
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron transport chain-inspired coordination polymers for macroscopic spatiotemporal scales of charge separation and transport in photocatalysis†
Lin
Ma
a,
Tiexin
Zhang
 *a,
Mochen
Li
a,
Xu
Zhang
a,
Lanqiao
Li
a,
Yusheng
Shi
b,
Rui
Cai
a,
Xueming
Yang
c and
Chunying
Duan
d
*a,
Mochen
Li
a,
Xu
Zhang
a,
Lanqiao
Li
a,
Yusheng
Shi
b,
Rui
Cai
a,
Xueming
Yang
c and
Chunying
Duan
d
aState Key Laboratory of Fine Chemicals, School of Chemistry, Frontier Science Center for Smart Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: zhangtiexin@dlut.edu.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
cState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
dState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210093, China
First published on 19th September 2024
Abstract
Classic homogeneous photocatalysis is limited by the temporal transience and the spatial proximity of photoinduced charge separation and transport. The electron transfer chain (ETC) in cellular respiration can mediate unidirectional and long-range electron transfer to isolate the oxidation and reduction centres. Inspired by this, we modified electron-accepting (A) viologen with π-extending thiazolothiazole and electron-donating (D) phenyl carboxylate into a D–A–π–A–D-type ligand and assembled segregated dye stacking in coordination polymer Cd−TzBDP for breaking the spatiotemporal limitation of single-molecule photocatalysis. The offset characteristics of D–A segregated stacking not only allowed the photoinduced-2e− transfer from the D-type carboxylate terminal to the spatially adjacent A-type viologen motif within 1 ps but also permitted the following delocalization of e− and h+ along stacked columns. These advantages endowed Cd−TzBDP with long-lived photochromic visualization of intermittent aerobic photooxidation steps, which enabled the bioinspired ETC-mediated aerobic respiration of mitochondria, achieving the continuous photocatalytic α-C(sp3)–H functionalization of tertiary amines with pharmaceutical interest. Enlightened by ETC-mediated electron leak in hypoxia, the coordination polymer was further employed in a photocatalytic membrane reactor, which visually illustrated the photo-driven cross-membrane long-range transfers of multiple electrons and protons from the hypoxic compartment to normoxic one, benefiting the distal photooxidation and photoreduction with biomimetic compartment selectivity.
Introduction
Visible-light-driven photoredox catalysis has become a valuable synthetic means for organic synthesis by utilization of solar energy.1,2 The photoinduced charge separation provides the origin of photoreduction and photooxidation, but the transient lifetime (ps to ns scale) and the spatially proximal distribution (Å to sub nm scale) of photoseparated charges lead to the sluggish diffusion-controlled charge transfer.3–5 Moreover, the uncontrolled thermal motions and collisions in the solution phase further cause intra-/inter-molecular back-electron transfer (BET) of photoseparated charges, causing mutual interference between photoreductive and photooxidative centres. These characteristics impose the inherent spatiotemporal limitation towards the practical photocatalytic applications under the realm of single-molecule photoredox.Naturally existing biological electron transfer is one of the key steps enabling virtually all cellular activities,6 of which the most fascinating characteristics are the broad length and time scales: from ps to ms and from nm to cm,7–10 providing unique perspectives for breaking through the limitations of artificial photocatalysis. For example, the electron transport chain (ETC)11–15 of cellular respiration is made up of the membrane-associated series connection of enzyme/cofactor redox shuttles with gradient electronic potentials, which mediates the unidirectional electron transfer from an electron-donating source, such as NADH (nicotinamide adenine dinucleotide), to a terminal electron sink like O2 (Scheme 1a), at the synchronous paces of other metabolisms with the macroscopic time scales. Merging the kinetically macroscopic spatiotemporal scales of biological ground-state electron transfers and the thermodynamically potent driving forces of artificial photoexcited states provides a unique perspective for breaking through the limitations of artificial single-molecule photocatalysis. Inspired by the biological ETC, it is desirable to temporally lengthen the duration of photoreductive and photooxidative centres and spatially isolate two events of photoreduction and photooxidation at distal positions,16,17 which, however, was difficult to realize by the transient and proximal photoseparated charges owing to the dearth of the toolbox in homogeneous mode.
 | ||
| Scheme 1 (a) ETC in biological aerobic respiration. (b) This work: biomimicking by dye stacking goes beyond single-molecule photocatalysis. (c) Ligand design. (d) Coordination environment between the viologen-based ligand and cadmium ion in Cd−TzBDP. (e) 3D framework of Cd−TzBDP showing unique segregated D/A stacking and charge-separation of Cd−TzBDP. The π⋯π interaction between the adjacent pyridinium, and Tz moiety with a distance of 3.29 Å. The blue, purple, and green columns represent the D, A, and π constituents. (f) The offset in segregated D/A stacking. The distances between the inter-ligand Ocarboxylate atom and Npyridinium atom are 3.68 Å (green dotted line) and 3.83 Å (purple dotted line). The distances between the intra-ligand Ocarboxylate atom and Npyridinium atom are 4.98 Å and 5.11 Å (black dotted line). The sets of (h) HOMO and (g) LUMO for TzBDP. The sets of (i) HOMO and (j) LUMO for truncated Cd−TzBDP, possibly involving the photoinduced inter-ligand charge separation from the carboxyl group to the pyridinium cation. | ||
Thinking outside the box of typical single-molecule photoredox, the supramolecular assemblies based on the modules of electron donor (D) and acceptor (A) might learn and simplify the function of biological ETC, which possibly tunes the direction and range of charge flow at ground and excited states. Especially, the segregated stackings of D⋯D⋯D and A⋯A⋯A can regulate the directional charge transport along the stacking column after photoinduced charge separation.18–20 Moreover, it is noteworthy that photochromic materials featuring the photoinduced charge-separation states of macroscopic lifetime scales can endow catalysis with “visualization” detected by the naked eye.21,22 Thus, combining the photochromic modules and the electronic complementary units in a D–A segregated stacking manner might simultaneously achieve the temporally and spatially larger scales for photoinduced charge separation and the anisotropic transport of charge carriers, respectively, providing a possible way to go beyond the spatiotemporal limitations of single-molecule photocatalysis (Schemes 1b and S1†). However, the mixed stacking of D/A modules (D⋯A⋯D⋯A), other than segregated stacking, tends to dominate the aggregation mode due to the electronic complementarity of D and A.
As a family of crystalline organic–inorganic hybrid materials with tunable structures, coordination polymers (CPs) have been recognized as promising candidates for the assembly of D–A-type photocatalysts.23–26 The coordination polymers can provide rigid environments and conformations for D and A motifs, further averting intermolecular quenching and decelerating intramolecular BET. The coordination-oriented structural coercions were believed to balance off the strong tendency of D–A mixed stacking. Thus, it is promising to realize the ETC-inspired design of photocatalysts by assembling the D and A into a segregated stacking manner within the coordination polymer.
