanoBRET NanoGlo substrate for 20 min (37 °C, 5% CO2). Subsequently, compounds were added at different concentrations and BRET signal was measured for 4 h. Values are shown as mean ± SEM (n = 3). | ||
In our previous work, we established that moderate intrinsic warhead reactivity is pivotal in the development of covalent chemical probes and potential clinical candidates.43 To determine compound-specific intrinsic reactivity (kchem), the newly synthesized covalent chemical probes (3–9), CNX1351, the first reported covalent inhibitor of PI3Kα58 and ibrutinib, a covalent inhibitor of BTK,34 were incubated at a concentration of 1 mM with β-mercaptoethanol (βME, 600 mM). LC-MS was used to monitor the reactions over time and βME-inhibitor adduct formation was confirmed by MS. Fitting the time-dependent adduct formation curves to pseudo-first-order reaction kinetics and subsequently calculating to the second-order reaction rate constant kchem confirmed that all seven covalent compounds (3–9) have intrinsic reactivities in the optimal range of 10−3 to 10−4 M−1 s−1 (Fig. 3c). Using KinTek Global Kinetic Explorer, we modelled covalent reactions with 10 nM of protein (PI3Kα), 100 nM of the indicated compound and 7 mM of intracellular free sulfhydryl. The latter has been reported to be mostly intracellular glutathione (GSH).59 Experimentally determined Ki and kinact values (see Table 1) were used to model each compound's reaction kinetics. The predicted >90% covalent target engagement for 9 was reached at <10 min, while compounds with a lower kinact/Ki values lagged behind (Fig. 3d).
Off-target reactions with free sulfhydryls were predicted to be negligible for all modelled compounds. Model calculations with elevated kchem values exemplify rapid off-target engagement with cellular sulfhydryls for kchem values >10−3 M−1 s−1 (Fig. 3e). The presented modeling with empirically determined compound parameters (kchem, Ki and kinact) illustrate clearly that full target engagement must be achieved with warheads with moderate intrinsic reactivity to avoid off-target reactions, which lead to neo-antigen formation and elimination of active covalent compound.
The covalent bonds between inhibitor 9 and the target Cys862 in PI3Kα were confirmed by X-ray crystallography (Fig. 3f). The electron density map of compound 9 showed a defined density over all atoms of the inhibitor. This contrasts with gaps appearing in electron density maps derived for 1 and 2 bound to PI3Kα (Fig. 3g), which point to increased local mobility and reduced ligand–protein interaction. This is further supported by covalent docking studies of compounds 3, 7, 8 and 9 to p110α X-ray co-crystal structure of compound 1 (PDB ID: 7R9V), which suggest an inverse correlation between target engagement (determined by kinact/Ki) and the distance between the linker and the protein surface using Thr856 and Gln859 as main reference points (for docking data and discussion see Fig. S4†).
Lipophilicity and diffusion coefficients
To correct for hydrophobic effects influencing cellular efficacy, we calculated lipophilic ligand efficiency (LipE) values to normalize compound potency relative to lipophilicity.60,61 Plotting pIC50 (negative Log10 of the IC50s for PKB phosphorylation) measured in SKOV3 cells (Table 1, see Methods for details) vs. LogD revealed that 3 to 9 have systematically increased cellular potency with a LogD in the range of 1.4 to 3.4 (Fig. 3h and Table 1). LipE calculations showed that compounds 3, 4, 8 and 9 pass the standard threshold value of ≥5,62 desired for high-quality chemical probes. When comparing covalent target engagement efficiency (kinact/Ki) and LipE, compound 9 stands out as the preferred covalent PI3Kα inhibitor of the series with favorable physicochemical properties (Fig. 3i).
To investigate whether this translates to faster and more efficient target engagement intracellularly, we utilized a NanoBRET tracer displacement assay.63 HEK293 cells transfected with a Nanoluc-p110α fusion construct were incubated with a cell-permeable NanoBRET target engagement tracer (200 nM Tracer K-3).52 In the presence of furimazine, a strong BRET signal was detected, which decreased over time after compound was added extracellularly to displace the tracer from Nanoluc-p110α. The BRET signal decay was observed after compound addition in a time- and concentration-dependent fashion (Fig. 3l). An equilibrium was reached at ca. 20 min for reversible inhibitors (9r and BYL719), while typical two-step binding kinetics were observed for all tested covalent compounds (CNX1351, 1, 2, 6, 7, 9). After diffusion into the cells and reversible target engagement, covalent bond formation with PI3Kα proceeds. The overall rate of this process is a convolution of on-target reactivity and rate of diffusion across the plasma membrane. A clear trend for faster tracer displacement was therefore observed for compounds with high kinact/Ki (>20 × 10−5 nM−1 s−1, 1, 2, 9), as compared to compounds with low kinact/Ki (<10 × 10−5 nM−1 s−1, 6, 7 and CNX1351). To deconvolute on-target reactivity and diffusion rates, kinetic nanoBRET data were fitted with a diffusion/tracer-displacement/reaction model using KinTek Global Kinetic Explorer. The emerging diffusion coefficients were normalized to the one of BYL719 to avoid distortion of comparisons by cellular volumes (see Methods for details).
CNX1351, 1 and 2 displayed slower cellular diffusion than BYL719; 6, 7r and 7 were comparable to BYL719, while 8, 9r and 9 showed 2- to 3-fold increase in cellular permeability (Fig. 3j). While the large differences in the diffusion coefficients between 1, 2 and 7, 8, 9 (up to 28×) can be explained by altered lipophilicities, 7 and 9 possess similar physicochemical traits, but 9 permeates ca. 2.8× faster into cells as compared to 7. To investigate if chameleon-like effects64 could explain this difference, we performed computational conformational searches for 1, 7 and 9 in water and octanol. Representative conformers of 1, 7 and 9 displayed significant conformational changes according to the polarity of the solvent (i.e., extended hydrophilic conformations in water, and compacted intramolecularly-bonded conformations in octanol, Fig. S6†). Nonetheless, no significant reduction of 3D PSA computed for each representative conformer of 7 and 9 (see Table S6†) was observed when transitioning from water to octanol. This shows that the computational conformational sampling herein performed doesn't predict a strong tendency of 7 or 9 for chameleon-like effects.
Taken together, these data demonstrate that compound 9 is a potent covalent PI3Kα inhibitor with a superior rate of intracellular covalent target engagement due to its high cellular permeability (Fig. 3k), and excellent cellular potency. Additionally, the biochemical and cellular assays presented here provide a workflow for covalent chemical probe characterization to meet high-quality standards towards improved cellular activity based on experimentally determined parameters.
Potency of covalent PI3Kα chemical probes in cancer cells
Advantages of covalent inhibition include exceptional efficiency, specificity and extended pharmacodynamic effects, which may provide a competitive edge over reversible inhibition of PI3Kα. To test this, the potential to abrogate growth of cancer cells was determined for covalent compound 9 and the three reversible inhibitors 9r, BYL719 (alpelisib) and GDC-0077 (inavolisib, currently in phase III). For GDC-0077 it was recently claimed that this compound also has the unique ability to degrade mutant PI3Kα.65The growth rate inhibition (GR) method and associated metric for potency, GR50,66 was used to quantify anti-proliferative activity in a panel of cancer cell lines with PI3K pathway activation. After 72 h incubation, we observed exceptional sensitivity to 9 in PI3Kα-mutant cell lines, yielding 2- to 273-fold more potent growth inhibition than reference compounds BYL719 and GDC-0077 (Fig. 4a and Table S7†). Notably, 9 was also 6- to 70-fold more potent than the reversible analogue 9r, indicating that the covalent engagement of 9 with PI3Kα is the main driver to significantly improve its anti-proliferative potential. Growth rate inhibition was related to G1 arrest in response to PI3K inhibition (Fig. S7†). In PTEN-deficient cell lines, 9 was only modestly more potent than 9r, with high concentrations required to sustain growth inhibition. Furthermore, BYL719 and GDC-0077 were ineffective in the tested concentration range, suggesting pan-PI3K inhibition is necessary for efficacy in these cell lines (Fig. S8† for dose–response curves and Table S7† for calculated GR50 values).
