
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom small changes to big gains: pyridinium-based tetralactam macrocycle for enhanced sugar recognition in water†
Canjia
Zhai
 a,
Ethan Cross
Zulueta
a,
Alexander
Mariscal
a,
Chengkai
Xu
a,
Yunpeng
Cui
a,
Xudong
Wang
b,
Huang
Wu
c,
Carson
Doan
a,
Lukasz
Wojtas
a,
Haixin
Zhang
d,
Jianfeng
Cai
a,
Libin
Ye
b,
Kun
Wang
de and
Wenqi
Liu
*a
a,
Ethan Cross
Zulueta
a,
Alexander
Mariscal
a,
Chengkai
Xu
a,
Yunpeng
Cui
a,
Xudong
Wang
b,
Huang
Wu
c,
Carson
Doan
a,
Lukasz
Wojtas
a,
Haixin
Zhang
d,
Jianfeng
Cai
a,
Libin
Ye
b,
Kun
Wang
de and
Wenqi
Liu
*a
aDepartment of Chemistry, University of South Florida, Tampa, FL 33620, USA. E-mail: wenqi@usf.edu
bDepartment of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, FL 33620, USA
cDepartment of Chemistry, The University of Hong Kong, Hong Kong SAR 999077, China
dDepartment of Physics, University of Miami, Coral Gables, Florida 33146, USA
eDepartment of Chemistry, University of Miami, Coral Gables, Florida 33146, USA
First published on 5th November 2024
Abstract
The complex distribution of functional groups in carbohydrates, coupled with their strong solvation in water, makes them challenging targets for synthetic receptors. Despite extensive research into various molecular frameworks, most synthetic carbohydrate receptors have exhibited low affinities, and their interactions with sugars in aqueous environments remain poorly understood. In this work, we present a simple pyridinium-based hydrogen-bonding receptor derived from a subtle structural modification of a well-known tetralactam macrocycle. This small structural change resulted in a dramatic enhancement of glucose binding affinity, increasing from 56 M−1 to 3001 M−1. Remarkably, the performance of our synthetic lectin surpasses that of the natural lectin, concanavalin A, by over fivefold. X-ray crystallography of the macrocycle–glucose complex reveals a distinctive hydrogen bonding pattern, which allows for a larger surface overlap between the receptor and glucose, contributing to the enhanced affinity. Furthermore, this receptor possesses allosteric binding sites, which involve chloride binding and trigger receptor aggregation. This unique allosteric process reveals the critical role of structural flexibility in this hydrogen-bonding receptor for the effective recognition of sugars. We also demonstrate the potential of this synthetic lectin as a highly sensitive glucose sensor in aqueous solutions.
Introduction
The design of synthetic receptors capable of recognizing hydrophilic substrates in water stands as a challenging topic in modern supramolecular chemistry.1–7 Water, as a good hydrogen bond donor and acceptor, competes heavily with the polar substrates and synthetic receptors, hindering the establishment of effective interactions.8–24 Additionally, the low solubility of most organic building blocks further complicates the development of water-soluble receptors. This issue is particularly pronounced in the design of synthetic receptors for carbohydrates. The structural complexity and hydrophilic nature of sugars make them tricky targets for both natural and synthetic receptors.25,26 Even lectins—proteins evolved specifically for carbohydrate recognition—often exhibit weak interaction with simple sugars, with binding affinities (Ka) typically below 103 M−1.26 The development of synthetic lectins capable of efficiently binding sugars in water remains a critical yet unsolved problem. Addressing this challenge is crucial for advancing a wide range of biomedical applications, including diabetes management, the development of synthetic antibodies for targeting cancer cells, anti-infective and anti-inflammatory therapies, as well as many diagnostics tools.27–34A promising strategy has involved leveraging hydrogen bonding and hydrophobic interactions within an intricately designed temple-shaped molecular framework.35–47 For example, a tricyclic molecular cage was initially designed48 for glucose binding with a binding affinity of 9.5 M−1 in water, achieved by combining the hydrophobic surfaces of two parallel biphenyl panels with eight amide hydrogen bonding residues. Subsequent structural optimizations have involved38–40,42,49–55 using larger aromatic panels, such as anthracene and pyrene, and variations in the number and location of hydrogen bonding residues. While these modifications have led to improvements in selectivity and affinity, the synthesis of these receptors remains complex, and their carbohydrate-binding affinities are generally weak. Moreover, the lack of single crystal structures of the synthetic carbohydrate receptors associated with sugars hinders our understanding of how synthetic receptors interact with carbohydrates in aqueous environments. To date, only one such structure has been reported, making it difficult to fully elucidate binding mechanisms and guide receptor design.56 Thus, there is a pressing need for general molecular design principles to develop synthetic lectins capable of more efficient carbohydrate recognition in water.45,53,55,57
In our quest to unravel the complexities of molecular recognition directed by hydrogen bonding in water, we introduced58 a dynamic approach to access a pyridinium-based tetralactam macrocycle with two parallel durene panels. This macrocycle can be easily synthesized through dynamic imine chemistry followed by a Pinnick oxidation reaction.59,60 Although the macrocycle exhibits a high affinity of 850 M−1 for glucose in water, the origin of this high affinity and its binding pattern with glucose remained unclear. In our current research, we report a synthetic lectin that is structurally similar to an anthracene macrocycle reported42 by the Davis group but shows significantly improved binding affinity from 56 M−1 to 3000 M−1. The performance of our synthetic lectin surpasses the natural lectin concanavalin A by more than five times. We obtained the X-ray single crystal structure of the macrocycle in complex with glucose. Compared to the Davis macrocycle, our structure reveals a distinct hydrogen bonding pattern that enables a larger overlapping surface area between the receptor and glucose, resulting in enhanced binding affinity. This significant improvement, driven by subtle structural variations, highlights the sensitivity of sugar binding to the microenvironment within the synthetic lectin's binding pocket, offering valuable insights for the future design of more effective synthetic lectins. Furthermore, our receptor is equipped with two allosteric binding sites for chloride ions, which initiate receptor aggregation and further rigidification of the binding pocket. This allosteric effect provides critical insights into the role of structural flexibility in this hydrogen-bonding receptor for effective sugar recognition in water. Additionally, we demonstrate the potential of our synthetic lectin as a highly sensitive glucose sensor in aqueous solutions. This research not only sheds light on the mechanisms of hydrogen bonding in water but also advances the development of practical applications for synthetic lectins.
Results and discussion
Structural design and synthesis
In our design, we introduced (Fig. 1a) BPAT2+·2Cl−, inspired by the synthetic lectins developed26,36,40,42,45,47,61 by the groups of Davis, Roelens, and Francesconi. This structure is characterized by two large anthracene walls, creating a hydrophobic cavity that provides a substantial contact surface for sugars. A noticeable structure characteristic of BPAT2+·2Cl− is its four convergent NH hydrogen bond donors, which converge towards the binding pocket as a result of steric hindrance between protons c and d.42,62,63 These features coalesce to form an amphiphilic binding pocket, blending polar hydrogen-bonding elements within a nonpolar, nanoconfined space.64 Furthermore, the integration of two pyridinium bridges enhances the design in several ways: firstly, the para C–H bonds, polarized by the pyridinium units, act as effective hydrogen bond donors, pointing convergently towards the binding pocket. Secondly, the receptor's association with the hydrophilic counterion Cl− ensures good water solubility.65–67 Lastly, the methyl group and ortho protons on the pyridinium ring potentially serve as an allosteric binding site, offering a pathway to modulate the substrate binding. | ||
| Fig. 1 (a) A comparison of structural formula of BPAT2+·2Cl− and Davis macrocycle. (b) Conventional (method I) and dynamic approaches (method II) to synthesizing BPAT2+·2Cl−. (c) DFT-optimized structure of BPAT2+. (d) A snapshot from MD simulations of the hydration of BPAT2+ showing the hydrogen bonds of cavity water molecules. | ||
A key precursor to the synthesis BPAT2+·2Cl− is the tetralactam macrocycle 3. This macrocycle can be obtained (Fig. 1b) through a conventional approach under high dilution conditions, leading to a modest yield of 13%.42,62,68,69 While this method benefits from the low solubility of 3, facilitating purification by simple solvent washing, it suffers from low yield. Consequently, an alternative synthetic strategy was explored. A dynamic approach employing the condensation of anthracene methylene diamine 2 with a pyridine 3,5-bisaldehyde precursor 4 provides imine macrocycle 5 in an excellent 88% yield. This intermediate can be readily converted to the desired tetralactam 3via a Pinnick oxidation with a satisfactory 59% yield.58,59 Subsequent alkylation of 3 with iodomethane, followed by ion exchange, furnishes the water-soluble BPAT2+·2Cl− in a good yield of 61%.