Viologen, as a coenzyme-like redox mediator and rigid electron-deficient A-type dye motif, has been widely used in electrochemical and photoelectrochemical processes.27,28 Viologen can undergo a stepwise two-electron redox process and exhibit three stable/semi-stable states (dication, radical cation, and neutral) with different colours. Light irradiation could trigger the chromism by photoinduced electron transfer (PET) from an external D to the viologen A moiety, and further exposure to the oxidative atmosphere like air led to decolouration.29 As the rigid aromatic unit with premium free charge carrier mobility,30 thiazolothiazole (Tz) was inserted into two pyridinium A sections, extending the π-conjugation structure to enhance the visible light harvesting ability of viologen derivatives.31 By further decorating two anionic phenyl carboxylates as D-type coordination terminals, we designed a viologen-derived ligand, 4,4′-(thiazolo[5,4-d]thiazole-2,5-diyl)bis(1-(3,5-dicarboxyphenyl)pyridin-1-ium) (denoted as TzBDP). Herein, a novel viologen-based coordination polymer with segregated dye stacking, Cd−TzBDP was constructed through the solvothermal reaction of the TzBDP ligand and cadmium (Cd) salt. Taking advantage of the charge complementarity between the π-section (thiazolothiazole) and A-section (pyridinium) within this D–A–π–A–D-type TzBDP ligand (Scheme 1c), the spontaneous inter-ligand aggregation of π⋯A⋯π⋯A⋯π was believed to bring structural glide factors into self-assembly (Scheme 1b and e). The links of non-conductive Cd-carboxylate clusters between redox-active ligands helped maintain the photoinduced charge-separation states. The multi-coordination mode of cadmium was conducive to forming interesting inter-cluster links,32 which together with the tendency of inter-ligand glide possibly provided the comprehensive structural coercions to forge the offset characteristics into the D–A segregated stacking. This distinctive dye-stacking mode within the coordination polymer provided the possibility of photoinduced charge separation between spatially adjacent ligands and still allowed the successive hopping of h+ and e− along the stacked columns of D and A, respectively (Scheme 1b), making up for the consumptive recombination of the intra-ligand separated charges.
As expected, a series of photoelectronic examinations revealed that Cd-TzBDP featured faster-photoinduced charge separation, more durable charge carriers, and more feasible charge delocalizations compared to ligand TzBDP. The coordination polymer further showed a persistent photochromism upon external light stimuli, favouring the “visualization” of the intermittent aerobic photooxidation of electron-donating amines. These advantages made Cd−TzBDP competent in the probe reaction of continuous photocatalytic α-C(sp3)–H functionalizations of tertiary amines with added values and pharmaceutical interest. After verifying the rapid photoinduced charge separation of the coordination polymer by transient absorption spectroscopy, the coordination polymer crystals were successfully employed in a photocatalytic membrane reactor that was enlightened by the ETC-mediated electron leak in the case of mitochondrial hypoxia, exhibiting the stronger driving force in the consecutive multiple electron oxidation than the membrane composed of ligands, of which the different colourations visually illustrated the advantages of the coordination polymer in the directional cross-membrane long-range transfers of multiple electrons and protons, benefiting the bioinspired photooxidation and photoreduction at the spatially distal positions with compartment selectivity.
Results and discussion
First, we report on synthesizing a novel D–A type viologen-based ligand, where the π-extended viologen core works as an electron-withdrawing A section, and two anionic phenyl carboxylates as D sections. Through the Zincke reaction of dimethyl 5-amino isophthalate with diquaternary pyridinium salt of the thiazolo[5,4-d]thiazole (Tz) moiety, followed by the successive hydrolysis (Scheme S2†), the D–A–π–A–D-type ligand TzBDP was obtained (Scheme 1c). The cadmium-based coordination polymer Cd−TzBDP was synthesized via a solvothermal reaction of ligand TzBDP and Cd(NO3)2·4H2O in the presence of CH3CN/H2O mixture solution at 130 °C within 2 days. Single crystal X-ray structural analysis revealed that the compound Cd−TzBDP adopts a three-dimensional (3D) framework and is crystallized in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). The phase purity of Cd−TzBDP was verified using powder X-ray diffraction (PXRD) patterns (Fig. S1†). According to thermogravimetric analysis (TGA), Cd−TzBDP is thermally stable up to 160 °C (Fig. S2†). The scanning electron microscopy (SEM) (Fig. S4†) and energy dispersive spectroscopy (EDS) mapping images (Fig. S5†) showed that Cd, S, and N elements were uniformly distributed in the block crystal of Cd−TzBDP.
(Table S1†). The phase purity of Cd−TzBDP was verified using powder X-ray diffraction (PXRD) patterns (Fig. S1†). According to thermogravimetric analysis (TGA), Cd−TzBDP is thermally stable up to 160 °C (Fig. S2†). The scanning electron microscopy (SEM) (Fig. S4†) and energy dispersive spectroscopy (EDS) mapping images (Fig. S5†) showed that Cd, S, and N elements were uniformly distributed in the block crystal of Cd−TzBDP.
Each CdII is seven-coordinated with five oxygen atoms from four carboxylic groups of the different TzBDP ligands and two oxygen atoms from one nitrate (Scheme 1d, Fig. S6 and S7†). The adjacent cadmium ions are linked by two μ2-O from the electron-donating (D) carboxylate terminals of different TzBDP ligands to obtain a dinuclear metallic cluster Cd2(O2C)6(O3N)2 (abbreviated as Cd2) with a Cd⋯Cd distance of 3.89 Å (Fig. S7 and S8†). The neighbouring Cd2 clusters are continuously bridged by two carboxylic acid groups, further constituting a “D”-type column in a series-connection manner (Scheme 1e and Fig. S8†). Cd−TzBDP holds the close aromatic stackings of ligand moieties (with a plane-to-plane distance of 3.4 Å, Fig. S9†) into the infinite columns along the 1D-chain of Cd-carboxylate (Fig. S10†), therein the charge delocalization of photoinduced e−/h+ pairs might be facilitated through the segregated D/A stacking columns (Scheme 1e). The inter-layer proximity (ca. 3.29 Å) between the “A”-type pyridinium motif and “π-extension”-type thiazolothiazole section from different ligands clearly shows the tight column stacking with glide features (Scheme 1e). The high free charge carrier mobility of π-conjugative thiazolothiazole promisingly endows the close packing of π⋯A⋯π⋯A⋯π an ideal pathway for electron transport. Interestingly, the inter-ligand nearest O⋯N distances between the electron-rich Ocarboxylate atom and electron-deficient Npyridinium atom are 3.68 and 3.83 Å, respectively (Scheme 1f), which are remarkably shorter than the intra-ligand O⋯N distances (ca. 4.98 and 5.11 Å). These noteworthy structural features reflected the offset characteristics of segregated stacking in Cd−TzBDP, providing the possible routes of through-space charge transfer from the spatially adjacent D to A sections of different ligands.33 According to the density functional theory (DFT) calculations, the highest occupied molecular orbital (HOMO) is localized on the electron-rich terminal of the phenyl carboxylate moiety of the TzBDP ligand, and the lowest unoccupied molecular orbital (LUMO) is mainly situated on the π-bridged bispyridinium motif (Scheme 1g and h).34 The coordination polymer Cd−TzBDP also demonstrated similar spatial isolation characteristics of frontier orbitals, but it is noteworthy that HOMO and LUMO sections of Cd−TzBDP were further separated by the non-variable valence Cd2+ site (Scheme 1i and j), which implied the possibility of inter-ligand charge separation and the retarded charge recombination compared with the case of free ligand (Scheme 1b and e).
