
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacilitating intrinsic delayed fluorescence of conjugated emitters by inter-chromophore interaction†
Yixuan
Gao‡
a,
Yingman
Sun‡
b,
Zilong
Guo
 a,
Guo
Yu
a,
Yaxin
Wang
a,
Yan
Wan
c,
Yandong
Han
d,
Wensheng
Yang
d,
Dongbing
Zhao
*b and
Xiaonan
Ma
*a
a,
Guo
Yu
a,
Yaxin
Wang
a,
Yan
Wan
c,
Yandong
Han
d,
Wensheng
Yang
d,
Dongbing
Zhao
*b and
Xiaonan
Ma
*a
aInstitute of Molecular Plus, Tianjin University, Tianjin 300072, P. R. China. E-mail: xiaonanma@tju.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: dongbing.chem@nankai.edu.cn
cCollege of Chemistry, Beijing Normal University, Beijing 100875, P. R. China
dEngineering Research Center for Nanomaterials, Henan University, Kaifeng 475004, P. R. China
First published on 20th September 2024
Abstract
Delayed fluorescence (DF) is a unique emitting phenomenon of great interest for important applications in organic optoelectronics. In general, DF requires well-separated frontier orbitals, inherently corresponding to charge transfer (CT)-type emitters. However, facilitating intrinsic DF for local excited (LE)-type conjugated emitters remains very challenging. Aiming to overcome this obstacle, we demonstrate a new molecular design strategy with a DF-inactive B,N-multiple resonance (MR) emitter as a model system. Without the necessity of doping with heavy atoms, we synthesized a co-facial dimer in which an excimer-like state (Sexc) was expected to facilitate efficient reverse intersystem crossing (RISC, T1 → Sexc) and intrinsic DF. Benefiting from greatly enhanced SOC and reduced ΔEST, the proof-of-concept emitter Np-2CzB exhibited kRISC up to 6.5 × 105 s−1 and intrinsic DF with >35% contribution (ΦDF/ΦF) in dilute solution. Further investigation indicated that Sexc state formation relies on an optimized co-facial distance (d = ∼4.7 Å), strong inter-chromophore interaction (Jcoul > 450 cm−1) and a rigid structure (ΓS1→S0 < 350 cm−1). Although our strategy was demonstrated with a B,N-MR emitter, it can be applicable to many LE-type conjugated emitters without intrinsic DF. By triggering potential DF emission, many classic emitters might play a more important role in optoelectronics.
Introduction
The thermally activated delayed fluorescence (TADF) of organic emitters refers to long-lived fluorescent emission occurring with a lifetime similar to phosphorescence, which is usually facilitated by efficient reverse intersystem crossing (RISC) from a populated triplet to a fluorescent singlet state.1–3 In the area of photophysics, TADF is actually a rather traditional concept. For instance, the unusually strong DF of C70 was successfully observed under appropriate conditions,4 while the DF of Cy-5 dye was demonstrated to be feasible with remarkable back isomerization of the T1 state.5 Benefitting from the pioneering work of Adachi et al., TADF emitters received intensive attention for application in the development of electroluminescent devices.6–8In devices, the recombination of injected electrons and holes creates 25% singlet and 75% triplet excitons, in which the TADF mechanism is a feasible way of harvesting generated dark triplet excitons.9,10 For organic light-emitting diode (OLED) devices, efficient triplet harvesting is regarded as a primary factor for achieving qualified external quantum efficiency (ηEQE), which can usually be expressed as a product of several contributing terms:11,12
| ηEQE = γ × ηEUE × ΦF × ηout | (1) |
However, for the majority of organic emitters, acquiring TADF emission remains challenging. In order to convert a T1 population to the S1 state, the RISC transition should be fast enough to compete kinetically with radiative and non-radiative T1 → S0 decay.23–26 According to Fermi's golden rule, the RISC rate (kRISC) is related to spin–orbit coupling (SOC) matrix elements and the S1–T1 energy gap (ΔEST):2,27,28
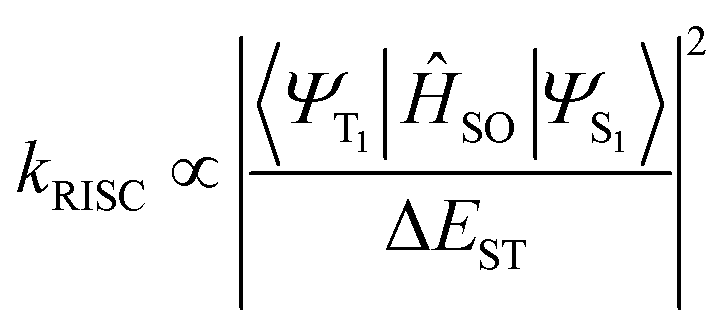 | (2) |
However, for organic emitters with the S1 state dominated by LE (π → π*) excitation, i.e. most conjugated emitters with a rigid polycyclic aromatic framework, an efficient RISC transition is usually inherently blocked. Firstly, the SOC Hamiltonian can be approximately described as:41,42
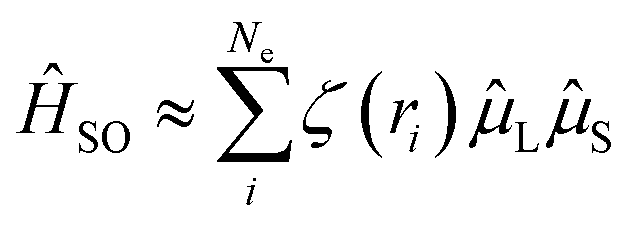 | (3) |
For instance, with typical conjugated frameworks, multiple resonance (MR) emitters have received enormous attention since the first report by Hatakeyama et al. in 2016.46–50 As B,N-doped conjugated chromophores, B,N-MR emitters exhibit an LE-type (π → π*) S1 state and low-frequency mode dominated vibronic coupling, leading to a high fluorescence quantum yield (ΦF) and color purity.51–54 However, many reported B,N-MR emitters were recognized as TADF-inactive in dilute solution (kRISC = 103–104 s−1) due to weak SOC and large ΔEST, leading to a nearly negligible contribution from intrinsic TADF, i.e. ΦDF/ΦF < 5%.46,55,56 Plenty of efforts have been made to overcome the inherent barrier, such as heavy atom (S, Se, etc.) doping57–59 and long-range CT mixing in the S1 state, by which the kRISC of B,N-MR emitters were promoted to 105–106 s−1.60
On the other hand, it was found that the RISC of many TADF-inactive B,N-MR emitters can be greatly boosted in doped films with a specific host, such as DABNA-1 in mCBP and TBN-TPA in 2,6-DCzppy.46,61 The underlying mechanism for this intriguing phenomenon was successfully uncovered by Chou and co-workers.62 In doped films, the S1 state of B,N-MR emitters can form an exciplex-like state with the host. As a result, the greatly changed electronic configuration of the emitting state (exciplex) leads to enhanced SOC, while ΔEST can be accordingly reduced as the exciplex is energetically lower than the S1 state. The landmark work of Chou et al. greatly inspired us to explore a new strategy for facilitating the intrinsic TADF of conjugated emitters. As an analog of the guest–host interaction, the intramolecular excimer-like state facilitated by delicate inter-chromophore interaction might lead to similarly enhanced SOC and reduced ΔEST, resulting in greatly boosted kRISC without the presence of a specific host, i.e. intrinsic TADF. More importantly, as a host is no longer included, this strategy may be useful for many conjugated emitters in addition to specific types of emitters. It is worth noting that the strategy of inter-chromophore interaction has been employed to improve the ηEQE of small-molecule (first-generation) OLED by the formation of excimer-like states,63–65 while controllable inter-chromophore interaction by rational molecular design has recently been used for emission switching that can rapidly respond to external stimuli in aggregated states.66 However, to the best of our knowledge, intrinsic TADF enabled by inter-chromophore interaction has rarely been reported.
In this work, we demonstrate a new strategy for facilitating the intrinsic TADF of conjugated emitters by inter-chromophore interaction. With a TADF-inactive B,N-MR emitter (CzB) as a model system, CzB dimers (BiPh-2CzB and Np-2CzB) with different linkers were synthesized. The fs-TA spectra indicated the formation of an excimer-like state (S1 → Sexc, ∼250 ps) in the S1 state decay of a proof-of-concept emitter Np-2CzB, leading to efficient RISC (T1 → Sexc) with kRISC up to 6.5 × 105 s−1 and intrinsic TADF with ΦDF/ΦF > 35% in dilute solution (10−5 M). Further investigation indicated that the Sexc formation of Np-2CzB is associated with at least three factors: (1) a suitable inter-chromophore distance (d = ∼4.7 Å); (2) strong electronic coupling for excimer formation (Jcoul > 450 cm−1); and (3) a rigid co-facial geometry (ΓS1→S0 < 350 cm−1). The formed Sexc state leads to a nearly degenerate Sexc/T1 state (ΔEST < 20 meV) and enhanced SOC due to the very different electronic configurations of the Sexc state, which all contribute to the intrinsic TADF of the proof-of-concept emitter Np-2CzB. Please note that the strategy described in this work should not be limited to B,N-MR emitters. In terms of the underlying photophysics, it can be generalized to any organic conjugated emitters with an S1 state dominated by LE (π → π*) excitation.