The structure (Fig. 1c) of BPAT2+ was optimized by DFT calculation at the BLYP-SVP-D3-gCP level. The macrocycle adopts a unique binding pocket formed by two parallel anthracene panels separated by a distance of 7.3 Å. Steric hindrance between the methylene protons and anthracene rings forces all four NH residues to converge toward the pocket. Using a two spherical probes method,70 the volume of the binding pocket was calculated (Fig. S109†) to be 138 Å3, which is slightly larger than cucurbit[6]uril (119 Å3) but smaller than cucurbit[7]uril (205 Å3).71
To understand the interaction between BPAT2+·2Cl− and water, a 10 ns molecular dynamics simulation under NPT ensemble at 300 K was performed.24,72 The result (Fig. 1d) shows the hydrated macrocycle accommodating an average of 8.7 water molecules within its binding pocket. The size-restricted binding pocket of BPAT2+·2Cl− compels some of these cavity water molecules to form [CH⋯π] interactions with the anthracene panels instead of hydrogen bonding with other water molecules. Consequently, these cavity water molecules only form an average of 2.96 hydrogen bonds, significantly less than the 3.62 observed in bulk water.72 Upon release to the bulk, these poorly hydrogen-bonded cavity water molecules are expected to dissociate from [CH⋯π] interactions and reform more hydrogen bonds with bulk water molecules, potentially providing a favorable change of enthalpy when guest molecules displace these cavity water molecules from the binding pocket.
Aggregation of BPAT2+·2Cl−
One distinctive property of BPAT2+·2Cl− that is not observed in previously reported Davis tetralactam macrocycles is its aggregation behavior in water.42,58 This characteristic is evidenced by the concentration-dependent behavior displayed (Fig. 2a) in 1H NMR spectra of BPAT2+·2Cl−. Below 25 μM, the macrocycle exists in its free state, characterized by a distinct set of peaks (red). As the concentration increases to 150 μM, new peaks (blue) gradually emerge in the upfield region, indicating the formation of aggregates. Above 150 μM, over 90% of the macrocycle adopts this aggregated form. The coexistence of both sets of peaks suggests a slow exchange between the free and aggregated states.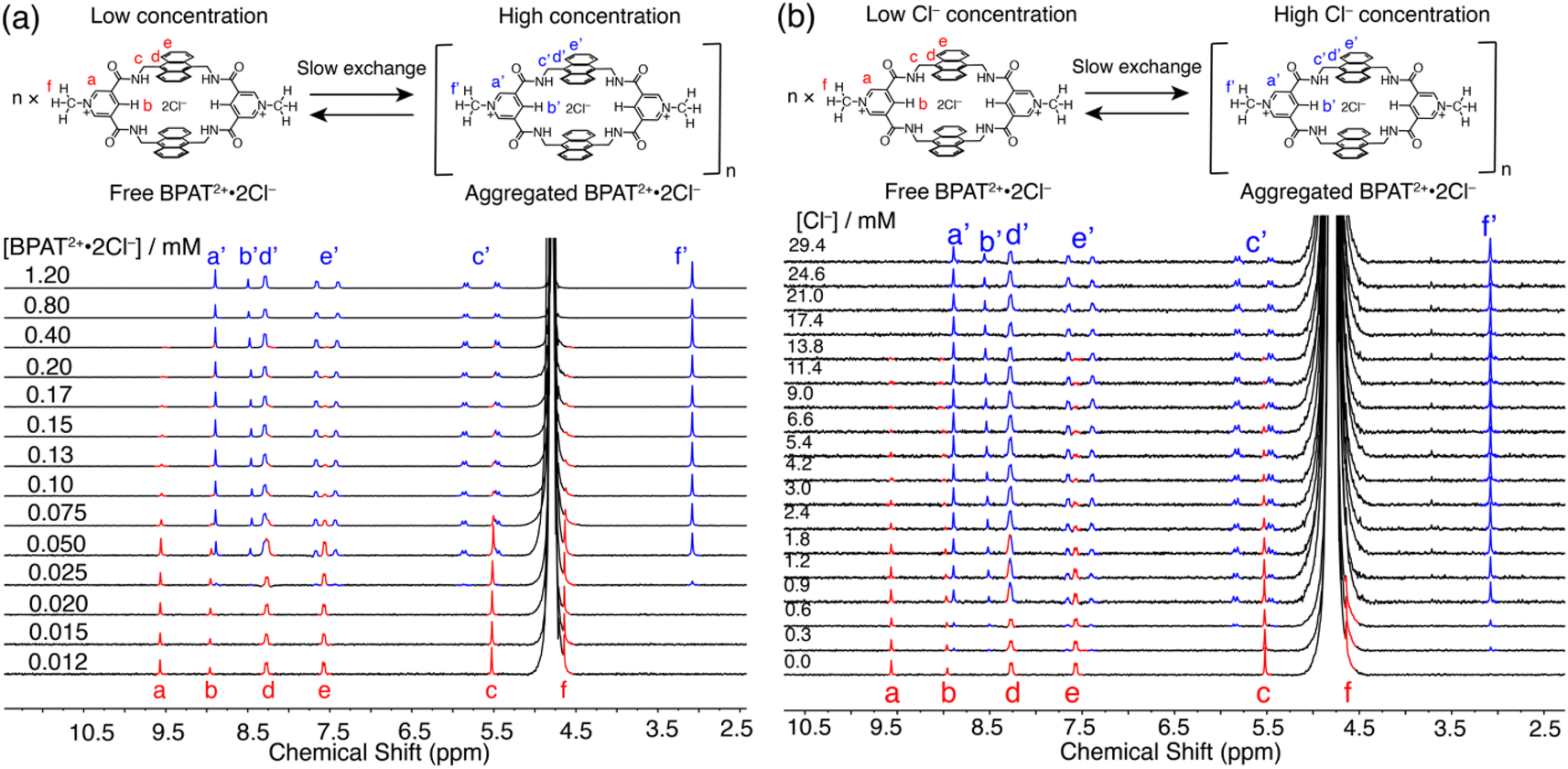 | ||
| Fig. 2 (a) 1H NMR (400 MHz, D2O) spectra of BPAT2+·2Cl− at increasing concentrations from 0.012 mM to 1.2 mM. (b) 1H NMR (400 MHz, D2O) spectra of BPAT2+·2Cl− (0.017 mM) titrated with Cl−. | ||
To further probe this aggregation behavior, diffusion-ordered spectroscopy (DOSY) experiments were performed on three BPAT2+·2Cl− samples in D2O at varying concentrations (0.02, 0.2, and 0.8 mM). The lowest concentration (0.02 mM) displayed (Fig. S21†) a diffusion coefficient of 2.31 × 106 cm2 s−1, corresponding to a hydrodynamic radius of 1.07 nm for a spherical hydrated BPAT2+·2Cl− cluster.73 Notably, at 0.2 mM, the diffusion coefficient dropped to 1.13 × 106 cm2 s−1, indicating a significantly larger hydrodynamic radius of 2.17 nm. The 0.8 mM sample revealed a slightly larger hydrodynamic radius of 2.27 nm compared to the 0.2 mM sample. The effect of concentration on the size of the aggregates was independently confirmed by atomic force microscopy (AFM) imaging. For the sample measured at a 0.02 mM concentration, particles with a molecular height of 0.7 to 1.1 nm were observed (Fig. S102†), matching the size of the macrocycle. At a concentration of 0.2 mM, larger particles with an average diameter of 2.1 nm were observed.