As shown in Fig. 1a, the solid-state absorption spectra of TzBDP exhibited a characteristic charge-transfer band from 400 to 500 nm, implying strong polarity of the D–A type ligand. A much broader charge-transfer absorption band (400–800 nm) was observed for coordination polymer Cd−TzBDP, which could be attributed to the great extent of partial charge transfer within the stacked columns. Photoluminescence spectroscopy was applied to estimate the charge separation and transfer of Cd−TzBDP. Under an inert Ar atmosphere, Cd−TzBDP exhibited the ca. 22 nm-red-shifted photoluminescence with a slightly weaker intensity compared with the case of TzBDP (Fig. 1b). More notably, Cd−TzBDP showed a nearly three times longer fluorescence decay lifetime (1.78 ns) than that of TzBDP (0.64 ns) (Fig. 1c). These phenomena implied the enhanced charge separation and delocalization of the coordination polymer after photoexcitation, which might be correlated with the offset segregated dye stacking (Scheme 1e).
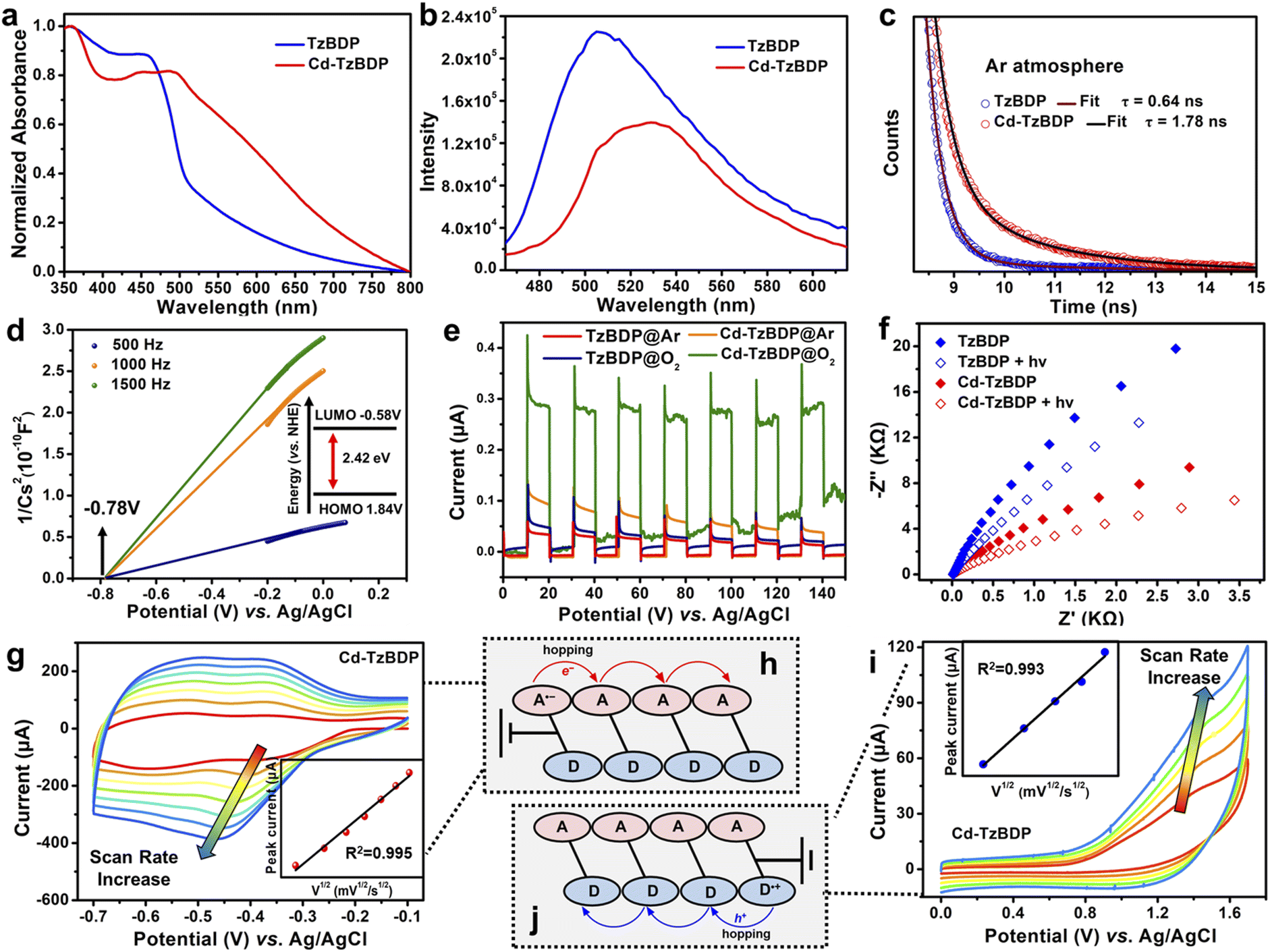 | ||
| Fig. 1 (a) UV-vis spectra, (b) photoluminescence spectra, (c) luminescence decays under an Ar atmosphere of TzBDP and Cd−TzBDP. (d) Mott–Schottky plots of Cd−TzBDP. (e) Photocurrent responses of TzBDP and Cd−TzBDP under an Ar and O2 atmosphere. (f) EIS plots of TzBDP and Cd-TzBDP before and after irradiation. Cyclic voltammetry curves for Cd−TzBDP in the negative potential range (g) with varying scan rates: 10, 20, 30, 40, 50, 60, and 70 mV s−1 and in the positive potential range (i) with varying scan rates: 10, 20, 30, 40 and 50 mV s−1. The inset shows the peak current to be linear with the square root of the scan rate (ν)1/2. Illustration of charge hopping of (h) e− and (j) h+. | ||
We conducted the Mott–Schottky measurements at the frequencies of 500, 1000, and 1500 Hz for TzBDP and Cd−TzBDP. The results showed positive slopes of the C−2 values, indicating their typical characteristics of n-type semiconductors. For Cd−TzBDP, the flat band position decided by the intersection was −0.78 V vs. Ag/AgCl (−0.58 V vs. NHE), approximately to the bottom of the conduction band (LUMO) (Fig. 1d). Combining with the optical band gap (2.42 eV) based on the Kubelka–Munk method from Tauc plots (Fig. S11†), the top of the valence band (HOMO) was then calculated to be 1.64 V vs. Ag/AgCl (1.84 V vs. NHE). In comparison, the HOMO and LUMO of ligand TzBDP were determined to be 2.13 V and −0.57 V vs. Ag/AgCl, respectively (Fig. S12†). The energy-band structure in Cd−TzBDP made it theoretically feasible for photoreduction of O2 to O2˙– (−0.33 eV vs. NHE) and photooxidation of organic substrates.35
Solid-state cyclic voltammograms (CVs) were recorded to clarify the electron transfer process. The processes in the cathodic region observed at −0.40 V and −0.66 V vs. Ag/AgCl for Cd−TzBDP can be assigned to two sequential one-electron reductions, which were comparable to the case of ligand (−0.36 V and −0.61 V vs. Ag/AgCl) (Fig. S13†). CV curves at different scan rates showed the linear relationship between the reduction and oxidation peak currents ipc and the square roots of the scan rate (ν)1/2 for Cd−TzBDP (Fig. 1g and i and insets). In comparison, the ligand TzBDP displayed a decreased linear relationship (Fig. S14 and S15†). These results implied the possible charge hopping of photogenerated e− and h+ along the stacked columns of Cd−TzBDP (Fig. 1h–j),36 providing the possibility of long-range charge delivery. Photocurrent measurements revealed the higher photocurrent response of Cd−TzBDP than TzBDP, indicating the more remarkable photoinduced-charge separation in the coordination polymer (Fig. 1e). Compared to the ligand TzBDP, a smaller resistance was observed for Cd−TzBDP in the electrochemical impedance spectroscopy (EIS) (Fig. 1f), indicative of better conductivity of the column stacking structure. After illumination, the EIS impedance of Cd−TzBDP decreased to a greater extent than that of the ligand, which further confirmed the enhanced photoinduced-charge separation.37
It is known that most viologen-based compounds present photochromic properties under light irradiation. Herein, when exposed to a 405 nm LED, the colour of Cd−TzBDP changed from brown to ash black. The absorption intensity around 500–800 nm was enhanced in UV-vis absorption spectra (Fig. 2a), and the differential spectrum clearly showed the band of the chromic viologen radical centred at 672 nm (Fig. 2c), which indicated the photoinduced-two electron (2e−) transfer from the metal–carboxylate cluster to the electron-deficient core of the ligand.30,38 Powder X-ray diffraction for Cd-TzBDP shows no significant change after irradiation (Fig. S16†), ruling out the possibility of photoinduced structural isomerization. The solid sample of ligand TzBDP also exhibited photochromic behaviour (Fig. 2b). Upon standing in air, Cd−TzBDP showed improved colouration time and moisture stability compared with the ligand.39 The structural characteristics such as offset segregated stacking of Cd−TzBDP were believed to maintain the long-lived charge separation and enhance the stability of chromogenic viologen radicals. As is shown in Fig. 2d, after photoirradiation of Cd−TzBDP, the asymmetric and symmetric vibrations of coordinated carboxylate groups (1580 cm−1 and 1380 cm−1) of infrared spectra (IR) diminished,40 implying that the carboxylate moiety within the node might be involved in the photochromic charge-separation process.