Results and discussion
Molecular design
Here we aim to develop a new strategy which can potentially facilitate the intrinsic TADF of conjugated emitters by inter-chromophore interaction and the corresponding formed excimer-like state (Sexc). We selected the B,N-MR emitter CzB, i.e. 2,6-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)boron, as a model chromophore, which was demonstrated to be lacking intrinsic TADF in solution (kRISC = ∼1.5 × 104 s−1).67,68 It is well known that an excimer can usually form between chromophores with co-facial geometry, while the co-facial distance (d) might be the primary factor for the formation of an Sexc state.69–72 Therefore, we designed two co-facial CzB dimers with different linkers. Please note that dimerization of B,N-MR frameworks has been widely employed in co-planar geometry to extend conjugation and in a helicene structure for chiroptical responses,73–75 but co-facial dimerization of B,N-MR emitters to facilitate an Sexc state has rarely been reported.As shown in Scheme 1, we employed biphenyl (Biph) and naphthalene (Np) as covalent linkers to construct co-facial CzB dimers with different co-facial distances (d). By using Suzuki coupling (ESI, Section S1†), CzB units were covalently linked to neighboring α sites of Biph and Np linkers, leading to dimers named Biph-2CzB and Np-2CzB, respectively. Preliminary DFT calculation confirmed the co-facial geometry of two CzB units in both Biph-2CzB and Np-2CzB (Table S1†). The co-facial distance (dB–B) in the S0 state was estimated to be 5.4 Å and 4.9 Å in Biph-2CzB and Np-2CzB, respectively, which are significantly larger than the corresponding distance between neighboring α sites on Biph (3.9 Å) and Np (3.6 Å) due to the non-parallel configuration of the two CzB units. The key role of geometry in inter-chromophore interaction will be discussed in the following sections.
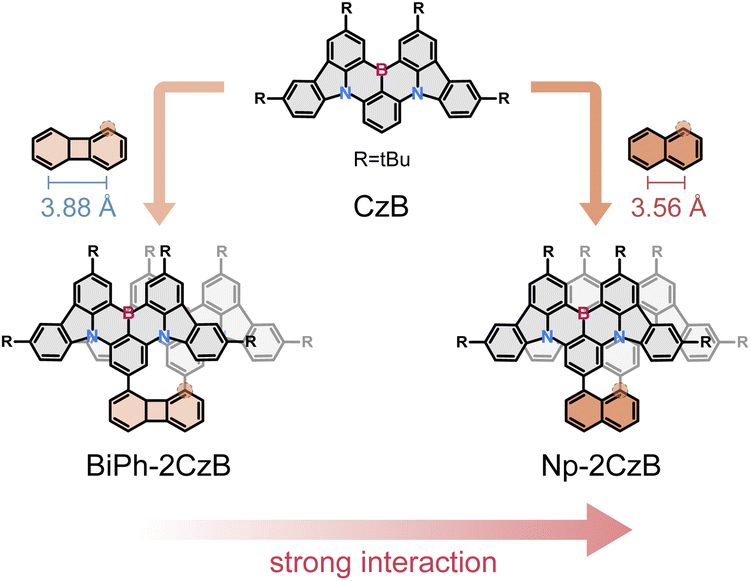 | ||
| Scheme 1 Molecular design of co-facial CzB dimers with biphenylene (BiPh) and naphthalene (Np) as covalent linkers. | ||
Delayed fluorescence
The steady-state UV/Vis absorption and fluorescence spectra of synthesized CzB, Biph-2CzB and Np-2CzB were measured in N2-saturated DCM solution (10−5 M) and PMMA doped films (0.2 mg g−1). As shown in Fig. 1a and b, Biph-2CzB exhibited fluorescence emission at 500 nm, which is nearly identical to the measured emission spectra of CzB, indicating the absence of inter-chromophore interaction in Biph-2CzB. Meanwhile, an extra-broad emission peak centered at 540 nm was observed for Np-2CzB (Fig. 1c), overlapping with CzB monomer emission at 500 nm. By measuring the concentration-dependent absorption spectra (Fig. S5†) and solvent-polarity-dependent fluorescence spectra (Fig. S6†) of Np-2CzB, the participation of inter-molecular interaction and long-range CT states can be ruled out. Thus, the observed broad emission of Np-2CzB at 540 nm was preliminarily assigned to the emission of the expected Sexc state. We further examined the potential TADF activity of synthesized CzB, Biph-2CzB and Np-2CzB emitters. By measuring the fluorescence spectra in air- (triplet states are quenched) and N2-saturated DCM solution, both CzB (Fig. 1d) and Biph-2CzB (Fig. 1e) were recognized as TADF inactive. However, pronounced TADF was observed for Np-2CzB (Fig. 1f), and the fluorescence spectra in N2-saturated solution were substantially stronger than in an air-saturated solution. Intriguingly, the observed TADF (green line in Fig. 1f) exhibited a single peak at 540 nm, different from the double-peak shape (500 nm and 540 nm) of the fluorescence spectra of Np-2CzB, indicating that TADF is associated only with the Sexc state.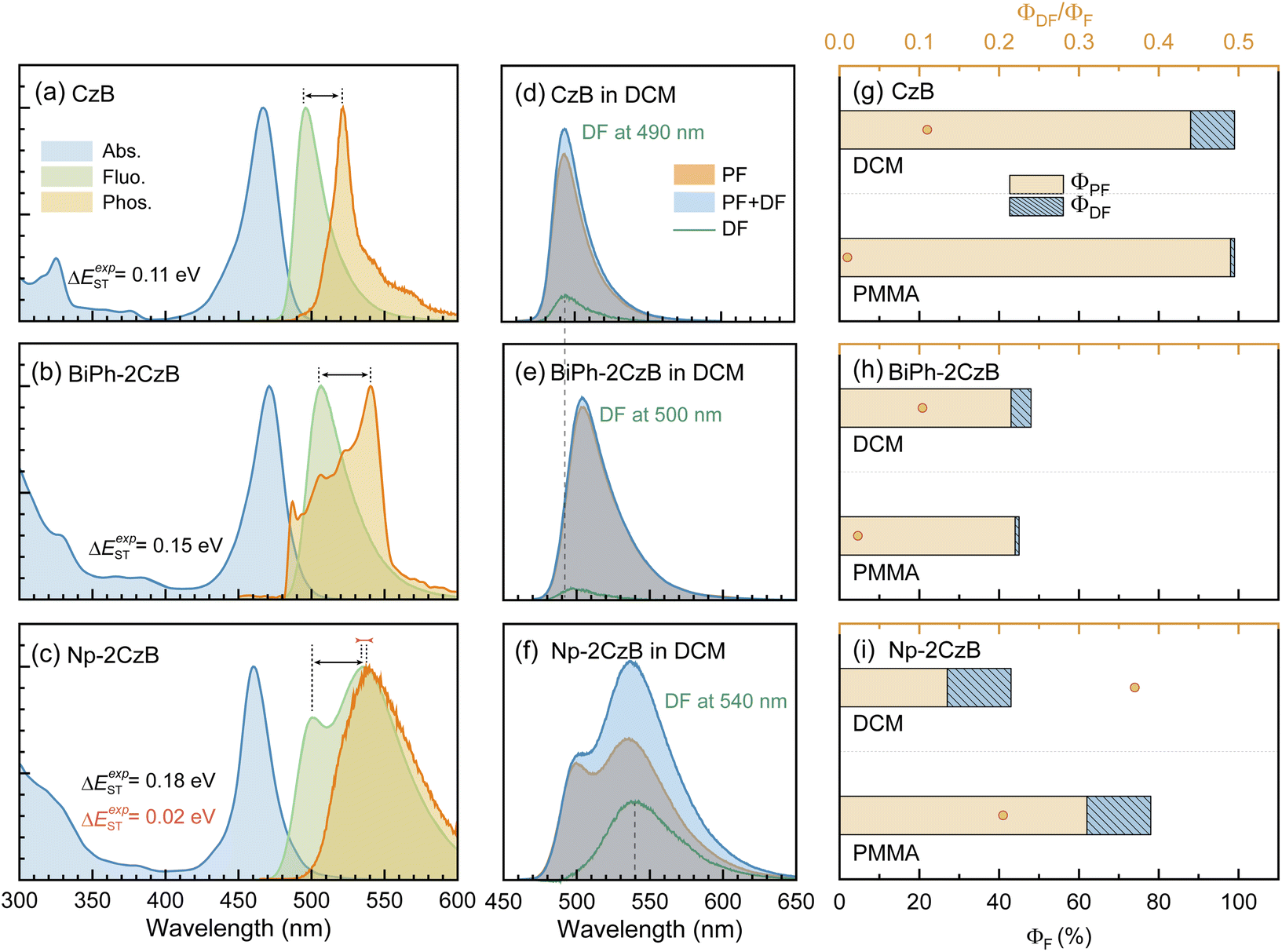 | ||
| Fig. 1 Measured absorption, fluorescence and phosphorescence (50 ms gated) spectra of CzB (a), BiPh-2CzB (b) and Np-2CzB (c) in N2-saturated DCM solution (1 × 10−5 M); comparison of fluorescence spectra of CzB (d), BiPh-2CzB (e) and Np-2CzB (f) in air- (gray bands) and N2- (blue bands) saturated DCM solution (1 × 10−5 M), the differential spectra (green lines) correspond to the DF component, measured fluorescence quantum yield (ΦF, ΦPF, ΦDF) and relative contribution of delayed fluorescence (ΦDF/ΦF) of CzB (g), BiPh-2CzB (h) and Np-2CzB (i) in N2-saturated DCM solution (1 × 10−5 M) and PMMA doped films (0.2 mg g−1). | ||
To quantify the contributions of the PF and DF components, we estimated the total fluorescence quantum yield (ΦF) and the contributions of PF and DF components, i.e. ΦPF, ΦDF and ΦF = ΦPF + ΦDF. As shown in Fig. 1g–i and Table 1, CzB exhibits a ΦF of ∼100% with a low DF contribution in both DCM solution (ΦDF/ΦF = 0.11) and PMMA doped films (ΦDF/ΦF = 0.01), which is consistent with the typical LE (π → π*) character of the S1 state.76–78 Furthermore, although Biph-2CzB is much less fluorescent (ΦF < 0.5) than CzB, its DF contributions in DCM solution (ΦDF/ΦF = 0.10) and PMMA doped films (ΦDF/ΦF = 0.02) are very consistent with the corresponding values for CzB, indicating similar RISC and different S1 state relaxation channels, such as faster non-radiative decay and slower radiative decay of Biph-2CzB compared with CzB. Intriguingly, Np-2CzB exhibited a considerable DF contribution (ΦDF/ΦF = 0.37) in DCM solution, indicating an efficient RISC channel predominantly associated with Sexc emission at 540 nm. We further measured the fluorescence decay traces of CzB, Biph-2CzB and Np-2CzB in DCM solution (Fig. 2) and PMMA doped films (Fig. S7†) to investigate the relaxation channels of the corresponding low-lying singlet and triplet states. As shown in Fig. 2a, CzB exhibited a PF lifetime of τPF = 6.30 ns and a barely observable DF component in DCM solution, which is comparable with the measured τPF = 9.00 ns of Biph-2CzB. Although a long-lived tail can be observed in the fluorescence decay traces of CzB and Biph-2CzB in DCM solution, its low contribution (<10 photon counts compared with 104 counts at the maximum) resulted in difficulty in the quantitative fitting of τDF. Thus, the lack of intrinsic TADF was confirmed for CzB and Biph-2CzB, corresponding to the reported slow RISC of CzB (kRISC = 1.5 × 104 s−1) in solution.67,68
| Φ PF | Φ DF | τ PF (ns) | τ DF (μs) | k Sr (107 s−1) | k Snr (106 s−1) | k ISC (106 s−1) | k RISC (105 s−1) | ||