The aggregation behavior of BPAT2+·2Cl− is markedly dependent on temperature, as shown by variable temperature (VT) 1H NMR spectra (Fig. S22†) of a 0.2 mM sample in D2O. At temperatures below 25 °C, we observe a predominant aggregation of the sample. From 35 °C to 55 °C, there is a gradual shift from the aggregated BPAT2+·2Cl− to its free form. Above 65 °C, the macrocycle fully dissociates, indicating a complete transition to the free state. Notably, the slow exchange between these two states on the 1H NMR timescale allows for the convenient determination of equilibrium constants for the aggregation process across these temperatures by comparing (Fig. S22 and Table S1†) their differences in integration numbers. The corresponding Van't Hoff plot analysis revealed the aggregation of BPAT2+·2Cl− is an enthalpy-driven process with a large entropy penalty, suggesting the electrostatic interactions between chloride ions and the macrocycles are essential for this distinct aggregation mechanism.
To validate our hypothesis that the chloride binding acts as the driving force governing the aggregation behavior, we conducted a chloride titration experiment using a 0.017 mM solution of BPAT2+·2Cl− in D2O. Intriguingly, the gradual addition of Cl− ions promoted (Fig. 2b) the formation of a second set of peaks, mirroring the aggregation patterns observed at higher BPAT2+·2Cl− concentrations. This aggregation at such a low macrocycle concentration suggests that electrostatic interactions between the positively charged BPAT2+ and Cl− are the critical driving force to trigger the aggregation. The formation of ion pairs effectively neutralizes the charge of BPAT2+, leading to a decrease in its water solubility. This reduction in solubility, when combined with the large hydrophobic surface of the anthracene segments, induces aggregation. The Cl−-triggered aggregation was independently verified by a UV-vis titration experiment (Fig. S96†), where adding NaCl to a BPAT2+·2Cl− solution (2 μM) resulted in decreased and red-shifted absorption peaks. Notably, the hydrophobic effect is another driving force for the aggregation. When we introduced over 10% v/v CD3CN to a D2O solution of BPAT2+·2Cl− (Fig. S70†), its 1H NMR spectrum changed from the aggregated form to the free form, suggesting that the aggregation was disrupted by mitigating the hydrophobic effect.
While there is no doubt that the macrocycle aggregates at high concentrations and in the presence of chloride anions, the precise structure of the aggregate remains less clear. On the one hand DOSY and AFM measurements suggest that aggregate size is variable. On the other the NMR suggests a well-defined structure in which all receptor molecules are in the same environment but internally desymmetrized. At present we cannot resolve the precise structure of the aggregate, but the dramatic downfield shift of the pyridinium methyl protons f′ suggests (Fig. 2) that the pyridinium units are likely sandwiched between the anthracene walls, which shield the chemical shift of methyl protons. The splitting pattern observed in the 1H NMR spectrum of the aggregated state, along with the chloride-dependent aggregation behavior, suggests that chloride ions likely reside near both the anthracene panels and the pyridinium units, as seen in the crystal structure of glucose ⊂ BPAT2+·2Cl− (Fig. S99–S101†).
Evaluation of sugars binding in water
The interaction between free BPAT2+·2Cl− and sugars in water was investigated using 1H NMR titration. A solution of BPAT2+·2Cl− (17 μM) in D2O was titrated with aliquots of glucose, and 1H NMR spectra were recorded after each addition. Notably, the stacked spectra in Fig. 3a reveal that only proton b in BPAT2+·2Cl− exhibited a downfield shift upon glucose binding, suggesting its key role in the binding process. Considering the inward orientation of proton b, this observation strongly suggests glucose binding within the binding pocket through [C–H⋯O] hydrogen bonding with the macrocycle. Furthermore, the downfield shift implies a stronger [C–H⋯O] interaction between the macrocycle and glucose compared to cavity water. We conducted a separate experiment by mixing BPAT2+·2Cl− (0.4 mM) and glucose in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 molar ratios. This experiment showed (Fig. S71†) upfield shifts in the glucose protons, suggesting that glucose is encapsulated within the binding cavity of the macrocycle. We also examined (Fig. S75†) the time-dependent behavior of the binding by comparing the 1H NMR spectra of a mixture of BPAT2+·2Cl− (0.017 μM) and glucose (0.3 mM) at 0 hours and 24 hours, both of which showed identical spectra. This result indicates that glucose binding occurs rapidly and that each titration spectrum was collected at the equilibrium state of the system.
:1 and 1:2 molar ratios. This experiment showed (Fig. S71†) upfield shifts in the glucose protons, suggesting that glucose is encapsulated within the binding cavity of the macrocycle. We also examined (Fig. S75†) the time-dependent behavior of the binding by comparing the 1H NMR spectra of a mixture of BPAT2+·2Cl− (0.017 μM) and glucose (0.3 mM) at 0 hours and 24 hours, both of which showed identical spectra. This result indicates that glucose binding occurs rapidly and that each titration spectrum was collected at the equilibrium state of the system.
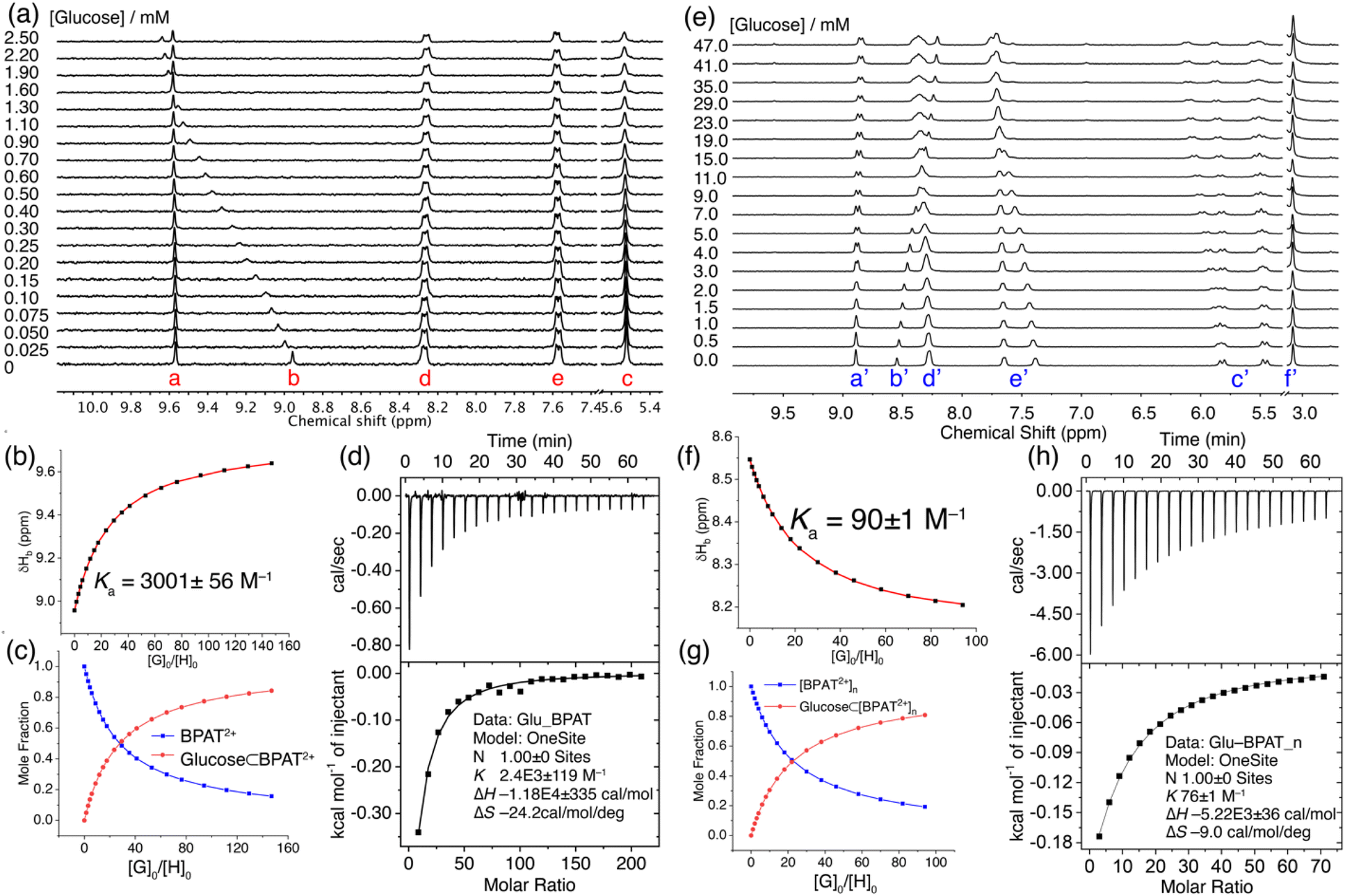 | ||
| Fig. 3 (a) 1H NMR (400 MHz, D2O) spectra of BPAT2+·2Cl− (17 μM, free state) titrated with glucose. (b) Changes in the chemical shift of proton b caused by the addition of glucose. The red trace represents non-linear fitting using a 1:1 receptor-substrate binding model. (c) Calculated changes in mole fraction of BPAT2+ and glucose ⊂ BPAT2+ in D2O as a function of the guest–host mole ratio. (d) ITC profile of BPAT2+·2Cl− (17 μM) for the binding of glucose. (e) 1H NMR (400 MHz, D2O) spectra of BPAT2+·2Cl− (0.5 mM, aggregated state) in the presence of NaCl (20 mM) titrated with glucose. (f) Changes in the chemical shift of proton b′ caused by the addition of glucose. The red trace represents non-linear fitting using a 1:1 receptor-substrate binding model. (g) Calculated changes in mole fraction of BPAT2+ and glucose ⊂ BPAT2+ in D2O as a function of the guest–host mole ratio. (h) ITC profile of BPAT2+·2Cl− (0.5 mM) in the presence of NaCl (20 mM) for the binding of glucose. | ||
Nonlinear fitting of the proton b's chemical shift changes against the guest/host ratio (Fig. 3b and c) revealed a remarkable binding affinity of (3001 ± 56) M−1.74,75 This result represents one of the highest reported affinities for synthetic lectins binding with simple sugars in water, a testament to the effectiveness of the BPAT2+·2Cl− design.36,38,39,42,52,58,76,77 Additionally, it's important to highlight that BPAT2+·2Cl− exhibits a binding affinity for glucose 5.8 times greater than that of the frequently employed lectin, concanavalin A, which possesses a binding constant (Ka) of 520 M−1.26 Compared to our previous durene-based macrocycle, replacing the smaller durene panels with larger anthracene walls impressively enhanced the affinity by a factor of 3.6.58 This significant improvement highlights the contribution of the extended π-system to the binding interaction.