 | ||
| Fig. 2 UV-vis spectra of (a) Cd-TzBDP and (b) TzBDP before and after irradiation. Inset: photochromic photographs before and after irradiation. (c) The difference spectra of Cd−TzBDP before and after irradiation. (d) IR spectra of TzBDP, Cd−TzBDP, and Cd−TzBDP after irradiation. XPS spectra of (e) O 1s, and (f) N 1s before and after irradiation. | ||
To further understand the photoinduced electron transfer processes, X-ray photoelectron spectra (XPS) and electron paramagnetic resonance (EPR) of Cd−TzBDP were recorded before and after irradiation, respectively. As shown in Fig. 2e, the XPS asymmetric peak of the O 1s core-level of Cd−TzBDP displayed two peaks at binding energies of 532.6 eV and 530.9 eV, assignable to nitrate and carboxylate, respectively. Upon irradiation, the peaks shifted to higher binding energies of 533.0 eV and 531.4 eV (Fig. 2e), which was consistent with the electron loss from oxygen atoms of the coordination polymer node upon irradiation.41 Correspondingly, the N 1s core-level spectrum exhibited three peaks, a signal of the nitrate N atom at 405.0 eV, and the other two at 402.2 eV and 399.3 eV attributed to pyridinium N and thiazole N, respectively (Fig. 2f). After irradiation, the peak intensity of pyridinium N at 402.1 eV decreased remarkably, and a new band of viologen radical N appeared at 401.5 eV, indicating the increase of electron density after accepting additional electrons.42–45 In comparison, the Cd 3d core-level bands remained unchanged after irradiation (Fig. S17†), suggesting that the Cd metal centre of the carboxylate node did not participate in photochromic charge transfer, which was as expected for the redox-innocent d10 ion.43–45 As depicted in Fig. 3d, a sharp single-line signal with g = 2.002 appeared in the EPR spectra after illumination of Cd−TzBDP,46 further confirming the generation of the viologen radical. These results comprehensively verified that the photochromism-related electron transfer was from the oxygen of the Cd-carboxylate cluster to the viologen moiety in the coordination polymer. Furthermore, as a “visualization” mode of photoinduced charge separation, the macroscopic photochromism of Cd−TzBDP possibly provided the thermodynamically competent reductive and oxidative reaction sites with a long duration for the ETC-inspired photoredox applications.
 | ||
| Fig. 3 (a) Time-resolved luminescence spectra of Cd−TzBDP suspension under Ar, O2 and by the addition of N,N-dimethylaniline 1a. (b) UV-vis spectra of Cd−TzBDP after N,N-dimethylaniline 1a addition and subsequent irradiation. (c) The difference spectra of Cd−TzBDP and 1a before and after irradiation. (d) Comparative EPR of the dark, the photoirradiated Cd−TzBDP, and the photoirradiated coordination polymer with DMPO added. (e) The intermittent photochromic photocatalysis by colour changes of Cd−TzBDP. (f) Optimization of reaction conditions of photocatalytic oxidative cyanation. Reaction conditions: N,N-dimethylaniline 1a (1 equiv., 0.2 mmol), TMSCN (0.6 mmol), Cd−TzBDP (5 mol% based on the ligand moiety) in DMF/CH3CN (2 mL/1 mL) under irradiation of white LEDs in an O2 atmosphere at room temperature (r.t.) for 24 h. Isolated yields are given. (g) Effect of scavengers on the optimal reaction conditions of photocatalysis. (h) Recycling experiments of photocatalyst Cd−TzBDP. (i) Comparative PXRD patterns of Cd−TzBDP before and after recycling experiments. | ||
Considering the strong electronic complementarity between viologen moieties and amine derivatives,33 an amine-typed inert C–H substate, N,N-dimethylaniline (1a), was chosen to examine the relationship between the host–guest photochromism and the photocatalytic cascade O2 utilization and C–H activation. Upon the addition of N,N-dimethylaniline 1a, the acetonitrile suspension of Cd−TzBDP showed decreased photoluminescence intensity and shortened fluorescence lifetime (Fig. 3a and S18†), indicating the PET from the amine-type substrate to the coordination polymer. Moreover, upon the addition of 1a to the solid sample of Cd−TzBDP, the intensified charge-transfer band covering 400–800 nm reflected their enzyme–substrate-like affinity and the ground-state partial electron transfer from the substrate to the framework (Fig. 3b). After the successive photoirradiation at 405 nm, the emergence of a typical viologen radical peak was observed (Fig. 3c), indicating the successive 2e− reduction of the viologen-based coordination polymer in the presence of 1a. To probe the ETC-inspired activity, we studied the O2 activation performance of coordination polymer Cd−TzBDP. Upon exposure to an O2 atmosphere, Cd−TzBDP exhibited a significantly increased photocurrent response (Fig. 1e), and the photoexcited states were partially quenched (Fig. 3a). EPR measurement was further carried out to identify reactive oxygen species (ROS) by adding 5,5-dimethyl-1-pyrroline N-oxide (DMPO) to the Cd−TzBDP sample that was irradiated in air. As is shown in Fig. 3d, the generation of the superoxide radical anion (O2˙–) was detected and the viologen radical signal was weakened, indicating the potential applicability of Cd−TzBDP in the photocatalytic aerobic oxidation. Based on the above results, we conducted intermittent photocatalysis experiments. After photoirradiating for ca. 2 min under an inert Ar atmosphere, the mixture of Cd−TzBDP and N,N-dimethylaniline 1a turned ash green, indicating the generation and accumulation of viologen radical species by the consecutive electron transfer from 1a. Following purging with O2, the ash green Cd−TzBDP faded to the original colour (Fig. 3e). This intermittent reaction process mimicked the unidirectional electron transfer, that started from NADH, mediated through a naturally existing ETC system, and finally delivered to the electron sink O2 in the same bio-compartment, which was empowered by artificial photoirradiation, more importantly, providing a possible “visualization” manner for photocatalysis.