|---|---|---|---|---|---|---|---|---|---|
| a Calculated with kSr = ΦPF/τPF. b Calculated with kSnr = kPF − kSr − kISC, kPF = 1/τPF. c Calculated with kISC = kPFΦISC = kPF × ΦDF/ΦF. d Calculated with kRISC = (kPF + kDF)/2 − [(kPF + kDF)2/2 − kPFkDF(1 + ΦDF/ΦPF)]0.5. e Collected from values reported in ref. 64. f τ PF of S1 state, i.e. CzB monomers in Np-2CzB. g τ PF of Sexc state. | |||||||||
| CzB | DCM | 0.88 | 0.11 | 6.30 | — | 14.0 | 1.41 | 17.6 | 0.15e |
| PMMA | 0.98 | 0.01 | 6.80 | 35.2 | 14.4 | 1.46 | 1.49 | 0.29 | |
| BiPh-2CzB | DCM | 0.43 | 0.05 | 9.00 | — | 4.78 | 51.8 | 11.6 | — |
| PMMA | 0.44 | 0.01 | 9.90 | 20.6 | 4.40 | 53.8 | 2.22 | 0.49 | |
| Np-2CzB | DCM | 0.27 | 0.16 | 5.60f | 2.50 | 0.55 | 7.25 | 7.53 | 6.45 |
| 49.4g | |||||||||
| PMMA | 0.62 | 0.16 | 5.20f | 1.25 | 1.26 | 3.56 | 4.18 | 1.01 | |
| 49.1g | |||||||||
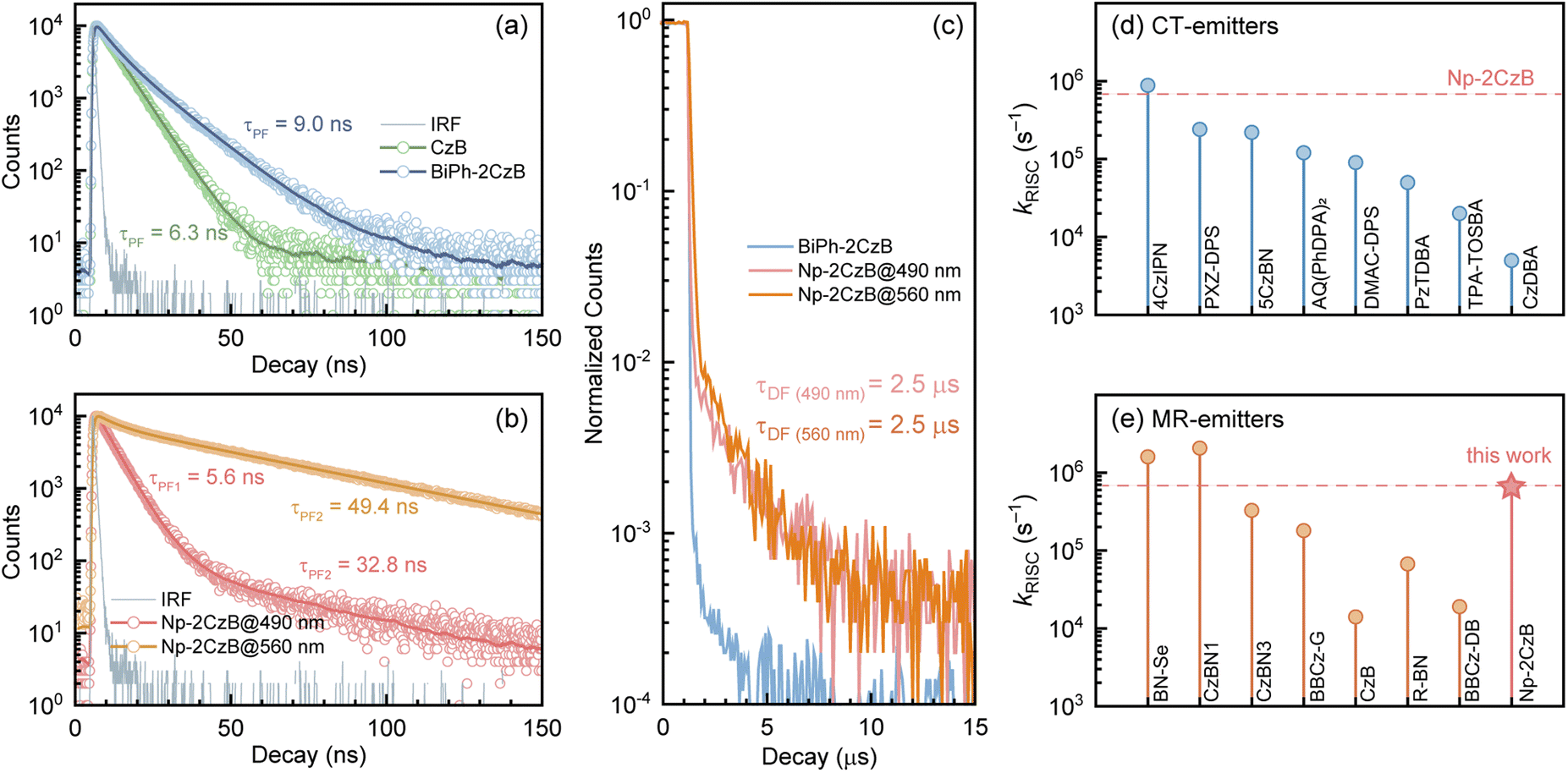 | ||
| Fig. 2 (a) The PF decay traces of CzB and Biph-2CzB in DCM solution; (b) the PF decay traces of Np-2CzB at emission wavelengths of 490 nm and 560 nm in DCM solution; (c) the DF decay traces of Biph-2CzB and Np-2CzB in DCM solution, in which the DF component of Biph-2CzB was unobservable; (d) reported kRISC values of several typical CT-type TADF emitters in dilute solution; (e) comparison between the measured kRISC value of our proof-of-concept emitter Np-2CzB with several reported B,N-MR emitters in dilute solution. | ||
However, as shown in Fig. 2b, Np-2CzB exhibited bi-exponential fluorescence decay in DCM solution (τPF1 = 5.6 ns and τPF2 = 49.4 ns) and PMMA doped films (τPF1 = 5.2 ns and τPF2 = 49.1 ns). More specifically, emission at 500 nm is dominated by fast decay (τPF1 = ∼5.6 ns), assigned to the S1 state lifetime, which is consistent with the measured τPF of CzB and Biph-2CzB. Meanwhile, slow decay (τPF2 = ∼49 ns) is associated with Sexc at 540 nm, which agrees with the prolonged lifetime of the excimer-like states in reported cases.79–82
Please note that the observed τPF2 must be distinguished from the TADF of Np-2CzB, which was observed with a lifetime of ∼2.5 μs (τDF, Fig. 2c) in DCM solution and is associated with Sexc emission at 540 nm. With the measured fluorescence quantum yield (ΦPF, ΦDF) and lifetime (τPF, τDF), we calculated the decay rate of each relaxation channel (ESI, Section S2†), i.e. kSr, kSnr, kISC and kRISC. As listed in Table 1, CzB exhibits highly efficient radiative decay to the S0 state (kSr = 1.4 × 108 s−1), which is one or two orders of magnitude faster than the corresponding kSnr and kISC, leading to close-to-unity ΦF. Please note that we can barely observe the DF components of CzB and Biph-2CzB in DCM solution (Fig. 2a), for which kRISC cannot be calculated. However, a kRISC of 1.5 × 104 s−1 was reported for CzB in dilute solution,67,68 which is half of our measured value (kRISC = 2.9 × 104 s−1) of CzB in PMMA doped films. Meanwhile, Biph-2CzB exhibited very similar RISC dynamics to CzB, i.e. unobservable RISC in DCM solution and kRISC = 4.9 × 104 s−1 in PMMA doped films. However, unlike CzB, the kSnr value (∼5.2 × 107 s−1 in DCM solution) of Biph-2CzB was fast enough to be competitive with kSr (∼4.8 × 107 s−1 in DCM solution), leading to ΦF < 0.5. Since the band gaps of CzB and Biph-CzB are similar to each other, the observation that the kSnr of Biph-CzB is nearly 50 times faster than that of CzB might be attributed to specific vibrational modes with a pronounced Huang–Rhys factor (Sk) and reorganization energy contribution (λk) to the S1 → S0 transition, which was described as the band-gap law in a weak coupling regime.24,25,31
Furthermore, the observed DF contribution (ΦDF/ΦF = 0.37) and ∼2.5 μs DF lifetime of Np-2CzB correspond to a surprisingly high kRISC = 6.5 × 105 s−1 in DCM solution. Quantitatively, the estimated kRISC = 6.5 × 105 s−1 in DCM solution is nearly 50 times faster than the reported kRISC of CzB (1.5 × 104 s−1), which is also comparable to reported selenium-doped B,N-MR emitters with symmetric (BN–SeSe, kRISC = 2.0 × 106 s−1) and asymmetric (BN–Se, kRISC = 1.6 × 106 s−1) frameworks.57,59 As illustrated in Fig. 2d and e, the measured kRISC of Np-2CzB is higher than those of many well-known CT-type TADF emitters and recently reported B,N-MR emitters in dilute solution; the details can be found in Tables S2 and S3.†
We then attempted to explore the plausible origin of the efficient RISC of Np-2CzB in dilute solution. As described in eqn (2), kRISC is highly dependent on ΔEST and the corresponding SOC matrix element 〈ΨT1|ĤSO|ΨS1〉.28,43,83 To estimate ΔEST, we measured the phosphorescence spectra of CzB, Biph-2CzB and Np-2CzB in DCM solution at 77 K (Fig. 1a–c), in which the PF component was screened by 50 ms time-gating. Combined with the corresponding fluorescence spectra, ΔEST of 0.11 eV (CzB) and 0.17 eV (Biph-2CzB) were estimated, which are much smaller than the TDDFT calculated values (Fig. S8†) and usually regarded as a thermally accessible gap at room temperature.37,84 Therefore, the RISC channels of CzB and Biph-2CzB might be mainly blocked by a low SOC matrix element, and were calculated as 0.044 cm−1 and 0.005 cm−1, respectively, using the linear-response method. As both S1 and T1 states of CzB feature an identical LE (π → π*) electronic configuration, the magnetic moment resulting from orbital angular momentum (μL) can be extremely low, leading to the TADF-inactive character of CzB and Biph-2CzB. For Np-2CzB, although 〈ΨT1|ĤSO|ΨS1〉 was also calculated to be low (<0.1 cm−1), the 〈ΨT1|ĤSO|ΨSexc〉 associated with RISC (T1 → Sexc) can be much higher due to the dramatically changed electronic configuration of the Sexc state, which explains the observed fact that the TADF of Np-2CzB is associated only with Sexc emission. Due to the limitation of electronic structure calculation of an excimer-like state, directly calculating the SOC matrix element involving the Sexc state remains challenging, but we still believe that the greatly enhanced SOC may be the main reason for the intrinsic TADF of Np-2CzB. Meanwhile, as the Sexc state is energetically lower than the S1 state of Np-2CzB, the resulting ΔEST (T1 → Sexc) < 0.02 eV may also contribute to the observed kRISC of 6.5 × 105 s−1 in DCM solution.