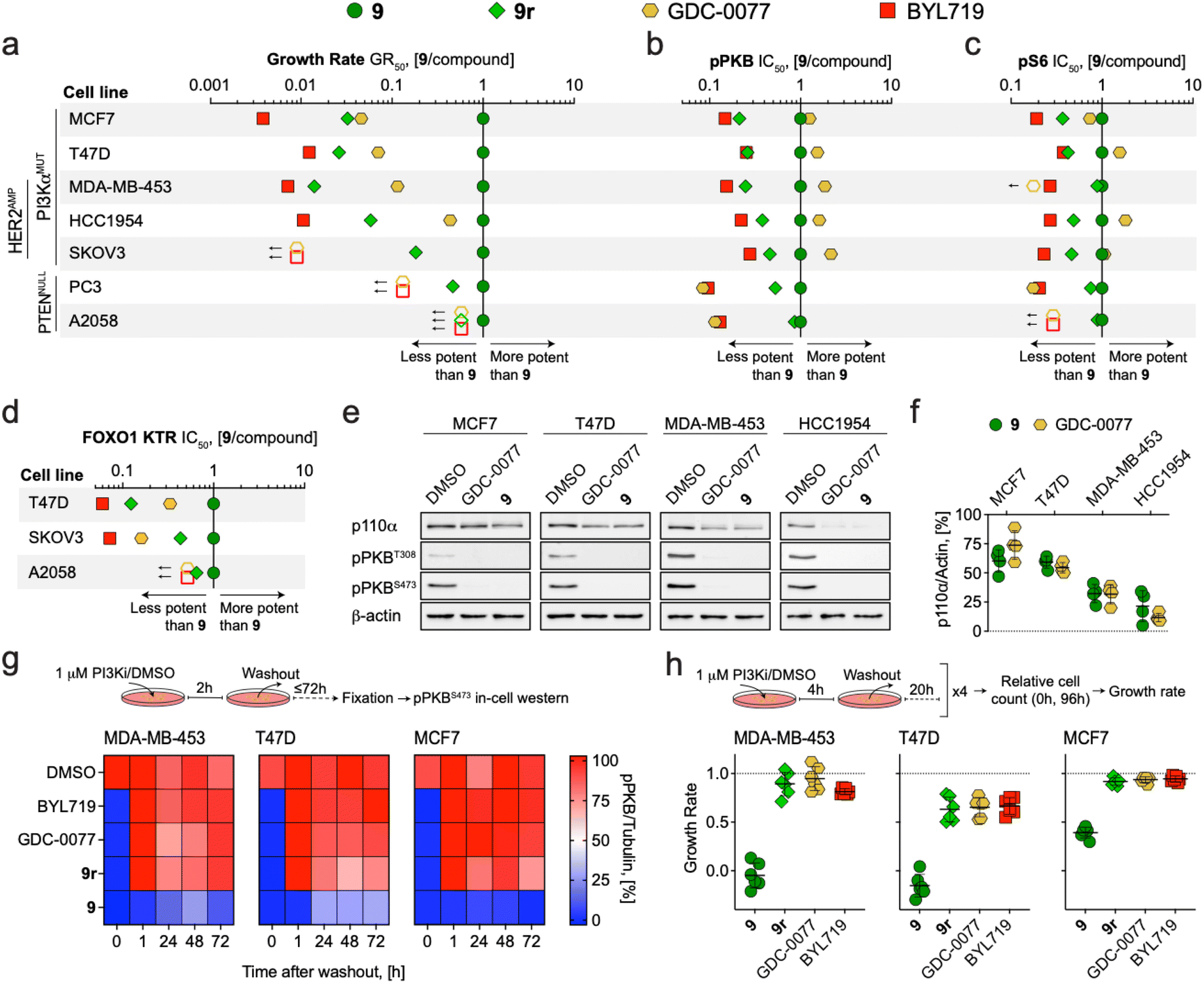 | ||
| Fig. 4 (a–d) Dot plots comparing the potency of covalent compound 9 to reversible PI3Kα inhibitors in PI3Kα or PTEN mutant cancer cell lines. (a) GR50 ratios for cell growth determined after 72 h incubation (see Table S6† for GR50 values). (b) IC50 ratios for PKB phosphorylation (pPKB, Ser473) determined after 2 h incubation (see Table S8† for pPKB IC50 values). (c) IC50 ratios for ribosomal protein S6 phosphorylation (pS6, Ser235/Ser236) determined after 2 h incubation. See Table S9† for calculated pS6 IC50 values. (d) IC50 ratios for FOXO1 KTR translocation (cytosolic/nuclear ratio) determined after 2 h incubation. Symbols show the ratio of GR50/IC50 values derived from at least n = 3 independent experiments. Where GR50/IC50 values were extrapolated, ratios were calculated using the highest assay concentration instead and represented as empty symbols (GR50 – 5 μM; pS6 IC50 – 3 μM). See Fig. S8† for dose–response curves. (e) Detection of p110α, phosphorylated PKB (pPKB, Thr308 and Ser473), and β-actin by western blot in cell lines treated with DMSO, 9 (1 μM), or GDC-0077 (1 μM) for 48 h prior to lysis (representative images are shown). (f) Quantification of remaining p110α protein levels relative to DMSO control (n = 4, mean ± SD). (g) Heatmaps showing pPKB (Ser473) levels in cell lines treated with DMSO or PI3Kα inhibitors (1 μM) for 2 h, followed by drug washout (twice for 10 min with fully supplemented growth medium), and further incubation in fully supplemented growth medium for the indicated times. pPKB levels were determined by in-cell western in n = 2 independent experiments (t = 0, inhibitor present). (h) Growth rate of cancer cell lines with mutated PI3Kα in response to intermittent inhibitor exposure (4 h per day followed by washout on 4 consecutive days; n = 2 independent experiments with each in technical triplicates, mean ± SD). | ||
Concentration-dependent inhibition of PI3K signaling was measured in the same cell line panel using in-cell western detection of phosphorylated PKB (pPKB, Ser473) and phosphorylated ribosomal protein S6 (pS6, Ser235/Ser236) after 2 h treatment. In this short-term assay, the differences in potency (IC50) between 9 and reference compounds were less evident (Fig. S8† for dose–response curves and Tables S8 and S9† for calculated IC50 values for pPKB and pS6, respectively). In PI3Kα mutated lines, 9 and GDC-0077 were nearly equipotent and achieved more potent inhibition of pPKB and pS6 than BYL719 and 9r (Fig. 4b and c). However, quantification of FOXO1 KTR translocation after 2 h incubation in 3 of the cancer cell lines highlighted 9 as most potently inhibiting downstream signaling through PKB (Fig. 4d, see Fig. S8† for dose–response curves and Table S10† for calculated FOXO1 KTR IC50 values). Together, these data suggest that covalent targeting of PI3Kα in cancer cell lines expressing mutant forms of PI3Kα significantly improves anti-proliferative activity compared to reversible inhibitors through long-lasting target engagement and persistently efficacious inhibition of downstream pathway signaling.
Both irreversible covalent inhibition and targeted protein degradation can be exploited to achieve a long lasting and durable signaling pathway inhibition. In both settings, target re-synthesis is required to rescue signaling. Degradation of mutated p110α was recently reported for GDC-0032 (taselisib) and GDC-0077, but seems limited to specific breast cancer cell lines, and is modulated through an undefined mechanism.65 We validated this in four PIK3CA mutated breast cancer lines exposed to PI3Kα inhibitors (1 μM for 48 h). In HCC1954 and MDA-MB-453 cells, GDC-0077 and 9 induced a prominent, indistinguishable depletion of p110α, which was insignificant in MCF7 and T47D cells (Fig. 4e and f).
The fact that the potency of GDC-0077 almost approached that of 9 in inhibiting growth of HCC1954 (Fig. 4a), fits with the notion that degradation of p110α can produce an extended pharmacodynamic action comparable to covalent engagement.
To further investigate the duration of action of 9 in comparison to reversible inhibitors, we incubated MDA-MB-453, T47D, and MCF7 breast cancer cells with mutated PI3Kα with inhibitors (1 μM for 2 h), followed by a drug washout, and monitored pPKB levels over time. After exposure to reversible inhibitors, including 9r, PKB phosphorylation recovered within <1 h, whereas 9 led to loss of detectable pPKB for up to 72 h (Fig. 4g). In an assay setup to simulate a peak and trough model of drug levels, cells were exposed to 1 μM of compounds for 4 h daily over 4 days and relative growth rates were measured as previously described.66 Intermittent exposure to 9 prevented (growth rate ≤0) or reduced (growth rate ≤0.5) proliferation of PI3Kα mutant cell lines. This was in contrast to reversible inhibitors, which showed very limited efficacy (Fig. 4h). The same transient treatment with 9 yielded negligible growth rate inhibition in PTEN-deficient cell lines (Fig. S7†), consistent with growth dependencies on other class IA PI3K isoform. Altogether, these results highlight that covalent inhibitors achieve a unique long-term pathway inhibition over the course of several days following a single administration. This decoupling of inhibitor exposure from inhibitor action may offer advantages for a highly selective targeting of PI3Kα in oncology.
Covalent PI3Kα chemical probes deconvolute receptor-PI3K isoform coupling
The ability to obtain pure, isoform-specific PI3Kα inhibition by exposure with 9 followed by drug washout (here referred to as 9-wo) was exploited to deconvolute class IA PI3K isoform coupling to upstream extracellular receptors in serum-starved breast MCF7 and ovarian SKOV3 cancer cell lines stimulated with various receptor ligands (Fig. 5). The PI3Kβ-selective inhibitor TGX221 was used after 9-wo, before ligand addition, to evaluate residual contribution of PI3Kβ to PI3K signaling. Ligands tested were EGF (engaging EGFR family RTKs), insulin (activating insulin RTK), and CXCL12 (triggering the CXCR4 and CXCR7 GPCRs). Signaling contribution from different class IA isoforms was calculated as remaining area under the pPKB (Ser473) signal curve (Fig. 5a). In the absence of inhibitors, addition of ligands resulted always in a time-dependent, transient increase in PKB phosphorylation (Fig. 5b and d).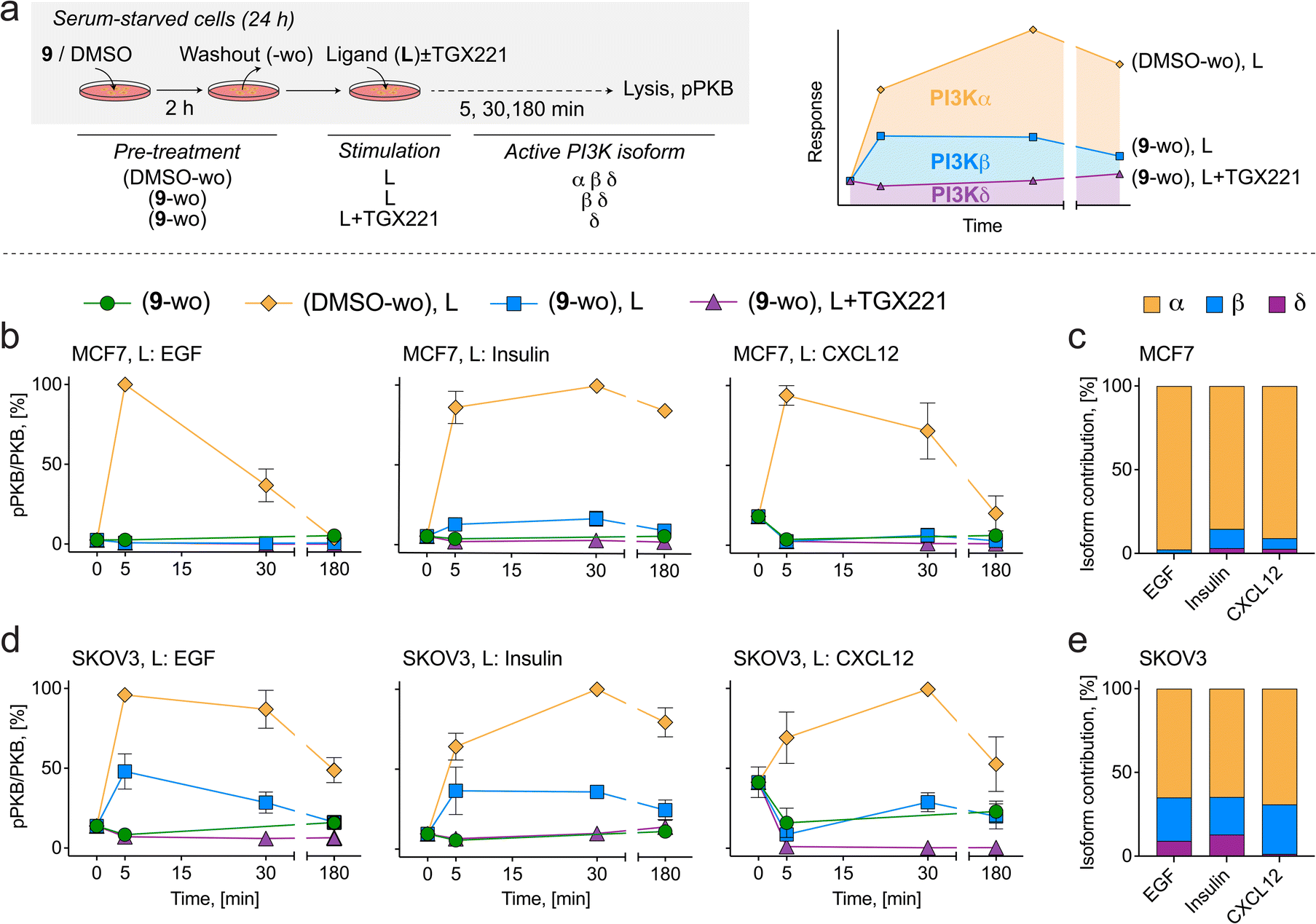 | ||
| Fig. 5 (a) Schematic diagram of PI3K pathway stimulation in cancer cell lines to deconvolute PI3Kα isoform contribution downstream of membrane receptors. Serum starved cells (24 h) were incubated with 1 μM 9 or DMSO for 2 h followed by washout (labelled 9-wo or DMSO-wo). Receptor ligands (L) were then added to the medium in the absence or presence of the PI3Kβ-selective inhibitor TGX221 (1 μM) for the indicated times (5, 30, 180 min) prior to lysis. Total PKB and pPKB (Ser473) levels were measured by western blot (Fig. S9†). All treatments and media changes were carried out using serum-free medium. Depicted in the schematic are the active and inactive (red line-crossed) PI3K isoforms resulting from inhibitor treatments. (b) PI3K signaling in MCF7 cells treated with inhibitors and stimulated with 10 ng mL−1 EGF (left), 10 μg mL−1 insulin (center) or 50 ng mL−1 CXCL12 (right). (c) Quantification of PI3K isoform contribution (area under curve) to PI3K signaling in response to EGF, insulin and CXCL12 in MCF7 cells. (d) PI3K signaling in SKOV3 cells treated with inhibitors and stimulated with 10 ng mL−1 EGF (left), 10 μg mL−1 insulin (center) or 50 ng mL−1 CXCL12 (right). (e) Quantification of PI3K isoform contribution (area under curve) to PI3K signaling in response to EGF, insulin and CXCL12 in SKOV3 cells. | ||
In MCF7 cells, PI3K signaling in response to EGF, insulin and CXCL12 was inhibited by 98%, 85%, and 91% after 9-wo, respectively, indicating that PI3Kα operates as the dominant PI3K isoform in this cell line (Fig. 5c). Addition of TGX221 suppressed the remaining PI3K signaling by 2%, 12% and 6%, respectively, suggesting a minor role for PI3Kβ and a negligible contribution of the PI3Kδ isoform.
In SKOV3 cells, PI3Kα relayed 65% of the PI3K signal triggered by EGF and insulin, and 69% in response to CXCL12, while the output from PI3Kβ accounted for 26%, 22% and 30%, respectively (Fig. 5e). By exclusion, the remaining PI3K signaling amounting to 9% for EGF and 13% for insulin can be attributed to the last class IA member, PI3Kδ, while the residual signaling in response to CXCL12 excludes a significant role of PI3Kδ downstream of CXCR4/7 receptors.
These findings corroborate previous work showing that PI3Kα is the dominant isoform relaying PI3K signaling downstream of ligands that activate RTKs. The major contribution of PI3Kα to GPCR-triggered PKB activation was, however, not anticipated, as a direct GPCR input would rather involve direct PI3Kβ activation. This result must be considered in the context of oncogenic PI3Kα mutations which may diminish specificity of the signal relayed by this isoform.67 Alternatively, PI3Kα activity could be triggered by GPCR-mediated transactivation of HER2.68
In parallel, the effect of specific PI3Kα inhibition on MAPK signaling in response to the above stimuli was investigated, since compensatory upregulation of MAPK signaling in response to PI3K inhibition has been reported in cancer cell lines with overexpressed HER2 or KRAS.69,70 In MCF7 cells, MAPK activation in response to EGF, insulin and CXCL12 was slightly reduced by 9-wo, indicating the existence of a feedforward loop where PI3Kα promotes MAPK signaling (Fig. S10†). In contrast, in SKOV3 cells, MAPK signaling was strongly activated in response to PI3Kα inhibition by 9-wo alone, presumably through compensatory activation of HER2 via the suppression of the mTORC1/Grb10 feedback loop.71
Altogether, this work illustrates a novel, straightforward approach to quantify isoform specific signaling through PI3Kα in response to external stimuli with the use of covalent PI3Kα inhibitors. Notably, it also shows that the impact of PI3Kα inactivation is context-specific and suggests that contributions from other class IA isoforms and prominent upregulation of MAPK signaling may dictate worse response to PI3Kα-selective targeting compounds, particularly in the context of oncology.
Conclusions
Compounds optimized to covalently target a single enzyme provide potent and selective inhibition. We previously established a workflow to design proximity-optimized covalent tool compounds targeting a remote cysteine in PI3Kα.43 Here, we present a novel set of cell-permeable, covalent PI3Kα targeting small molecule chemical probes developed by varying linker composition with the aim of modulating physicochemical properties, including polarity, lipophilicity and flexibility, and tailoring interactions with the protein surface. Through a series of structured biochemical and cell-based assays, we validated the covalent mode of PI3Kα inhibition engaging Cys862 and determined the compounds' cellular activity profiles. The covalent chemical probes generated from this work are characterized by low intrinsic reactivity and high reaction rates for covalent bond formation with Cys862 of PI3Kα, leading to rapid and efficacious on-target engagement with negligible off-target reactions. This has also been confirmed testing 9's interaction in a kinase panel (KinomeScan, Fig. 6), where 9 showed insignificant binding to protein kinases, except for minor interactions with the JAK1 JH2 domain-pseudokinase and CSF1R (autoinhibited). | ||
| Fig. 6 Interactions of compound 9 at 1 μM with protein and lipid kinases visualized as kinome TREEspot™ plot. Kinases that bind to 9 are marked by red circles (larger circles indicate higher-affinity; see free kinase as % of control). Kinases are organized according to phylogeny and kinase families, including atypical kinases, lipid kinases, mutated kinases and some pathogen-derived kinases: AGC: PKA, PKG and PKC kinases; CAMK: calcium-calmodulin-dependent protein kinases; CK1: casein kinase 1; CMGC: CDK, MAPK, GSK3, CLK family kinases; STE: homologs of yeast sterile 7, sterile 11, sterile 20 kinases; TK: tyrosine kinases; TKL: tyrosine kinase-like. Precise% values for 9 are in Table S11,† together with previously reported kinase interaction data for 1 and BYL719 (alpelisib),43 PQR514,44 PQR309, GDC0980, and PKI-057.72 Extended TREEspot™ in Fig. S11;† selectivity scores are depicted in Table S12.† | ||
Together with the improved cellular diffusion, this contributes to the exceptional potency of compound 9 in cellular assays measuring PI3K pathway activation and cell proliferation. Furthermore, with the aid of compound 9, we were able to demonstrate that, in the case of PI3Kα targeting, prolonged target engagement and durable pathway inhibition inherently result in superior anti-cancer cell activity relative exceeding the action of reversible inhibitors.
The small molecule chemical probes generated in this study have unprecedented pharmacological properties for PI3Kα inhibition. Due to the high affinity ATP-pocket binding module, all class I PI3K isoforms can be inhibited reversibly in vitro, but only specific inhibition of PI3Kα remains after removal of unbound compound by washout. Under these assay conditions, the establishment of PI3Kα isoform signaling is simple and absolute, and provides quantitative links between ligated membrane receptors and PI3K isoform activation.
When extrapolating the above findings to a potential in vivo pharmacology, prolonged and specific inhibition of PI3Kα will persist until protein re-synthesis, and inhibition of other isoforms would be relieved as unreacted compound undergoes clearance. Therefore, therapeutic covalent PI3Kα inhibitors have the potential to minimize adverse effects associated with reversible pan-PI3K inhibitors, and pave the way for new PI3Kα selective inhibitors for targeted cancer therapy.
Methods
Compound synthesis and characterization
The final compounds were synthesized in a purity higher than 95%, determined by high performance liquid chromatography (HPLC) analyses on an Ultimate 3000SD System from ThermoFisher. 1H, 19F and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer. NMR spectra were obtained in deuterated solvents, namely CDCl3 or (CD3)2SO. The chemical shift (δ values) are reported in ppm and corrected to the signal of the deuterated solvents [7.26 ppm (1H NMR) and 77.16 ppm (13C NMR) for CDCl3; and 2.50 ppm (1H NMR) and 39.52 ppm (13C NMR) for (CD3)2SO]. 19F NMR spectra are calibrated relative to CFCl3 (δ = 0 ppm) as external standard. For detailed methodology see ESI.†Inhibitor binding and determination of dissociation and kinetic constants by TR-FRET
Time-Resolved Förster's Resonance Energy Transfer (TR-FRET) assays were performed as previously described.73 Briefly, a kinase–antibody–tracer complex was formed by combining His6-tagged p110α recombinant protein (5 nM, PV4789, Life Technologies), biotinylated anti-His6-tag antibody (2 nM, PV6089), europium-labeled streptavidin (2 nM, PV5899) and an AlexaFluor647-labelled kinase Tracer (20 nM, PV6087, Tracer 314) in TR-FRET assay buffer (50 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) pH 7.5, 10 mM MgCl2, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 0.01% (v/v) Brij-35). DMSO (controls) and compounds were rapidly dispensed using an I.DOT One dispenser (Dispendix). TR-FRET was measured on a Synergy Neo2 plate reader (BioTek instruments; emission filters: 665/8 nm and 620/10 nm; excitation at 330/80 nm using a xenon flash lamp; 100 μs delay, 200 μs data collection; 37 °C; dichroic mirror 400 nm).The dissociation constants (Ki) and the rate of covalent bond formation (kinact) were obtained by globally fitting the time- and concentration-dependent changes in FRET emission ratios over time (Fig. S5†) using KinTek Global Kinetic Explorer Software.74,75 For further details and data analysis, see ESI.†
Determination of cellular PKB/Akt and ribosomal protein S6 phosphorylation
Phosphorylation of PKB/Akt and ribosomal protein S6 were measured using in-cell western (ICW) assays as previously described.73 Briefly, cells seeded into 96-well CellCarrier black plates with optically clear bottom (PerkinElmer) were exposed to inhibitors for 2 h unless otherwise stated, fixed with 4% paraformaldehyde (PFA) in PBS, permeabilized and blocked with 5% goat serum, 1% BSA, 0.1% Triton X-100 in PBS for 30 min at room temperature, and stained with rabbit monoclonal anti-phospho-Ser473 PKB/Akt (Cell Signaling Technology, #4058) or anti-phospho-Ser235/236 S6 ribosomal protein (Cell Signaling Technology, #4856) and anti-α-tubulin (Sigma, #T9026) antibodies overnight at 4 °C. Secondary antibodies (IRDye680-conjugated goat anti-mouse [LICOR, #926-68070] and IRDye800-conjugated goat anti-rabbit [LICOR, #926-32211]) were added and incubated for 90 min with shaking in the dark. Fluorescence was recorded on an Odyssey CLx infrared imaging scanner (LICOR). For additional experimental details and data analysis, refer to the ESI.†NanoBRET target engagement and tracer displacement assays
NanoBRET assays were performed as described previously.43 In brief, expression vectors encoding Nanoluc fused the N-terminus of class IA PI3K catalytic subunit isoforms and a PI3Kα mutant were purchased from Promega (PI3Kα wild type, #NV3901; PI3Kα C862S; custom vector; PI3Kβ, #NV4011; and PI3Kδ, #NV4021). Together with a vector encoding the regulatory p85α subunit (Promega, #NV4031), 2 × 106 HEK293 cells were transiently transfected (JetPEI Polyplus transfection, #101B-010N) with a total of 10 μg plasmid DNA and a mass ratio of regulatory:catalytic subunit of 10:1. For tracer displacement assays, cells were incubated with Tracer K-3 (200 nM, Promega, #N2602) for 20 min before dispensing inhibitors using an I.DOT One dispenser (Dispendix). Immediately after dispensing, BRET measurements were performed. Filtered luminescence was measured sequentially on a Berthold Mithras2 LB 943 Plate Reader luminometer at 25 °C equipped with 460 nm BP filter (donor) and 600 nm LP filter (acceptor), using 0.5 s integration time for the duration of the assay. For target engagement assays, cells were incubated with inhibitors (3 μM) for 2 h. After incubation, the medium containing inhibitors was gently aspirated and cells were washed twice with inhibitor-free Opti-MEM (Gibco, #11058021) (10 min at 37 °C and 5% CO2) and Tracer K-3 (200 nM) was dispensed using an I.DOT One dispenser (Dispendix). BRET measurements were performed as described above. For further details on data analysis and experimental setup, see ESI.†
FOXO1 kinase translocation reporter
Cell lines (A2058, SKOV3, T47D) stably expressing a FOXO1-Clover fusion protein56 were seeded in 96-well CellCarrier black plates with optically clear bottom (PerkinElmer) the day before inhibitor treatment. To generate dose–response curves, cells were exposed to compounds for 1 h unless otherwise stated, fixed with 4% PFA for 30 min at room temperature, permeabilized and stained with Hoechst33324 (10 μg mL−1) diluted in 1% BSA, 0.1% Triton-X in PBS for 30 min at room temperature in the dark. Fluorescent microscopy images were acquired on an Operetta high content analysis system (PerkinElmer) and batch analyzed using Cell Profiler version 4.2.4 (ref. 76) (Broad Institute, http://www.cellprofiler.org). Briefly, nuclei were identified using the nuclear stain fluorescence and cells were identified by extending the nuclear segmentation to create a ring around the nucleus based on the FOXO1 Clover reporter fluorescence (secondary object). Subsequently, the nuclear region was subtracted from the cellular region to give a cytosolic ring (“cytoring”). The average ratio of the median fluorescence intensity of the cytoring/nucleus was calculated for each inhibitor concentration or DMSO control well and used to quantify FOXO1 localization.Live cell imaging studies were performed in phenol red-free medium with 2% FCS and nuclei were stained with SiR Hoechst (http://spirochrome.com) for 1 h prior to imaging using a Leica DMi8 microscope (Leica) with a stage top incubator maintained at 37 °C, and in 95% air, 5% CO2. Cells were exposed to vehicle or compounds (3 μM) for 2 h followed by washout and imaging was continued for a further 10 hours. Cells were imaged every minute until 1 h following washout and at later time points every 30 min using a 20× HC Plan-Apo objective (N.A. 0.8), and a GFP filter cube (excitation band, 470/40 nm; emission band, 525/50 nm) and a Cy5 filter cube (excitation band, 620/60 nm; emission band, 700/72 nm). Image analysis was performed as above. Further details on cell line generation, image acquisition and data analysis are provided in ESI.†
Compound washout experiments
For assays with timed removal of compounds, addition of compounds was carried out at the indicated final concentrations and incubated for 2 h unless otherwise stated. Medium containing compounds was gently aspirated and cells were washed twice with inhibitor-free medium (10 min at 37 °C and 5% CO2) and further incubated for the indicated times. Values at t = 0 represent measurements before washout (compounds present).Intrinsic warhead reactivity (kchem) determination using β-mercaptoethanol (βME)
Compounds (1 mM) were reacted with βME (600 mM) at 37 °C in a 3:1 DMSO/PBS mixture. Compound loss and compound + βME adduct formation was monitored every 20 min for the first three hours and every hour for up to eight hours. Compound + βME adduct formation was confirmed by MALDI-ToF mass spectrometry obtained on a Voyager-DeTM Pro (measured in m/z, see ESI† for MALDI-MS spectra). Percentages of compound against compound + βME adduct was determined by measuring the area under the curve in HPLC (Ultimate 3000SD System, ThermoFisher) chromatograms. For further details on kchem calculations, see ESI.†
Modeling of on- and off-target reactions
The modeling of on- and off-target reactions were carried out with KinTek Global Kinetic Explorer modeling software. The reaction parameters were determined experimentally by TR-FRET for each compound. For details on the enzymatic reaction model and parameters, see ESI.†Crystallography
Crystals were generated as described previously.43 Initial apo p110α crystals were obtained from a grid of 1 μL hanging drops containing 0.5 μL of protein at 5.8 mg mL−1 (in 50 mM Tris pH 8.0, 100 mM NaCl, 2% EG, 1 mM TCEP pH 7.5) mixed 1:1 with 0.5 μL reservoir (poly(ethylene glycol)6000 (PEG6000) 6–12%, 0.6 M sodium formate, 0.1 M N-cyclohexyl-2-aminoethanesulfonic acid (CHES) pH 9.1–9.7, 5 mM TCEP pH 7.5) grown for several days at 18 °C. For co-crystallization of 9, protein at 5.8 mg mL−1 was initially incubated for 20 hours on ice with a 2-fold molar excess of inhibitor. Crystals collected for diffraction were obtained from 1 μL hanging drops containing 0.5 μL of inhibitor-bound protein mixed with 0.4 μL reservoir (6–10% PEG6000, 0.6 M Na formate, 0.1 M CHES pH 9.5, 5 mM TCEP pH 7.5) and 0.1 μL of 1/1000 diluted microseeds crushed from a drop of the original apo crystals. Co-crystals were flash-frozen in liquid nitrogen after transferring to 1 μL of reservoir solution containing 25% (v/v) glycerol as cryoprotectant. Diffraction data for PI3K crystals were collected at beamline 08ID-1 of the Canadian Light Source. Data was collected at 13.0 keV (CLS) for 9. Data were processed using XDS.77 Phases were initially obtained by molecular replacement using Phaser78 with the structure of truncated PI3Kα (PDB ID: 4TUU).79 Iterative model building and refinement were performed in COOT80 and phenix.refine.81 Refinement was carried out with rigid body refinement, followed by translation/libration/screw B-factor and xyz refinement. The final model was verified in Molprobity82 for the absence of both Ramachandran and rotamer outliers. The structure of 9 had 1.0% Ramachandran outliers and 0.6% rotamer outliers. Data collection and refinement statistics are shown in Table S3.†
Viability assay and growth rate calculations
Cells seeded in 96-well CellCarrier black plates (PerkinElmer) in growth medium supplemented with 10% FCS and 2 mM L-glutamine were allowed to adhere overnight (37 °C, 5% CO2) before compounds were added to the wells. Relative live cells numbers were measured using the resazurin assay using 0.3 mg mL−1 resazurin solution prepared from resazurin sodium salt (Sigma, #R7017) dissolved in Dulbecco's PBS. Cell numbers were measured at the time of inhibitor addition and at the end of the treatment period by adding resazurin solution (10% of cell culture medium volume) and incubating for 4 hours (37 °C, 5% CO2). Fluorescence (565/590 nm excitation/emission) was recorded on a Synergy 4 plate reader (BioTek). Background fluorescence values (medium only) were subtracted from raw test values, and growth rates (GR) and GR50 values were calculated as previously described.66 For detailed experimental method and data analysis see ESI.†Immunodetection by western blot
Whole cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes (see ESI†). Primary antibodies against pSer473-PKB/Akt (Cell Signaling Technology, #4058), pThr308-PKB/Akt (Cell Signaling Technology, #9275), pan-PKB/Akt (Cell Signaling Technology, #2920), p44/42 MAPK (Erk1/2) (Cell Signaling Technology, #4695), pThr202/Tyr204-p44/42 MAPK (Erk1/2) (Cell Signaling Technology, #4370), β-actin (Sigma, #A5441) and p110α U3A83 were incubated overnight. Membranes were probed with HRP-conjugated secondary antibodies (Sigma) and imaged using Immobilon western HRP substrate (Millipore) on a Fusion FX (Vilber Lourmat) imaging system. Levels of phosphorylated and total protein signals were quantified using ImageJ.Data availability
The coordinates of compound 9 covalently bound to the PI3Kα catalytic subunit p110α have been deposited at https://www.pdb.org and https://www.rcsb.org with PDB ID code 8TWY.Author contributions
Authors have contributed as follows to conceptualization: TAC, LR, CB, VB, TB, MPW; data curation: LB, TAC, EK, JEB, CB, MPW; formal analysis: all authors; funding acquisition: JEB, MPW; investigation: all authors; methodology: LB, TAC, EK, RS, JEB, CB, MPW; project administration: MPW; modelling and equations: LB, EK, VB, MPW; resources: MG, JEB, MPW; supervision: LR, MG, JEB, CB, MPW; validation: LB, EK, LR, JEB, MPW; visualization: LB, TAC, EK, LR, IBB, JEB, CB, MPW; writing – original draft: LB, TAC, LR, CB, MPW; writing – review & editing: LB, TAC, JEB, CB, MPW.Conflicts of interest
MPW is founder and shareholder of Akylox Therapeutics ApS.Acknowledgements
This work was supported by grants from the Swiss National Science Foundation (SNSF) 200021_204602, 310030_189065, 316030_198526 and 320030-231361, from Innosuisse 108.486 IP-LS, from the Swiss Cancer League KFS-5442-08-2021, the Stiftung für Krebsbekämpfung grant 341 and Novartis Foundation 14B095 (to MPW), and the Peter and Traudl Engelhorn Foundation (to RS). JEB was supported by a Michael Smith Foundation for Health Research (MSFHR) Scholar award (17686), and a Cancer Research Society operating grant (CRS-1052949), and MG was supported by EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant 875510). The authors thank the Department of Biomedicine Microscopy Core Facility and in particular Thomas Bürglin for help with the establishment of the FOXO1-KTR assay, Lalita Oparija-Rogenmozere for advice and proof-reading, and Paul Hebeisen for discussions of synthetic chemistry. X-ray crystallography research described in this paper was performed using beamline CMCF-ID at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.References
- J. Blagg and P. Workman, Choose and Use Your Chemical Probe Wisely to Explore Cancer Biology, Cancer Cell, 2017, 32, 9–25, DOI:10.1016/j.ccell.2017.06.005.
- V. Vu, M. M. Szewczyk, D. Y. Nie, C. H. Arrowsmith and D. Barsyte-Lovejoy, Validating Small Molecule Chemical Probes for Biological Discovery, Annu. Rev. Biochem., 2022, 91, 61–87, DOI:10.1146/annurev-biochem-032620-105344.
- R. M. Garbaccio and E. R. Parmee, The Impact of Chemical Probes in Drug Discovery: A Pharmaceutical Industry Perspective, Cell Chem. Biol., 2016, 23, 10–17, DOI:10.1016/j.chembiol.2015.11.011.
- M. E. Bunnage, E. L. Chekler and L. H. Jones, Target validation using chemical probes, Nat. Chem. Biol., 2013, 9, 195–199, DOI:10.1038/nchembio.1197.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, The promise and peril of chemical probes, Nat. Chem. Biol., 2015, 11, 536–541, DOI:10.1038/nchembio.1867.
- J. Sterling, J. R. Baker, A. McCluskey and L. Munoz, Systematic literature review reveals suboptimal use of chemical probes in cell-based biomedical research, Nat. Commun., 2023, 14, 3228, DOI:10.1038/s41467-023-38952-1.
- M. M. Mader, J. Rudolph, I. V. Hartung, D. Uehling, P. Workman and W. Zuercher, Which Small Molecule? Selecting Chemical Probes for Use in Cancer Research and Target Validation, Cancer Discovery, 2023, 13, 2150–2165, DOI:10.1158/2159-8290.CD-23-0536.
- M. P. Wymann, M. Zvelebil and M. Laffargue, Phosphoinositide 3-kinase signalling-which way to target, Trends Pharmacol. Sci., 2003, 24, 366–376, DOI:10.1016/S0165-6147(03)00163-9.
- A. Berndt, S. Miller, O. Williams, D. D. Le, B. T. Houseman, J. I. Pacold, F. Gorrec, W.-C. Hon, P. Ren and Y. Liu, The p110δ structure: mechanisms for selectivity and potency of new PI (3) K inhibitors, Nat. Chem. Biol., 2010, 6, 117–124, DOI:10.1038/nchembio.293.
- B. Vanhaesebroeck, M. W. D. Perry, J. R. Brown, F. André and K. Okkenhaug, PI3K inhibitors are finally coming of age, Nat. Rev. Drug Discovery, 2021, 20, 741–769, DOI:10.1038/s41573-021-00209-1.
- L. R. Stephens, K. T. Hughes and R. F. Irvine, Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils, Nature, 1991, 351, 33–39, DOI:10.1038/351033a0.
- P. T. Hawkins, T. R. Jackson and L. R. Stephens, Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase, Nature, 1992, 358, 157–159, DOI:10.1038/358157a0.
- M. P. Wymann and C. Schultz, The chemical biology of phosphoinositide 3-kinases, Chembiochem, 2012, 13, 2022–2035, DOI:10.1002/cbic.201200089.
- M. P. Wymann and R. Schneiter, Lipid signalling in disease, Nat. Rev. Mol. Cell Biol., 2008, 9, 162–176, DOI:10.1038/nrm2335.
- D. A. Fruman, H. Chiu, B. D. Hopkins, S. Bagrodia, L. C. Cantley and R. T. Abraham, The PI3K Pathway in Human Disease, Cell, 2017, 170, 605–635, DOI:10.1016/j.cell.2017.07.029.
- J. J. Zhao, H. Cheng, S. Jia, L. Wang, O. V. Gjoerup, A. Mikami and T. M. Roberts, The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16296–16300, DOI:10.1073/pnas.0607899103.
- Z. A. Knight, B. Gonzalez, M. E. Feldman, E. R. Zunder, D. D. Goldenberg, O. Williams, R. Loewith, D. Stokoe, A. Balla, B. Toth, T. Balla, W. A. Weiss, R. L. Williams and K. M. Shokat, A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling, Cell, 2006, 125, 733–747, DOI:10.1016/j.cell.2006.03.035.
- H. Kurosu, T. Maehama, T. Okada, T. Yamamoto, S. Hoshino, Y. Fukui, M. Ui, O. Hazeki and T. Katada, Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide, J. Biol. Chem., 1997, 272, 24252–24256, DOI:10.1074/jbc.272.39.24252.
- S. Sampattavanich, B. Steiert, B. A. Kramer, B. M. Gyori, J. G. Albeck and P. K. Sorger, Encoding Growth Factor Identity in the Temporal Dynamics of FOXO3 under the Combinatorial Control of ERK and AKT Kinases, Cell Syst., 2018, 6, 664–678, DOI:10.1016/j.cels.2018.05.004.
- L. C. Foukas, M. Claret, W. Pearce, K. Okkenhaug, S. Meek, E. Peskett, S. Sancho, A. J. Smith, D. J. Withers and B. Vanhaesebroeck, Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation, Nature, 2006, 441, 366–370, DOI:10.1038/nature04694.
- A. Molinaro, B. Becattini, A. Mazzoli, A. Bleve, L. Radici, I. Maxvall, V. R. Sopasakis, A. Molinaro, F. Bäckhed and G. Solinas, Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kα and PI3Kβ Activities and Is Promoted by RAS, Cell Metab., 2019, 29, 1400–1409, DOI:10.1016/j.cmet.2019.03.010.
- B. Zhang, C. Luk, J. Valadares, C. Aronis and L. C. Foukas, Dominant Role of PI3K p110α over p110β in Insulin and β-Adrenergic Receptor Signalling, Int. J. Mol. Sci., 2021, 22, 12813, DOI:10.3390/ijms222312813.
- Y. Samuels, Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, High frequency of mutations of the PIK3CA gene in human cancers, Science, 2004, 304, 554, DOI:10.1126/science.1096502.
- G. A. R. N. Cancer, J. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. Shaw, B. A. Ozenberger, K. Ellrott, I. Shmulevich, C. Sander and J. M. Stuart, The Cancer Genome Atlas Pan-Cancer analysis project, Nat. Genet., 2013, 45, 1113–1120, DOI:10.1038/ng.2764.
- R. Arafeh and Y. Samuels, PIK3CA in cancer: The past 30 years, Semin. Cancer Biol., 2019, 59, 36–49, DOI:10.1016/j.semcancer.2019.02.002.
- Y. Zhang, P. Kwok-Shing Ng, M. Kucherlapati, F. Chen, Y. Liu, Y. H. Tsang, G. de Velasco, K. J. Jeong, R. Akbani, A. Hadjipanayis, A. Pantazi, C. A. Bristow, E. Lee, H. S. Mahadeshwar, J. Tang, J. Zhang, L. Yang, S. Seth, S. Lee, X. Ren, X. Song, H. Sun, J. Seidman, L. J. Luquette, R. Xi, L. Chin, A. Protopopov, T. F. Westbrook, C. S. Shelley, T. K. Choueiri, M. Ittmann, C. Van Waes, J. N. Weinstein, H. Liang, E. P. Henske, A. K. Godwin, P. J. Park, R. Kucherlapati, K. L. Scott, G. B. Mills, D. J. Kwiatkowski and C. J. Creighton, A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations, Cancer Cell, 2017, 31, 820–832, DOI:10.1016/j.ccell.2017.04.013.
- B. D. Hopkins, C. Pauli, X. Du, D. G. Wang, X. Li, D. Wu, S. C. Amadiume, M. D. Goncalves, C. Hodakoski, M. R. Lundquist, R. Bareja, Y. Ma, E. M. Harris, A. Sboner, H. Beltran, M. A. Rubin, S. Mukherjee and L. C. Cantley, Suppression of insulin feedback enhances the efficacy of PI3K inhibitors, Nature, 2018, 560, 499–503, DOI:10.1038/s41586-018-0343-4.
- F. Janku, T. A. Yap and F. Meric-Bernstam, Targeting the PI3K pathway in cancer: are we making headway, Nat. Rev. Clin. Oncol., 2018, 15, 273–291, DOI:10.1038/nrclinonc.2018.28.
- D. Juric, J. Rodon, J. Tabernero, F. Janku, H. A. Burris, J. H. M. Schellens, M. R. Middleton, J. Berlin, M. Schuler, M. Gil-Martin, H. S. Rugo, R. Seggewiss-Bernhardt, A. Huang, D. Bootle, D. Demanse, L. Blumenstein, C. Coughlin, C. Quadt and J. Baselga, Phosphatidylinositol 3-Kinase α-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study, J. Clin. Oncol., 2018, 36, 1291–1299, DOI:10.1200/JCO.2017.72.7107.
- A. B. Hanker, V. Kaklamani and C. L. Arteaga, Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors, Cancer Discovery, 2019, 9, 482–491, DOI:10.1158/2159-8290.CD-18-1175.
- F. André, E. Ciruelos, G. Rubovszky, M. Campone, S. Loibl, H. S. Rugo, H. Iwata, P. Conte, I. A. Mayer, B. Kaufman, T. Yamashita, Y. S. Lu, K. Inoue, M. Takahashi, Z. Pápai, A. S. Longin, D. Mills, C. Wilke, S. Hirawat and D. Juric, SOLAR-1, S. G. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer, N. Engl. J. Med., 2019, 380, 1929–1940, DOI:10.1056/NEJMoa1813904.
- S. Dent, J. Cortés, Y. H. Im, V. Diéras, N. Harbeck, I. E. Krop, T. R. Wilson, N. Cui, F. Schimmoller, J. Y. Hsu, J. He, M. De Laurentiis, S. Sousa, P. Drullinsky and W. Jacot, Phase III randomized study of taselisib or placebo with fulvestrant in estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced breast cancer: the SANDPIPER trial, Ann. Oncol., 2021, 32, 197–207, DOI:10.1016/j.annonc.2020.10.596.
- P. L. Bedard, M. K. Accordino, A. Cervantes, V. Gambardella, E. P. Hamilton, A. Italiano, D. Juric, K. Kalinsky, I. E. Krop, M. Oliveira, C. Saura, P. Schmid, N. C. Turner, A. Varga, N. Shankar, J. Schutzman, S. Royer-Joo, M. V. Martin and K. L. Jhaveri, Long-term safety of inavolisib (GDC-0077) in an ongoing phase 1/1b study evaluating monotherapy and in combination (combo) with palbociclib and/or endocrine therapy in patients (pts) with PIK3CA-mutated, hormone receptor-positive/HER2-negative (HR+/HER2-) metastatic breast cancer (BC), J. Clin. Oncol., 2022, 40, 1052, DOI:10.1200/JCO.2022.40.16_suppl.1052.
- Z. Pan, H. Scheerens, S. J. Li, B. E. Schultz, P. A. Sprengeler, L. C. Burrill, R. V. Mendonca, M. D. Sweeney, K. C. Scott, P. G. Grothaus, D. A. Jeffery, J. M. Spoerke, L. A. Honigberg, P. R. Young, S. A. Dalrymple and J. T. Palmer, Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase, ChemMedChem, 2007, 2, 58–61, DOI:10.1002/cmdc.200600221.
- P. Ghia, A. Pluta, M. Wach, D. Lysak, T. Kozak, M. Simkovic, P. Kaplan, I. Kraychok, A. Illes, J. de la Serna, S. Dolan, P. Campbell, G. Musuraca, A. Jacob, E. Avery, J. H. Lee, W. Liang, P. Patel, C. Quah and W. Jurczak, ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia, J. Clin. Oncol., 2020, 38, 2849–2861, DOI:10.1200/JCO.19.03355.
- J. R. Brown, B. Eichhorst, P. Hillmen, W. Jurczak, M. Kaźmierczak, N. Lamanna, S. M. O'Brien, C. S. Tam, L. Qiu, K. Zhou, M. Simkovic, J. Mayer, A. Gillespie-Twardy, A. Ferrajoli, P. S. Ganly, R. Weinkove, S. Grosicki, A. Mital, T. Robak, A. Osterborg, H. A. Yimer, T. Salmi, M. D. Wang, L. Fu, J. Li, K. Wu, A. Cohen and M. Shadman, Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia, N. Engl. J. Med., 2023, 388, 319–332, DOI:10.1056/NEJMoa2211582.
- V. A. Miller, V. Hirsh, J. Cadranel, Y. M. Chen, K. Park, S. W. Kim, C. Zhou, W. C. Su, M. Wang, Y. Sun, D. S. Heo, L. Crino, E. H. Tan, T. Y. Chao, M. Shahidi, X. J. Cong, R. M. Lorence and J. C. Yang, Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial, Lancet Oncol., 2012, 13, 528–538, DOI:10.1016/S1470-2045(12)70087-6.
- A. Chan, S. Delaloge, F. A. Holmes, B. Moy, H. Iwata, V. J. Harvey, N. J. Robert, T. Silovski, E. Gokmen, G. von Minckwitz, B. Ejlertsen, S. K. L. Chia, J. Mansi, C. H. Barrios, M. Gnant, M. Buyse, I. Gore, J. Smith, G. Harker, N. Masuda, K. Petrakova, A. G. Zotano, N. Iannotti, G. Rodriguez, P. Tassone, A. Wong, R. Bryce, Y. Ye, B. Yao and M. Martin, ExteNET Study Group Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial, Lancet Oncol., 2016, 17, 367–377, DOI:10.1016/S1470-2045(15)00551-3.
- J. C. Soria, Y. Ohe, J. Vansteenkiste, T. Reungwetwattana, B. Chewaskulyong, K. H. Lee, A. Dechaphunkul, F. Imamura, N. Nogami, T. Kurata, I. Okamoto, C. Zhou, B. C. Cho, Y. Cheng, E. K. Cho, P. J. Voon, D. Planchard, W. C. Su, J. E. Gray, S. M. Lee, R. Hodge, M. Marotti, Y. Rukazenkov and S. S. Ramalingam, FLAURA Investigators Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer, N. Engl. J. Med., 2018, 378, 113–125, DOI:10.1056/NEJMoa1713137.
- Y. L. Wu, Y. Cheng, X. Zhou, K. H. Lee, K. Nakagawa, S. Niho, F. Tsuji, R. Linke, R. Rosell, J. Corral, M. R. Migliorino, A. Pluzanski, E. I. Sbar, T. Wang, J. L. White, S. Nadanaciva, R. Sandin and T. S. Mok, Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial, Lancet Oncol., 2017, 18, 1454–1466, DOI:10.1016/S1470-2045(17)30608-3.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors, J. Med. Chem., 2020, 63, 52–65, DOI:10.1021/acs.jmedchem.9b01180.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer, J. Med. Chem., 2020, 63, 6679–6693, DOI:10.1021/acs.jmedchem.9b02052.
- C. Borsari, E. Keles, J. A. McPhail, A. Schaefer, R. Sriramaratnam, W. Goch, T. Schaefer, M. De Pascale, W. Bal, M. Gstaiger, J. E. Burke and M. P. Wymann, Covalent Proximity Scanning of a Distal Cysteine to Target PI3Kα, J. Am. Chem. Soc., 2022, 144, 6326–6342, DOI:10.1021/jacs.1c13568.
- C. Borsari, D. Rageot, F. Beaufils, T. Bohnacker, E. Keles, I. Buslov, A. Melone, A. M. Sele, P. Hebeisen, D. Fabbro, P. Hillmann and M. P. Wymann, Preclinical Development of PQR514, a Highly Potent PI3K Inhibitor Bearing a Difluoromethyl-Pyrimidine Moiety, ACS Med. Chem. Lett., 2019, 10, 1473–1479, DOI:10.1021/acsmedchemlett.9b00333.
- I. V. Hartung, J. Rudolph, M. M. Mader, M. P. C. Mulder and P. Workman, Expanding Chemical Probe Space: Quality Criteria for Covalent and Degrader Probes, J. Med. Chem., 2023, 66(14), 9297–9312, DOI:10.1021/acs.jmedchem.3c00550.
- P. Furet, V. Guagnano, R. A. Fairhurst, P. Imbach-Weese, I. Bruce, M. Knapp, C. Fritsch, F. Blasco, J. Blanz, R. Aichholz, J. Hamon, D. Fabbro and G. Caravatti, Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation, Bioorg. Med. Chem. Lett., 2013, 23, 3741–3748, DOI:10.1016/j.bmcl.2013.05.007.
- C. O. Ndubaku, T. P. Heffron, S. T. Staben, M. Baumgardner, N. Blaquiere, E. Bradley, R. Bull, S. Do, J. Dotson, D. Dudley, K. A. Edgar, L. S. Friedman, R. Goldsmith, R. A. Heald, A. Kolesnikov, L. Lee, C. Lewis, M. Nannini, J. Nonomiya, J. Pang, S. Price, W. W. Prior, L. Salphati, S. Sideris, J. J. Wallin, L. Wang, B. Wei, D. Sampath and A. G. Olivero, Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity, J. Med. Chem., 2013, 56, 4597–4610, DOI:10.1021/jm4003632.
- E. J. Hanan, M. G. Braun, R. A. Heald, C. MacLeod, C. Chan, S. Clausen, K. A. Edgar, C. Eigenbrot, R. Elliott, N. Endres, L. S. Friedman, E. Gogol, X. H. Gu, R. H. Thibodeau, P. S. Jackson, J. R. Kiefer, J. D. Knight, M. Nannini, R. Narukulla, A. Pace, J. Pang, H. E. Purkey, L. Salphati, D. Sampath, S. Schmidt, S. Sideris, K. Song, S. Sujatha-Bhaskar, M. Ultsch, H. Wallweber, J. Xin, S. Yeap, A. Young, Y. Zhong and S. T. Staben, Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kα, J. Med. Chem., 2022, 65(44), 16589–16621, DOI:10.1021/acs.jmedchem.2c01422.
- W. T. Jauslin, M. Schild, T. Schaefer, C. Borsari, C. Orbegozo, L. Bissegger, S. Zhanybekova, D. Ritz, A. Schmidt, M. Wymann and D. Gillingham, A high affinity pan-PI3K binding module supports selective targeted protein degradation of PI3Kα, Chem. Sci., 2024, 15, 683–691, 10.1039/d3sc04629j.
- M. P. Hall, J. Unch, B. F. Binkowski, M. P. Valley, B. L. Butler, M. G. Wood, P. Otto, K. Zimmerman, G. Vidugiris, T. Machleidt, M. B. Robers, H. A. Benink, C. T. Eggers, M. R. Slater, P. L. Meisenheimer, D. H. Klaubert, F. Fan, L. P. Encell and K. V. Wood, Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate, ACS Chem. Biol., 2012, 7, 1848–1857, DOI:10.1021/cb3002478.
- N. C. Dale, E. K. M. Johnstone, C. W. White and K. D. G. Pfleger, NanoBRET: The Bright Future of Proximity-Based Assays, Front. Bioeng. Biotechnol., 2019, 7, 56, DOI:10.3389/fbioe.2019.00056.
- J. D. Vasta, C. R. Corona, J. Wilkinson, C. A. Zimprich, J. R. Hartnett, M. R. Ingold, K. Zimmerman, T. Machleidt, T. A. Kirkland, K. G. Huwiler, R. F. Ohana, M. Slater, P. Otto, M. Cong, C. I. Wells, B. T. Berger, T. Hanke, C. Glas, K. Ding, D. H. Drewry, K. V. M. Huber, T. M. Willson, S. Knapp, S. Müller, P. L. Meisenheimer, F. Fan, K. V. Wood and M. B. Robers, Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement, Cell Chem. Biol., 2018, 25, 206–214, DOI:10.1016/j.chembiol.2017.10.010.
- S. P. Jackson, S. M. Schoenwaelder, I. Goncalves, W. S. Nesbitt, C. L. Yap, C. E. Wright, V. Kenche, K. E. Anderson, S. M. Dopheide, Y. Yuan, S. A. Sturgeon, H. Prabaharan, P. E. Thompson, G. D. Smith, P. R. Shepherd, N. Daniele, S. Kulkarni, B. Abbott, D. Saylik, C. Jones, L. Lu, S. Giuliano, S. C. Hughan, J. A. Angus, A. D. Robertson and H. H. Salem, PI 3-kinase p110beta: a new target for antithrombotic therapy, Nat. Med., 2005, 11, 507–514, DOI:10.1038/nm1232.
- B. J. Lannutti, S. A. Meadows, S. E. Herman, A. Kashishian, B. Steiner, A. J. Johnson, J. C. Byrd, J. W. Tyner, M. M. Loriaux, M. Deininger, B. J. Druker, K. D. Puri, R. G. Ulrich and N. A. Giese, CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability, Blood, 2011, 117, 591–594, DOI:10.1182/blood-2010-03-275305.
- A. J. Lam, F. St-Pierre, Y. Gong, J. D. Marshall, P. J. Cranfill, M. A. Baird, M. R. McKeown, J. Wiedenmann, M. W. Davidson, M. J. Schnitzer, R. Y. Tsien and M. Z. Lin, Improving FRET dynamic range with bright green and red fluorescent proteins, Nat. Methods, 2012, 9, 1005–1012, DOI:10.1038/nmeth.2171.
- S. M. Gross and P. Rotwein, Akt signaling dynamics in individual cells, J. Cell Sci., 2015, 128, 2509–2519, DOI:10.1242/jcs.168773.
- S. M. Gross, M. A. Dane, E. Bucher and L. M. Heiser, Individual Cells Can Resolve Variations in Stimulus Intensity along the IGF-PI3K-AKT Signaling Axis, Cell Syst., 2019, 9, 580–588, DOI:10.1016/j.cels.2019.11.005.
- M. Nacht, L. Qiao, M. P. Sheets, T. St Martin, M. Labenski, H. Mazdiyasni, R. Karp, Z. Zhu, P. Chaturvedi, D. Bhavsar, D. Niu, W. Westlin, R. C. Petter, A. P. Medikonda and J. Singh, Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kα, J. Med. Chem., 2013, 56, 712–721, DOI:10.1021/jm3008745.
- D. Montero, C. Tachibana, J. Rahr Winther and C. Appenzeller-Herzog, Intracellular glutathione pools are heterogeneously concentrated, Redox Biol., 2013, 1, 508–513, DOI:10.1016/j.redox.2013.10.005.
- M. D. Shultz, The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations, Bioorg. Med. Chem. Lett., 2013, 23, 5992–6000, DOI:10.1016/j.bmcl.2013.08.030.
- T. W. Johnson, R. A. Gallego and M. P. Edwards, Lipophilic Efficiency as an Important Metric in Drug Design, J. Med. Chem., 2018, 61, 6401–6420, DOI:10.1021/acs.jmedchem.8b00077.
- P. D. Leeson and B. Springthorpe, The influence of drug-like concepts on decision-making in medicinal chemistry, Nat. Rev. Drug Discovery, 2007, 6, 881–890, DOI:10.1038/nrd2445.
- M. B. Robers, M. L. Dart, C. C. Woodroofe, C. A. Zimprich, T. A. Kirkland, T. Machleidt, K. R. Kupcho, S. Levin, J. R. Hartnett, K. Zimmerman, A. L. Niles, R. F. Ohana, D. L. Daniels, M. Slater, M. G. Wood, M. Cong, Y. Q. Cheng and K. V. Wood, Target engagement and drug residence time can be observed in living cells with BRET, Nat. Commun., 2015, 6, 10091, DOI:10.1038/ncomms10091.
- V. Poongavanam, L. H. E. Wieske, S. Peintner, M. Erdélyi and J. Kihlberg, Molecular chameleons in drug discovery, Nat. Rev. Chem, 2023, 8, 45–60, DOI:10.1038/s41570-023-00563-1.
- K. W. Song, K. A. Edgar, E. J. Hanan, M. Hafner, J. Oeh, M. Merchant, D. Sampath, M. A. Nannini, R. Hong, L. Phu, W. F. Forrest, E. Stawiski, S. Schmidt, N. Endres, J. Guan, J. J. Wallin, J. Cheong, E. G. Plise, G. D. Lewis Phillips, L. Salphati, T. P. Heffron, A. G. Olivero, S. Malek, S. T. Staben, D. S. Kirkpatrick, A. Dey and L. S. Friedman, RTK-Dependent Inducible Degradation of Mutant PI3Kα Drives GDC-0077 (Inavolisib) Efficacy, Cancer Discovery, 2022, 12, 204–219, DOI:10.1158/2159-8290.CD-21-0072.
- M. Hafner, M. Niepel, M. Chung and P. K. Sorger, Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs, Nat. Methods, 2016, 13, 521–527, DOI:10.1038/nmeth.3853.
- R. R. Madsen, A. Le Marois, O. Mruk, M. Voliotis, S. Yin, J. Sufi, X. Qin, S. J. Zhao, J. Gorczynska, D. Morelli, L. Davidson, E. Sahai, V. I. Korolchuk, C. J. Tape and B. Vanhaesebroeck, Oncogenic PIK3CA corrupts growth factor signaling specificity, bioRxiv, 2023, 12(23), 573207, DOI:10.1101/2023.12.23.573207.
- M. Neves, V. Marolda, F. Mayor and P. Penela, Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells, Int. J. Mol. Sci., 2022, 23, 11887, DOI:10.3390/ijms231911887.
- V. Serra, M. Scaltriti, L. Prudkin, P. J. Eichhorn, Y. H. Ibrahim, S. Chandarlapaty, B. Markman, O. Rodriguez, M. Guzman, S. Rodriguez, M. Gili, M. Russillo, J. L. Parra, S. Singh, J. Arribas, N. Rosen and J. Baselga, PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer, Oncogene, 2011, 30, 2547–2557, DOI:10.1038/onc.2010.626.
- M. Toulany, M. Minjgee, M. Saki, M. Holler, F. Meier, W. Eicheler and H. P. Rodemann, ERK2-dependent reactivation of Akt mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition, Cancer Biol. Ther., 2014, 15, 317–328, DOI:10.4161/cbt.27311.
- P. P. Hsu, S. A. Kang, J. Rameseder, Y. Zhang, K. A. Ottina, D. Lim, T. R. Peterson, Y. Choi, N. S. Gray, M. B. Yaffe, J. A. Marto and D. M. Sabatini, The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling, Science, 2011, 332, 1317–1322, DOI:10.1126/science.1199498.
- F. Beaufils, N. Cmiljanovic, V. Cmiljanovic, T. Bohnacker, A. Melone, R. Marone, E. Jackson, X. Zhang, A. Sele, C. Borsari, J. Mestan, P. Hebeisen, P. Hillmann, B. Giese, M. Zvelebil, D. Fabbro, R. L. Williams, D. Rageot and M. P. Wymann, 5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (PQR309), a Potent, Brain-Penetrant, Orally Bioavailable, Pan-Class I PI3K/mTOR Inhibitor as Clinical Candidate in Oncology, J. Med. Chem., 2017, 60, 7524–7538, DOI:10.1021/acs.jmedchem.7b00930.
- T. Bohnacker, A. E. Prota, F. Beaufils, J. E. Burke, A. Melone, A. J. Inglis, D. Rageot, A. M. Sele, V. Cmiljanovic, N. Cmiljanovic, K. Bargsten, A. Aher, A. Akhmanova, J. F. Díaz, D. Fabbro, M. Zvelebil, R. L. Williams, M. O. Steinmetz and M. P. Wymann, Deconvolution of Buparlisib's mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention, Nat. Commun., 2017, 8, 14683, DOI:10.1038/ncomms14683.
- K. A. Johnson, Fitting enzyme kinetic data with KinTek Global Kinetic Explorer, Methods Enzymol., 2009, 467, 601–626, DOI:10.1016/S0076-6879(09)67023-3.
- K. A. Johnson, Z. B. Simpson and T. Blom, Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data, Anal. Biochem., 2009, 387, 20–29, DOI:10.1016/j.ab.2008.12.024.
- C. McQuin, A. Goodman, V. Chernyshev, L. Kamentsky, B. A. Cimini, K. W. Karhohs, M. Doan, L. Ding, S. M. Rafelski, D. Thirstrup, W. Wiegraebe, S. Singh, T. Becker, J. C. Caicedo and A. E. Carpenter, CellProfiler 3.0: Next-generation image processing for biology, PLoS Biol., 2018, 16, e2005970, DOI:10.1371/journal.pbio.2005970.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132, DOI:10.1107/S0907444909047337.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2007, 40, 658–674, DOI:10.1107/S0021889807021206.
- P. Chen, Y. L. Deng, S. Bergqvist, M. D. Falk, W. Liu, S. Timofeevski and A. Brooun, Engineering of an isolated p110α subunit of PI3Kα permits crystallization and provides a platform for structure-based drug design, Protein Sci., 2014, 23, 1332–1340, DOI:10.1002/pro.2517.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501, DOI:10.1107/S0907444910007493.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367, DOI:10.1107/S0907444912001308.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21, DOI:10.1107/S0907444909042073.
- A. Klippel, J. A. Escobedo, M. Hirano and L. T. Williams, The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity, Mol. Cell. Biol., 1994, 14, 2675–2685, DOI:10.1128/mcb.14.4.2675.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05459h |
| ‡ Equal contribution. |
| § Current address: Department of Pharmaceutical Sciences, University of Milan, Via Mangiagalli 25, 20133, Milano, Italy. |
| This journal is © The Royal Society of Chemistry 2024 |