Then, the above-mentioned photocatalysis was examined in a continuous reaction manner. Considering the importance of aerobic activation and direct functionalization of α-C(sp3)–H bonds in the pharmaceutical field, such as accessing the bioactive α-amino nitriles,47,48 the photocatalytic oxidative cyanation of aryl tertiary amines was chosen as the model reaction. Upon white-light irradiation of a DMF/CH3CN solution of N,N-dimethylaniline 1a and trimethylsilyl cyanide (TMSCN) in the presence of 5 mol% Cd−TzBDP photocatalyst, the desired product 2a was obtained in an isolated yield of 86% (Fig. 3f, entry 1). In comparison, simple salt Cd(NO3)2·4H2O provided no formation of the desired product (entry 2), and much lower yields of 24% and 33% were afforded when using mere TzBDP ligand (entry 3) and the mixture of Cd salt and ligand (entry 4) as photocatalysts, respectively. Control experiments delivered no generation of target products in the absence of photocatalyst, light irradiation, or O2 (entries 5–7), indicating the indispensability of these reaction parameters.
To understand the key intermediates in photocatalytic oxidative cyanation, we further performed control experiments with some scavengers (Fig. 3g). Upon addition of hole scavenger KI and electron scavenger AgNO3, the conversions below 20% were obtained (16% and 12%), implying that both e− and h+ in the charge-separated state played crucial roles in the whole transformation. In the case of adding O2˙– scavenger p-benzoquinone to the optimized condition, a significant decrease in yield (20%) was observed, but no obvious change in reaction efficiency (83%) could be detected when employing the 1O2 scavenger triethylenediamine (DABCO). These results verified O2˙– as the dominant ROS in the oxidation step. Moreover, the recycling performance of photocatalyst Cd−TzBDP was also investigated. As shown in Fig. 3h, no remarkably decreased reactivity was found after three rounds of iterative tests. The maintained PXRD patterns of the recovered coordination polymer indicated the structural integrity after reactions (Fig. 3i).
With the optimized conditions of the photooxidative cyanation reaction in hand, we explored the substrate scope (Fig. 4a). The aryl-substituted N,N-dimethylaniline derivatives bearing electron-donating groups like methyl, or electron-withdrawing chloro and bromo groups generated the corresponding α-C–H cyanation products in good yields (Fig. 4a, 2b–2e). Besides the open-chain N,N-dimethyl groups of the amine moiety, the substrates bearing 5- and 6-membered close-ring N-substituents, such as N-phenylpyrrolidine (2f), N-phenylpiperidine (2g), N-phenylmorpholine (2h), and N-phenyltetrahydroisoquinoline (2i) were also applicable, furnishing the products in good to high yields. Notably, using 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as the raw material, the cyano derivative of bioactive corydaline (2j) was efficiently synthesized. The natural alkaloid product gramine also could be converted to the corresponding α-aminonitrile 2k in 78% yield.
 | ||
| Fig. 4 (a) Substrate scope of photooxidative α-cyanation of tertiary amine. Reaction conditions: N,N-dimethylaniline derivatives (1 equiv., 0.2 mmol), TMSCN (0.6 mmol), Cd−TzBDP (5 mol%) in DMF/CH3CN (2 mL/1 mL) under irradiation of white LEDs in an O2 atmosphere at r.t. for 24 h. Isolated yields. #77% yield was obtained after 48 h for a 10 mmol scale reaction using 2.5 mol% catalyst. (b) The substrate scope of photocatalytic miscellaneous α-functionalization. Reaction conditions: N-aryl tetrahydroisoquinoline (1 equiv., 0.2 mmol), nitroalkane (1.0 mL) or other nucleophiles (2 mmol), Cd−TzBDP (5 mol%) in CH3OH (1.0 mL) under irradiation of white LEDs in an O2 atmosphere at r.t. for 24 h. Isolated yields are given. | ||
Based on the above results, a plausible mechanism for this photocatalytic oxidative cyanation reaction was proposed (Fig. S19†).49 Upon irradiation, Cd−TzBDP can be excited for the e−/h+ separation. Then, the electrons in the LUMO section reduced oxygen to generate O2˙–, while the holes in the HOMO section of Cd-carboxylate cluster oxidized substrates like N,N-dimethylaniline 1a to afford 1a˙+ (Eox(1a/1a˙+) = +0.74 V vs. SCE).50 Next, the radical cationic aniline 1a˙+ was subjected to hydrogen-atom abstraction by O2˙–, leading to the possible formation of the iminium intermediate, which was attacked by the nucleophile CN− that released from TMSCN, affording the target product α-aminonitrile 2a. Compared with the above-mentioned N,N-dimethylaniline 1a, another kind of amine substrate like N-phenyl tetrahydroisoquinoline 3a possessed more positive oxidation potential (Eox(3a/3a˙+) = +1.04 V vs. SCE)51 which was theoretically still within the scope of photooxidation capability of Cd-TzBDP (HOMO, 1.84 V vs. NHE). The fluorescence quenching experiment of Cd−TzBDP by 3a implied the PET between them (Fig. S20 and S21†). After illuminating the reaction mixture of Cd−TzBDP and 3a under an O2 atmosphere, the electrospray ionization mass spectroscopy (ESI-MS) detected an intense peak at m/z = 208 assignable to the corresponding iminium ion of 3a (Fig. S22†).52 These iminium ion intermediates could be further attacked by different nucleophiles to explore the versatility of this protocol in miscellaneous direct α-C–H functionalization (Fig. S23†).
As expected, with the addition of CH3NO2 as a nucleophile, N-phenyl tetrahydroisoquinoline 3a could be transformed into the targeted aerobic cross-dehydrogenative coupling product 4a in an excellent yield of 94%, in the presence of 5 mol% Cd−TzBDP and photoirradiation. Under the optimized conditions (Table S2,† entry 1), a series of N-aryl tetrahydroisoquinoline derivatives and nucleophiles were applied to this methodology (Fig. 4b). The starting materials with electron-donating substituents were converted into the products in excellent yields of 90–99% (Fig. 4b, 4b, 4c, and 4h).
While changing to electron-withdrawing substituents (F, Cl, Br, and CF3), the direct α-C–H functionalization reactions furnished the products in good to excellent yields (70–99%) (Fig. 4b, 4d–4g). This photocatalysis also worked well with other carbon-centred nucleophiles like acetone (4j), dimethyl malonate (4k), indole (4l), and the heteroatom-centred nucleophile-like diethyl phosphite (4m). Notably, the protocol could be further extended to access the nitromethyl decoration of bioactive corydaline at the α-amino position (4i), showing the potential pharmaceutical interest.