Inter-chromophore interaction
We successfully observed the intrinsic TADF of Np-2CzB, which is associated with the Sexc state facilitated by inter-chromophore interaction. The formation of the Sexc state relies on interaction between a chromophore (M*) with a localized S1 state and the neighboring chromophore (M) on the S0 state, i.e. |1(S1S0)〉 = (1/2)0.5[|M*M〉 ± |MM*〉].85,86 For Np-2CzB, the formed Sexc state exhibited red-shifted emission and efficient RISC (kRISC = 6.5 × 105 s−1) in DCM solution. However, Biph-2CzB exhibited nearly identical emission to CzB, indicating very weak inter-chromophore interaction. We performed reduced density gradient (RDG) analysis for Biph-2CzB and Np-2CzB (Fig. 3a and b).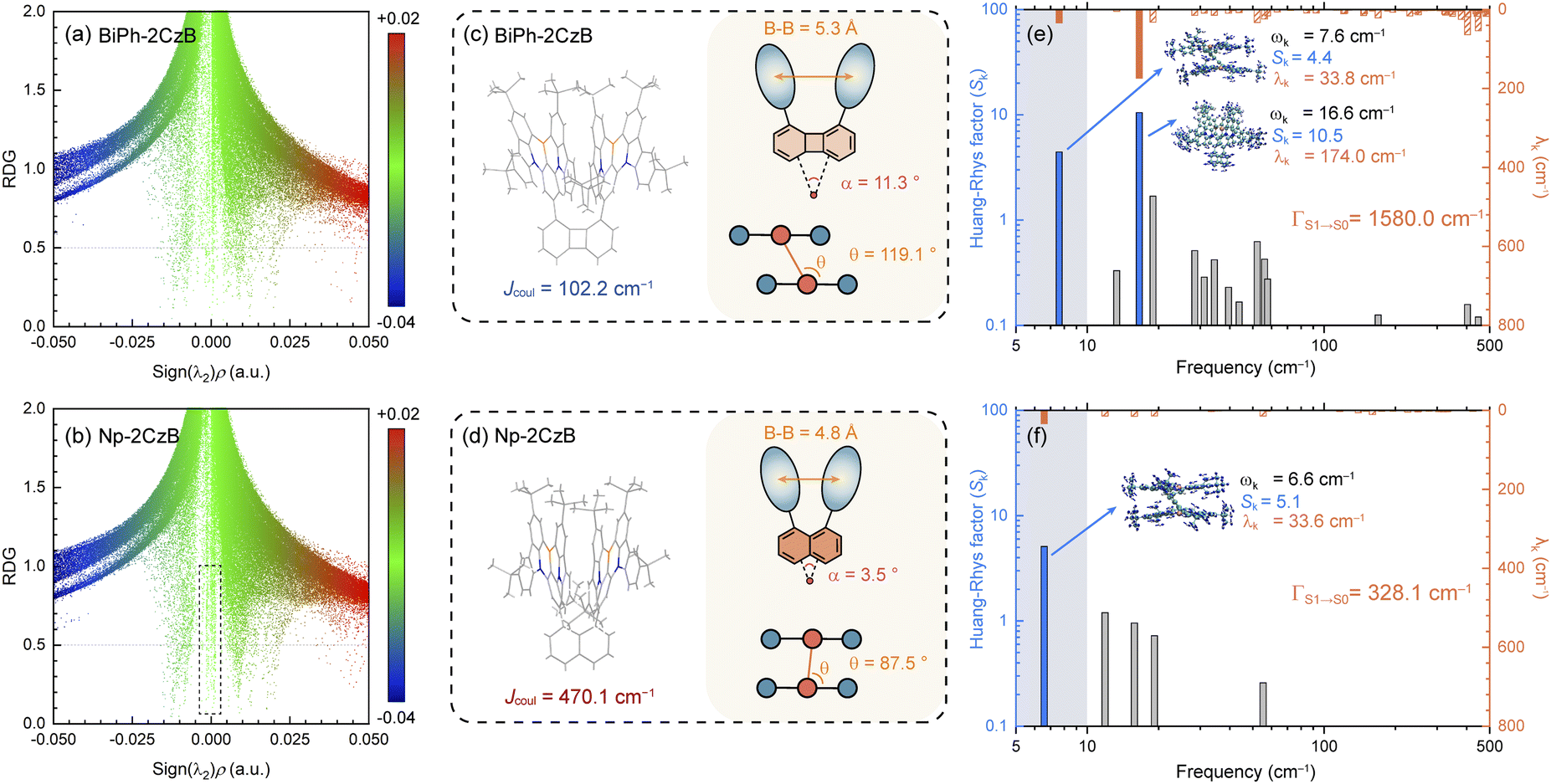 | ||
| Fig. 3 Calculated reduced density gradient (RDG) scattering diagrams based on the S1 state geometry of BiPh-2CzB (a) and Np-2CzB (b); the TDDFT (B3LYP/6-31g*) optimized geometry of BiPh-2CzB (c) and Np-2CzB (d); calculated Huang–Rhys factor (Sk) and reorganization energy contribution (λk) of each vibrational mode of BiPh-2CzB (e) and Np-2CzB (f) for the S1 → S0 transition. | ||
The calculated RDG scattering diagram of Np-2CzB exhibited several spikes in the low-density and low-gradient region (highlighted by the dashed box in Fig. 3b), indicating the presence of pronounced inter-chromophore interaction, which can also be visualized by the corresponding RDG iso-surface displayed in Fig. S9.† However, such features can barely be observed on the RDG scattering diagram and iso-surface of Biph-2CzB, implying the absence of inter-chromophore interaction, which is consistent with its observed fluorescence spectra being similar to CzB. We further attempted to explore the structural and energetic origin of the different inter-chromophore interactions within the two CzB dimers with different linkers.