Furthermore, the cationic pyridinium units are crucial in the observed high affinity. A structurally similar anionic macrocycle with an isophthalamide unit (Fig. 1a) only registered an affinity of 56 M−1, demonstrating the importance of the polarized C–H bonds provided by the pyridinium groups in BPAT2+·2Cl−.42 These additional hydrogen bond donors significantly enhance the complex stability, contributing to the high binding affinity for glucose.
The strong affinity between BPAT2+·2Cl− and glucose was further corroborated by isothermal titration calorimetry (ITC) experiments (Fig. 3d). The ITC data revealed strong exothermic peaks upon glucose binding, indicating favorable enthalpic interactions. Nonlinear fitting of the binding isotherm using a 1:1 binding model yielded a binding constant of (2400 ± 119) M−1, consistent with the 1H NMR titration results. Notably, the binding process was characterized by a strong binding enthalpy (ΔH) of −11.8 kcal mol−1, accompanied by a substantial entropy penalty (TΔS) of −7.2 kcal mol−1.
This enthalpy-driven binding suggests that the interaction is primarily governed by comprehensive hydrogen bonding between glucose and the macrocycle. The enthalpic contribution likely arises from two factors: stronger hydrogen bonds between glucose and the macrocycle compared to those involving cavity water and the release of cavity water molecules that can subsequently form additional hydrogen bonds with bulk water.72 These energetically favorable interactions outweigh the entropic cost associated with restricted conformational and translational freedom of the macrocycle and glucose within the complex.
A separate ITC experiment conducted at a lower concentration (10 μM) of BPAT2+·2Cl− yielded (Fig. S76†) a similar binding constant of (2690 ± 495) M−1, indicating that the measured binding constant is independent of the macrocycle concentration when below 20 μM. This finding also suggests that the macrocycle can remain in a free state and avoid aggregation at low micromolar concentrations. Moreover, the results confirm that the measured signal reflects glucose binding rather than any shifts in the aggregation state of the macrocycle in water.78
The binding of sugars by BPAT2+·2Cl− was examined (Table 1) through 1H NMR titration experiments. Notably, glucose exhibits the highest binding affinity. This pronounced preference underscores the vital role played by the unique arrangement of hydroxyl groups on the glucose ring, which aligns well with the binding pocket of BPAT2+·2Cl−. Methyl-β-glucoside, structurally analog to glucose but differentiated by a methyl group at the C1 position, shows a diminished affinity of (1189 ± 16) M−1. This variation implies that even subtle alterations in the sugar structure can profoundly influence binding affinity. A more striking example is methyl-α-glucoside, where the orientation of the C1 hydroxyl group is altered to an axial position, resulting in a binding affinity that drops by more than 12-fold. This observation emphasizes the criticality of the equatorial positioning of hydroxyl groups for optimal interaction with the macrocycle.26 Exploring sugars beyond glucose, including mannose, galactose, and psicose, which do not possess an all-equatorial hydroxyl group arrangement, revealed significantly weaker binding affinities. This pattern agrees with the observation that the binding pocket of BPAT2+·2Cl− is intricately tailored to accommodate the equatorial hydroxyl configuration specific to glucose. Interestingly, even cellobiose, a disaccharide comprised of two β-glucose units, only demonstrates a moderate affinity of (592 ± 16) M−1, suggesting that the binding pocket of BPAT2+·2Cl− is too small to accommodate larger sugar molecules. Similarly, maltose mirrors that of cellobiose, attributable to its glucose unit.
:1 complexes of BPAT2+·2Cl− with sugars in D2O or H2O as determined by 1H NMR titrations and ITC at 23 °C
| Carbohydrates | Free state Ka (M−1) | Aggregated state Ka (M−1) | ||
|---|---|---|---|---|
| NMR | ITC | NMR | ITC | |
| a N.D.: not determined as a result of low affinities or low heat formation. | ||||
| Glucose | 3001 ± 56 | 2410 ± 119 | 90 ± 1 | 76 ± 1 |
| Methyl-β-glucoside | 1189 ± 16 | 1470 ± 101 | 129 ± 7 | 137 ± 3 |
| Methyl-α-glucoside | 94 ± 2 | N.Da | 20 | N.Da |
| Mannose | 16 ± 1 | N.Da | 6 ± 1 | N.Da |
| GlcNAc | 171 ± 3 | N.Da | <5 | N.Da |
| Galactose | 62 ± 2 | N.Da | 7 | N.Da |
| Psicose | 71 ± 17 | 126 ± 15 | <5 | 15 ± 1 |
| Fructose | 16 ± 1 | N.Da | <5 | N.Da |
| Maltose | 568 ± 47 | 476 ± 40 | 55 ± 2 | 38 ± 2 |
| Cellobiose | 592 ± 16 | 209 ± 29 | 41 ± 1 | 23 ± 1 |
Complementary insights were gained from ITC experiments, which revealed a similar trend in binding affinities and selectivity to those observed in the 1H NMR titration. Notably, the binding of glucose exhibited a significantly higher enthalpy compared to methyl-β-glucoside. This substantial difference in binding enthalpy suggests that the macrocycle's selectivity for glucose over methyl-β-glucoside is due to the formation of a more extensive hydrogen bonding network. This aspect will be further discussed in the crystal structure analysis section. Collectively, these findings delineate the extraordinary selectivity of BPAT2+·2Cl− for glucose. The precise complementarity between the hydroxyl groups of the sugar and the macrocycle's binding pocket, coupled with the pronounced sensitivity to structural variations, highlights the sophisticated nature of this molecular recognition mechanism.
We further investigated the glucose binding properties of BPAT2+·2Cl− in its aggregated state. These studies were conducted at a high concentration of BPAT2+·2Cl− (0.5 mM), supplemented with 20 mM NaCl to enhance the stability of the aggregate.79 Our observations revealed (Fig. 3e) that the glucose binding behavior in the aggregated state of BPAT2+·2Cl− is notably different from its behavior in the free state. Specifically, we noted that the inward-facing protons b′ exhibited an upfield shift upon adding glucose. This shift suggests a decrease in hydrogen bonding strength with glucose compared to their interaction with cavity water. Concurrently, one set of anthracene protons (e′) exhibited a downfield shift, while one set of methylene signals from proton c′ split into two distinct sets of peaks. The remaining peaks from protons e′ and c′ remained almost unchanged during the entire titration process. These observations suggest that, under the conditions of our titration experiment, BPAT2+·2Cl− maintains its aggregated form while engaging in glucose binding without compromising its structural integrity.