It can be seen from the above that, at the macroscopic time scales, the photochromism enhanced the practical intermittent and continuous photocatalyses, reflecting the advantages of coordination polymers with the specific dye stacking manner. In comparison, the photoelectronic behaviour and structure–activity relationship of Cd−TzBDP at the microscopic time scale were intriguing and still elusive. Thus, the femtosecond transient absorption spectroscopy (fs-TAs) was conducted to elucidate the photoinduced charge separation (Fig. 5). Upon excitation at 400 nm, the transient absorption spectra of ligand TzBDP displayed the excited-state absorption (ESA) band covering a broad range of 500–600 nm, and the relatively weak band around 700 nm can be ascribed to the characteristic absorption of diradical viologen species that formed after a 2e− intra-ligand charge separation from the phenyl carboxylate terminal to the electron-deficient core of the ligand.30,38 For coordination polymer Cd−TzBDP, the ESA band was much weaker than the case of ligand, but the ground state bleach (GSB) of the D–A charge-transfer band at ca. 480 nm was more remarkable. Moreover, the profound viologen diradical signal of Cd−TzBDP appeared within 1 ps. These phenomena implied that, in comparison to the photoinduced intra-ligand charge separation of single-molecule TzBDP, the more facile route of charge separation probably exists in the offset-segregated D–A stacking of coordination polymer. Subsequently, the decay kinetics at 700 nm were compared, and the corresponding time constants were 739.5 ps and 290.5 ps for Cd−TzBDP and TzBDP, respectively, exhibiting a nearly three-fold lengthened decay lifetime of charge-separation state in the coordination polymer. The much-retarded charge recombination here, the similar near three-fold fluorescence lifetime of Cd−TzBDPvs.TzBDP (Fig. 1c), and the electrochemical charge hopping in Cd−TzBDP were noticeable (Fig. 1g–j). These clues comprehensively corresponded to the possible delocalization of carriers along the stacked D/A columns after the rapid photoinduced charge separation (Scheme 1b),53 which implied the potential bridging role of Cd−TzBDP from the transient and local photoexcitation to the charge separation and transport at the macroscopic spatiotemporal scales, making Cd−TzBDP competent in the ETC-inspired anisotropic and consecutive electron transfer under photoirradiation.
 | ||
| Fig. 5 The comparative fs-TA analyses of Cd−TzBDP and TzBDP. The 400 nm laser excited fs-TA spectra of (a) Cd−TzBDP and (b) TzBDP at the indicated delay times. The corresponding kinetic traces were measured at 700 nm for (c) Cd−TzBDP and (d) TzBDP. | ||
The mitochondrial ETC-mediated production of ROS such as O2˙– and H2O2 is important in cell signalling and disease, which is contributed by the intra-mitochondria oxidation of substrates and the cross-membrane electron leak to reduce O2 outside of mitochondria (Fig. 6a).54,55 Inspired by the electron leak of mitochondria under hypoxia, we utilized a handmade nested double-layered photocatalytic membrane reactor with the outer layer normoxic and the intra-layer hypoxic compartments that were separated by the composite membrane of TzBDP@Nafion or Cd−TzBDP@Nafion (Fig. 6b–e and S24–S26†). Based on the above-mentioned intermittent or continuous photocatalytic reaction in the same compartment (Fig. 3e and f), the electron and proton-donating 3,3′,5,5′-tetramethylbenzidine (TMB) was selected as an NADH-mimicking substrate to examine light-driven multi-electron transfer in the multi-compartment system confined by biology-like membranes (Scheme S3†).56,57 UV-vis spectroscopy was used to monitor the absorption change58 during the photooxidation process of TMB in the hypoxic chamber. When using TzBDP@Nafion as the photocatalytic composite membrane, TMB was converted to 1e− oxidation product TMBox1+ under photoirradiation with a 405 nm LED, showing the typical green-coloured solution and the absorptions at 378 nm and 654 nm (Fig. 6g). In contrast, Cd−TzBDP@Nafion gave rise to the dominant formation of 2e− oxidation product TMBox2+, which was evidenced by the yellow colour of solution and the representative band of TMBox2+ at 456 nm (Fig. 6h), visually illustrating the stronger driving force of the coordination polymer-based membrane than the one composed of ligand in the successive multi-electron transfer. In the outer normoxic compartment without TMB, the peroxide-type ROS could be detected using KI/starch testing paper after the photocatalytic TMB oxidation (Fig. 6f and S25†), mimicking the ETC-mediated cell signalling under the mitochondrial hypoxic stress, which implied the cross-membrane long-range diffusion of released electrons and protons from the interior hypoxic side to the outer normoxia, benefiting the bioinspired photooxidation and photoreduction at the spatially distal positions. These results reflected the advantages of offset segregated D–A stacking in the photo-driven charge separation and anisotropic multi-charge transportation from the microscopic perspectives to the macroscopic applications.
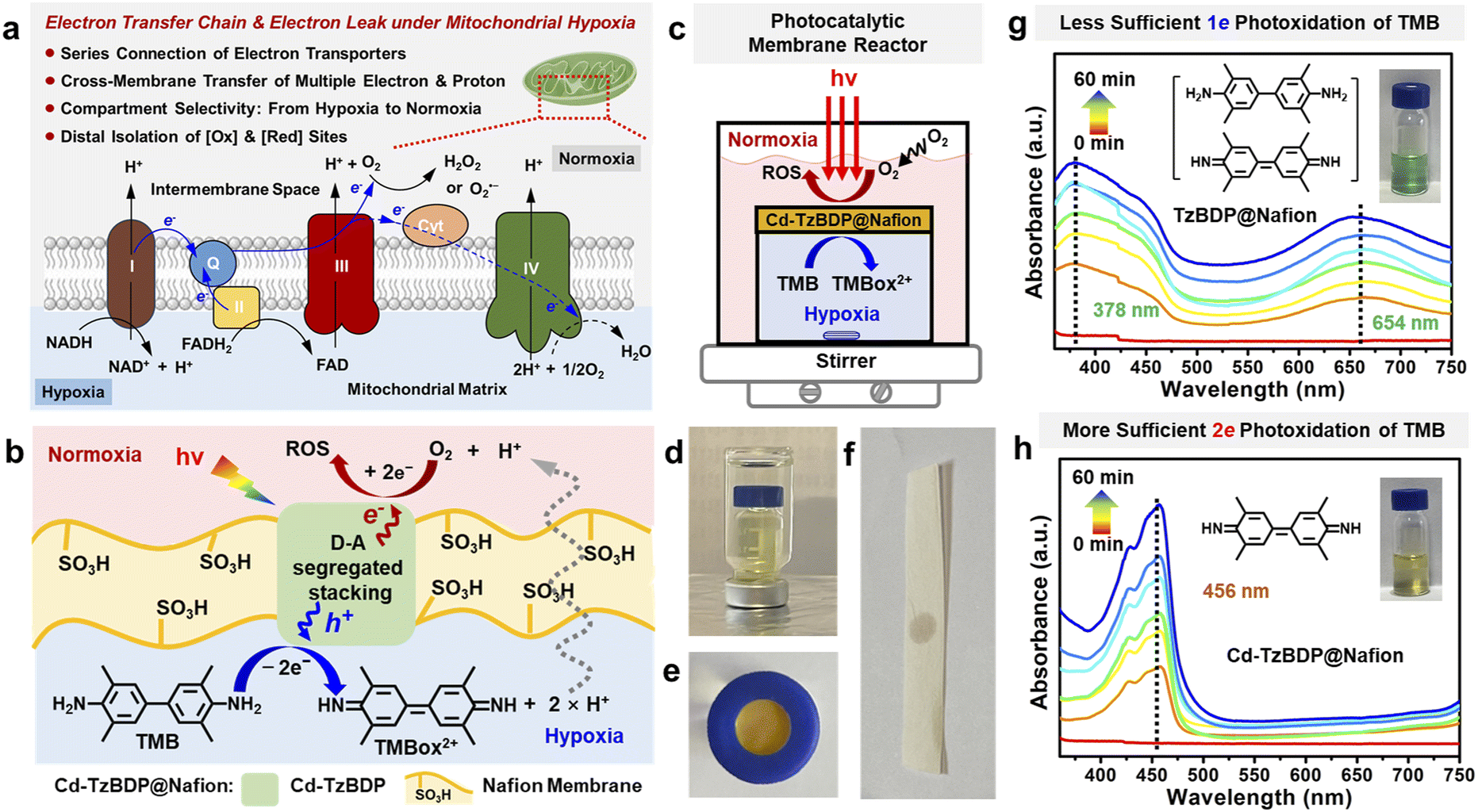 | ||