Firstly, Sexc formation relies on co-facial geometry with a suitable co-facial distance (d = 3.0–5.0 Å),87–89 for which we optimized the S1 state structure of BiPh-2CzB and Np-2CzB by the TDDFT approach; the corresponding structural parameters are listed in Table 2. As illustrated in Fig. 3c and d, Np-2CzB exhibited a co-facial (B–B) distance of d = 4.8 Å in the S1 state, which is 0.5 Å shorter than that of BiPh-2CzB (d = 5.3 Å). Meanwhile, the S1 state geometry of Np-2CzB exhibited a dihedral angle of α = 3.5° and a slipping angle of θ = 87.5° between the two CzB units. As a result, Np-2CzB exhibited improved co-facial configuration in the S1 state compared to BiPh-2CzB (α = 11.3°, θ = 119.1°), i.e. the two CzB units are more spatially overlapped in Np-2CzB than in BiPh-2CzB, leading to a favorable configuration for Sexc formation. Furthermore, the calculated S0 and S1 state geometries also revealed the different S1/S0 excited-state structural relaxation (ES-SR) of BiPh-2CzB and Np-2CzB. As listed in Table 2, BiPh-2CzB exhibited pronounced CzB twisting (β1/β2) and bending (γ1/γ2) angles with up to 30% change between S1 and S0 states, which might lead to two-step S1/S0 ES-SR, as we reported previously.31,55 Intriguingly, the twisted S1/S0 ES-SR of Np-2CzB (∼7% S1/S0 changing) is less pronounced than that of BiPh-2CzB. Meanwhile, the two CzB units in Np-2CzB exhibited a significantly planarized (bending angles γ1/γ2 = 5.1°) feature in the S1 state compared to CzB (γ = 18.5°) or BiPh-2CzB (γ1/γ2 = 11.3°/15.7°), which might also contribute to the Sexc state formation of Np-2CzB. Furthermore, the formation of an Sexc state also relies on appreciable inter-chromophore electronic coupling (J), which has been widely discussed in the framework of the exciton theory.90–92 In general, electronic coupling includes contributions from Coulomb (Jcoul) and charge transfer terms (JCT), while Sexc state formation is known to require considerable Jcoul and weak CT coupling.70,93 The low value of JCT in both BiPh-2CzB and Np-2CzB can be ensured by their barely observable solvatochromism (Fig. S6†), while Jcoul can usually be estimated with:85,93,94
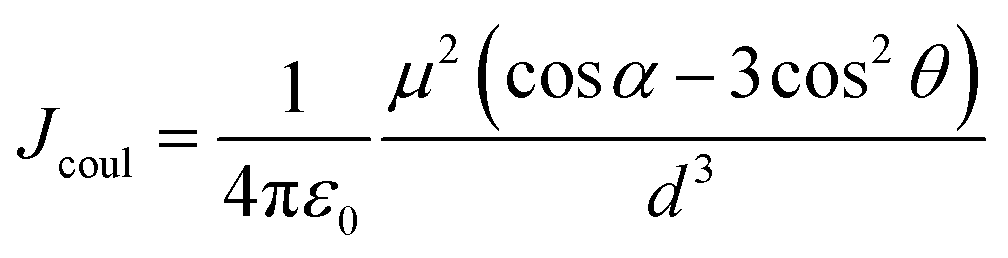 | (4) |
| BiPh-2CzB | Np-2CzB | CzB | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| a Dihedral angle between two CzB units. b Slipping angle of two CzB units. c Distance between two CzB units. d Twisting angle of two CzB units with BiPh/Np linker. e Bending angle of two CzB units. | ||||||
| α (°)a | 11.2 | 11.3 | 4.8 | 3.5 | ||
| θ (°)b | 118.5 | 119.1 | 91.3 | 87.5 | ||
| J coul (cm−1) | 102.2 | 470.1 | ||||
| B–B (Å)c | 5.4 | 5.3 | 4.9 | 4.8 | ||
| N1–N1 (Å)c | 5.2 | 5.1 | 4.7 | 4.7 | ||
| N2–N2 (Å)c | 5.2 | 5.1 | 4.7 | 4.7 | ||
| β 1/β2 (°)d | 41.7/45.6 | 30.0/31.9 | 52.1/52.1 | 48.4/48.4 | ||
| γ 1/γ2 (°)e | 16.6/13.7 | 11.3/15.7 | 9.3/9.3 | 5.1/5.1 | 19.9 | 18.5 |
| Γ S1→S0 (cm−1) | 1579.9 | 328.1 | 480.6 | |||
| Γ S0→S1 (cm−1) | 1661.8 | 423.2 | 410.4 | |||
Finally, with suitable co-facial geometry and electronic coupling, the formation of the Sexc state can still be disrupted by vibrational motion and S1/S0 ES-SR of the S1 state, for which we further performed vibrational analysis on BiPh-2CzB and Np-2CzB. By performing vibrational analysis, we estimated the total internal reorganization energy of the S1 → S0 transition (ΓS1→S0), which can be employed to generalize the S1/S0 ES-SR. It can be seen that the ΓS1→S0 of BiPh-2CzB (1579.9 cm−1) is almost 5 times that of CzB (480.6 cm−1) (Fig. S10†), indicating the highly flexible structure of BiPh-2CzB in the S1 state, which can potentially disrupt the formation of the Sexc state. In contrast, the ΓS1→S0 of Np-2CzB (328.1 cm−1) is even lower than that of CzB, indicating a rigid structure without pronounced vibrational motion in the S1 state, which might also be critical for Sexc formation. We further calculated the Huang–Rhys factor (Sk) and reorganization energy contribution (λk) of each vibrational mode and attempted to find the key modes that can promote or disrupt Sexc formation.
It is known that vibrational modes with considerable Sk (i.e. promoting modes) are heavily involved in the vibronic coupling of the S1 state, while the λk of promoting modes is associated with either S1/S0 ES-SR or the vibrational motion of the S1 state,97–100 which can disrupt Sexc formation. As shown in Fig. 3e, the ΓS1→S0 of BiPh-2CzB is mainly contributed by two promoting modes in the low-frequency regime, i.e. mode 1 (Sk = 4.4) at ωk = 7.6 cm−1 with λk = 33.8 cm−1 and mode 2 (Sk = 10.5) at ωk = 16.6 cm−1 with λk = 174.0 cm−1. As illustrated in Fig. S11,† mode 1 corresponds to the twisting motion of two CzB units along with a Biph linker, associated with the fluctuation of β1/β2 and θ angles with respect to the equilibrium position in the S1 state. Meanwhile, mode 2 corresponds to bending of the CzB units, i.e. the fluctuation of γ1/γ2 angles. Intriguingly, for Np-2CzB, mode 1 was found to be identical to that of BiPh-2CzB (Fig. 3f), but mode 2 of Np-2CzB at ωk = 15.8 cm−1 was one order of magnitude lower at λk (15.1 cm−1) than that of BiPh-2CzB, indicating that the bending motion of CzB units is greatly suppressed in the S1 state of Np-2CzB. Thus, we deduced that the bending mode (ωk = ∼16 cm−1) of CzB can potentially disrupt Sexc formation in co-facial dimers, while reducing the λk of such a mode might be critical for formation of the Sexc state.
The vibrational analysis can also explain the excitation (λex)-dependent emission of Np-2CzB in DCM solution (Fig. 4a) and PMMA doped films (Fig. 4b). By performing multi-Gaussian fitting on the emission spectra of Np-2CzB (Fig. S12 and S13†), the contributions of S1 and Sexc state emission can be quantified. For convenience, we defined the indicator δ = Aexc/AS1, in which Aexc and AS1 stand for the peak areas of Sexc and S1 emission spectra, respectively. In DCM solution, the δ indicator exhibits pronounced λex-dependence, i.e. visible excitation (λex > 410 nm) resulting in Sexc dominated emission (δ > 12), while Sexc might be disrupted (δ < 6) upon UV excitation (λex < 350 nm). As illustrated in Fig. 4c, the UV excitation populated high-lying singlet states (Sn, n > 1) can rapidly decay to a vibrationally hot S1 state. As a result, the excessive vibrational energy (probably on the bending mode of CzB units) can disrupt Sexc formation, leading to reduced δ values, while a cold S1 state populated by visible excitation leads to a more favourable condition for Sexc formation with reduced vibrational motion in the S1 state. However, in PMMA doped films, the vibrational motion of the corresponding mode might be greatly confined, leading to λex-independent emission. Meanwhile, due to external structural restraint on S1/S0 ES-SR, the CzB units in the S1 state might not be fully relaxed to an optimal configuration (bending angles γ1/γ2 = 5.1°) that is favorable for Sexc formation. As a result, the Sexc emission remains at a low level (δ < 8) upon all tested λex. Vibrational analysis and λex-dependent emission spectra of Np-2CzB further confirmed that Sexc formation in Np-2CzB can be disrupted by the bending mode of CzB units, which is strongly coupled with the S1 → S0 transition. However, the formation dynamics of the Sexc state in Np-2CzB still remains unknown, because of which we performed fs-TA measurement on CzB, BiPh-2CzB and Np-2CzB in both DCM solution and PMMA doped films.
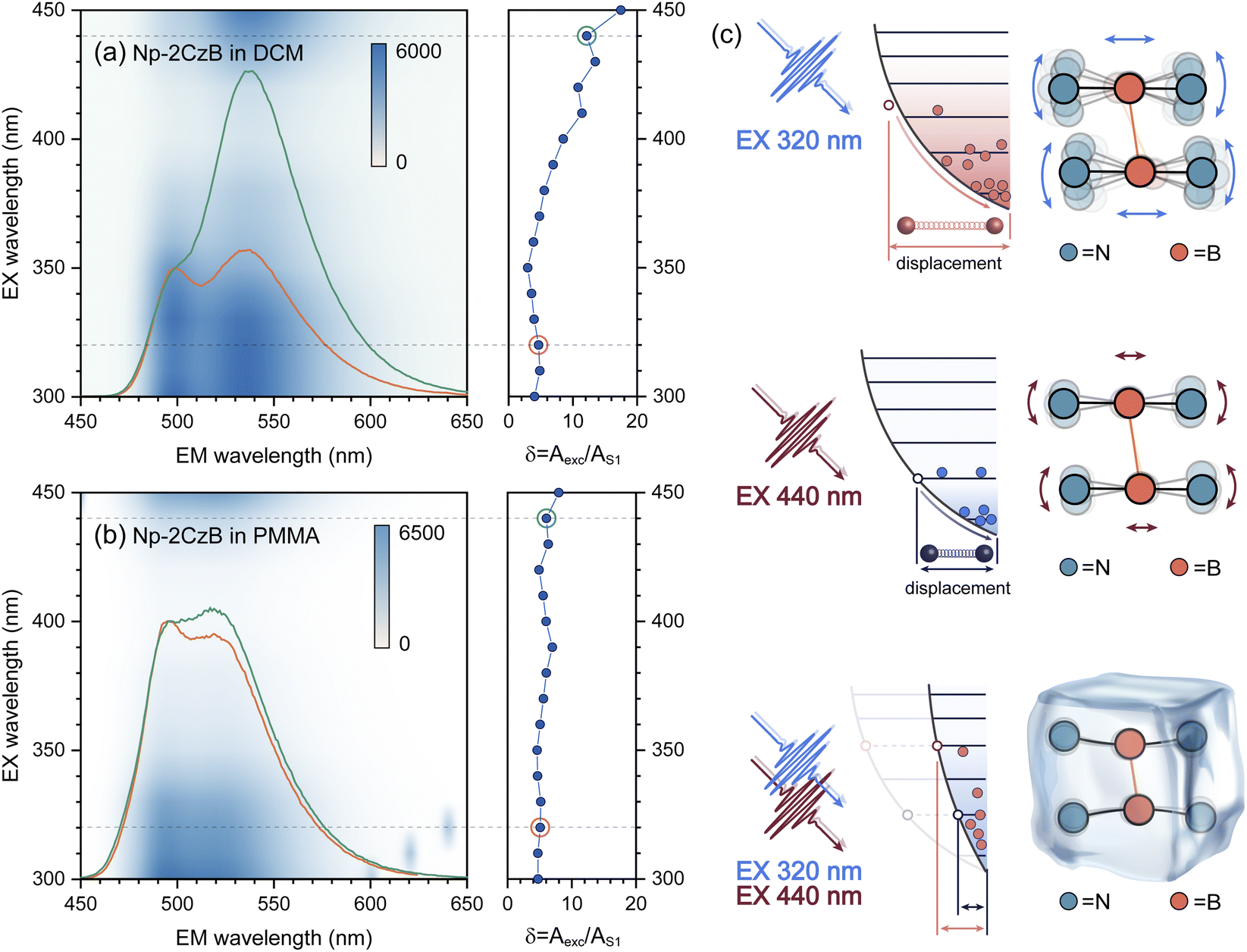 | ||
| Fig. 4 The measured excitation–emission spectra of Np-2CzB in DCM solution (a) and PMMA doped films (b); corresponding excitation dependence of the contribution ratio (Aexc/AS1) of Sexc and S1 emission, as well as emission spectra upon excitation at 320 nm (orange lines) and 440 nm (green lines); (c) illustrative sketch of Sexc emission upon 320 nm (upper) and 440 nm excitation (middle) in solution and in PMMA doped films (bottom). | ||
Excited-state dynamics
To further resolve the excited-state relaxation of CzB, BiPh-2CzB and Np-2CzB emitters, especially to explore the formation dynamics of the Sexc state, the fs-TA response of the corresponding emitters in both DCM solution and PMMA doped films were recorded upon UV excitation.As shown in Fig. S14,† the fs-TA spectra of CzB exhibited pure decay without reshaping of the fs-TA spectra at a probe wavelength of λpr = 350–750 nm. With minimized S1/S0 ES-SR (Table 2), S1 state decay is dominated by the radiative channel, leading to close-to-unity ΦF (Table 1). For BiPh-2CzB, pronounced S1/S0 ES-SR leads to highly efficient (kSnr ≈ 5 × 107 s−1) non-radiative S1 → S0 decay. However, the fs-TA spectral shape of BiPh-2CzB (Fig. S15†) remains nearly unchanged in a time window up to 7 ns, indicating that fs-TA in the UV/Vis regime is insensitive to S1/S0 ES-SR of these emitters. Thus, observation of S1/S0 ES-SR dynamics relies on quantitative fitting of fs-TA. Intriguingly, the fs-TA spectra of Np-2CzB exhibit a negative band at λpr = ∼465 nm in both DCM solution and PMMA doped films (Fig. S16†), which is clearly blue shifted in comparison with CzB (λpr = ∼475 nm) and BiPh-2CzB (λpr = ∼478 nm). Considering that the observed negative band is comprised of ground state bleaching (GSB) and stimulated emission (SE) of the S1 state, the blue-shifted negative fs-TA band of Np-2CzB can be explained by the lack of an SE (S1) band, while the replaced Sexc state might not be capable of having an SE.
