
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking metal–ligand cooperative catalytic photochemical benzene carbonylation: a mechanistic approach†
Francesco
Crisanti
 a,
Michael
Montag
b,
David
Milstein
b,
Julien
Bonin
*ac and
Niklas
von Wolff
*ac
a,
Michael
Montag
b,
David
Milstein
b,
Julien
Bonin
*ac and
Niklas
von Wolff
*ac
aUniversité Paris Cité, Laboratoire d’Electrochimie Moléculaire, CNRS, F-75013, Paris, France. E-mail: julien.bonin@sorbonne-universite.fr; niklas.von_wolff@sorbonne-universite.fr
bDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 7610001, Israel
cSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, F-75005, Paris, France
First published on 3rd October 2024
Abstract
A key challenge in green synthesis is the catalytic transformation of renewable substrates at high atom and energy efficiency, with minimal energy input (ΔG ≈ 0). Non-thermal pathways, i.e., electrochemical and photochemical, can be used to leverage renewable energy resources to drive chemical processes at well-defined energy input and efficiency. Within this context, photochemical benzene carbonylation to produce benzaldehyde is a particularly interesting, albeit challenging, process that combines unfavorable thermodynamics (ΔG° = 1.7 kcal mol−1) and the breaking of strong C–H bonds (113.5 kcal mol−1) with full atom efficiency and the use of renewable starting materials. Herein, we present a mechanistic study of photochemical benzene carbonylation catalyzed by a rhodium-based pincer complex that is capable of metal–ligand cooperation. The catalytic cycle, comprising both thermal and non-thermal steps, was probed by NMR spectroscopy, UV-visible spectroscopy and spectrophotochemistry, and density functional theory calculations. This investigation provided us with a detailed understanding of the reaction mechanism, allowing us to unlock the catalytic reactivity of the Rh–pincer complex, which represents the first example of a metal–ligand cooperative system for benzene carbonylation, exhibiting excellent selectivity.
Introduction
For the chemical sector to achieve the goal of net-zero carbon emission, it is crucial to increase the energetic efficiency of chemical processes, to improve their atom economy, and to make extensive use of renewable resources, in accordance with green chemistry principles and projected roadmaps.1–3 In an ideal scenario, a desired product would be generated catalytically from renewable feedstocks at full atom efficiency, using renewable sources of energy with minimal input (ΔG ≈ 0). In this respect, both electrochemical and photochemical approaches are desirable, as they allow for a controlled, measurable and sustainable energy input (via the applied potential or chosen wavelength). Over the last decades, electrocatalysis has evolved as a tool to transform renewable substrates (e.g., CO2) at minimal overpotential,4–8 while catalytic photochemical processes have been used to leverage highly atom-efficient C–H bond activation for the construction of molecular complexity.9–15 In recent years, it has been shown that both electrochemical and photochemical approaches can produce CO from CO2 at high rates and with excellent selectivity,16–18 making it a valuable and renewable C1-building block. While a plethora of carbonylative C–H transformations exist,19–24 we are interested in applying photo- and electro-chemical approaches to these reactions, to enable otherwise endergonic processes via the use of these renewable energy inputs. Within this context, the photochemical carbonylation of benzene to produce benzaldehyde is particularly interesting, as it combines two renewable25,26 and non-functionalized substrates, namely, benzene and CO, into an important platform chemical at full atom efficiency.27 Nevertheless, this reaction is challenging, due to its unfavorable thermodynamics (ΔG° = 1.7 kcal mol−1) and the high dissociation energy of the benzene C–H bond (113.5 kcal mol−1), and can thus serve as a testing ground for novel non-thermal catalytic approaches. However, since the initial reports on rhodium-catalyzed photochemical benzene carbonylation, little progress has been made in this field.28–33 In recent work, we showed that non-thermal, electrochemical pathways can be used to activate metal–ligand cooperative (MLC)34–36 catalysts for endergonic dehydrogenation reactions at room temperature.37,38 This prompted us to explore whether non-thermal MLC chemistry could also be leveraged for catalytic benzene carbonylation. Applying a mechanistic approach, we seek to identify, understand and overcome bottlenecks in photochemical carbonylative C–H activation promoted by MLC systems, with the goal of providing novel alternatives for catalytic benzene carbonylation.Despite the elegance of benzene carbonylation, which allows benzaldehyde to be synthesized in a single step from simple and renewable feedstocks,25 the widespread application of this process is hampered by unfavorable thermodynamics and the low productivity of most catalysts employed for this reaction. Early work by Kunin and Eisenberg showed that a rhodium-based Vaska-type complex, trans-[Rh(CO)Cl(PPh3)2], can catalyze this reaction, but exhibits low activity [turnover number (TON) of 2; see ESI,† Tables S1 and S2].33 In their report, the authors demonstrated the role of light in overcoming kinetic barriers, and the system was also found to be very competent in promoting the reverse process, i.e., photochemical benzaldehyde decarbonylation. Nevertheless, although the forward reaction could proceed at room temperature under irradiation, the system was limited by the low equilibrium concentration of benzaldehyde. Using a similar system, the complex trans-[Rh(CO)Cl(PMe3)2], the groups of Tanaka39 and Goldman40 demonstrated that the thermodynamic limitation could be overcome, albeit with reduced selectivity, leading to numerous side-products (e.g., benzophenone, benzyl alcohol and biphenyl), and only under high irradiation power (500 W; see ESI,† Tables S1 and S2). Thus, changing the phosphine ligand clearly affects the behavior of the catalyst under light, and it appears that under irradiation the PMe3 ligand enables to leverage a sufficient driving force to overcome the thermodynamic constraints. The photochemical activation of these systems is somewhat reminiscent of recent developments involving the use of electrochemical activation to surmount thermodynamic and kinetic barriers in alcohol dehydrogenation by MLC systems.41,42 We were thus intrigued to know whether this approach could be extended to the photochemical activation of MLC complexes in order to provide an alternative means of photochemical benzene carbonylation.
Results and discussion
In 2016, Milstein and coworkers reported the photochemical activation of a PNP–rhodium pincer complex, as part of a sequence of reactions that converted benzene and CO2 into benzaldehyde (Scheme 1).43 In the initial step, a PNP–rhodium(I) hydride complex was shown to participate in a formal reverse water–gas shift reaction upon treatment with CO2, generating a dearomatized rhodium(I) carbonyl complex (1) and water. In this reaction, a pincer side-arm C–H proton, together with the hydride ligand, are involved in the cleavage of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of CO2. Interestingly, under UV irradiation (λmax = 320 nm), complex 1 was found to promote the C–H activation of benzene via metal–ligand cooperativity, to form the corresponding acyl complex 3. Treatment of the latter with tosylic acid released benzaldehyde, with concomitant formation of the tosylate complex 4. Addition of base and H2 to this complex regenerated the initial Rh(I)–hydride complex, thereby closing a stoichiometric cycle for benzaldehyde formation from benzene and CO2. Stoichiometric carbonylation of benzene was also achieved by Huang and coworkers under thermal conditions, using an MLC complex of rhodium bearing a different PNP-type ligand, with benzaldehyde release being promoted by hydrochloric acid.44 Given the previously-observed photochemical activation of benzene by 1, and the possibility of releasing benzaldehyde, albeit under acidic conditions, we attempted direct benzene carbonylation under UV irradiation using 1 as catalyst, with limited success (TON = 1.3 after 120 h). Inspired by this preliminary result, we sought to understand the mechanism of this reaction in greater detail, in order to determine its limiting steps, and probe the conditions under which light can serve as a driving force for significant product formation and catalytic turnover. This could open the door for the use of MLC systems for catalytic benzene carbonylation, which has yet to be achieved. To this end, several aspects of a potential catalytic cycle have to be understood: (i) What is the role of light in the C–H activation step? (ii) Is benzene carbonylation by MLC complexes an associative process (as in the work of Tanaka and Goldman) or a dissociative one (as proposed by Kunin and Eisenberg)? (iii) How can benzaldehyde be released from the catalyst in order to close the catalytic cycle?
