
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bond cleavage in diazirines by a cyclic diborane(4) compound†
N bond cleavage in diazirines by a cyclic diborane(4) compound†
Minling
Zhong
a,
Jie
Zhang
 b and
Zuowei
Xie
*a
b and
Zuowei
Xie
*a
aShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: xiezw@sustech.edu.cn
bDepartment of Chemistry and State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin, N. T., Hong Kong, China
First published on 7th October 2024
Abstract
Reactions of o-carborane-fused diborane(4) with 3H-diazirines led to the complete cleavage of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, with one nitrogen atom being incorporated into the B–B bond. The molecular and electronic structures of the resultant borylnitrogen compounds were confirmed through single-crystal X-ray analyses and computational studies. The related reaction mechanism was investigated using DFT calculations.
N bond, with one nitrogen atom being incorporated into the B–B bond. The molecular and electronic structures of the resultant borylnitrogen compounds were confirmed through single-crystal X-ray analyses and computational studies. The related reaction mechanism was investigated using DFT calculations.
Diaziridines are highly strained three-membered rings composed of one carbon and two nitrogen atoms, with a double bond between the nitrogen atoms.1 Upon activation with light, heat,2 or ultrasonication,3 they can be transformed into carbenes by breaking the C–N bonds and liberating dinitrogen. Consequently, they have been extensively studied as carbene precursors in organic chemistry4,5 and find wide applications in photoaffinity labelling (PAL)6–9 and polymer crosslinkers.10–12
In striking contrast, the complete cleavage of the NN bond in diazirines has rarely been reported. In 1987, the Kisch group showed that Cp2Ti(CO)2 had an unprecedented capability to activate diazo compounds, exposure of which to 3H-diazirines led to oxidative addition of the NN double bond and the generation of Cp2Ti(NCO)(NCR2) (Scheme 1a).13 Except for transition metals, certain main-group species such as Grignard reagents,14 lithium reagents,14,15 and phosphines16,17 have also been reported to cleave the NN double bonds of the halodiazirines through nucleophilic substitution processes (Scheme 1b and c). However, it is only in recent years that low-valent boron-containing species (e.g., borylenes, boron radicals) have been employed to activate the enthalpically strong NN or N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds.18–21 In 2022, the Stephan group reported the reactions of phosphine/borane frustrated Lewis pairs with chlorodiazirines, resulting in the reduction of the NN double bond (Scheme 1d).22 In 2023, the Kinjo group demonstrated that triboraazabutenynes bearing an extremely polarized BB bond reacted with phenyldiazirine, leading to the cleavage of the NN bond and the incorporation of a single nitrogen atom into the BB bond (Scheme 1e).23
N bonds.18–21 In 2022, the Stephan group reported the reactions of phosphine/borane frustrated Lewis pairs with chlorodiazirines, resulting in the reduction of the NN double bond (Scheme 1d).22 In 2023, the Kinjo group demonstrated that triboraazabutenynes bearing an extremely polarized BB bond reacted with phenyldiazirine, leading to the cleavage of the NN bond and the incorporation of a single nitrogen atom into the BB bond (Scheme 1e).23
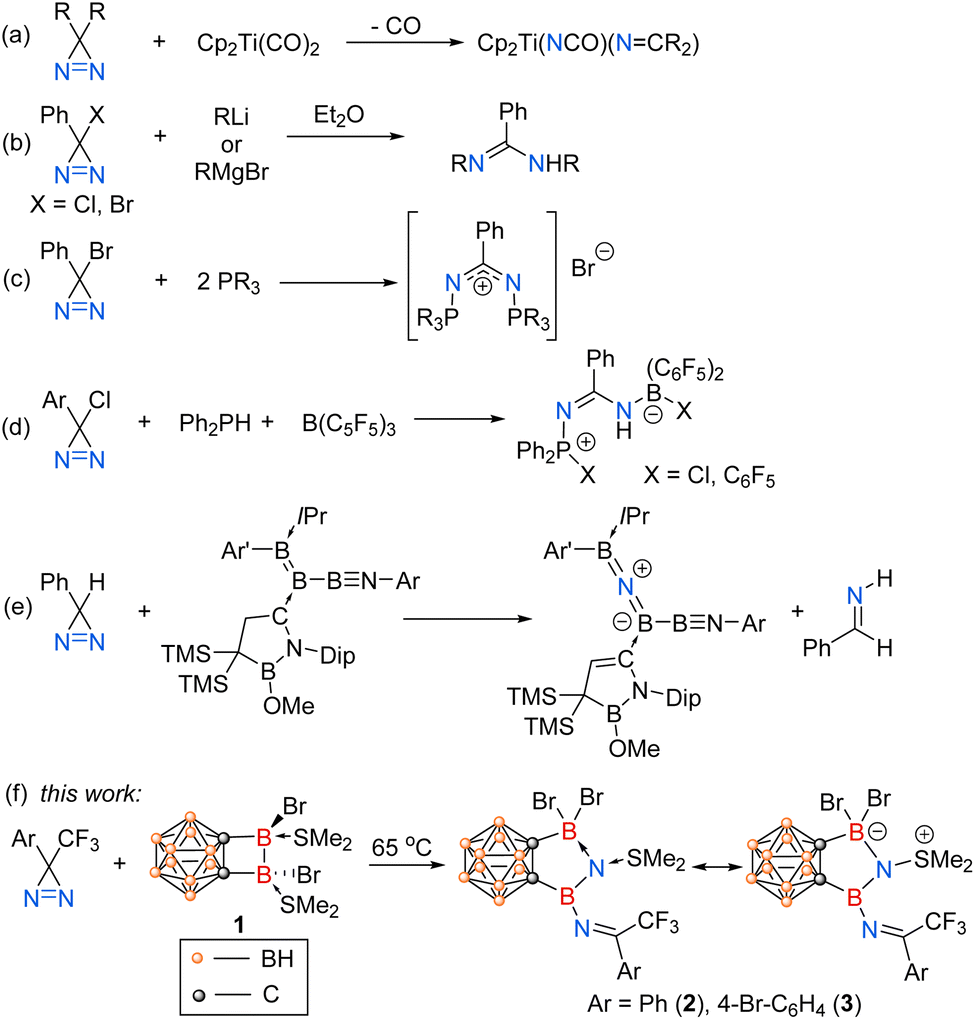 | ||
| Scheme 1 Examples of NN bond cleavage of 3H-diazirines. | ||
We recently reported that o-carborane-fused diboracycle (1) can serve as a “strain-release” reagent for catalyst-free diboration of alkenes and alkynes.24 During our reactivity studies of compound 1, we serendipitously discovered that o-carborane-fused diborane(4) completely cleaved the strong NN bond in 3H-diazirines, resulting in the incorporation of one nitrogen atom into the B–B bond and the subsequent formation of borylnitrogen compounds (Scheme 1f). These results are reported in this paper.
Treatment of o-carborane-fused diboracycle 1 with 1.5 equiv. of 3-phenyl-3-(trifluoromethyl)-3H-diazirine in benzene at 65 °C overnight gave compound 2 as colourless crystals in 49% isolated yield (Scheme 2). Its 11B NMR spectrum exhibited two singlets at 29.13 and −0.25 ppm for two exo-polyhedral boron atoms. The obvious down-field shift of one boron relative to the starting material 1 (δ = −3.04 ppm)24 indicated the formation of a three-coordinate boron center in 2. The 19F NMR spectrum displayed one singlet at −66.65 ppm attributable to the CF3 group in 2. In its 1H NMR, the two methyl groups of SMe2 showed two singlets at 2.99 and 2.92 ppm, which were magnetically inequivalent due probably to the restricted rotation caused by the adjacent bromide atoms.