Target analysis was further performed to acquire quantitative information about the excited-state relaxation of BiPh-2CzB and Np-2CzB, in which formation of an Sexc state might be entangled with S1/S0 ES-SR motion. By including two or three sequential processes, the measured fs-TA data can be well reproduced by the extracted decay-associated spectra (DAS, Fig. 5a–d and S17†) of each decay process and the concentration evolution of each transient species (Fig. 5e, f and S18†).
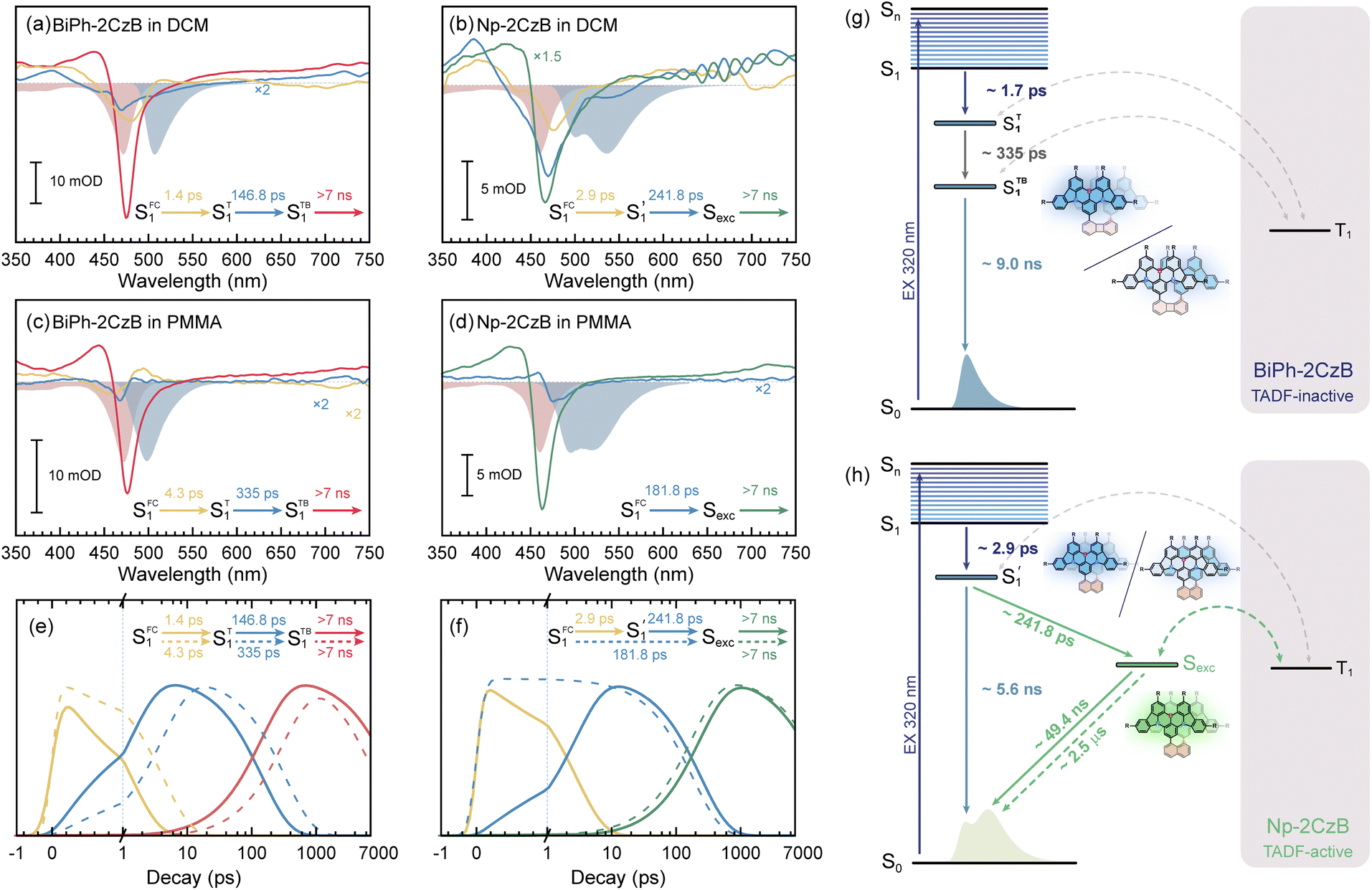 | ||
| Fig. 5 Target analysis extracted decay-associated spectra (DAS) of fs-TA data of BiPh-2CzB (a) and Np-2CzB (b) in DCM solution, as well as BiPh-2CzB (c) and Np-2CzB (d) in PMMA doped films; the corresponding steady absorption (red filled bands) and fluorescence spectra (blue filled bands) overlap; concentration evolution of transient species extracted from the fs-TA of BiPh-2CzB (e) and Np-2CzB (f) in DCM solution (solid lines) and PMMA doped films (dashed lines); simplified excited-state relaxation path of BiPh-2CzB (g) and Np-2CzB (h). | ||
For CzB, an ultrafast process 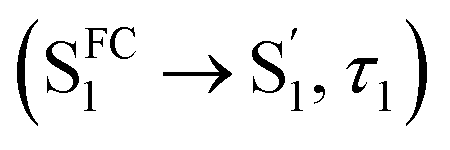 might correspond to the slight S1/S0 ES-SR revealed by TDDFT calculation, which becomes slower (∼6.4 ps) in PMMA doped film than in DCM solution (∼2.0 ps) due to external structural restraint. The subsequent process (∼5 ns) accompanied by the S1 state SE at ∼490 nm corresponds to the S1 state lifetime
might correspond to the slight S1/S0 ES-SR revealed by TDDFT calculation, which becomes slower (∼6.4 ps) in PMMA doped film than in DCM solution (∼2.0 ps) due to external structural restraint. The subsequent process (∼5 ns) accompanied by the S1 state SE at ∼490 nm corresponds to the S1 state lifetime 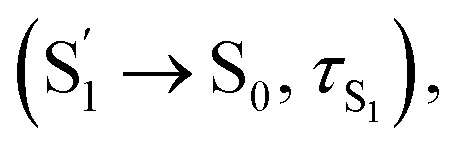 which is comparable to the measured fluorescence lifetime (τPF).
which is comparable to the measured fluorescence lifetime (τPF).
For BiPh-2CzB, target analysis revealed two-step relaxation with 2–3 times slower time constants in PMMA doped films than in DCM solution, corresponding to two-step S1/S0 ES-SR, as previously reported.31,55 As discussed in the sections above, S1/S0 ES-SR of BiPh-2CzB features simultaneous changing of CzB twisting (β1 and β2 angles) and their own bending motion (γ1 and γ2 angles). The fast step of S1/S0 ES-ER (τ1 = 1–4 ps) corresponds to CzB twisting along with the biphenyl linker (noted as SFC1 → ST1), while the slow step (τ2 = 150–350 ps) might originate from the bending motion of CzB frameworks (ST1 → STB1). Furthermore, the structurally relaxed S1 state (STB1) exhibits decay with >7 ns time constants, which is comparable to the measured τPF = 9 ns, corresponding to the S1 state lifetime (τS1). The corresponding DAS of τS1 feature a pronounced negative peak at λpr = 475–480 nm, contributed by the GSB and SE of relaxed STB1. Meanwhile, the τS1 of BiPh-2CzB is also similar to the τPF (∼7 ns) of CzB. Therefore, it is clear that the S1 state of BiPh-2CzB is dominated by CzB units without inter-chromophore interaction. Since the S1/T1 states of CzB units are dominated by similar π → π* character, the RISC of T1 → S1 is actually forbidden, leading to TADF-inactive Biph-2CzB.