| Fig. 6 (a) ETC-mediated electron leak of mitochondria under hypoxic stress. (b) Illustration of the transfer of electrons and protons across the Cd−TzBDP@Nafion membrane and the distal photooxidation and photoreduction in different compartments. (c) Schematic illustration of the setup for the photocatalytic membrane reactor. (d) A nested double-layered photoreactor consisting of an interior vial (hypoxic CH3CN solution of TMB) and an outer sealed ampoule (normoxic CH3CN). (e) Cap of the vial inlaid with the Cd−TzBDP@Nafion membrane. (f) Detecting H2O2 from the reaction mixture of the outer ampoule using KI/starch testing paper after photocatalysis over the Cd−TzBDP@Nafion membrane. UV-vis absorbance for monitoring TMB oxidation with (g) TzBDP@Nafion and (h) Cd−TzBDP@Nafion under light irradiation. | ||
Conclusions
In summary, we report the biological ETC-inspired design and synthesis of a D–A type coordination polymer Cd−TzBDP with offset characteristics for mimicking the biological ETC. This stacking mode of dyes in the framework facilitated the photoinduced inter-ligand charge separation and the following delocalization of e− and h+ along the stacked D/A columns, which suppressed the recombination of separated carriers, spatially isolated reductive and oxidative centres, achieving the visualization of photocatalytic steps by the long-lived photochromism of the coordination polymer. Cd−TzBDP was proved to be capable of photomediating the aerobic respiration-like electron transfer from NADH-like electron donor to O2 within the same compartment, realizing the photocatalytic aerobic activation of α-C(sp3)–H bonds of tertiary amines to achieve the direct α-functionalization with added values and pharmaceutical interest. Cd−TzBDP was successfully employed in a photocatalytic membrane reactor inspired by the ETC-mediated cross-compartment electron leak of hypoxic mitochondria, which intuitively exhibited the advantages of directional and consecutive long-range transfer of multiple electrons, benefiting the distal photooxidation and photoreduction with compartment selectivity. This protocol showcased a supramolecular stacking paradigm to well-compromise the microscopic mechanism of transient and local excitations with the macroscopic perspective of biology-like temporally long-term and spatially long-range redox behaviours, which went beyond the limitation of single-molecule photocatalysis, paving the way to bioinspiration design empowered by the penetrative outer-field light and more.Data availability
The data supporting this work are available in the ESI.†Author contributions
T. X. Zhang conceived the project, designed the experiments and supervised the work. L. Ma carried out the main experiments, and collected and interpreted the data. M. C. Li solved and refined the X-ray single crystal structures. X. Zhang contributed to data collection and analysis of fs-TA. L. Q. Li contributed to theoretical calculations. R. Cai contributed to experiments and analyses of fluorescence and ground-state UV/vis absorption. Y. S. Shi, X. M. Yang and C. Y. Duan assisted in funding acquisition and reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the National Natural Science Foundation of China (No. 21971031, 22301028, 21820102001, 21890381, and 21231003), Liaoning Province Science and Technology Plan Joint Project (No. 2023JH2/101700297), Foreign Youth Talent Program (No. QN2023127001L), the China Postdoctoral Science Foundation (No. 2022M710577), and Central University Basic Research Business Funding Project (No. DUT24YG123). Our special thanks are due to Dr Rui Cai, Dr Dan Wang, and Dr Liyan Zhang at the Instrumental Analysis Center and State Key Laboratory of Fine Chemicals, Dalian University of Technology, for the assistance with experiments and analyses of fluorescence, EPR, and ground-state UV/vis/IR absorption, respectively.Notes and references
- K. A. Ryu, C. M. Kaszuba, N. B. Bissonnette, R. C. Oslund and O. O. Fadeyi, Nat. Rev. Chem., 2021, 5, 322–337 CrossRef CAS PubMed.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- C. Wang, H. Li, T. H. Bürgin and O. S. Wenger, Nat. Chem., 2024, 16, 1151–1159 CrossRef CAS PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- C. Ye, D.-S. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Cent. Sci., 2024, 10, 529–542 CrossRef CAS PubMed.
- J. Blumberger, Chem. Rev., 2015, 115, 11191–11238 CrossRef CAS PubMed.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796–802 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- C. Pfeffer, S. Larsen, J. Song, M. Dong, F. Besenbacher, R. L. Meyer, K. U. Kjeldsen, L. Schreiber, Y. A. Gorby, M. Y. El-Naggar, K. M. Leung, A. Schramm, N. Risgaard-Petersen and L. P. Nielsen, Nature, 2012, 491, 218–221 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Q. Rev. Biophys., 1999, 36, 341–372 CrossRef PubMed.
- S. Cogliati, J. L. Cabrera-Alarcón and J. A. Enriquez, Biochem. Soc. Trans., 2021, 49, 2655–2668 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed.
- F. Kracke, I. Vassilev and J. O. Krömer, Front. Microbiol., 2015, 6, 575 Search PubMed.
- T. K. Baikie, L. T. Wey, J. M. Lawrence, H. Medipally, E. Reisner, M. M. Nowaczyk, R. H. Friend, C. J. Howe, C. Schnedermann, A. Rao and J. Z. Zhang, Nature, 2023, 615, 836–840 CrossRef CAS PubMed.
- A. T. Castner, B. A. Johnson, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2021, 143, 7991–7999 CrossRef CAS PubMed.
- R. Yanagi, T. Zhao, D. Solanki, Z. Pan and S. Hu, ACS Energy Lett., 2022, 7, 432–452 CrossRef CAS.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- R. Ramakrishnan, M. A. Niyas, P. M. Lijina and M. Hariharan, Acc. Chem. Res., 2019, 52, 3075–3086 CrossRef CAS PubMed.