O bond of CO2. Interestingly, under UV irradiation (λmax = 320 nm), complex 1 was found to promote the C–H activation of benzene via metal–ligand cooperativity, to form the corresponding acyl complex 3. Treatment of the latter with tosylic acid released benzaldehyde, with concomitant formation of the tosylate complex 4. Addition of base and H2 to this complex regenerated the initial Rh(I)–hydride complex, thereby closing a stoichiometric cycle for benzaldehyde formation from benzene and CO2. Stoichiometric carbonylation of benzene was also achieved by Huang and coworkers under thermal conditions, using an MLC complex of rhodium bearing a different PNP-type ligand, with benzaldehyde release being promoted by hydrochloric acid.44 Given the previously-observed photochemical activation of benzene by 1, and the possibility of releasing benzaldehyde, albeit under acidic conditions, we attempted direct benzene carbonylation under UV irradiation using 1 as catalyst, with limited success (TON = 1.3 after 120 h). Inspired by this preliminary result, we sought to understand the mechanism of this reaction in greater detail, in order to determine its limiting steps, and probe the conditions under which light can serve as a driving force for significant product formation and catalytic turnover. This could open the door for the use of MLC systems for catalytic benzene carbonylation, which has yet to be achieved. To this end, several aspects of a potential catalytic cycle have to be understood: (i) What is the role of light in the C–H activation step? (ii) Is benzene carbonylation by MLC complexes an associative process (as in the work of Tanaka and Goldman) or a dissociative one (as proposed by Kunin and Eisenberg)? (iii) How can benzaldehyde be released from the catalyst in order to close the catalytic cycle?
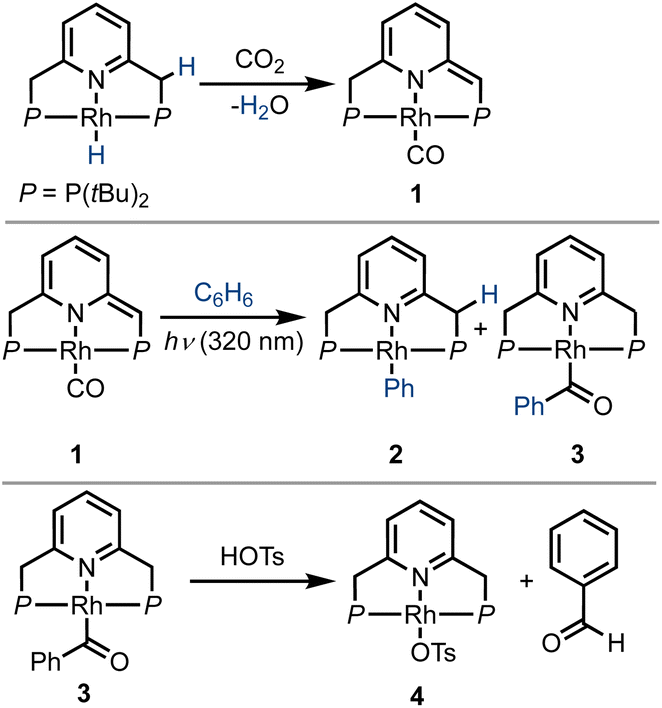 | ||
| Scheme 1 Reaction of a PNP–rhodium–hydride pincer complex with CO2, its subsequent activation of a benzene C–H bond under UV irradiation, and acid-induced release of benzaldehyde, as previously reported by Milstein and coworkers.43 | ||
We first investigated the behavior of different PNP–rhodium pincer complexes towards benzene C–H activation in the absence of carbon monoxide gas (Scheme 2). Complex 1, bearing the strongly-coordinated CO ligand trans to the lutidine core, does not activate benzene C–H bonds in the dark, i.e., it slowly decomposes in neat benzene over the course of two weeks at room temperature, but does not form phenyl complex 2 or acyl complex 3. By contrast, dearomatized PNP–rhodium complexes analogous to 1, which bear more weakly-coordinated ligands, namely, N2,45 chloride (Cl−)46 or triflate (OTf−),45 react with benzene in the dark, with the time required to complete this reaction decreasing from 4 d to 9 h to 1 h, respectively. The reactivity of these complexes towards C–H activation is thus inversely correlated with metal–ligand bonding strength (spectrochemical series and ESI,† Table S2), i.e., CO > N2 > Cl− > OTf−. Based on this observation, our initial hypothesis was that a transiently formed three-coordinate 14e Rh(I) species is responsible for C–H activation, as proposed for the system reported by Kunin and Eisenberg.29 In complex 1, the strongly bonded CO ligand would prevent the formation of such a transient, coordinatively unsaturated species in the dark, hence explaining its lack of reactivity in the absence of light.
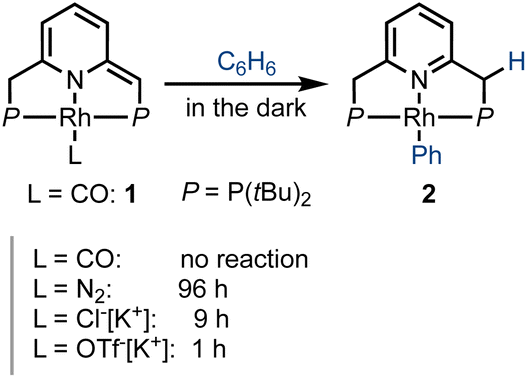 | ||
| Scheme 2 Rate of benzene C–H activation by different neutral and anionic PNP–rhodium(I) pincer complexes, as reflected in the approximate time to reaction completion. | ||
To probe this hypothesis, we examined the CO self-exchange rates of complex 1 in the dark and under irradiation. In a sealed NMR tube, a 2 mM solution of unlabeled complex 1 [1(12CO)] in 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 n-heptane:cyclohexane-d12 was placed under 1 atm of 13CO, and the CO ligand exchange was monitored by 31P{1H} NMR spectroscopy. In the dark, at room temperature, 13CO incorporation was observed within a few minutes, as clearly evidenced by the appearance of 31P–13C coupling (2JP–C = 12.7 Hz) in the corresponding NMR peaks (Fig. 1a). The same experiment was repeated at −40 °C (Fig. 1b), showing roughly 75% 13CO incorporation after 4 h. These results show that CO lability is high in the absence of irradiation on the timescale of photocatalysis and that incorporation of 13CO takes place readily in the dark. However, this does not rule out the involvement of a three-coordinate intermediate responsible for C–H activation. Having shown that putative three-coordinate species can quickly generate the rhodium phenyl complex 2 (Scheme 2), we wanted to know whether this complex could be an intermediate en route to the acyl species 3, potentially leading to the release of benzaldehyde. When a benzene solution of 2 was placed under 1 atm of CO, this complex converted into 3 within several minutes at room temperature, in the dark, as observed by 31P{1H} NMR spectroscopy (Fig. 2). CO insertion into the Rh–Ph bond is thus a facile, non-photochemical process. While it was initially thought that externally-added acid is necessary for the release of benzaldehyde from 3, we wanted to understand whether this could instead be triggered by light or CO coordination.40,42
:1 n-heptane:cyclohexane-d12 was placed under 1 atm of 13CO, and the CO ligand exchange was monitored by 31P{1H} NMR spectroscopy. In the dark, at room temperature, 13CO incorporation was observed within a few minutes, as clearly evidenced by the appearance of 31P–13C coupling (2JP–C = 12.7 Hz) in the corresponding NMR peaks (Fig. 1a). The same experiment was repeated at −40 °C (Fig. 1b), showing roughly 75% 13CO incorporation after 4 h. These results show that CO lability is high in the absence of irradiation on the timescale of photocatalysis and that incorporation of 13CO takes place readily in the dark. However, this does not rule out the involvement of a three-coordinate intermediate responsible for C–H activation. Having shown that putative three-coordinate species can quickly generate the rhodium phenyl complex 2 (Scheme 2), we wanted to know whether this complex could be an intermediate en route to the acyl species 3, potentially leading to the release of benzaldehyde. When a benzene solution of 2 was placed under 1 atm of CO, this complex converted into 3 within several minutes at room temperature, in the dark, as observed by 31P{1H} NMR spectroscopy (Fig. 2). CO insertion into the Rh–Ph bond is thus a facile, non-photochemical process. While it was initially thought that externally-added acid is necessary for the release of benzaldehyde from 3, we wanted to understand whether this could instead be triggered by light or CO coordination.40,42
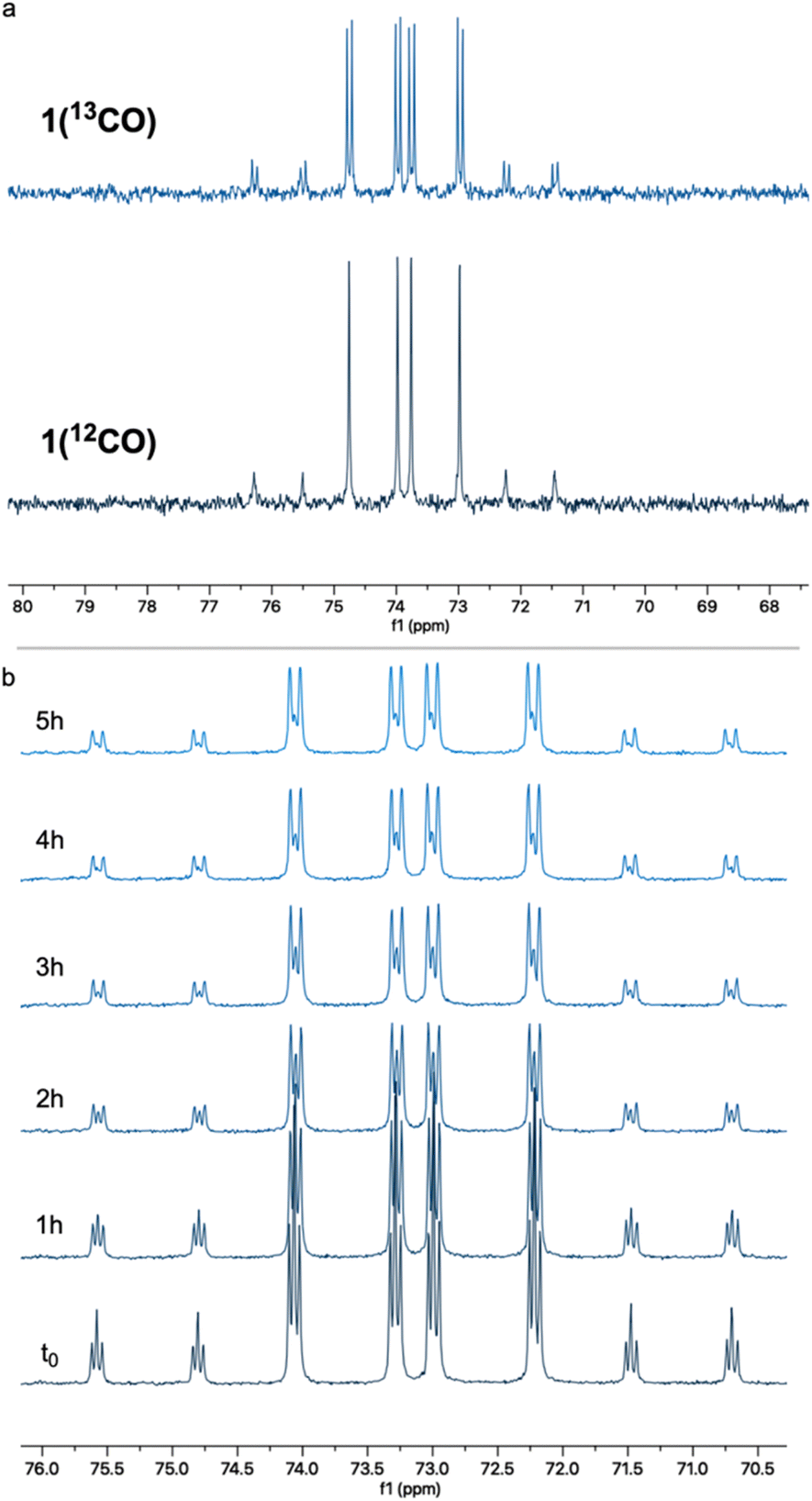 | ||
| Fig. 1 (a) CO self-exchange in a 2 mM solution of 1(12CO) in 7:1 n-pentane/cyclohexane-d12 under 1 atm of 13CO, as observed by 31P{1H} NMR spectroscopy in the dark at room temperature. (b) Low temperature (−40 °C) 31P{1H} NMR monitoring of such CO self-exchange in the same solvent mixture [all spectra represent mixtures of 1(12CO) and 1(13CO) at varying ratios]. | ||
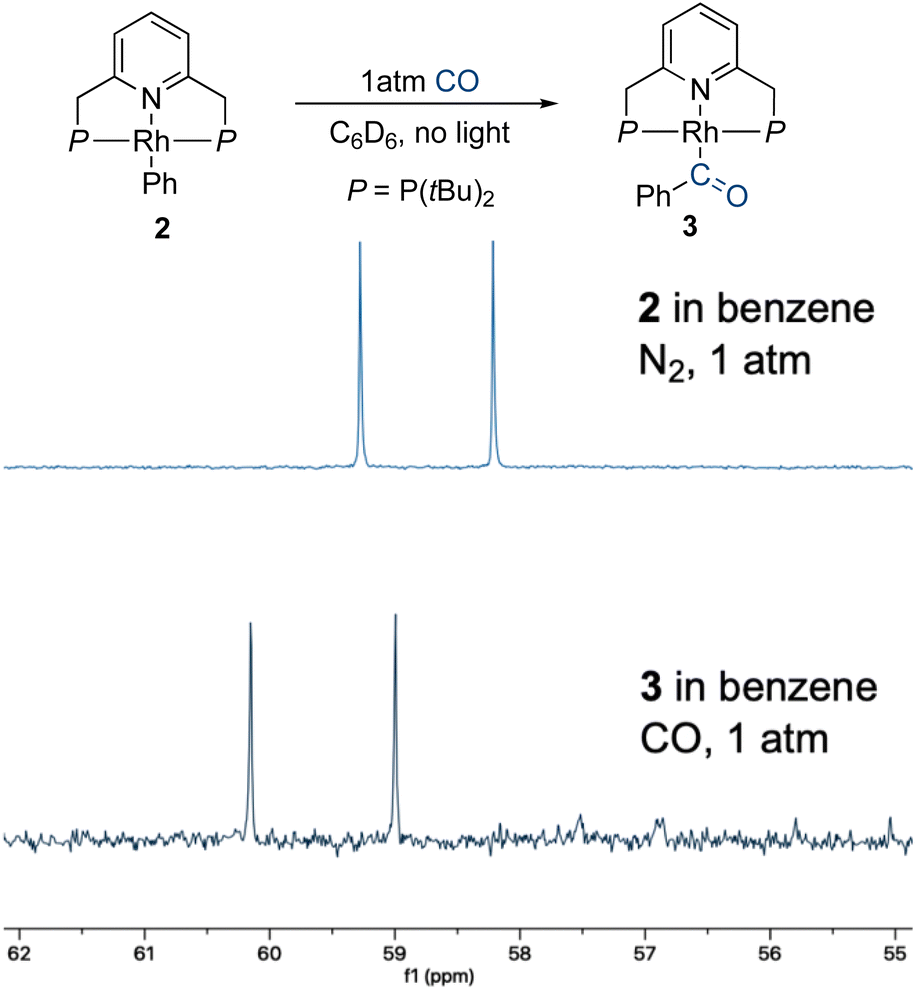 | ||
| Fig. 2 CO insertion into the Rh–Ph bond of 2 to form 3 in the dark, and the consequent changes in the 31P{1H} NMR spectrum. | ||
To explore potential CO-induced benzaldehyde release, we used UV-visible absorption spectroscopy to monitor the reaction of a 30 μM solution of 3 under CO at room temperature (Fig. 3), employing pentane as solvent in order to avoid benzene C–H activation as a side-reaction. After 55 min under 1 atm of CO, the reaction mixture showed the characteristic absorption bands of complex 1, with spectral deconvolution giving an excellent fit to a mixture of 1 and 3 (Fig. 4). 1H NMR spectroscopy revealed the concomitant formation of benzaldehyde under these conditions (see ESI,† Section 2.8), thereby confirming the successful elimination of the product in the presence of CO. These results clearly indicate that CO can promote benzaldehyde release from 3, while regenerating 1 through a non-photochemical pathway. Spectral deconvolution over time allowed us to calculate an approximate pseudo-first-order rate constant of 9.6 × 10−5 s−1 for the reaction 3 + CO → 1 + PhCHO (see ESI,† Section 8), in line with a thermally activated rate-determining step at room temperature. Having established that benzaldehyde release and concurrent formation of 1 can be thermally facilitated by CO, thus potentially closing the catalytic cycle, we turned our attention to the role of light in the initial C–H activation step. Importantly, our aim was to determine whether this process is associative or dissociative, and to probe the effect of light on its thermodynamics and kinetics.
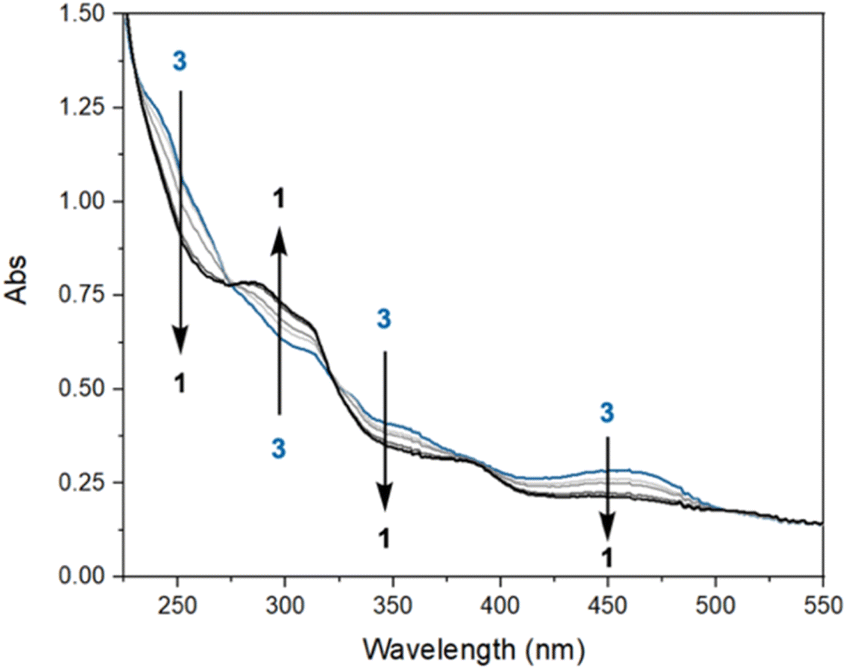 | ||
| Fig. 3 UV-visible spectral evolution of a 30 μM solution of 3 in pentane under 1 atm of CO, at room temperature in the absence of additional external irradiation, over the course of 1 h. | ||
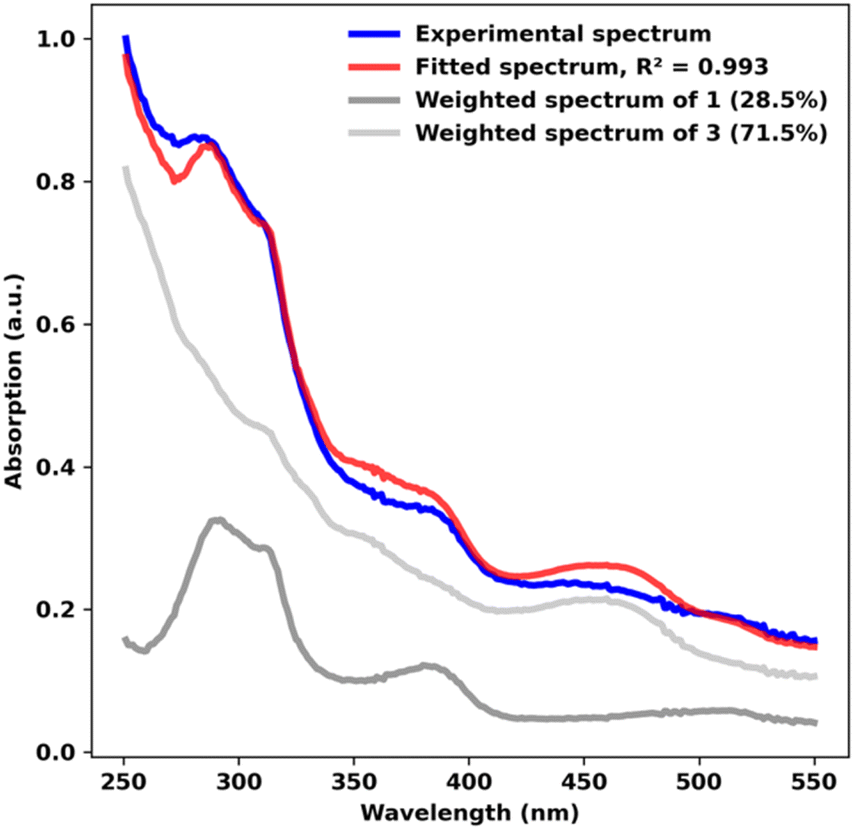 | ||
| Fig. 4 Spectral deconvolution of the UV-visible spectrum of 3 in pentane under 1 atm of CO, recorded after 55 min at room temperature in the absence of additional external irradiation. The individual UV-visible spectra of 1 and 3 are shown for reference. | ||
The UV-visible spectrum of 1 in pentane shows three discernible bands in the UV range: one centered around 280 nm, one around 300 nm and one at roughly 390 nm (Fig. 5), as well as one in the visible region (520 nm). Using time-dependent density functional theory (TD-DFT) calculations, we sought to gain insight into the electronic transitions involved in these absorptions. The absorption at ∼300 nm seems to be associated with a mixed metal–ligand-based orbital transition, but the one at ∼390 nm involves a metal-to-ligand charge transfer (MLCT) transition, with the donor orbital being the Rh dz2 and the acceptor orbital having a Rh–CO non-bonding character (Fig. 5). This latter transition resembles the one involved in the mechanism proposed by Goldman and coworkers for trans-[Rh(CO)Cl(PMe3)2], in which irradiation depopulates the dz2 orbital, thereby decreasing unfavorable electron–electron repulsions between the rhodium center and benzene substrate.40
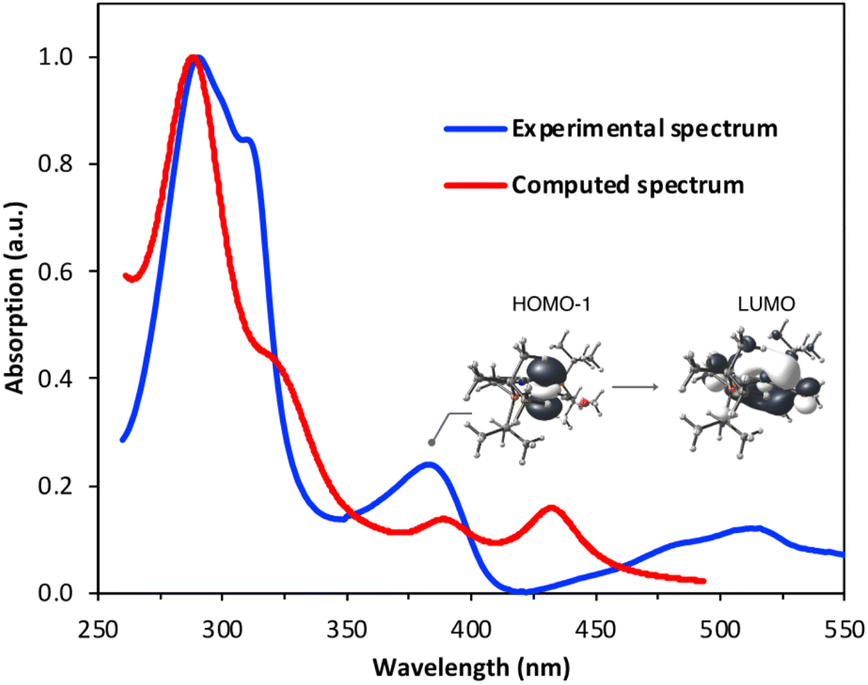 | ||
| Fig. 5 Overlay of the experimental and TD-DFT-derived UV-visible spectra of complex 1 in pentane (see ESI† for details). The band at ∼390 nm can be assigned to a HOMO−1 (dz2) → LUMO metal–ligand charge transfer transition. | ||
In the initial work on C–H activation by 1, reported by Milstein and coworkers, a light source with λmax = 320 nm was used.43 We wanted to investigate whether selectively promoting the MLCT transition at 390 nm could lead to a more active system, by increasing Lewis acidity on the metal center and Lewis basicity on the ligand framework. Starting from 1, we performed spectrophotochemistry at different temperatures to learn more about the nature of the C–H activation step. A thermostated quartz cuvette containing a 30 μM solution of 1 in benzene under N2 was placed in a UV-visible spectrophotometer, and was irradiated at 390 nm using a LED light source positioned at a 90° angle with respect to the spectrophotometer beam path, while maintaining the temperature at 20 °C. Under these conditions, the characteristic absorption bands of 1 decreased over time, giving way to the absorption bands of 2 (Fig. 6a), with typical isosbestic points, indicating full conversion of 1 into 2, with concomitant release of CO, over the course of 3 h.
 | ||
| Fig. 6 (a) UV-visible spectral evolution during photochemical C–H activation of benzene by 1 in the absence of added CO (30 μM of 1 in benzene under N2 at 20 °C, irradiated for 3 h at 390 nm at 90° relative to the spectrophotometer beam). (b) Corresponding Eyring plot and derived activation parameters for the C–H activation. | ||
Discriminating between associative and dissociative mechanisms is difficult when fitting the data to kinetic rate laws, since first order kinetics are expected in both cases (see ESI,† Section 7). Therefore, the reaction kinetics were studied at different temperatures, in order to extract the activation enthalpy and entropy from the corresponding Eyring plot (Fig. 6b). It is important to stress that although the Eyring approximation is generally applied to strictly thermal systems, both Eyring himself,47 as well as others,48,49 have shown that it can be successfully applied to photochemical systems. Based on our kinetic data, negative values were obtained for both ΔH‡ and ΔS‡, i.e., −4.7 kcal mol−1 and −90.5 cal mol−1 K−1, and these are in line with an associative or exchange mechanism (SN2-type), followed by CO release. It should be noted that relatively small values of ΔH‡, and the large negative value of ΔS‡ measured for the C–H activation step with 1, are usually linked to an exchange mechanism.50 Moreover, negative entropy values of this magnitude have been observed experimentally,51–53 and are generally explained by entropy-governed processes.54 These mechanistic attributes seem to be corroborated by the apparent absence of intermediates in the C–H activation reaction (Fig. 6a), although it is possible that their concentrations fall below the detection limit or time-resolution of our UV-visible spectrophotometer.
In order to understand how photoactivation of MLC complexes such as 1 leads to bond activation, we also investigated the photochemical step computationally, using DFT calculations (Fig. 7 and ESI,† Section 9). In line with Goldman's proposition that photoexcitation of Rh(I) species can lead to dz2 depopulation, which is also supported by our TD-DFT calculations (Fig. 5), we were able to locate an associative transition state for C–H activation (TS1–2, ΔGcalc = 55.4 kcal mol−1). This transition state, which involves metal–ligand cooperation, should be thermally easily accessible from the excited state of 1 (approximated here by its triplet state, at ΔGcalc = 45.6 kcal mol−1). Notably, the observed reaction barrier, ΔG‡exp = 21.9 kcal mol−1 (Fig. 6), is higher than the computed one, ΔG‡calc(TS1–2) = 9.8 kcal mol−1 (relative to the triplet state of 1; Fig. 7). This could be explained by rovibronic relaxation, as well as triplet-to-singlet intersystem crossing, both of which must take place in the real system before reaching the computed transition state, but neither of which has been considered in our TD-DFT calculations. After CO dissociation, the formation of 2 is strongly downhill in energy (as is the formation of 1 and 3; see ESI,† Section 9.2), and is thus consistent with our observation that 2 is exclusively formed under irradiation in the absence of externally-added CO (Fig. 6a). Alternatively, photoexcitation of 1 could lead to CO dissociation prior to its reaction with benzene, opening the way for C–H oxidative addition pathways. Indeed, we located an oxidative addition transition state (TS1–5, ΔGcalc = 59.2 kcal mol−1) leading to the Rh(III) species 5 (ΔGcalc = 46.9 kcal mol−1), with the former being roughly 4 kcal mol−1 higher in energy than TS1–2. Product 5 could convert into 2 through intramolecular proton transfer from the metal center to the olefinic pincer side-arm. As noted above, our experimental Eyring analysis reveals a strongly negative entropy of activation, which is consistent with our computational results for the associative pathway, i.e., TS1–2 exhibits ΔS‡calc = −41.7 cal mol−1 K−1, whereas the oxidative addition (dissociative) pathway shows a much smaller entropy of activation, at only −2.2 cal mol−1 K−1. Taken together, our experimental and DFT findings indicate that C–H bond activation by complex 1 proceeds through an associative photochemical mechanism.
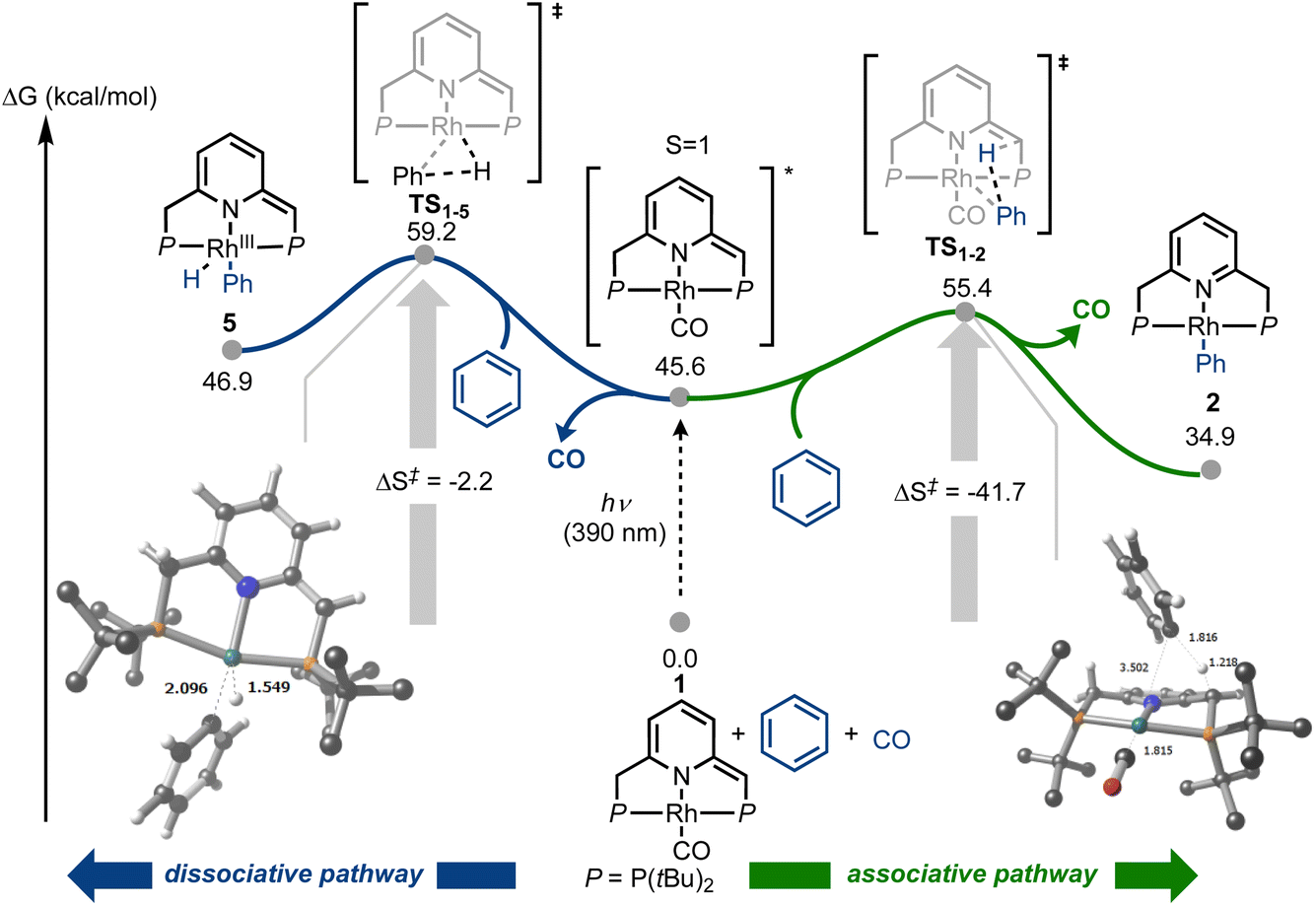 | ||
| Fig. 7 Potential energy surface for the photochemical carbonylative C–H bond activation of benzene promoted by 1, showing two alternative reaction pathways. All structures were calculated at the M06-L/def2VP/W06/GD3/SMD//ωB97M-V/def2TZVPP/RIJCOSX/SMD level of theory and energies are standard-state corrected. t-Butyl protons omitted for clarity. Distances in Å. | ||
The DFT calculations are also helpful in rationalizing the rapid CO self-exchange that occurs in the absence of light. The pentacoordinate dicarbonyl complex 1·CO, obtained upon coordination of a second CO ligand to the metal center of 1, was calculated to be only 9.0 kcal mol−1 higher in energy than the latter, providing a fast associative process for CO self-exchange (see ESI,† Section 9.2, Tables S3 and S4). Such CO exchange pathways involving pentacoordinate rhodium(I) species have been previously observed in similar systems.40,41
Having gained a deeper understanding of the benzene carbonylation mechanism, we set to examine whether the mechanistic analysis, as well as identification of the absorption band associated with C–H activation (390 nm), can be translated into higher catalytic activity. As shown previously,43 irradiation of a benzene solution of complex 1 at 320 nm led to a TON of only 1.3 after 120 h (Table 1, entry 1). In our experiments, we employed different light sources and filters in order to selectively apply the irradiation band responsible for catalytic turnover. Using a solar simulator equipped with a 400 nm bandpass filter, it was found that visible light is insufficient to promote the desired reaction (entry 2) and that the absorption band centered around 520 nm does not promote C–H activation (Fig. 5). Nevertheless, when the same light source was operated without a bandpass filter, benzaldehyde production was observed (TON ≈ 4; entry 3). Finally, in order to focus on the UV band that we have associated with C–H activation, we selectively irradiated the sample at 390 nm, using a LED light source (entry 4). Under these conditions, we were able to generate benzaldehyde at an even higher TON of ∼14, and with no observable side-products. Thus, by employing 1 as catalyst, and irradiating the reaction mixture at 390 nm, it is possible to drive the carbonylation of benzene beyond the low equilibrium concentration of benzaldehyde, i.e., ∼2 mM in CO-saturated benzene at room temperature. To the best of our knowledge, no catalytic MLC-promoted benzene carbonylation has been reported thus far, with the only available examples of such reactions having been achieved in a stoichiometric fashion.43,44
| Entry | Light source | Power | Wavelength (nm) | TONd |
|---|---|---|---|---|
| a 20 mM of 1 in benzene, 1 atm of CO, irradiation in an NMR tube for 120 h under 10 × 8 W fluorescent LZC-UVB lamps. b 1 mM of 1 in 3 mL of benzene, 1 atm of CO, irradiation for 72 h under 100 W Xe lamp in a quartz cuvette with a 3 mL headspace. c Kessil LED lamp (λmax = 390 nm) instead of Xe lamp. d Error corresponds to repeated (>2) runs. | ||||
| 1 (ref. 43) | UVBa | 80 W | λ max = 320 | 1.3 |
| 2 | Solar simulatorb | 100 W | λ > 400 | 0 |
| 3 | Solar simulatorb | 100 W | 300 < λ < 1000 | 4.2 ± 0.3 |
| 4 | LEDc | 52 W | λ max = 390 | 14.1 ± 1.5 |
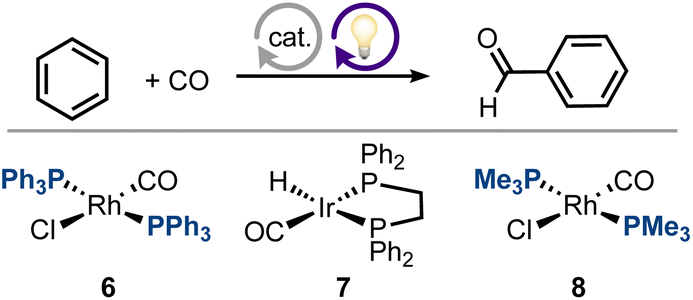
Our results suggest that under carefully chosen conditions, an MLC framework can enable excellent selectivity at high activity and low irradiation power (e.g., 52 vs. 500 W; see Table 2, entries 2–4). This contrasts with previously-reported Vaska-type complexes,39 which operate via oxidative addition/reductive elimination pathways, and show a trade-off between activity (Table 2, entries 2 and 3) and selectivity (Table 2, entries 1 and 2), possibly due to the forcing reaction conditions required in such systems (i.e., high temperature and irradiation power; see ESI,† Section 4, Table S1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Power | Wavelength (nm) | TONa | Selectivity | Complex |
| a Reaction run at 25 °C and 1 atm of CO at 1 mM catalyst in neat benzene, if not stated otherwise. Turnovers correspond to benzaldehyde formation. b 0.2 atm of CO, 7 mM 6, 40 h. c 0.9 atm of CO, 3.6 mM 7, 22 h. d 0.7 mM 8, 33 h. e 7 mM 8, 24 h, TON = 27 for benzyl alcohol, TON = 19 for benzophenone, TON = 3 for biphenyl. f This work. | |||||
| 1 (ref. 33) | 200 W | λ < 366 | 2.1b | 100% | 6 |
| 2 (ref. 33) | 200 W | λ < 366 | 1.3c | 100% | 7 |
| 3 (ref. 39) | 500 W | λ > 290 | 72d | 54% | 8 |
| 4 (ref. 40) | 500 W | λ > 290 | 52 (50 °C)e | 51% | 8 |
| 5f | 52 W | λ max = 390 | 14.1 ± 1.5 | 100% | 1 |
Conclusions
Our successful application of MLC catalysis for photochemical benzene carbonylation shows that, like electrochemical approaches, non-thermal activation pathways can provide the thermodynamic driving force necessary to promote endergonic reactions in a clean and sustainable fashion, without the need for highly reactive co-substrates. The light source is employed as a monodirectional thermodynamic pump that populates high-energy intermediates.The results of our mechanistic experiments involving pincer complex 1 in benzene are consistent with a light-driven C–H activation step, followed by a series of thermal steps responsible for product release and catalytic cycle closure (see Fig. 8). Importantly, CO was shown to promote benzaldehyde release in this system, whereas similar previously-described MLC systems necessitated the use of strong acids to accomplish this step.44 The presence of a strong acid in an MLC system would ultimately quench the basicity of the ligand, thus preventing successful C–H activation and accounting for the lack of catalytic turnover in such systems. It should also be noted that although intermittent CO release from catalyst 1 is possible, the C–H activation step is associative, indicating that increased CO pressure in a suitable reactor could be beneficial. Such a high-pressure approach could also counter the low solubility of CO in most organic solvents, including benzene (<10 mM at 1 atm).
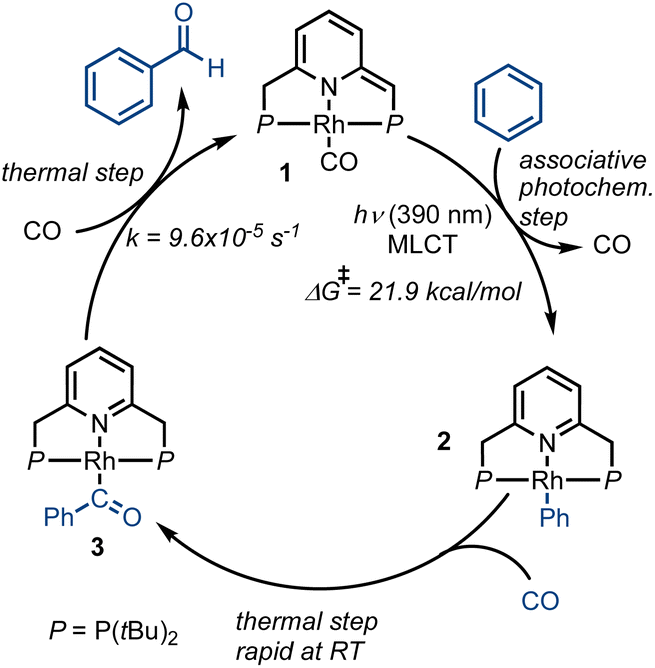 | ||
| Fig. 8 Proposed catalytic cycle for the photochemical benzene carbonylation promoted by 1. | ||
In summary, photochemical benzene carbonylation via C–H activation is a challenging process that has seen little advancement over the last decades, from both a performance and mechanistic point of view. Herein, we demonstrate the unprecedented implementation of MLC catalysis to successfully promote this reaction. Unlocking the catalytic carbonylative C–H activation of benzene using MLC catalysts demonstrates that excellent selectivity can be achieved under mild conditions (LED irradiation at room temperature). Indeed, preliminary studies conducted in our research group have shown that benzene is not the only substrate that can be functionalized using this type of chemistry, and we hope that our findings would spur the development of new generations of catalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization and funding acquisition: N. v. W. Experiments: F. C. DFT calculations: N. v. W. Supervision: M. M., D. M., J. B., N. v. W. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. C. warmly acknowledges the IdEx Université de Paris 2021 program for PhD funding, and the Pôle Collège des Ecoles Doctorales of Université Paris Cité for the Bourse Doctorale de Mobilité Internationale that funded his stay at the Weizmann Institute of Science. Computations were performed using HPC resources from GENCI-CINES (Grants AD010812061R2 and AD010812061R3).Notes and references
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2009, 39, 301–312 RSC.
- Chemicals – Analysis, https://www.iea.org/reports/chemicals, accessed November 24, 2022 Search PubMed.
- M. C. Leech and K. Lam, Nat. Rev. Chem, 2022, 6, 275–286 CrossRef PubMed.
- J. E. Nutting, J. B. Gerken, A. G. Stamoulis, D. L. Bruns and S. S. Stahl, J. Org. Chem., 2021, 86, 15875–15885 CrossRef CAS PubMed.
- A. M. Appel and M. L. Helm, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- C. Costentin, ACS Catal., 2021, 5678–5687 CrossRef CAS.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS.
- J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS.
- M. W. Campbell, M. Yuan, V. C. Polites, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 3901–3910 CrossRef CAS.
- M. Oliva, G. A. Coppola, E. V. Van der Eycken and U. K. Sharma, Adv. Synth. Catal., 2021, 363, 1810–1834 CrossRef CAS.
- B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585–2634 CAS.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367 CrossRef CAS PubMed.
- Z. Guo, G. Chen, C. Cometto, B. Ma, H. Zhao, T. Groizard, L. Chen, H. Fan, W.-L. Man, S.-M. Yiu, K.-C. Lau, T.-C. Lau and M. Robert, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- A. Kaiser and B. A. Arndtsen, in The Chemical Transformations of C1 Compounds, John Wiley & Sons, Ltd, 2022, pp. 533–565 Search PubMed.
- T. N. Allah, L. Ponsard, E. Nicolas and T. Cantat, Green Chem., 2021, 23, 723–739 RSC.
- L. Lukasevics and L. Grigorjeva, Org. Biomol. Chem., 2020, 18, 7460–7466 RSC.
- D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 3133–3142 RSC.
- C. Le Berre, P. Serp, P. Kalck and G. P. Torrence, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2014, pp. 1–34 Search PubMed.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 4534 CrossRef CAS.
- M. Machas, G. Kurgan, A. K. Jha, A. Flores, A. Schneider, S. Coyle, A. M. Varman, X. Wang and D. R. Nielsen, J. Chem. Technol. Biotechnol., 2019, 94, 38–52 CrossRef CAS.
- F. Brühne and E. Wright, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed.
- R. Eisenberg, Isr. J. Chem., 2017, 57, 932–936 CrossRef CAS.
- A. J. Kunin and R. Eisenberg, J. Am. Chem. Soc., 1986, 108, 535–536 CrossRef CAS.
- S. E. Boyd, L. D. Field and M. G. Partridge, J. Am. Chem. Soc., 1994, 116, 9492–9497 CrossRef CAS.
- E. M. Gordon and R. Eisenberg, J. Mol. Catal., 1988, 45, 57–71 CrossRef CAS.
- W. Kläui, D. Schramm and W. Peters, Eur. J. Inorg. Chem., 2001, 2001, 3113–3117 CrossRef.
- A. J. Kunin and R. Eisenberg, Organometallics, 1988, 7, 2124–2129 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- D. Tocqueville, F. Crisanti, J. Guerrero, E. Nubret, M. Robert, D. Milstein and N. von Wolff, Chem. Sci., 2022, 13, 13220–13224 RSC.
- S. Kasemthaveechok, P. Gérardo and N. von Wolff, Chem. Sci., 2023, 14, 13437–13445 RSC.
- T. Sakakura, T. Sodeyama, K. Sasaki, K. Wada and M. Tanaka, J. Am. Chem. Soc., 1990, 112, 7221–7229 CrossRef CAS.
- G. P. Rosini, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1994, 116, 9498–9505 CrossRef CAS.
- E. A. McLoughlin, B. D. Matson, R. Sarangi and R. M. Waymouth, Inorg. Chem., 2020, 59, 1453–1460 CrossRef CAS.
- S. P. Annen, V. Bambagioni, M. Bevilacqua, J. Filippi, A. Marchionni, W. Oberhauser, H. Schönberg, F. Vizza, C. Bianchini and H. Grützmacher, Angew. Chem., Int. Ed., 2010, 49, 7229–7233 CrossRef CAS.
- A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9941–9950 CrossRef CAS PubMed.
- C. Zhou, J. Hu, Y. Wang, C. Yao, P. Chakraborty, H. Li, C. Guan, M.-H. Huang and K.-W. Huang, Org. Chem. Front., 2019, 6, 721–724 RSC.
- S. K. Hanson, D. M. Heinekey and K. I. Goldberg, Organometallics, 2008, 27, 1454–1463 CrossRef CAS.
- L. Schwartsburd, M. A. Iron, L. Konstantinovski, E. Ben-Ari and D. Milstein, Organometallics, 2011, 30, 2721–2729 CrossRef CAS.
- H. Eyring, Trans. Faraday Soc., 1938, 34, 41–48 RSC.
- N. Hoffmann, H. Buschmann, G. Raabe and H.-D. Scharf, Tetrahedron, 1994, 50, 11167–11186 CrossRef CAS.
- H. Buschmann, H. D. Scharf, N. Hoffmann, M. W. Plath and J. Runsink, J. Am. Chem. Soc., 1989, 111, 5367–5373 CrossRef CAS.
- F. J. Monlien, L. Helm, A. Abou-Hamdan and A. E. Merbach, Inorg. Chem., 2002, 41, 1717–1727 CrossRef CAS PubMed.
- M. T. H. Liu and R. Bonneau, J. Am. Chem. Soc., 1992, 114, 3604–3607 CrossRef CAS.
- R. A. Moss, W. Lawrynowicz, N. J. Turro, I. R. Gould and Y. Cha, J. Am. Chem. Soc., 1986, 108, 7028–7032 CrossRef CAS.
- R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811–3826 CrossRef CAS PubMed.
- K. N. Houk and N. G. Rondan, J. Am. Chem. Soc., 1984, 106, 4293–4294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photochemical reaction setups, DFT calculations and product characterization. See DOI: https://doi.org/10.1039/d4sc05683c |
| This journal is © The Royal Society of Chemistry 2024 |