In comparison, Np-2CzB exhibited very different excited-state dynamics. In DCM solution, two-step relaxation of the initially populated SFC1 of Np-2CzB was observed (Fig. 5b). Although the fast step (τ1 = 2.9 ps) can be assigned to S1/S0 CzB twisting 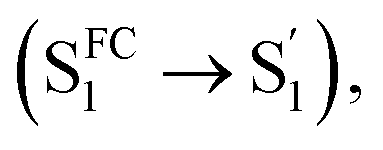 the slow step (τ2 = 240 ps) can hardly be attributed to S1/S0 ES-ER as it became faster (∼180 ps) in PMMA doped films. With external structural restraint in doped films, the promoting modes that can disrupt Sexc formation, such as the bending mode of CzB units in Np-2CzB, can be further suppressed. As a result, unlike S1/S0 ES-ER, Sexc formation can be even faster in PMMA doped films than in solution, which is what we observed for Np-2CzB. Therefore, the observed 180–240 ps process was assigned to the formation of the Sexc state
the slow step (τ2 = 240 ps) can hardly be attributed to S1/S0 ES-ER as it became faster (∼180 ps) in PMMA doped films. With external structural restraint in doped films, the promoting modes that can disrupt Sexc formation, such as the bending mode of CzB units in Np-2CzB, can be further suppressed. As a result, unlike S1/S0 ES-ER, Sexc formation can be even faster in PMMA doped films than in solution, which is what we observed for Np-2CzB. Therefore, the observed 180–240 ps process was assigned to the formation of the Sexc state 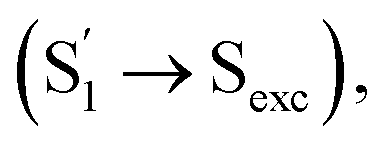 which is consistent with the reported formation time of an intramolecular excimer of ∼250 ps in sandwich-linked dicarbazolyl compounds (m-DCzPe) and of ∼140 ps in Ant-(SiMe2)n-Ant, in which two anthracene groups are linked by disilane.101,102 Meanwhile, it is found that the corresponding DAS of further relaxation of Sexc features a negative peak at λpr = 460–465 nm, i.e. ∼700 cm−1 blue shifted compared to the corresponding peak of Biph-2CzB, which can be explained by the reduced SE (S1) signal due to the S1 → Sexc transition and the lack of SE for the Sexc state. Therefore, the observed long-lived species on fs-TA of can be largely attributed to the formed Sexc state.
which is consistent with the reported formation time of an intramolecular excimer of ∼250 ps in sandwich-linked dicarbazolyl compounds (m-DCzPe) and of ∼140 ps in Ant-(SiMe2)n-Ant, in which two anthracene groups are linked by disilane.101,102 Meanwhile, it is found that the corresponding DAS of further relaxation of Sexc features a negative peak at λpr = 460–465 nm, i.e. ∼700 cm−1 blue shifted compared to the corresponding peak of Biph-2CzB, which can be explained by the reduced SE (S1) signal due to the S1 → Sexc transition and the lack of SE for the Sexc state. Therefore, the observed long-lived species on fs-TA of can be largely attributed to the formed Sexc state.
Intriguingly, the S1/S0 ES-SR associated with CzB bending motion was missing in the fs-TA of Np-2CzB, which can be explained by two aspects: (1) CzB bending in the S1 state takes place in a similar timescale to Sexc formation, because of which the two parallel processes cannot be distinguished from each other; (2) CzB bending might be too weak to be detected, which is consistent with the low contribution of reorganization energy (λk = 15.1 cm−1) of the corresponding vibrational mode at ωk = 15.8 cm−1. Actually, as discussed above, since the pronounced bending motion of CzB units might be able to disrupt Sexc formation, the greatly suppressed bending motion of S1/S0 ES-SR might be the key factor for Sexc formation in Np-2CzB. On the other hand, please note that although external structural restraint in PMMA doped films leads to the kinetically faster formation of the Sexc state, there might be a higher ratio at which Np-2CzB cannot fully relax to the optimized structure of the S1 state for Sexc formation. As a result, we observed a significantly higher DF ratio of Np-2CzB in DCM solution (ΦDF/ΦF = 0.37) than in PMMA doped films (ΦDF/ΦF = 0.20).
Unlike Biph-2CzB, the delocalized nature of the Sexc state ensures that its electronic configuration is very different from the LE (π → π*) nature of the S1 and T1 states, leading to the greatly enhanced 〈ΨSexc|ĤSO|ΨT1〉 of Np-2CzB. Meanwhile, the nearly degenerate excitation energy of the Sexc and T1 states leads to ΔEST < 0.02 eV. As a result, an efficient RISC channel (T1 → Sexc) with kRISC = 6.5 × 105 s−1 can be facilitated, leading to intrinsic TADF with τDF = 2.50 μs and ΦDF/ΦF = 0.37 in DCM solution. Through co-facial dimerization, we successfully converted a TADF-inactive chromophore (CzB, kRISC = 1.5 × 104 s−1) into a TADF-active emitter (Np-2CzB, kRISC = 6.5 × 105 s−1) with ∼50 times promotion of kRISC.
Conclusions
To summarize, we demonstrated a general strategy of molecular design, which can potentially facilitate the intrinsic TADF of conjugated emitters with LE (π → π*) dominated S1 and T1 states. With a TADF-inactive B,N-MR emitter as a model system, we synthesized a co-facial dimer with suitable structure and electronic coupling, in which an excimer-like state (Sexc) was facilitated with ∼250 ps formation time. Relying on enhanced SOC (Sexc–T1) and reduced ΔEST, an intrinsic TADF with kRISC = 6.5 × 105 s−1 and ΦDF/ΦF > 37% was successfully demonstrated in dilute solution. Further investigation indicated that a suitable co-facial distance (d = ∼4.7 Å) and inter-chromophore electronic coupling (Jcoul > 450 cm−1) are essential for Sexc formation, while suppressing a key vibrational mode (CzB bending) can be critical to avoid disrupting the inter-chromophore interaction. For the majority of conjugated organic emitters, the lack of intrinsic TADF greatly limits their application in emerging areas such as OLED and EPOL devices. Although the present strategy was demonstrated with a B,N-MR emitter as a model system, we believe that it can be regarded as a general strategy for other types of organic emitter, as long as they feature a rigid conjugated framework and localized π → π* character of S1 and T1 states. By activating the intrinsic DF emission, many traditional organic emitters might play more important roles in a wide range of applications, for which our work might provide inspiration for the community.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yixuan Gao: conceptualization, methodology, investigation, data curation, formal analysis, visualization, and writing – original draft; Yingman Sun: synthesis the compounds; Zilong Guo: investigation, visualization, project administration, and supervision; Guo Yu: participated in the discussion of the inter-chromophore interaction; Yaxin Wang: methodology, data curation, formal analysis, and validation; Yan Wan: methodology and resources; Yandong Han and Wensheng Yang: resources and project administration; Dongbing Zhao: supervision, resources, and funding acquisition; Xiaonan Ma: conceptualization, formal analysis, funding acquisition, supervision, and writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2020YFA0714603 and 2020YFA0714604). Y. G. is grateful to the Excellent Doctoral Thesis Cultivating Grant of Tianjin University. We thank Prof. Tao Xue (Analytical and Testing Centre of Tianjin University) for his valuable support on TCSPC measurements.Notes and references
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- M. N. Berberan-Santos and J. M. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391–9394 CrossRef CAS.
- Z. Huang, D. Ji, A. Xia, F. Koberling, M. Patting and R. Erdmann, J. Am. Chem. Soc., 2005, 127, 8064–8066 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- Z. Huang, B. Lei, D. Yang, D. Ma, Z. Bin and J. You, Angew. Chem., Int. Ed., 2022, 134, e202213157 CrossRef.
- L. J. Rothberg and A. J. Lovinger, J. Mater. Res., 1996, 11, 3174–3187 CrossRef CAS.
- M. Pope, H. P. Kallmann and P. Magnante, J. Chem. Phys., 1963, 38, 2042–2043 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- D.-H. Kim, A. D'Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- T. Zhang, Z. Zhou, X. Liu, K. Wang, Y. Fan, C. Zhang, J. Yao, Y. Yan and Y. S. Zhao, J. Am. Chem. Soc., 2021, 143, 20249–20255 CrossRef CAS PubMed.
- Z. Zhou, C. Qiao, K. Wang, L. Wang, J. Liang, Q. Peng, Z. Wei, H. Dong, C. Zhang, Z. Shuai, Y. Yan and Y. S. Zhao, Angew. Chem., Int. Ed., 2020, 59, 21677–21682 CrossRef CAS PubMed.
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 18630–18638 CrossRef CAS PubMed.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed.
- D. M. Mayder, R. Hojo, W. L. Primrose, C. M. Tonge and Z. M. Hudson, Adv. Funct. Mater., 2022, 32, 2204087 CrossRef CAS.
- M. Zhao, M. Li, W. Li, S. Du, Z. Chen, M. Luo, Y. Qiu, X. Lu, S. Yang, Z. Wang, J. Zhang, S. Su and Z. Ge, Angew. Chem., Int. Ed., 2022, 134, e202210687 CrossRef.
- C. J. Christopherson, N. R. Paisley, Z. Xiao, W. R. Algar and Z. M. Hudson, J. Am. Chem. Soc., 2021, 143, 13342–13349 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- Y. Wei, B. Chen, R. Ye, H. Huang, J. Su, C. Lin, J. Hodgkiss, L. Hsu, Y. Chi, K. Chen, C. Lu, S. Yang and P. Chou, Angew. Chem., Int. Ed., 2023, 62, e202300815 CrossRef CAS PubMed.