In further analysis, we fitted the change in chemical shift for proton b′ against the guest/host ratio using a 1:1 binding model, as illustrated in Fig. 3f and g. This analysis yielded an affinity of (90 ± 1) M−1. This finding was corroborated by ITC experiments, which indicated (Fig. 3h) a binding affinity of (76 ± 1) M−1. The binding isotherm from ITC also revealed a binding enthalpy of −5.2 kcal mol−1, which is 6.6 kcal mol−1 weaker than the glucose binding observed in the free state of BPAT2+·2Cl−. The glucose binding of BPAT2+·2Cl− in its aggregated state still suffers from an entropy penalty (TΔS) of −2.6 kcal mol−1. This penalty is much lower than that observed during glucose binding in the free state of BPAT2+·2Cl−. The general trend of sugar binding by BPAT2+·2Cl− in its aggregated state mirrors its free state, albeit binding affinities reduced by factors ranging from 4–30. This result indicates that the aggregation of BPAT2+·2Cl− does not alter its glucose selectivity compared to other sugars. Computational analysis, including DFT calculations and conformational sampling, indicates (Fig S110†) that in its aggregated state, BPAT2+·2Cl− becomes rigidified due to aromatic stacking between the pyridinium units and anthracene panels. This structural rigidity compromises the macrocycle's ability to adapt its conformation to match the complex functional group distributions of sugars, lowering its binding affinity for sugars. A more detailed discussion on computational analysis of the binding in free and aggregated states can be found in ESI.† These findings suggest that when designing hydrogen-bonding receptors for substrates with multiple binding sites, incorporating a certain level of structural flexibility is crucial, even while aiming for a preorganized binding pocket.
The formation of a complex between glucose and BPAT2+·2Cl− was further solidified by two additional experiments. Nuclear Overhauser Effect Spectroscopy (NOESY) provided crucial spatial information, revealing several through-space correlation peaks between protons b, d, and e on the macrocycle and C–H protons on glucose (Fig. S19†). This observation directly confirms the proximity between the two molecules in D2O. High-resolution mass spectrometry (HRMS) provided further validation, with a peak at m/z 979.3444 matching the theoretical m/z of 979.3433 for the [BPAT2+·glucose·Cl−]+ adduct (Fig. S4 and S5†) with a molecular formula of [C54H52ClN6O10]+. These findings conclusively demonstrate the stoichiometry and formation of the complex.
Structure elucidation by X-ray crystallography
Single crystals of glucose ⊂ BPAT2+·2Cl− were successfully grown by slowly evaporating an aqueous solution containing glucose (50 mM), BPAT2+·2Cl− (0.5 mM), and NaCl (20 mM) over three months. In this structure (Fig. 4a), each glucose molecule forms eight hydrogen bonds with the inward-facing CH and NH hydrogen bond donors of the macrocycle. Additionally, the glucose's axial C–H bonds are sandwiched between two parallel anthracene panels spaced 7.9 Å apart, an optimal distance for establishing multiple [C–H⋯π] interactions.21,36,80,81 This spacing, which extended from 7.3 Å to 7.9 Å, highlights the binding pocket's flexibility in BPAT2+·2Cl−. By comparing this crystal structure with the one reported56 by Davis, we discovered (Fig. 4d–i) that our receptor exhibits a different binding pattern with glucose, despite the structural similarity between the two macrocycles. First, we observed that one side of our macrocycle forms hydrogen bonds with the C3 and C4 hydroxyl groups on glucose, while the other side forms hydrogen bonds with the C6 hydroxyl group and the pyranose oxygen. In contrast, Davis's macrocycle only forms hydrogen bonds with the hydroxyl groups on the C1, C2, C3, and C4 carbons. Second, our macrocycle covers 91% of the van der Waals surface of the glucose, whereas Davis's macrocycle covers only 76% of the glucose surface area.65,82 This difference in the hydrogen bonding pattern allows our macrocycle to create a larger contact surface, which we believe is the primary reason for the higher affinities observed in our system. This result also suggests that the binding of carbohydrates is highly sensitive to slight modifications in the receptor's structure, which is an important consideration when designing new receptors for sugars.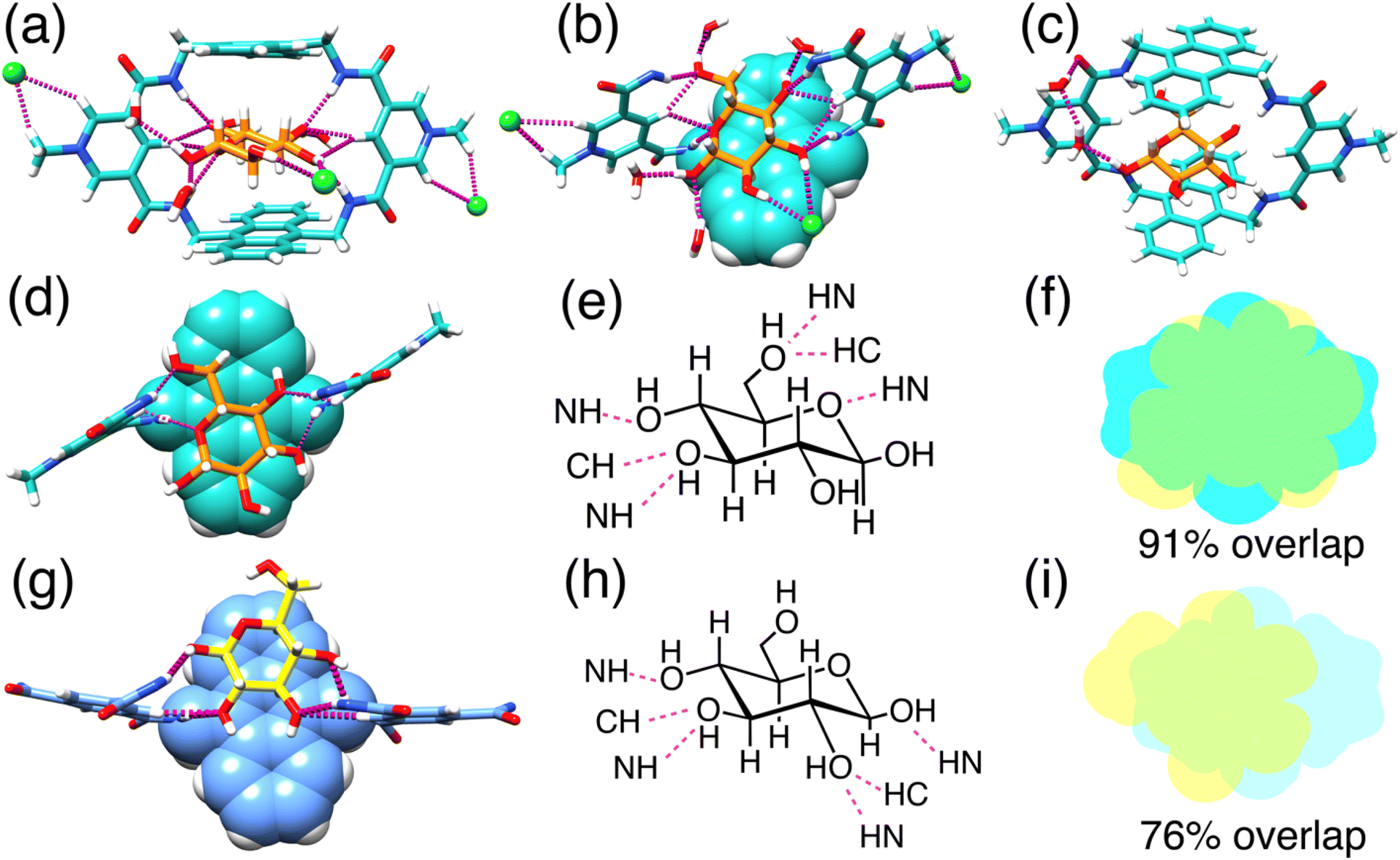 | ||
| Fig. 4 X-ray single crystal structures. Front view (a) and top truncated view (b) of glucose encapsulated in the binding cavity of BPAT2+·2Cl− with two chloride anions binding at the allosteric sites. (c) Indirect interaction between the 1-OH group of the glucose and the carbonyl group of the BPAT2+ through hydrogen bonding over two water molecules as a bridge. A truncated view (d) of the single crystal structure of glucose ⊂ BPAT2+ and the structural formula (e) indicating the detailed binding pattern. Overlapped map (f) of glucose and the anthracene panel in the complex of glucose ⊂ BPAT2+. A truncated view (g) of the single crystal structure of glucose ⊂ Davis macrocycle and the structural formula (h) indicating the detailed binding pattern. Overlapped map (i) of glucose and the anthracene panel in the complex of glucose ⊂ Davis macrocycle. | ||
Notably, the glucose remains hydrated within the hydrophobic binding pocket of our macrocycle, engaging in hydrogen bonding with four external water molecules. The 2 and 3-hydroxyl groups of the glucose also interact with a chloride anion via hydrogen bonding. In total (Fig. 4b and S108†), each glucose substrate is involved in 14 hydrogen bonds, encompassing interactions with the macrocycle, chloride ions, and water molecules. A particularly intriguing finding (Fig. 4c) is an indirect interaction of the 1-hydroxyl group of glucose with a carbonyl group from the macrocycle, mediated by two water bridges. This observation suggests a novel role for solvent water in receptor-substrate binding, facilitating indirect hydrogen bonding. This additional hydrogen bonding could be responsible for the higher glucose selectivity over the methyl-β-glucoside.
Each macrocycle engages (Fig. 4a) in ion pairing with two Cl− anions. These ion pairs are sustained (Fig. S107†) by [C–H⋯Cl−] hydrogen bonds formed between the chloride anion and the protons of both the methyl group and the ortho protons on the pyridinium units, further stabilized by electrostatic interactions. These chloride binding sites, situated externally to the main binding pocket, act as allosteric regulators, influencing the macrocycle's affinity for glucose.
Glucose sensing
To explore the potential of BPAT2+·2Cl− as a glucose-responsive material, we assessed its sensitivity to variations in glucose concentrations in both free and aggregated states. In its free state, BPAT2+·2Cl− reaches saturation at a 2 mM concentration due to its high affinity for glucose, as shown in Fig. 5a. As a result, within the physiological glucose concentration range of 4 to 15 mM, further increases in glucose levels cause negligible changes in the circular dichroism (CD) signal. Conversely, in its aggregated state, BPAT2+·2Cl− exhibits lower affinity, leading (Fig. 5b) to a noticeable variation in the induced CD signals within the physiological range of 5–10 mM glucose concentrations. This characteristic of showing a continuous response to glucose concentration fluctuations within the physiological range underscores its value in the development of glucose-responsive materials based on synthetic receptors. Thus, despite its lower affinity, aggregated BPAT2+·2Cl− offers distinct advantages in responding to changes in physiological glucose levels compared to the free BPAT2+·2Cl−, which shows stronger binding for glucose.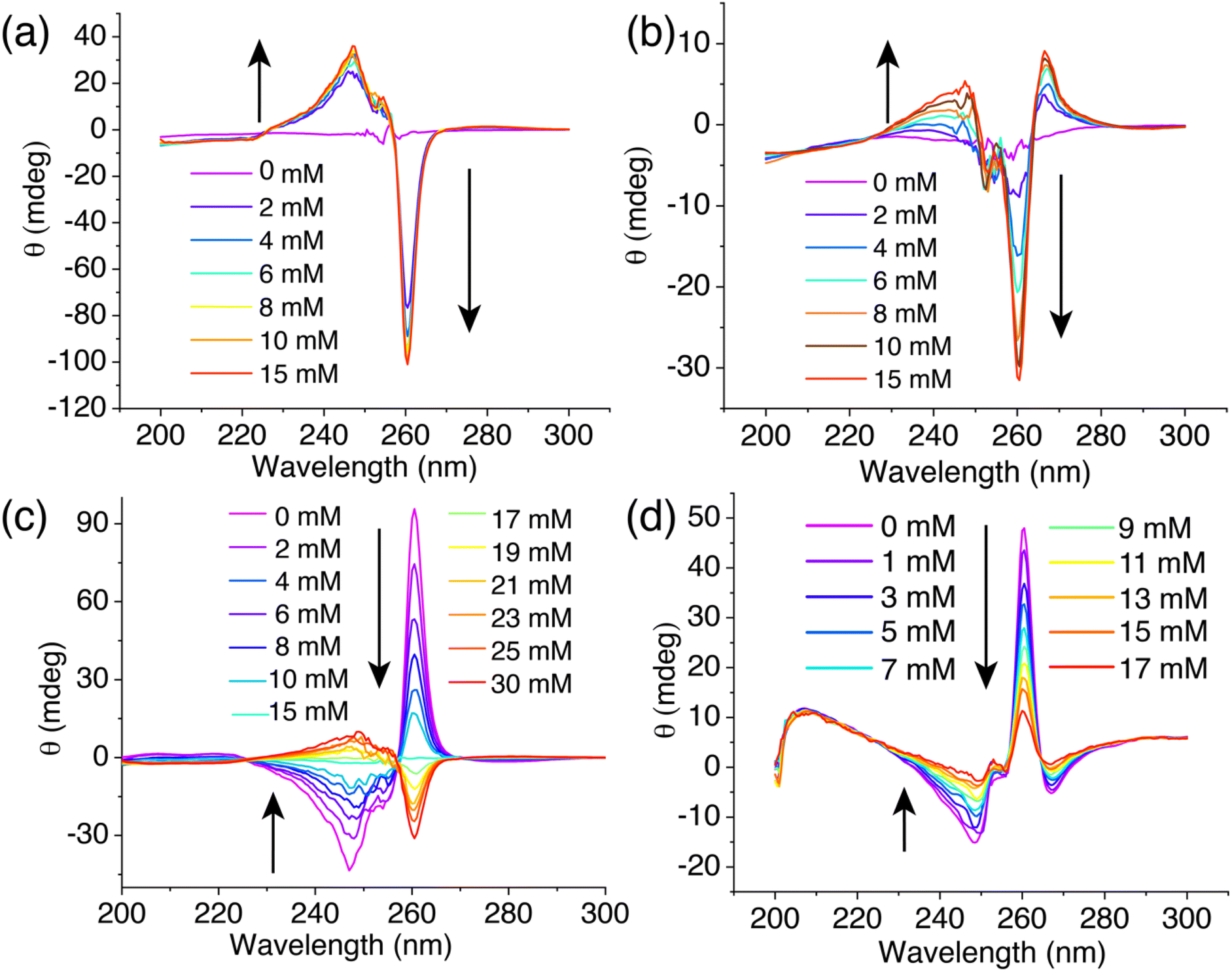 | ||
| Fig. 5 (a) CD spectra of BPAT2+·2Cl− (10 μM) in its free state with increasing glucose concentrations. (b) CD spectra of BPAT2+·2Cl− (10 μM) in its aggregated state in an aqueous solution of NaCl (20 mM) with increasing glucose concentration. (c) CD spectra of BPAT2+·2Cl− (10 μM) and L-glucose (15 mM) in H2O titrated with D-glucose ranging from 0 to 30 mM. (d) CD spectra of BPAT2+·2Cl− (10 μM) and L-glucose (50 mM) in a 1× PBS buffer titrated with D-glucose from 0 to 17 mM. | ||
To further demonstrate the potential of BPAT2+·2Cl− as a glucose sensor, we introduced L-glucose as both an indicator and competitor. Notably, L-glucose induced (Fig. S97†) a reverse cotton effect in the CD spectrum of the complex. This approach effectively addresses the saturation issue of free BPAT2+ at physiological glucose concentrations.35 In an experiment, a mixture of BPAT2+·2Cl− (10 μM) and L-glucose (15 mM) in water produced a positive induced CD signal at 260 nm. The subsequent addition of D-glucose gradually displaced (Fig. 5c) the L-glucose, reducing the CD signal to zero when an equal amount of D-glucose (15 mM) was added. Adding more D-glucose further decreased the CD signal, which then shifted into the negative region. This displacement method allows for adjusting the sensitivity across a broad dynamic range by controlling L-glucose concentration.
We also tested this glucose sensing approach in more complex media, specifically in 1× PBS buffer with high salt concentrations (NaCl: 137 mM, KCl: 2.7 mM, Na2HPO4: 10 mM, NaH2PO4: 1.8 mM). To prevent (Fig. S69†) aggregation of BPAT2+·2Cl−, we first prepared a mixture of L-glucose (50 mM) and BPAT2+·2Cl− (10 μM) in water, then lyophilized it and redissolved it in 1× PBS buffer. CD spectra were recorded following the addition of D-glucose. We observed (Fig. 5c) a continuous decrease in the induced CD signal at 260 nm as the D-glucose concentration gradually increased from 0 to 17 mM. This finding indicates the efficacy of BPAT2+·2Cl− in glucose sensing within physiologically relevant concentrations in the competitive environment of PBS buffer.
Conclusion
In conclusion, our study introduces a hydrogen-bonding receptor that effectively binds sugars in water, demonstrating a remarkable binding affinity of 3001 M−1 for glucose. The remarkable improvement in carbohydrate binding performance compared to a structurally similar macrocycle underscores that carbohydrate binding is highly sensitive to slight modifications in the receptor's structure. This insight is an important consideration when designing new receptors for sugars. This receptor exhibits distinctive aggregation behavior triggered by chloride binding at the allosteric sites, which, while enhancing structural rigidity, leads to reduced sugar binding affinities. These findings suggest that when designing hydrogen-bonding receptors for substrates with multiple binding sites, it is essential to balance structural flexibility with preorganization to ensure effective binding. Moreover, our synthetic lectin showcases exceptional sensitivity as a glucose sensor in various aqueous media, including water and PBS buffer, underlining its potential for broad biomedical applications, from glucose-responsive devices to sensors and therapeutic agents.27,28,30–32,83,84 Future efforts will focus on optimizing the receptor to eliminate aggregation, facilitating the development of advanced glucose-responsive materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. Z. and W. L. conceived and designed the project and co-wrote the manuscript. C. Z., with assistance from E. Z., A. M., C. X., Y. C., H. W., C. D., J. C., X. W., and L. Y., conducted most of the experiments and analyzed the data. DOSY experiments were performed and analyzed by X. W. and L. Y. H. Z. and K. W. conducted the AFM studies and analyzed the results. Computational analysis was carried out by A. M. and W. L. L. W. solved the crystal structure.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
W.L acknowledges the financial support from the University of South Florida start-up funding and the National Science Foundation (Award No. CHE-2337419). K. W. acknowledges financial support from U.S. Department of Energy, Basic Energy Sciences (Award No. DE-SC0024924) that enabled the scanning probe microscopy studies. This research used the XRAY and CPAS Core facilities at the University of South Florida. The computational resources provided by the CIRCE research cluster facility at the University of South Florida partially supported the research.Notes and references
- J. Hatai and C. Schmuck, Acc. Chem. Res., 2019, 52, 1709–1720 CrossRef CAS.
- C. Schmuck, Coord. Chem. Rev., 2006, 250, 3053–3067 CrossRef CAS.
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS.
- L. P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 CrossRef CAS PubMed.
- C. L. Schreiber and B. D. Smith, Nat. Rev. Chem, 2019, 3, 393–400 CrossRef CAS.
- W. Liu, S. K. Samanta, B. D. Smith and L. Isaacs, Chem. Soc. Rev., 2017, 46, 2391–2403 RSC.
- M. Giese, J. Niemeyer and J. Voskuhl, ChemPlusChem, 2020, 85, 985–997 CrossRef CAS.
- C. Schmuck and M. Schwegmann, J. Am. Chem. Soc., 2005, 127, 3373–3379 CrossRef CAS.
- C. Schmuck and L. Geiger, J. Am. Chem. Soc., 2005, 127, 10486–10487 CrossRef CAS.
- C. Schmuck and L. Geiger, J. Am. Chem. Soc., 2004, 126, 8898–8899 CrossRef CAS PubMed.
- J. Samanta, M. Tang, M. Zhang, R. P. Hughes, R. J. Staples and C. Ke, J. Am. Chem. Soc., 2023, 145, 21723–21728 CrossRef CAS PubMed.
- Y. Chen, G. Wu, L. Chen, L. Tong, Y. Lei, L. Shen, T. Jiao and H. Li, Org. Lett., 2020, 22, 4878–4882 CrossRef CAS.
- Y. Wu, C. Zhang, S. Fang, D. Zhu, Y. Chen, C. Ge, H. Tang and H. Li, Angew. Chem., Int. Ed., 2022, 61, e202209078 CrossRef CAS.
- F.-Y. Chen, W.-C. Geng, K. Cai and D.-S. Guo, Chin. Chem. Lett., 2023, 109161 Search PubMed.
- R. Fu, Q. Zhao, H. Han, W. Li, F. Chen, C. Tang, W. Zhang, S. Guo, D. Li, W. Geng, D. Guo and K. Cai, Angew. Chem., Int. Ed., 2023, 135, e202315990 CrossRef.
- R. Wang, W. Li, J. Deng, H. Han, F. Chen, D. Li, L. Jing, Z. Song, R. Fu, D. Guo and K. Cai, Angew. Chem., Int. Ed., 2023, 136, e202317402 CrossRef.
- J. M. Yang, Y. Q. Chen, Y. Yu, P. Ballester and J. Rebek, J. Am. Chem. Soc., 2021, 143, 19517–19524 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- S. C. Patrick, P. D. Beer and J. J. Davis, Nat. Rev. Chem, 2024, 8, 256–276 CrossRef PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS.
- C. S. Webster, F. Balduzzi and A. P. Davis, Org. Biomol. Chem., 2022, 21, 525–532 RSC.
- G. B. Huang, S. H. Wang, H. Ke, L. P. Yang and W. Jiang, J. Am. Chem. Soc., 2016, 138, 14550–14553 CrossRef CAS PubMed.
- L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed.
- H. Yao, H. Ke, X. Zhang, S. J. Pan, M. S. Li, L. P. Yang, G. Schreckenbach and W. Jiang, J. Am. Chem. Soc., 2018, 140, 13466–13477 CrossRef CAS.
- Z. Sun, B. Fan and M. J. Webber, ChemSystemsChem, 2023, 5, e202200050 CrossRef CAS.
- A. P. Davis, Chem. Soc. Rev., 2020, 49, 2531–2545 RSC.
- D. H. C. Chou, M. J. Webber, B. C. Tang, A. B. Lin, L. S. Thapa, D. Deng, J. V. Truong, A. B. Cortinas, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 2401–2406 CrossRef CAS PubMed.
- M. A. VandenBerg and M. J. Webber, Adv. Healthcare Mater., 2019, 8, 1801466 CrossRef.
- S. Yu, S. Xian, Z. Ye, I. Pramudya and M. J. Webber, J. Am. Chem. Soc., 2021, 143, 12578–12589 CrossRef CAS.
- J. Wang, J. Yu, Y. Zhang, X. Zhang, A. R. Kahkoska, G. Chen, Z. Wang, W. Sun, L. Cai, Z. Chen, C. Qian, Q. Shen, A. Khademhosseini, J. B. Buse and Z. Gu, Sci. Adv., 2019, 5, eaaw4357 CrossRef CAS PubMed.
- J. Yu, J. Wang, Y. Zhang, G. Chen, W. Mao, Y. Ye, A. R. Kahkoska, J. B. Buse, R. Langer and Z. Gu, Nat. Biomed. Eng., 2020, 4, 499–506 CrossRef CAS.
- Y. Zeng, J. Wang, Z. Gu and Z. Gu, Med. Drug Discovery, 2019, 3, 100010 CrossRef.
- S. Tommasone, F. Allabush, Y. K. Tagger, J. Norman, M. Köpf, J. H. R. Tucker and P. M. Mendes, Chem. Soc. Rev., 2019, 48, 5488–5505 RSC.
- O. Francesconi and S. Roelens, ChemBioChem, 2019, 20, 1329–1346 CrossRef CAS.
- R. A. Tromans, S. K. Samanta, A. M. Chapman and A. P. Davis, Chem. Sci., 2020, 11, 3223–3227 RSC.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed.
- P. Stewart, C. M. Renney, T. J. Mooibroek, S. Ferheen and A. P. Davis, Chem. Commun., 2018, 54, 8649–8652 RSC.
- P. Ríos, T. J. Mooibroek, T. S. Carter, C. Williams, M. R. Wilson, M. P. Crump and A. P. Davis, Chem. Sci., 2017, 8, 4056–4061 RSC.
- P. Rios, T. S. Carter, T. J. Mooibroek, M. P. Crump, M. Lisbjerg, M. Pittelkow, N. T. Supekar, G. J. Boons and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 3387–3392 CrossRef CAS PubMed.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS PubMed.
- T. S. Carter, T. J. Mooibroek, P. F. N. Stewart, M. P. Crump, M. C. Galan and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 9311–9315 CrossRef CAS.
- C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef CAS PubMed.
- F. Milanesi, N. Burrini, G. Corti, S. Roelens and O. Francesconi, Chem.–Eur. J., 2024, e202401771 CrossRef CAS.
- F. Milanesi, L. Unione, A. Ardá, C. Nativi, J. Jiménez-Barbero, S. Roelens and O. Francesconi, Chem.–Eur. J., 2023, 29, e202203591 CrossRef CAS.
- O. Francesconi, F. Milanesi, C. Nativi and S. Roelens, Chem.–Eur. J., 2021, 27, 10456–10460 CrossRef CAS PubMed.
- O. Francesconi, F. Cicero, C. Nativi and S. Roelens, ChemPhysChem, 2020, 21, 257–262 CrossRef CAS.
- O. Francesconi, M. Martinucci, L. Badii, C. Nativi and S. Roelens, Chem.–Eur. J., 2018, 24, 6828–6836 CrossRef CAS PubMed.
- E. Klein, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2005, 44, 298–302 CrossRef CAS.
- N. P. Barwell, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 7673–7676 CrossRef CAS PubMed.
- Y. Ferrand, E. Klein, N. P. Barwell, M. P. Crump, J. Jiménez-Barbero, C. Vicent, G. Boons, S. Ingale and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 1775–1779 CrossRef CAS PubMed.
- B. Sookcharoenpinyo, E. Klein, Y. Ferrand, D. B. Walker, P. R. Brotherhood, C. Ke, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2012, 51, 4586–4590 CrossRef CAS PubMed.
- H. Destecroix, C. M. Renney, T. J. Mooibroek, T. S. Carter, P. F. N. Stewart, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2057–2061 CrossRef CAS.
- B. J. J. Timmer, A. Kooijman, X. Schaapkens and T. J. Mooibroek, Angew. Chem., Int. Ed., 2021, 60, 16178–16183 CrossRef CAS.
- B. J. J. Timmer and T. J. Mooibroek, Eur. J. Org Chem., 2021, 2021, 4218–4223 CrossRef CAS.
- X. Schaapkens, R. N. van Sluis, E. O. Bobylev, J. N. H. Reek and T. J. Mooibroek, Chem.–Eur. J., 2021, 27, 13719–13724 CrossRef CAS PubMed.
- P. K. Mandal, B. Kauffmann, H. Destecroix, Y. Ferrand, A. P. Davis and I. Huc, Chem. Commun., 2016, 52, 9355–9358 RSC.
- T. Hayashi, Y. Ohishi, H. Abe and M. Inouye, J. Org. Chem., 2020, 85, 1927–1934 CrossRef CAS PubMed.
- C. Zhai, C. Xu, Y. Cui, L. Wojtas and W. Liu, Chem.–Eur. J., 2023, 29, e202300524 CrossRef CAS PubMed.
- J. C. Lauer, A. S. Bhat, C. Barwig, N. Fritz, T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2022, 28, e202201527 CrossRef CAS.
- K. G. Andrews, T. K. Piskorz, P. N. Horton and S. J. Coles, J. Am. Chem. Soc., 2024, 146, 17887–17897 CrossRef CAS PubMed.
- O. Francesconi, F. Milanesi, C. Nativi and S. Roelens, Angew. Chem., Int. Ed., 2021, 133, 11268–11272 CrossRef.
- J. J. Gassensmith, E. Arunkumar, L. Barr, J. M. Baumes, K. M. DiVittorio, J. R. Johnson, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2007, 129, 15054–15059 CrossRef CAS.
- R. S. Das, D. Maiti, S. Kar, T. Bera, A. Mukherjee, P. C. Saha, A. Mondal and S. Guha, J. Am. Chem. Soc., 2023, 145, 20451–20461 CrossRef CAS PubMed.
- E. M. Peck, W. Liu, G. T. Spence, S. K. Shaw, A. P. Davis, H. Destecroix and B. D. Smith, J. Am. Chem. Soc., 2015, 137, 8668–8671 CrossRef CAS.
- W. Liu, S. Bobbala, C. L. Stern, J. E. Hornick, Y. Liu, A. E. Enciso, E. A. Scott and J. Fraser Stoddart, J. Am. Chem. Soc., 2020, 142, 3165–3173 CrossRef CAS.
- W. Liu, C. Lin, J. A. Weber, C. L. Stern, R. M. Young, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 8938–8945 CrossRef CAS.
- W. Liu, Y. Tan, L. O. Jones, B. Song, Q. H. Guo, L. Zhang, Y. Qiu, Y. Feng, X. Y. Chen, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 15688–15700 CrossRef CAS.
- R. S. Das, A. Mukherjee, S. Kar, T. Bera, S. Das, A. Sengupta and S. Guha, Org. Lett., 2022, 24, 5907–5912 CrossRef CAS PubMed.
- A. Martinez-Cuezva, L. V. Rodrigues, C. Navarro, F. Carro-Guillen, L. Buriol, C. P. Frizzo, M. A. P. Martins, M. Alajarin and J. Berna, J. Org. Chem., 2015, 80, 10049–10059 CrossRef CAS PubMed.
- J. B. Maglic and R. Lavendomme, J. Appl. Crystallogr., 2022, 55, 1033–1044 CrossRef CAS PubMed.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577 CrossRef CAS.
- F. Biedermann, W. M. Nau and H. J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS.
- S. Ivanova, P. Adamski, E. Köster, L. Schramm, R. Fröhlich and F. Beuerle, Chem.–Eur. J., 2023, e202303318 Search PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 829–835 CrossRef CAS PubMed.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew Chem. Int. Ed. Engl., 1994, 33, 2207–2209 CrossRef.
- C. M. Renney, G. Fukuhara, Y. Inoue and A. P. Davis, Chem. Commun., 2015, 51, 9551–9554 RSC.
- A separate titration of BPAT2+·2Cl− at 0.2 mM using glucose without NaCl revealed (Fig. S68†) that part of the aggregated BPAT2+·2Cl−dissociated into its free state upon glucose binding. The apparent binding affinity under this condition was determined to be (783 ± 62) M−1 using ITC (Fig. S94†). Over the course of 24 hours, glucose binding gradually disrupted the aggregation of BPAT2+·2Cl− (Fig. S72†), leading to the formation of individual glucose ⊂ BPAT2+ complexes. The 1H NMR spectrum of these complexes matches the spectrum measured at a low concentration (0.017 mM) of BPAT2+·2Cl−, where it exists in its free state. In contrast, no dissociation of the aggregation was observed (Fig. S73 and S74†) over 25 days in the presence of NaCl, suggesting NaCl can significantly stabilize the aggregate..
- E. Jiménez-Moreno, A. M. Gómez, A. Bastida, F. Corzana, G. Jiménez-Oses, J. Jiménez-Barbero and J. L. Asensio, Angew. Chem., Int. Ed., 2015, 54, 4344–4348 CrossRef PubMed.
- J. L. Asensio, A. Ardá, F. J. Cañada and J. Jiménez-Barbero, Acc. Chem. Res., 2013, 46, 946–954 CrossRef CAS PubMed.
- E. J. Dale, N. A. Vermeulen, A. A. Thomas, J. C. Barnes, M. Juríček, A. K. Blackburn, N. L. Strutt, A. A. Sarjeant, C. L. Stern, S. E. Denmark and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 10669–10682 CrossRef CAS.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- Y. Xiang, S. Xian, R. C. Ollier, S. Yu, B. Su, I. Pramudya and M. J. Webber, J. Controlled Release, 2022, 348, 601–611 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2322713. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06190j |
| This journal is © The Royal Society of Chemistry 2024 |