- K. P. Vermeulen, D. Goetz, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065–3076 RSC.
- T.-T. Ma, G.-Z. Huang, X.-H. Wang, Y. Liang, R.-H. Li, B. Wang, S.-J. Yao, J.-P. Liao, S.-L. Li, Y. Yan and Y.-Q. Lan, Natl. Sci. Rev., 2024, 11, nwae177 CrossRef PubMed.
- A. Kubacka and M. Fernández-García, Nat. Catal., 2018, 1, 643–644 CrossRef CAS.
- R. Haldar, A. Ghosh and T. K. Maji, Chem. Commun., 2023, 59, 1569–1588 RSC.
- G. A. Leith, A. A. Berseneva, A. Mathur, K. C. Park and N. B. Shustova, Trends Chem., 2020, 2, 367–382 CrossRef CAS.
- S. A. A. Razavi, W. Chen, H.-C. Zhou and A. Morsali, Coord. Chem. Rev., 2024, 517, 216004 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- H. Noh, C.-W. Kung, K.-i. Otake, A. W. Peters, Z. Li, Y. Liao, X. Gong, O. K. Farha and J. T. Hupp, ACS Catal., 2018, 8, 9848–9858 CrossRef CAS.
- J.-L. Zhao, M.-H. Li, Y.-M. Cheng, X.-W. Xu, Y. Zhao, Z.-Y. Cao, M.-H. You and M.-J. Lin, Coord. Chem. Rev., 2023, 475, 214918 CrossRef CAS.
- A. N. Woodward, J. M. Kolesar, S. R. Hall, N.-A. Saleh, D. S. Jones and M. G. Walter, J. Am. Chem. Soc., 2017, 139, 8467–8473 CrossRef CAS PubMed.
- X.-H. Zhou, Y. Fan, W.-X. Li, X. Zhang, R.-R. Liang, F. Lin, T.-G. Zhan, J. Cui, L.-J. Liu, X. Zhao and K.-D. Zhang, Chinese Chem. Lett., 2020, 31, 1757–1767 CrossRef CAS.
- W.-X. Yin, Y.-T. Liu, Y.-J. Ding, Q. Lin, X.-M. Lin, C.-L. Wu, X.-D. Yao and Y.-P. Cai, CrystEngComm, 2015, 17, 3619–3626 RSC.
- S. L. Li, M. Han, Y. Zhang, G. P. Li, M. Li, G. He and X. M. Zhang, J. Am. Chem. Soc., 2019, 141, 12663–12672 CrossRef CAS PubMed.
- P. Li, X.-M. Yin, L.-L. Gao, S.-L. Yang, Q. Sui, T. Gong and E.-Q. Gao, ACS Appl. Nano Mater., 2019, 2, 4646–4654 CrossRef CAS.
- Y. Qian, D. Li, Y. Han and H. L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464–2472 CrossRef CAS PubMed.
- L. Zeng, T. Zhang, R. Liu, W. Tian, K. Wu, J. Zhu, Z. Wang, C. He, J. Feng, X. Guo, A. I. Douka and C. Duan, Nat. Commun., 2023, 14, 4002 CrossRef CAS PubMed.
- T. J. Adams, N. F. Tumpa, M. Acharya, Q. H. Nguyen, N. Baliukonis, M. Shuchi, S. E. Starnes, T. Hofmann and M. G. Walter, ACS Appl. Opt. Mater., 2024, 2, 704–713 CrossRef CAS PubMed.
- C.-M. Yu, P.-H. Wang, Q. Liu, L.-Z. Cai and G.-C. Guo, Cryst. Growth Des., 2021, 21, 1323–1328 CrossRef CAS.
- X. Li, Y. Li, X. Yang, C. Yan, K. Zhang, X. Liang, J. Zhang, Y. Gai and K. Xiong, Chem. Commun., 2021, 57, 12496–12499 RSC.
- K.-Q. Hu, Z.-W. Huang, X.-B. Li, Y. Cheng, X.-H. Kong, L. Mei, L.-W. Zeng, Z.-H. Zhang, J.-P. Yu, J. K. Gibson, Z.-F. Chai, H.-Z. Kou and W.-Q. Shi, Adv. Funct. Mater., 2023, 33, 2213039 CrossRef CAS.
- Z. Ming, T. Zhang, W. Tian, J. Li, Z. Liu, R. Liu, Z. Liu and C. Duan, Chem. Commun., 2021, 57, 12812–12815 RSC.
- H.-Y. Li, X. Hua, T. Fu, X.-F. Liu and S.-Q. Zang, Chem. Commun., 2022, 58, 7753–7756 RSC.
- P. Li, M.-Y. Guo, X.-M. Yin, L. L. Gao, S.-L. Yang, R. Bu, T. Gong and E.-Q. Gao, Inorg. Chem., 2019, 58, 14167–14174 CrossRef CAS PubMed.
- Y. Luo, J.-P. Liu, L.-K. Li and S.-Q. Zang, Inorg. Chem., 2023, 62, 14385–14392 CrossRef CAS PubMed.
- C. Chen, J.-K. Sun, Y.-J. Zhang, X.-D. Yang and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14458–14462 CrossRef CAS PubMed.
- M. D'Incalci and C. M. Galmarini, Mol. Cancer Ther., 2010, 9, 2157–2163 CrossRef PubMed.
- L. Zhang, X. Gu, P. Lu and Y. Wang, Tetrahedron, 2016, 72, 2359–2363 CrossRef CAS.
- A. M. Nauth, E. Schechtel, R. Dören, W. Tremel and T. Opatz, J. Am. Chem. Soc., 2018, 140, 14169–14177 CrossRef CAS PubMed.
- D. Nicewicz, H. Roth and N. Romero, Synlett, 2016, 27, 714–723 CrossRef.
- S.-S. Zhu, Y. Liu, X.-L. Chen, L.-B. Qu and B. Yu, ACS Catal., 2021, 12, 126–134 CrossRef.
- H. Li, Y. Yang, C. He, L. Zeng and C. Duan, ACS Catal., 2019, 9, 422–430 CrossRef CAS.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS PubMed.
- S. Lee, E. Tak, J. Lee, M. A. Rashid, M. P. Murphy, J. Ha and S. S. Kim, Cell Res., 2011, 21, 817–834 CrossRef CAS PubMed.
- H.-S. Wong, P. A. Dighe, V. Mezera, P.-A. Monternier and M. D. Brand, J. Biol. Chem., 2017, 292, 16804–16809 CrossRef CAS PubMed.
- R. M. Young, S. C. Jensen, K. Edme, Y. Wu, M. D. Krzyaniak, N. A. Vermeulen, E. J. Dale, J. F. Stoddart, E. A. Weiss, M. R. Wasielewski and D. T. Co, J. Am. Chem. Soc., 2016, 138, 6163–6170 CrossRef CAS PubMed.
- S. Apichai, P. Kummuntakoon, T. Pattananandecha, J. Julsrigival, K. Sawangrat, F. Ogata, N. Kawasaki, K. Grudpan and C. Saenjum, Molecules, 2022, 27, 4841 CrossRef CAS PubMed.
- S. Gu, S. Risse, Y. Lu and M. Ballauff, ChemPhysChem, 2020, 21, 450–458 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2355405. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05592f |
| This journal is © The Royal Society of Chemistry 2024 |