- G. Chen, J. R. Swartzfager and J. B. Asbury, J. Am. Chem. Soc., 2023, 145, 25495–25504 CrossRef CAS PubMed.
- V. Lawetz, G. Orlandi and W. Siebrand, J. Chem. Phys., 1972, 56, 4058–4072 CrossRef CAS.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- H. Narita, H. Min, N. Kubo, I. Hattori, T. Yasuda and S. Yamaguchi, Angew. Chem., Int. Ed., 2024, 63, e202405412 CrossRef CAS PubMed.
- Y. Gao, Y. Wang, Z. Guo, Y. Wan, Z. Xue, Y. Han, W. Yang and X. Ma, Chem. Sci., 2024, 15, 6410–6420 RSC.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- K. Stavrou, L. G. Franca, T. Böhmer, L. M. Duben, C. M. Marian and A. P. Monkman, Adv. Funct. Mater., 2023, 33, 2300910 CrossRef CAS.
- L. G. Franca, A. Danos, R. Saxena, S. Kuila, K. Stavrou, C. Li, S. Wedler, A. Köhler and A. P. Monkman, J. Phys. Chem. Lett., 2024, 15, 1734–1740 CrossRef CAS PubMed.
- A. K. Narsaria, F. Rauch, J. Krebs, P. Endres, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Finze, J. Nitsch, F. M. Bickelhaupt and T. B. Marder, Adv. Funct. Mater., 2020, 30, 2002064 CrossRef CAS PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- G. Zhao, D. Liu, P. Wang, X. Huang, H. Chen, Y. Zhang, D. Zhang, W. Jiang, Y. Sun and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202212861 CrossRef CAS PubMed.
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 133, 23827–23832 CrossRef.
- S. Koseki, M. S. Gordon, M. W. Schmidt and N. Matsunaga, J. Phys. Chem., 1995, 99, 12764–12772 CrossRef CAS.
- S. Koseki, M. W. Schmidt and M. S. Gordon, J. Phys. Chem. A, 1998, 102, 10430–10435 CrossRef CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- W. Zhang, S. Li, Y. Gong, J. Zhang, Y. Zhou, J. Kong, H. Fu and M. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202404978 CrossRef CAS PubMed.
- Y. Shi, H. Ma, Z. Sun, W. Zhao, G. Sun and Q. Peng, Angew. Chem., Int. Ed., 2022, 61, e202213463 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- B. Lei, Z. Huang, S. Li, J. Liu, Z. Bin and J. You, Angew. Chem., Int. Ed., 2023, 62, e202218405 CrossRef CAS PubMed.
- Y. Xu, Q. Wang, X. Cai, C. Li, S. Jiang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202312451 CrossRef CAS PubMed.
- X. Cai, Y. Pu, C. Li, Z. Wang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304104 CrossRef CAS PubMed.
- Y. Wang, Y. Tian, Y. Gao, Z. Guo, Z. Xue, Y. Han, W. Yang and X. Ma, J. Phys. Chem. Lett., 2023, 14, 9665–9676 CrossRef CAS PubMed.
- P. Li, W. Li, Y. Zhang, P. Zhang, X. Wang, C. Yin and R. Chen, ACS Mater. Lett., 2024, 6, 1746–1768 CrossRef CAS.
- X. Cai, J. Xue, C. Li, B. Liang, A. Ying, Y. Tan, S. Gong and Y. Wang, Angew. Chem., Int. Ed., 2022, 134, e202200337 CrossRef.
- H. Jiang, J. Jin and W. Wong, Adv. Funct. Mater., 2023, 33, 2306880 CrossRef CAS.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 20498–20503 CrossRef CAS PubMed.
- Y. Gao, Y. Wang, Z. Guo, Y. Wan, C. Li, B. Yang, W. Yang and X. Ma, J. Phys. Chem. B, 2022, 126, 2729–2739 CrossRef CAS PubMed.
- J.-M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC.
- X. Cao, K. Pan, J. Miao, X. Lv, Z. Huang, F. Ni, X. Yin, Y. Wei and C. Yang, J. Am. Chem. Soc., 2022, 144, 22976–22984 CrossRef CAS PubMed.
- Z. Chen, D. Liu, M. Li, Y. Jiao, Z. Yang, K. Liu and S. Su, Adv. Funct. Mater., 2024, 2404278 CrossRef CAS.
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu, X. Cao and C. Yang, Nat. Photonics, 2022, 16, 803–810 CrossRef CAS.
- Z. Huang, H. Xie, J. Miao, Y. Wei, Y. Zou, T. Hua, X. Cao and C. Yang, J. Am. Chem. Soc., 2023, 145, 12550–12560 CrossRef CAS PubMed.
- T. Xu, X. Liang and G. Xie, Front. Chem., 2021, 9, 691172 CrossRef CAS PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
- J.-Y. Hu, Y.-J. Pu, G. Nakata, S. Kawata, H. Sasabe and J. Kido, Chem. Commun., 2012, 48, 8434 RSC.
- D. Thirion, M. Romain, J. Rault-Berthelot and C. Poriel, J. Mater. Chem., 2012, 22, 7149 RSC.
- J.-Y. Hu, Y.-J. Pu, Y. Yamashita, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 3871 RSC.
- Q. Liao, A. Li, A. Huang, J. Wang, K. Chang, H. Li, P. Yao, C. Zhong, P. Xie, J. Wang, Z. Li and Q. Li, Chem. Sci., 2024, 15, 4364–4373 RSC.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- S. Xu, Q. Yang, Y. Zhang, H. Li, Q. Xue, G. Xie, M. Gu, J. Jin, L. Huang and R. Chen, Chin. Chem. Lett., 2021, 32, 1372–1376 CrossRef CAS.
- E. Sebastian, J. Sunny and M. Hariharan, Chem. Sci., 2022, 13, 10824–10835 RSC.
- M. P. Lijina, A. Benny, E. Sebastian and M. Hariharan, Chem. Soc. Rev., 2023, 52, 6664–6679 RSC.
- P. Panthakkal Das, A. Mazumder, M. Rajeevan, R. S. Swathi and M. Hariharan, Phys. Chem. Chem. Phys., 2024, 26, 2007–2015 RSC.
- Y. Hong, W. Kim, T. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, Angew. Chem., Int. Ed., 2022, 61, e202114474 CrossRef CAS PubMed.
- W. Guo, W. Zhao, K. Tan, M. Li and C. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401835 CrossRef CAS PubMed.
- F. Zhang, F. Rauch, A. Swain, T. B. Marder and P. Ravat, Angew. Chem., Int. Ed., 2023, 62, e202218965 CrossRef CAS PubMed.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS PubMed.
- G. Jones, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- C.-W. Ju, B. Li, L. Li, W. Yan, C. Cui, X. Ma and D. Zhao, J. Am. Chem. Soc., 2021, 143, 5903–5916 CrossRef CAS PubMed.
- U. Müller, L. Roos, M. Frank, M. Deutsch, S. Hammer, M. Krumrein, A. Friedrich, T. B. Marder, B. Engels, A. Krueger and J. Pflaum, J. Phys. Chem. C, 2020, 124, 19435–19446 CrossRef.
- R. Jing, Y. Li, K. Tajima, Y. Wan, N. Fukui, H. Shinokubo, Z. Kuang and A. Xia, J. Phys. Chem. Lett., 2024, 15, 1469–1476 CrossRef CAS PubMed.
- P. Roy, G. Bressan, J. Gretton, A. N. Cammidge and S. R. Meech, Angew. Chem., Int. Ed., 2021, 60, 10568–10572 CrossRef CAS PubMed.
- Y. Wang, Z. Guo, Y. Gao, Y. Tian, Y. Deng, X. Ma and W. Yang, J. Phys. Chem. Lett., 2022, 13, 6664–6673 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- T. Azumi, A. T. Armstrong and S. P. McGlynn, J. Chem. Phys., 1964, 41, 3839–3852 CrossRef CAS.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- N. J. Hestand and F. C. Spano, J. Chem. Phys., 2015, 143, 244707 CrossRef PubMed.
- S. Kang, T. Kim, Y. Hong, F. Würthner and D. Kim, J. Am. Chem. Soc., 2021, 143, 9825–9833 CrossRef CAS PubMed.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2022, 14, 786–793 CrossRef CAS PubMed.
- E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2021, 143, 13769–13781 CrossRef CAS PubMed.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo and Y. Ma, Adv. Opt. Mater., 2021, 9, 2001845 CrossRef CAS.
- Z. Wang, R. Jing, Y. Li, D. Song, Y. Wan, N. Fukui, H. Shinokubo, Z. Kuang and A. Xia, J. Phys. Chem. Lett., 2023, 14, 8485–8492 CrossRef CAS PubMed.
- H.-M. Pan, C.-C. Wu, C.-Y. Lin, C.-S. Hsu, Y.-C. Tsai, P. Chowdhury, C.-H. Wang, K.-H. Chang, C.-H. Yang, M.-H. Liu, Y.-C. Chen, S.-P. Su, Y.-J. Lee, H. K. Chiang, Y.-H. Chan and P.-T. Chou, J. Am. Chem. Soc., 2023, 145, 516–526 CrossRef CAS PubMed.
- S. Jiang, Y. Yu, D. Li, Z. Chen, Y. He, M. Li, G. Yang, W. Qiu, Z. Yang, Y. Gan, J. Lin, Y. Ma and S. Su, Angew. Chem., Int. Ed., 2023, 62, e202218892 CrossRef CAS PubMed.
- H. Masuhara, N. Tamai, N. Mataga, F. C. De Schryver and J. Vandendriessche, J. Am. Chem. Soc., 1983, 105, 7256–7262 CrossRef CAS.
- T. Karatsu, T. Shibata, A. Nishigaki, A. Kitamura, Y. Hatanaka, Y. Nishimura, S. Sato and I. Yamazaki, J. Phys. Chem. B, 2003, 107, 12184–12191 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05494f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |