
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/NHC sequentially catalyzed atroposelective synthesis of planar-chiral macrocycles†
Gongming
Yang
 ,
Shangde
Liu
,
Shujie
Ji
,
Xingsen
Wu
and
Jian
Wang
*
,
Shangde
Liu
,
Shujie
Ji
,
Xingsen
Wu
and
Jian
Wang
*
School of Pharmaceutical Sciences, Key Laboratory of Bioorganic Phosphorous Chemistry and Chemical Biology, Ministry of Education, Tsinghua University, Beijing, 100084, China. E-mail: wangjian2012@tsinghua.edu.cn
First published on 4th November 2024
Abstract
Planar-chiral macrocycles play a pivotal role in host–guest chemistry and drug discovery. However, compared with the synthesis of other types of chiral compounds, the asymmetric construction of planar-chiral macrocycles still remains a forbidding challenge. Herein, we report a sequential palladium and N-heterocyclic carbene catalysis to build planar-chiral macrocycles. This protocol features broad scope and good functional group tolerance, and allows a rapid assembling of planar-chiral macrocycles with excellent enantioselectivities.
Planar-chiral macrocycles, as a class of atropisomers, have been widely used in asymmetric synthesis,1 materials science,2 and host–guest chemistry,3 and are also widely present in natural products.4 For instance, chrysophaentin A5 inhibits the activity of methicillin-resistant Staphylococcus aureus (MRSA); cylindrocyclophane A6 inhibits the activity of KB and LoVo tumor cells; and FVIIIa7 promotes coagulant activity (Fig. 1A); therefore, the asymmetric preparation of planar-chiral macrocyclic skeletons has attracted sustainable attention from chemists.8 However, the enantioselective synthesis of planar-chiral macrocycles seems to have encountered difficulties. The investigative results show that only a very limited number of methods have been used to prepare optically active planar-chiral macrocycles by far. These disclosed methods are mainly divided into four categories. (i) Intramolecular macrocyclisation:9 elegant studies involve the Ru-catalyzed C–S coupling9i and the phase transfer catalyst-catalyzed SNAr macrocycloaddition9c reported by Cai, respectively. Recently, our group disclosed the NHC-catalyzed atroposelective macrocycloaddition for the synthesis of planar-chiral indoles/pyrroles;10 (ii) intramolecular arene formation:11 the Tanaka group disclosed an enantioselective intramolecular [2 + 2 + 2] cycloaddition for the construction of macrocycles via chiral Rh-catalysis;11a (iii) modification of rings:12 Shibata and co-workers reported an asymmetric Sonogashira coupling to carry out such ring modification.12a In 2022, the Yang group reported the chiral phosphoric acid-catalyzed enantioselective electrophilic aromatic amination to deliver such macrocycles;12d (iv) intermolecular ring closure: in this context, the investigations of intermolecular ring closure are much less studied. In 2020, Collins and co-workers13 reported the only example of biocatalytic and enantioselective intermolecular macrocyclization of diacids and diols to deliver planar-chiral macrocycles. Although the above strategies have been widely utilized to assemble enantioenriched planar-chiral macrocycles, most of the methods require pre-assembly of chains in the substrate, which significantly limits the diversity of planar-chiral macrocyclic products. Therefore, the development of a catalytic methodology that enables the modular assembly of structurally diverse enantioenriched macrocycles from simple starting materials is a demanding yet highly desirable objective.
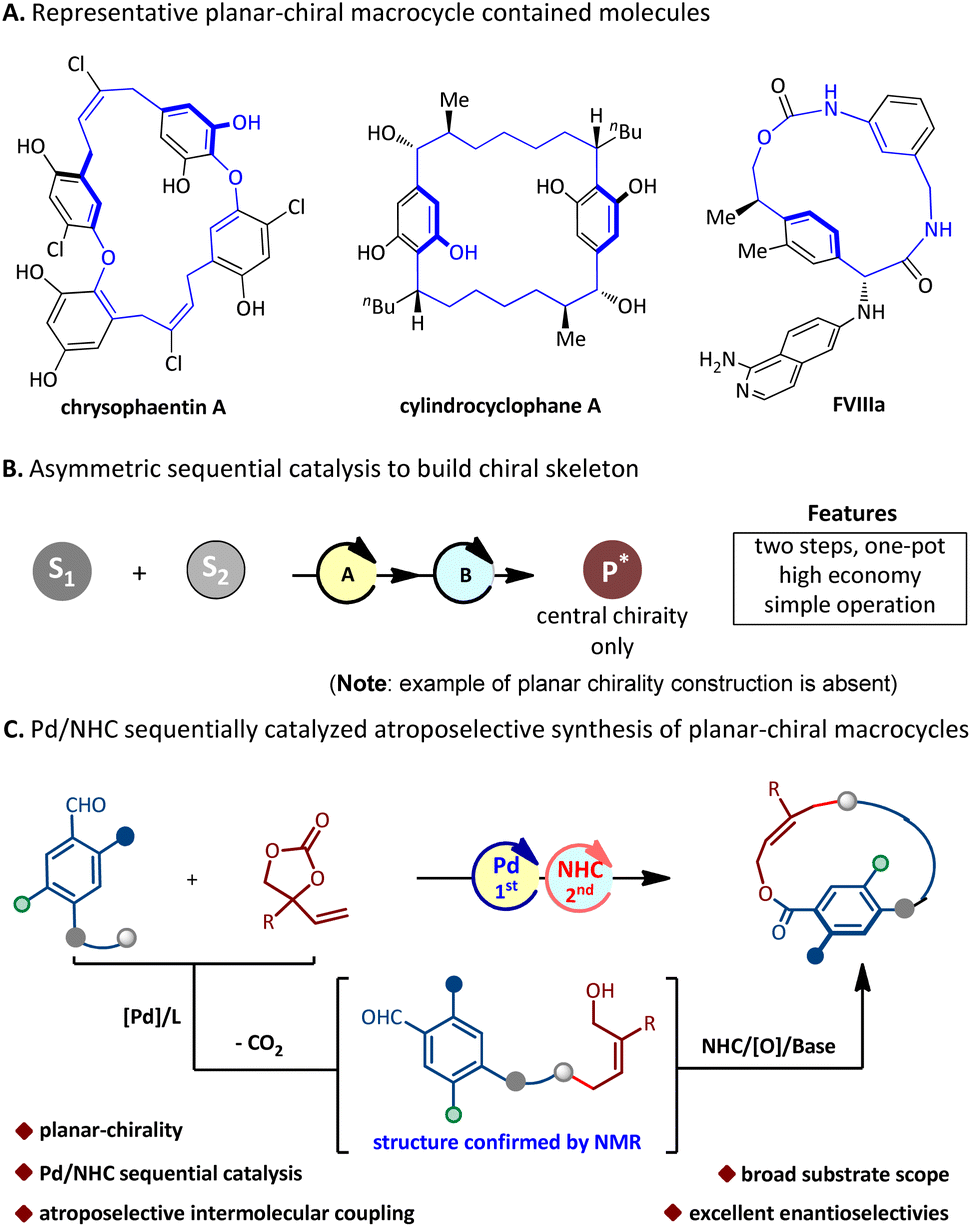 | ||
| Fig. 1 Background and our design. (A) Representative planar-chiral macrocycle-containing molecules. (B) Asymmetric sequential catalysis. (C) Our design. | ||
Asymmetric sequential catalysis, as an intriguing and effective strategy to promote efficient chemical synthesis, has enabled assembling valuable chiral molecules with a complex structure from readily available starting materials in a one-pot multi-step fashion of reducing time, costs, and waste generation. Therefore, it has received extensive attention from chemists and significant progress has been made.14 However, it is important to note that these reports have still focused on the construction of backbones with central chirality only. The enantioselective synthesis of planar chiral compounds via the strategy of sequential catalysis remains yet to be explored to date (Fig. 1B).15
N-Heterocyclic carbene (NHC) asymmetric catalysis, due to its unique advantages in the field of rapid construction of complex chiral scaffolds, has achieved significant developments in recent years.16 However, the development of sequential catalysis involving the NHC catalyst is still in its infancy.17 Meanwhile, our laboratory is highly interested in exploring NHC catalysis for the rapid assembling of atropisomeric molecules.18 Very recently, we disclosed the first NHC-catalyzed intramolecular atroposelective macrocyclization for the assembly of planar-chiral indoles/pyrroles.10 Herein, we present an unprecedented intermolecular reaction of aldehydes and vinyl ethylene carbonates via the Pd/NHC sequential catalysis, exhibiting a wide substrate scope and good functional group tolerance, which can rapidly deliver optically pure planar-chiral macrocycles (Fig. 1C).
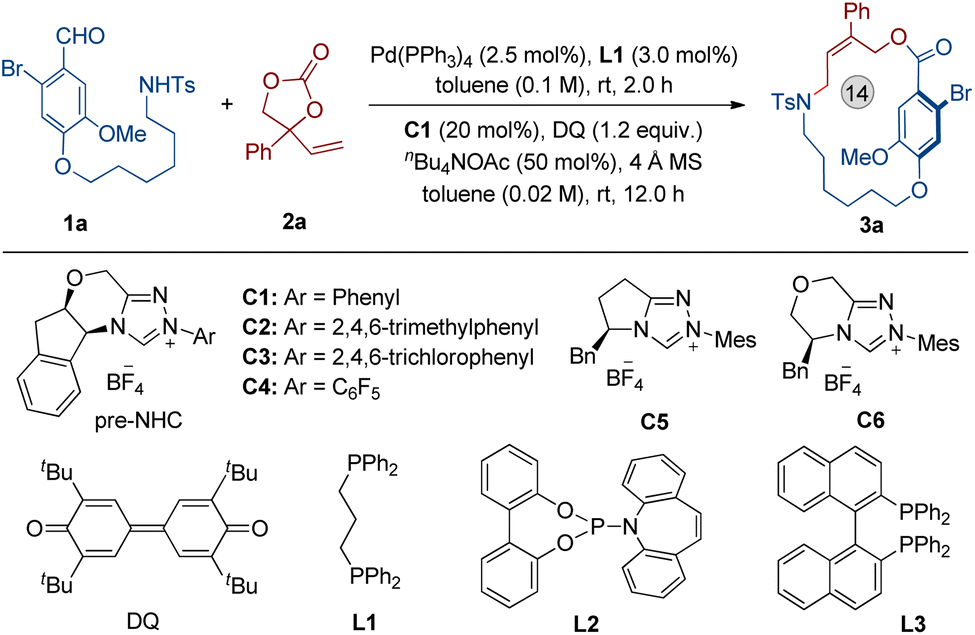
We chose the 6-bromovanillin derivative (1a) and phenyl vinylethylene carbonate (2a) as the model substrates, and DQ as the oxidant. The results revealed that the optimal reaction conditions are a combination of toluene as the solvent, nBu4NOAc as the base, Pd(PPh3)4 as the metal catalyst, DPPP (L1) as the ligand, and NHC C1 as the organocatalyst, which provided the planar-chiral macrocycle 3a in 72% yield and with excellent enantioselectivity (Table 1, entry 1, 96% ee). NHC catalyst screening showed that the triazolium derived carbene precursors C2, C5 and C6 afforded 3a in diminished yields and with very low enantioselectivities (entries 2, 4 and 5). In addition, the carbene precursors C3 and C4 were also found to be ineffective in the reaction (entry 4). When L2 or L3 replaced L1, the yield of 3a decreased and the absence of L1 led to a little loss in yield (entries 6–8). Notably, replacing nBuN4OAc with K2CO3, KOAc or Et3N resulted in a severe decrease in the yield of 3a (entries 9–11). The results of reactions performed in THF or CH2Cl2 were inferior compared to standard conditions (toluene as solvent) (entries 12 and 13). In the absence of 4 Å MS, 3a was still obtained with excellent enantioselectivity, but the yield was partially reduced (entry 14). Additionally, increasing the reaction concentration led to a decrease in the isolated yield of product 3a (entry 15).
|
|
|||
|---|---|---|---|
| Entrya | Variation of standard conditions | Yieldb (%) | eec (%) |
| a Conditions: 1a (0.10 mmol), 2a (0.15 mmol), Pd(PPh3)4 (2.5 mol%) and L1 (3.0 mol%) in 1.0 mL of toluene were allowed to stir at room temperature for 2 h. The solution was then transferred into a mixture of pre-NHC catalyst C1 (20 mol%), nBu4NOAc (50 mol%), DQ (0.12 mmol) and 4 Å MS 50 mg in toluene (4.0 mL). The reaction mixture was allowed to stir at room temperature for another 12 h under Ar. b Isolated yield after flash column chromatography. c Determined by HPLC analysis using a chiral stationary phase. | |||
| 1 | None | 72 | 96 |
| 2 | C2 instead of C1 | 65 | 30 |
| 3 | C3 or C4 instead of C1 | <5 | — |
| 4 | C5 instead of C1 | 30 | −16 |
| 5 | C6 instead of C1 | 46 | −22 |
| 6 | L2 instead of L1 | 60 | 96 |
| 7 | L3 instead of L1 | 65 | 96 |
| 8 | Without of L1 | 68 | 96 |
| 9 | K2CO3 instead of nBu4NOAc | 28 | 94 |
| 10 | KOAc instead of nBu4NOAc | 35 | 96 |
| 11 | Et3N instead of nBu4NOAc | 30 | 92 |
| 12 | THF instead of toluene | 52 | 90 |
| 13 | CH2Cl2 instead of toluene | 68 | 93 |
| 14 | Without 4 Å MS | 67 | 96 |
| 15 | Toluene (0.1 M) was used | 62 | 96 |
With optimal conditions in hand, we turned our attention to examine the scope of this sequential catalytic reaction for the synthesis of functional planar-chiral macrocycles. The vinyl ethylene carbonates (VECs) 2 were examined first (Fig. 2). A range of VECs bearing various substituted aryl groups have proved to be suitable substrates, affording the corresponding products 3a–3o at 95–98% ee. In addition, heteroaryl-substituted VECs also well participated and generated the planar-chiral macrocycles 3p and 3q in good yields and with excellent enantioselectivities (Fig. 2, 70% and 71% yield; 98% and 96% ee, respectively). The absolute configuration of product 3q was determined by X-ray single crystal analysis (CCDC: 2347510), and other structures were assigned by analogy. Pleasingly, varied alkyl-substituted VECs were also tolerated and delivered the corresponding planar-chiral macrocycles 3r–3t over longer reaction times with excellent enantioselectivities, albeit with a slightly decrease in yield.
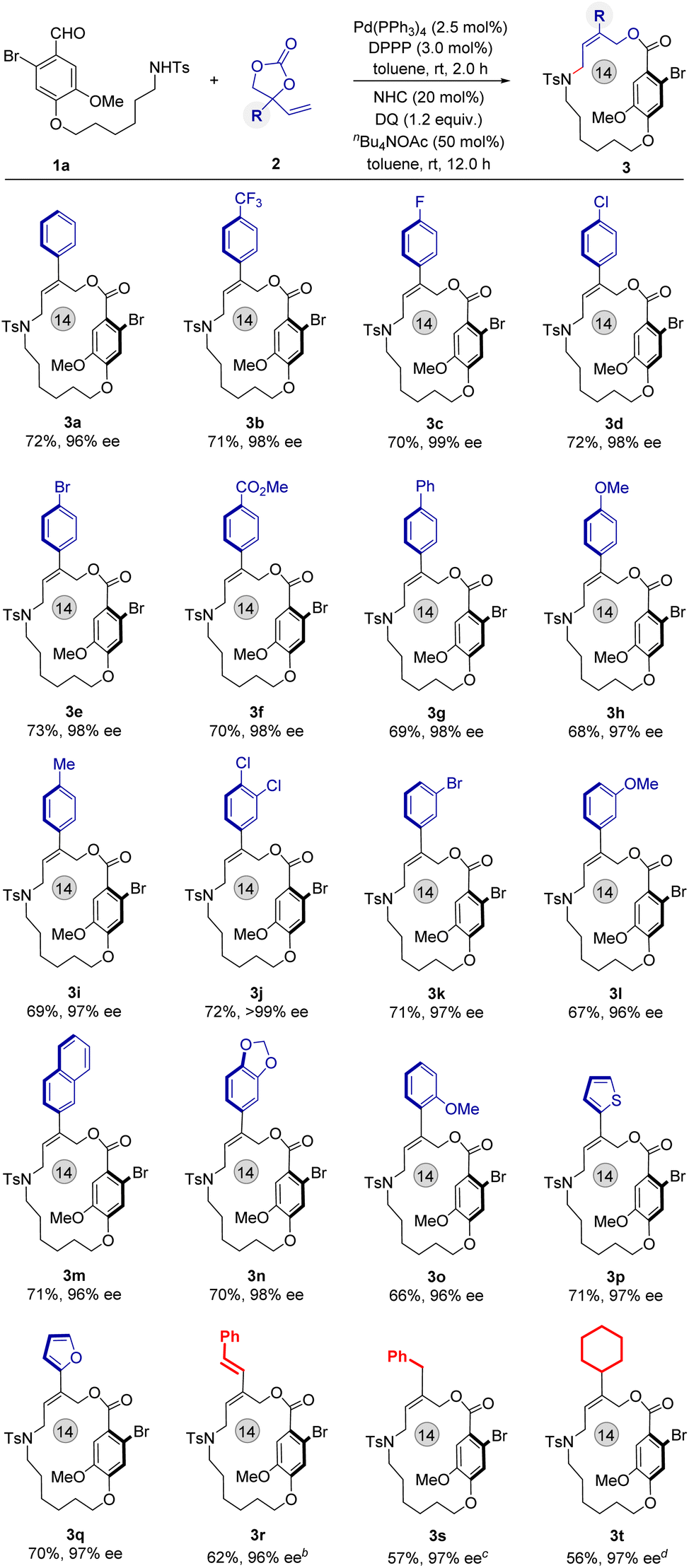 | ||
| Fig. 2 Scope of vinyl ethylene carbonate 2. aReaction conditions: see Table 1, entry 1. b12 h for the first step, 12 h for the second step. c24 h for the first step, 12 h for the second step.48 h. d36 h for the first step, 12 h for the second step. | ||
Encouraged by the success in the variation of VECs, we then focused on the scope of aryl aldehyde 1. As shown in Fig. 3, a range of aryl aldehyde substrates bearing either an electron-withdrawing group or electron-donating group at the 4-position generated their corresponding products smoothly with excellent enantioselectivities (90–>99% ee). In addition, when the bromine at the 2-position of aryl aldehyde became small chlorine, the corresponding planar-chiral products were still obtained and with excellent enantioselectivities (4f, 97% ee and 4g, 96% ee, respectively). Furthermore, the 2-phenyl-substituted substrate generated the desired planar-chiral macrocycle 4h in 74% yield and with >99% ee under optimal conditions. Pleasingly, varied five-membered aromatic heterocycle substituted substrates were also tolerated and delivered the corresponding planar-chiral macrocycles with high to excellent enantioselectivities (4i–4k).
 | ||
| Fig. 3 Scope of aryl aldehyde 1. aReaction conditions: see Table 1, entry 1. | ||
Subsequently, the modification of the O-substituted amide chain was conducted. Amide chains containing ester or ether groups were well tolerated under optimal conditions and provided their corresponding planar-chiral products with high enantioselectivities (Fig. 3, 4l and 4m). Next, the effect of length of the chain was also investigated. When the length of the amide chain was reduced, the yield of the corresponding planar-chiral macrocycle decreased dramatically (4n, n = 13). At the same time, substrates with extended chains were also tested and found to produce the desired product (4o, n = 15) in good yield and with excellent enantioselectivity. Notably, a further increase in amide chain length led to a loss of planar chirality (4p, n = 16), which clearly indicated that planar chirality is highly dependent on the size of the ring on the macrocycle.
To get insight into the thermal stability of the macrocyclic planar-chirality, a series of racemization experiments were performed.19 As shown in Fig. 4, planar-chiral macrocycle 3a or 4n was stirred at 140 °C in toluene under sealed conditions for 2 h and no racemization was observed (see ESI,† S43). For compound 4o, the ee value was maintained below 60 °C, indicating that the rotation of the ansa chain was restricted (Fig. 4A). On the other hand, the configuration stability studies revealed that the rotation barrier of 4o is 31.1 kcal mol−1, and the t1/2 of racemization is 19.3 h at 100 °C (Note: At 25 °C, the t1/2 of racemization is calculated to be 33.5 years) (Fig. 4B and C).
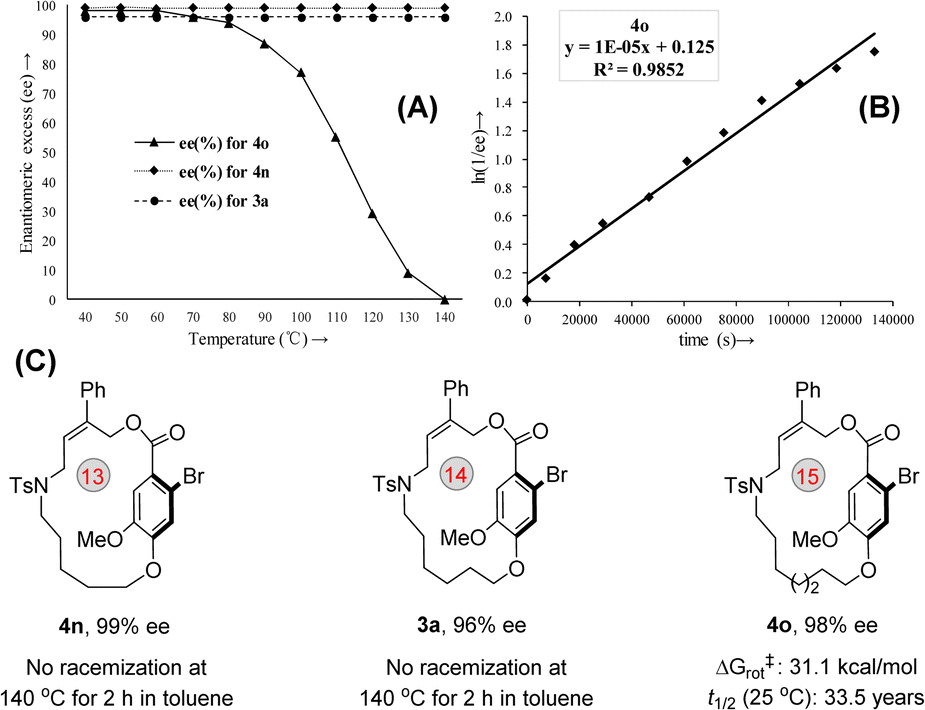 | ||
| Fig. 4 Determination of the racemization barrier. (A) Effect of temperature on the stability of macrocyclic planar chirality. (B) Plot for calculating the racemization barrier of 4o. (C) Racemization barriers of 3a, 4n and 4o. | ||
Before illustrating the utility of this method, a gram-scale synthesis of 3a was conducted under optimal conditions, producing the desired product with promising yield and enantioselectivity (Fig. 5A). Next, two follow-up transformations of 3a were carried out individually. The phenylethynyl-substituted planar chiral macrocycle 5 was prepared smoothly via a one-step Sonogashira coupling of 3a (Fig. 5B). Importantly, the multiple stereogenic enantioenriched compound 6, featuring both central and macrocyclic planar chirality, was obtained efficiently by epoxidation of 3a in the presence of meta-chloroperbenzoic acid (m-CPBA) (Fig. 5C).
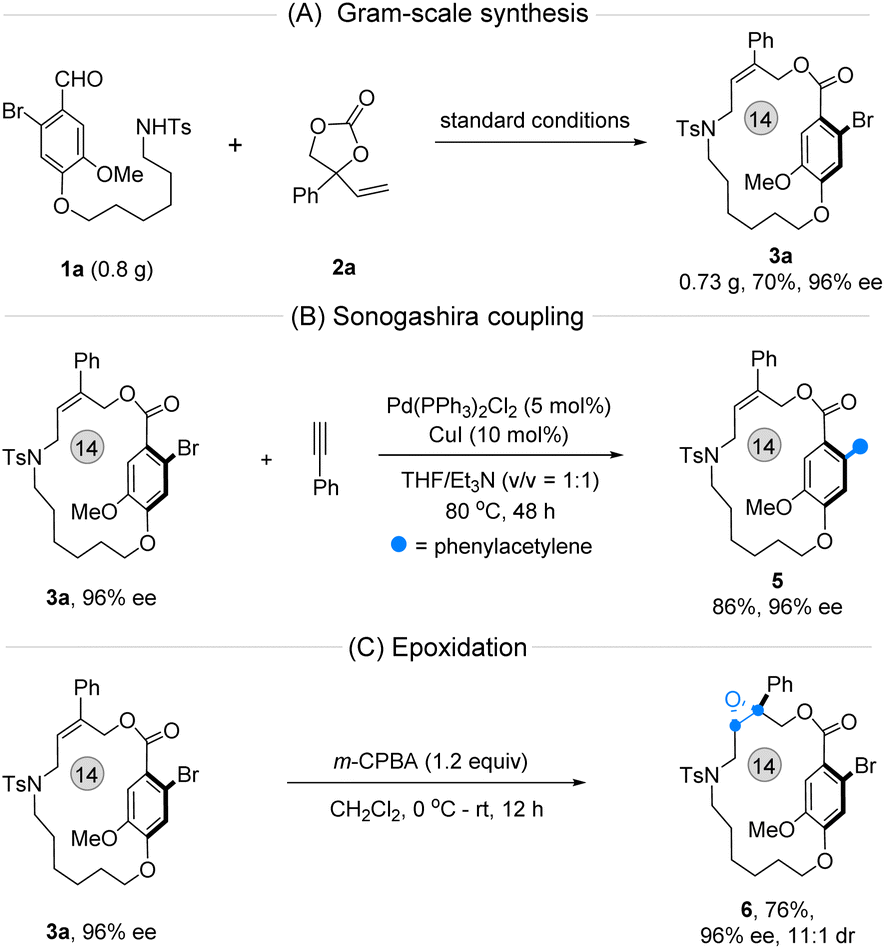 | ||
| Fig. 5 Gram-synthesis and synthetic transformations. (A) Gram-scale synthesis. (B) Sonogashira coupling. (C) Epoxidation reaction. | ||
To understand the pathway, we performed the reaction in a stepwise manner, expecting to obtain key intermediates to further elucidate the mechanism (Fig. 6, see ESI,† S52). The alkyl alcohol chain linked aryl aldehyde 3a′ (confirmed by NMR) was obtained from 1a and 2a under the conditions of Pd(PPh3)4 (2.5 mol%) and DPPP (3 mol%). Following the addition of C1 (20 mol%), DQ (1.2 equiv.) and nBuN4OAc (50 mol%), the desired product 3a was achieved in 69% yield with 96% ee. Similar results were compared between the stepwise reaction and the one-pot reaction (Table 1), suggesting that the reaction process most likely occurred via a sequential catalytic process.
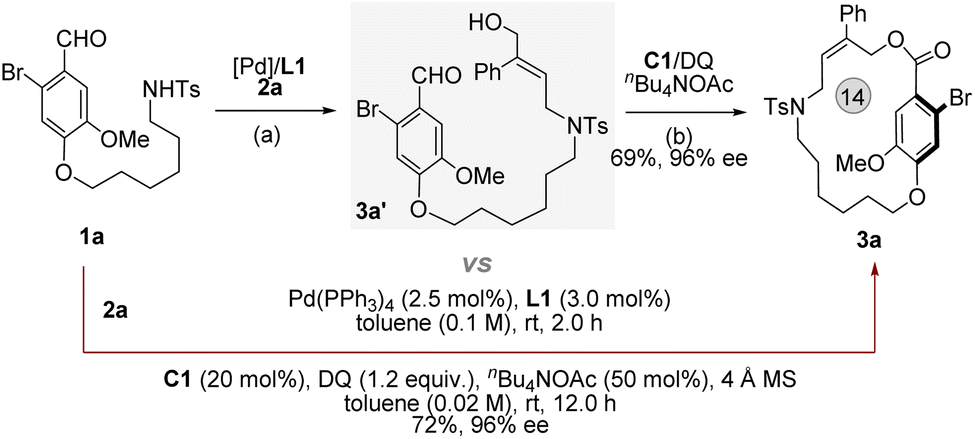 | ||
| Fig. 6 Mechanistic studies: (a) Pd(PPh3)4 (2.5 mol%) and L1 (3 mol%) in toluene (1.0 mL), 2 h. (b) C1 (20 mol%), DQ (1.2 equiv.) and nBuN4OAc in toluene (4.0 mL), room temperature for 12 h. | ||
In summary, we have developed a Pd/NHC sequential catalytic intermolecular atroposelective macrocyclization for the preparation of various planar-chiral macrocycles with high to excellent enantioselectivities under mild conditions. This protocol shows a broad scope and functional group tolerance. Multiple stereogenic macrocycles featuring both central and planar chirality have proven the synthetic utility of the present study. Control experiments reveal that these transformations possibly occurred via a Pd/NHC sequential catalytic process.
Data availability
All data supporting the findings of this study are available within the article and its ESI file.†Author contributions
G. M. Y. conducted main experiments; S. D. L., S. J. J. and X. S. W. prepared several starting materials, including substrates. J. W. conceptualized and directed the project, and drafted the manuscript with the assistance from co-authors. All authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support for this work is provided by: the National Natural Science Foundation of China (21871160, 21672121, and 22071130), the China Postdoctoral Science Foundation (2022M721804), and the Bayer Investigator fellow, the fellowship of Tsinghua-Peking centre for life sciences (CLS).Notes and references
- Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC.
- (a) D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158–4168 RSC; (b) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- (a) M. Quan, X.-Y. Pang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202201258 CrossRef CAS PubMed; (b) H. Zhu, L. Chen, B. Sun, M. Wang, H. Li, J. F. Stoddart and F. Huang, Nat. Rev. Chem, 2023, 7, 768–782 CrossRef CAS PubMed.
- (a) I. V. Smolyar, A. K. Yudin and V. G. Nenajdenko, Chem. Rev., 2019, 119, 10032–10240 CrossRef CAS PubMed; (b) J. A. Swain, S. R. Walker, M. B. Calvert and M. A. Brimble, Nat. Prod. Rep., 2022, 39, 410–443 RSC.
- (a) A. Plaza, J. L. Keffer, G. Bifulco, J. R. Lloyd and C. A. Bewley, J. Am. Chem. Soc., 2010, 132, 9069–9077 CrossRef CAS PubMed; (b) C. R. Fullenkamp, Y.-P. Hsu, E. M. Quardokus, G. Zhao, C. A. Bewley, M. VanNieuwenhze and G. A. Sulikowski, J. Am. Chem. Soc., 2020, 142, 16161–16166 CrossRef CAS PubMed.
- (a) H. Nakamura, E. E. Schultz and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 916–921 CrossRef CAS PubMed; (b) K.-Y. Chen, H.-Q. Wang, Y. Yuan, S.-B. Mou and Z. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202307602 CrossRef CAS.
- M. M. Aleman, S. Jindal, N. Leksa, R. Peters and J. Salas, Blood, 2018, 132, 2461 CrossRef.
- (a) Y. Wang and M. M. Joullié, Chem. Rec., 2021, 21, 906–923 CrossRef CAS PubMed; (b) R. López and C. Palomo, Angew. Chem., Int. Ed., 2022, 61, e202113504 CrossRef; (c) G. Yang and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202412805 CAS.
- (a) M. E. Layton, C. A. Morales and M. D. Shair, J. Am. Chem. Soc., 2002, 124, 773–775 CrossRef CAS PubMed; (b) M. Q. Salih and C. M. Beaudry, Org. Lett., 2013, 15, 4540–4543 CrossRef CAS; (c) Q. Ding, Q. Wang, H. He and Q. Cai, Org. Lett., 2017, 19, 1804–1807 CrossRef CAS PubMed; (d) K. Mori, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 5638–5641 CrossRef CAS; (e) S. Yu, G. Shen, F. He and X. Yang, Chem. Commun., 2022, 58, 7293–7296 RSC; (f) S. Wei, L.-Y. Chen and J. Li, ACS Catal., 2023, 13, 7450–7456 CrossRef CAS; (g) J. Wang, M. Wang, Y. Wen, P. Teng, C. Li and C. Zhao, Org. Lett., 2024, 26, 1040–1045 CrossRef CAS; (h) X. Lv, F. Su, H. Long, F. Lu, Y. Zeng, M. Liao, F. Che, X. Wu and Y. R. Chi, Nat. Commun., 2024, 15, 958 CrossRef CAS; (i) K. Tanaka, T. Hori, T. Osaka, K. Noguchi and M. Hirano, Org. Lett., 2007, 9, 4881–4884 CrossRef CAS PubMed.
- G. Yang, Y. He, T. Wang, Z. Li and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316739 CrossRef CAS PubMed.
- (a) K. Tanaka, H. Sagae, K. Toyoda, K. Noguchi and M. Hirano, J. Am. Chem. Soc., 2007, 129, 1522–1523 CrossRef CAS PubMed; (b) T. Araki, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2013, 52, 5617–5621 CrossRef PubMed.
- (a) K. Kanda, K. Endo and T. Shibata, Org. Lett., 2010, 12, 1980–1983 CrossRef CAS PubMed; (b) Z. Dong, J. Li, T. Yao and C. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202315603 CrossRef CAS; (c) D. Zhu, T. Mu, Z.-L. Li, H.-Y. Luo, R.-F. Cao, X.-S. Xue and Z.-M. Chen, Angew. Chem., Int. Ed., 2024, 63, e202318625 CrossRef CAS; (d) D. Wang, Y.-B. Shao, Y. Chen, X.-S. Xue and X. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201064 CrossRef CAS; (e) J. Li and C. Zhao, ACS Catal., 2023, 13, 14155–14162 CrossRef CAS.
- C. Gagnon, É. Godin, C. Minozzi, J. Sosoe, C. Pochet and S. K. Collins, Science, 2020, 367, 917–921 CrossRef CAS.
- (a) J. Zhou, Chem.–Asian J., 2010, 5, 422–434 CrossRef CAS; (b) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 RSC.
- (a) X.-P. Yin, X.-P. Zeng, Y.-L. Liu, F.-M. Liao, J.-S. Yu, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13740–13745 CrossRef CAS; (b) C. Romano, D. Fiorito and C. Mazet, J. Am. Chem. Soc., 2019, 141, 16983–16990 CrossRef CAS; (c) G. Yang, Y.-M. Ke and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12775–12780 CrossRef CAS; (d) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef.
- (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (b) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC; (c) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696–707 CrossRef CAS; (d) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (e) C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583–2596 CrossRef CAS; (f) X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS PubMed; (g) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS.
- (a) L. Dell'Amico, G. Rassu, V. Zambrano, A. Sartori, C. Curti, L. Battistini, G. Pelosi, G. Casiraghi and F. Zanardi, J. Am. Chem. Soc., 2014, 136, 11107–11114 CrossRef; (b) K. Namitharan, T. Zhu, J. Cheng, P. Zheng, X. Li, S. Yang, B.-A. Song and Y. R. Chi, Nat. Commun., 2014, 5, 3982 CrossRef CAS; (c) L. Zhou, X. Wu, X. Yang, C. Mou, R. Song, S. Yu, H. Chai, L. Pan, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2020, 59, 1557–1561 CrossRef CAS PubMed; (d) J. Jiang, X. Wang, S. Liu, S. Zhang, B. Yang, Y. Zhao and S. Lu, Angew. Chem., Int. Ed., 2022, 61, e202115464 CrossRef CAS PubMed.
- (a) C. Zhao, D. Guo, K. Munkerup, K.-W. Huang, F. Li and J. Wang, Nat. Commun., 2018, 9, 611 CrossRef; (b) J. Bie, M. Lang and J. Wang, Org. Lett., 2018, 20, 5866–5871 CrossRef CAS; (c) G. Yang, S. Sun, Z. Li, Y. Liu and J. Wang, Commun. Chem., 2021, 4, 144 CrossRef CAS PubMed; (d) G. Yang, D. Guo, D. Meng and J. Wang, Nat. Commun., 2019, 10, 3062 CrossRef; (e) G. Yang, Z. Li, Y. Liu, D. Guo, X. Sheng and J. Wang, Org. Lett., 2021, 23, 8109–8113 CrossRef CAS PubMed; (f) D. Guo, Q. Peng, B. Zhang and J. Wang, Org. Lett., 2021, 23, 7765–7770 CrossRef CAS PubMed; (g) J. Wang, C. Zhao and J. Wang, ACS Catal., 2021, 11, 12520–12531 CrossRef CAS.
- S. Shee, S. Shree Ranganathappa, M. S. Gadhave, R. Gogoi and A. T. Biju, Angew. Chem., Int. Ed., 2023, 62, e202311709 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05482b |
| This journal is © The Royal Society of Chemistry 2024 |