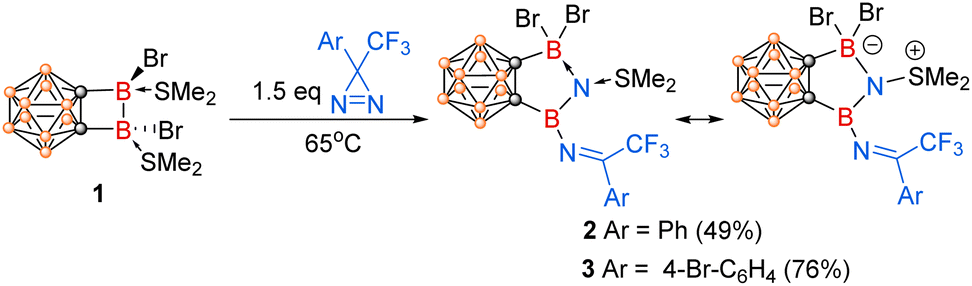 | ||
| Scheme 2 NN bond cleavage of 3H-diazirines by the cyclic diborane(4) compound 1. | ||
In the same manner, reaction of 1 with 1.5 equiv. of 3-(4-bromophenyl)-3-(trifluoromethyl)-3H-diazirine in dichloro-methane at 65 °C overnight afforded another analogous compound 3 in 76% yield. Its NMR spectra were very similar to those observed in 2, suggesting that they may have very similar molecular structure.
The solid-state structure of 2 was unambiguously confirmed by single-crystal X-ray analyses, showing a coplanar five-membered C2B2N ring (Fig. 1). Both the N1 and B14 centers adopt a trigonal-planar geometry as evidenced by the sum of the bond angles around N1 (360.1(9))° and B14 (359.9(2))°, respectively. The B14–N1 distance of 1.434(6) Å is much shorter than that of 1.531(6) Å for B13–N1 bond. On the other hand, the B14–N2 bond length of 1.384(6) Å is significantly shorter than that of B14–N1 one, indicating the presence of electron donation from the lone pair on N2 to the empty p orbital of B14. The S1–N1 distance (1.653(3) Å) lies within the range of the reported S–N dative bonds in the sulfiimines (1.610–1.669 Å).25–27
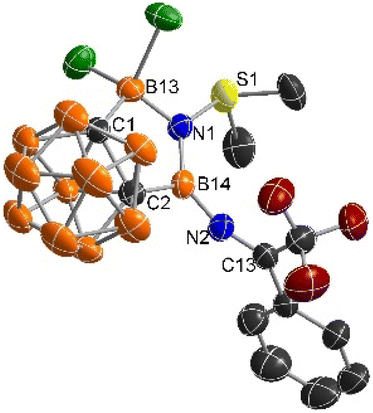 | ||
| Fig. 1 Molecular structure of 2 (thermal ellipsoids are set at the 50% probability level, and all hydrogen atoms are omitted for clarity). | ||
On the other hand, reaction of 3 with 1.1 equiv. of PMe3 resulted in the formation of boronium cation 4 in 75% yield via a nucleophilic displacement of Br− by PMe3 (Scheme 3). Treatment of excess PMe3 did not result in the replacement of the coordinated SMe2, suggesting very strong interactions between SMe2 and N1 centre. Compound 4 was not thermally stable under ambient conditions as evidenced by NMR spectra (see Fig. S1 in the ESI†). Its solid-state structure was confirmed by single-crystal X-ray analysis (Fig. 2), which also supports the formation of 3. The structural parameters of the N1 centre in 4 parallel those in 2, with the B14–N1, B13–N1, and S1–N1 bond lengths of 1.418(15) Å, 1.577(13) Å and 1.662(7) Å, respectively. The newly formed B13–P1 bond distance of 1.990(10) Å is comparable to those reported in phosphine-stabilized borenium or boronium species (1.89–1.97 Å).28–30
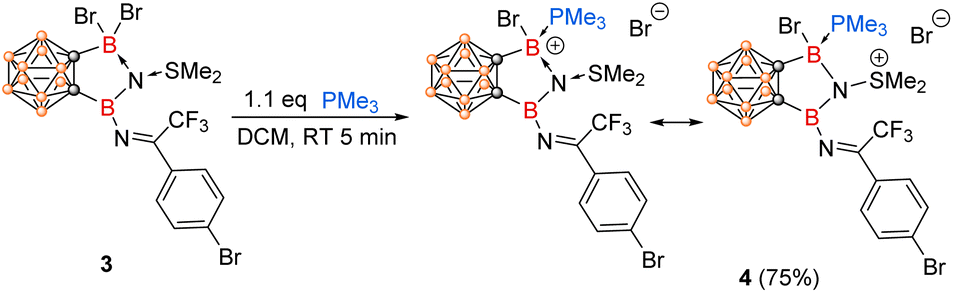 | ||
| Scheme 3 Reaction of 3 with PMe3. | ||
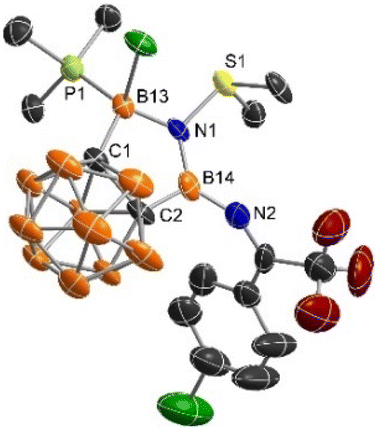 | ||
| Fig. 2 Molecular structure of the cation in 4 (thermal ellipsoids are set at the 35% probability level, all hydrogen atoms are omitted for clarity). | ||
To gain further insight into the electronic structure of compound 2, density functional theory (DFT) calculations were conducted at the B3LYP/6-311+G(d,p) level of theory. The molecular orbital analyses show that the LUMO corresponds to π* anti-bonding of the phenylimine moiety, whilst the HOMO primarily represents the lone pair orbital of the bromine groups (Fig. 3a). The natural bond orbital (NBO) analyses indicate that N1 has two lone pairs with the lone pair occupancy of LP(σ) and LP(π) is 1.74 and 1.68, respectively (see Table S2 in the ESI†). Second-order perturbation analyses show the donor–acceptor interactions from either lone pair orbital on N1 to the LP* orbitals of B13 or B14 atoms (Fig. 3b). The hyperconjugation donation from LP(σ) of N1 to LP*(σ) of B13 (175.45 kcal mol−1) is significantly stronger than that from LP(π) of N1 to LP*(π) of B14 (52.80 kcal mol−1) (see Table S3 in the ESI†).
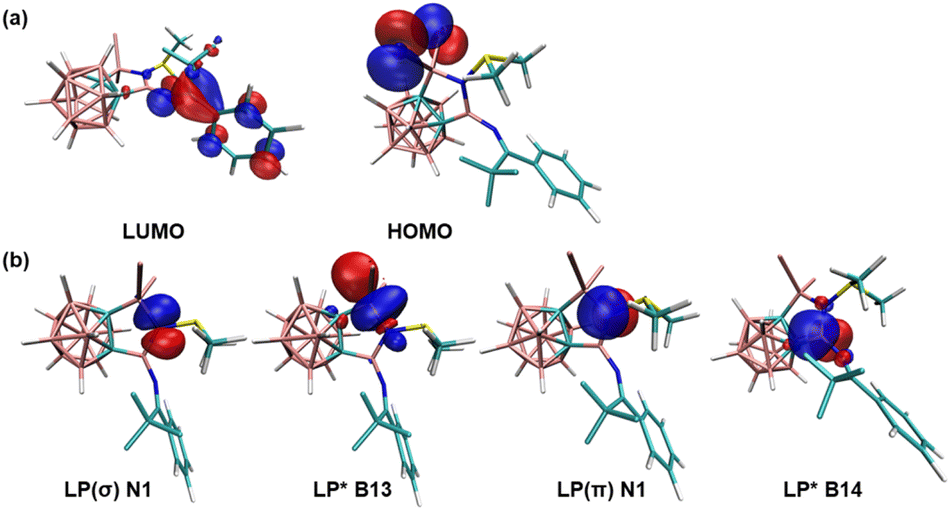 | ||
| Fig. 3 Plots of selected (a) MOs and (b) NBOs of 2 calculated at the B3LYP-D3/6-311+G(d,p) level of theory (isovalue 0.04 a.u.). | ||
The calculated natural population analyses (NPA) show that the N1 carries a considerably large negative charge (−1.18), while the N1-bonded boron and sulfur atoms carry positive charges (B13: 0.51; B14: 1.12; S1: 1.12) (Fig. 4). The calculated Wiberg bond indices (WBI) provide further insights into the bonding situation, indicating a B14–N1 covalent bond (WBI = 0.834), a rather strong S1–N1 dative bond (WBI = 0.741), and a weak N1–B13 dative bond (WBI = 0.583). The electronic structure and bonding analyses support 2 as a stabilized borylnitrogen compound, where the N donates its lone pairs to the neighboring boron centres and accepts electrons from the SMe2 ligand. It is salient to highlight that the Lewis acid–base interactions play a key role in the stabilization of this borylnitrogen species. Noted that matrix-free nonmetallic nitrenes have been rarely reported thus far.31–34
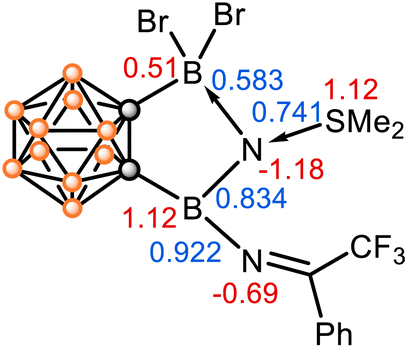 | ||
| Fig. 4 Natural population analysis (NPA) charge distribution of selected atoms (in red) and Wiberg bond indices (WBI) values of selected bonds in 2 (in blue). | ||
To understand the reaction mechanism for the formation of compound 2, DFT calculations were undertaken at the M06-2X/6-311G(d,p) level of theory in C6H6 solution (Scheme 4). The proposed mechanism involves several key steps. Initially, one SMe2 group is dissociated, followed by the migration of bromide, resulting in the formation of an unsymmetric intermediate BviaTS1 with an energy barrier of 23.5 kcal mol−1, which is the rate-determining step. Upon coordination of 3H-diazirine to the electron-deficient boron center, the B–B bond in C undergoes facile heterolytic cleavage, leading to the formation of a borylene intermediate DviaTS2. The coordination of 3H-diazirine moiety to the other boron center results in the formation of a six-membered intermediate E. Subsequent insertion of the borylene into the NN bond gives rise to the ring-contracted intermediate FviaTS3. Following the liberation of SMe2 from boron, and a nucleophilic attack of SMe2 on the nitrogen occurs, leading to N–N bond cleavage and the formation of intermediate IviaTS4. Finally, electron redistribution within the four-membered BN2C ring occurs, resulting in the formation of compound 2. The large energy gain in this step (ΔGI–2 = −99.1 kcal mol−1) may be attributed to the release of the ring strain.
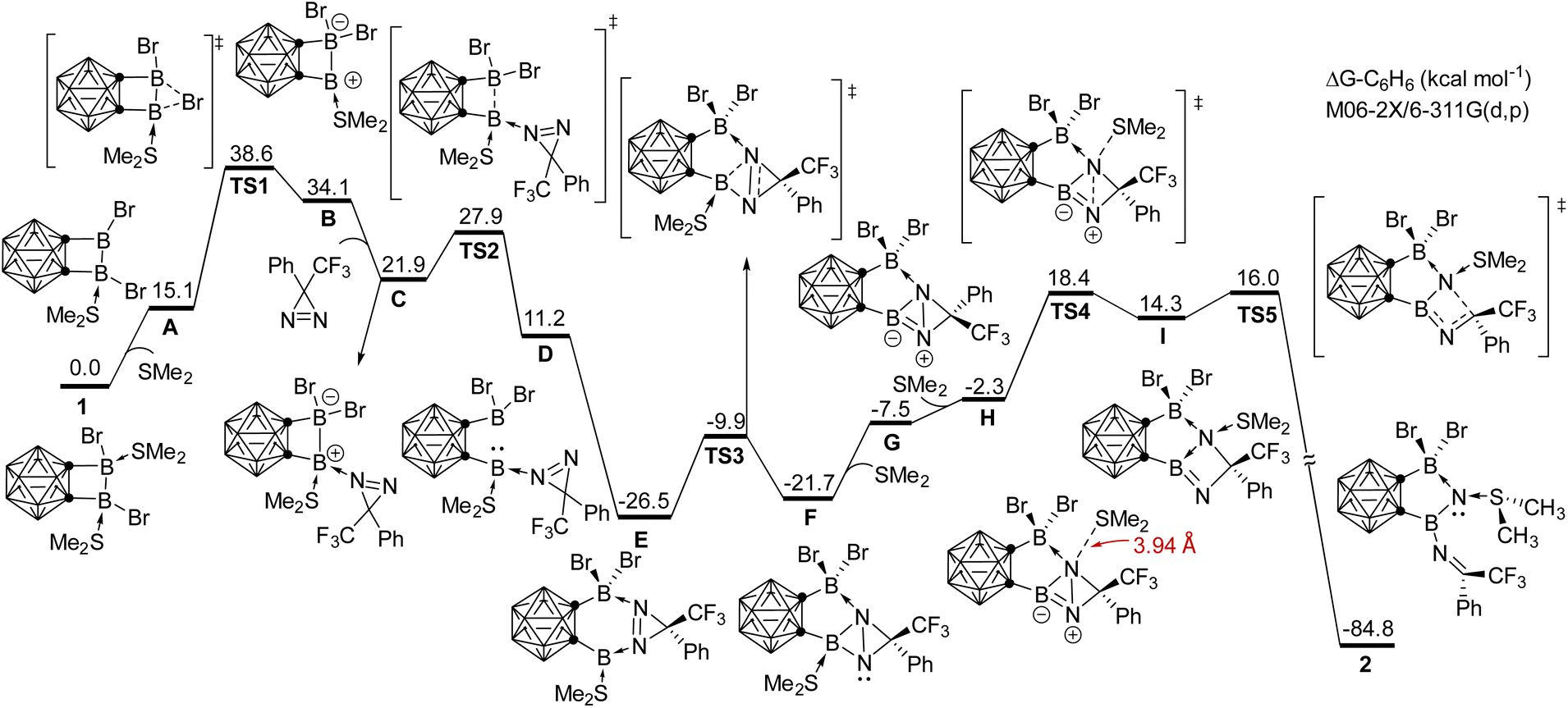 | ||
| Scheme 4 Energy profile for the DFT-based mechanism for the reaction of 1 with 3H-diazirine calculated at the M06-2X/6-311G(d,p) level of theory. The relative Gibbs free energies are given in kcal mol−1. | ||
Conclusions
In conclusion, this work demonstrated the first example of NN bond cleavage of 3H-diazirines by o-carborane-fused diborane(4) species 1. The electronic structure and bonding analyses of the resulted compounds 2 & 3 revealed that the nitrogen centre N1 bears two lone pairs of electrons, which was stabilized through Lewis acid/base interactions. DFT calculations indicated that the reaction proceeded through a key intermediate, borylene D, followed by its insertion into the NN double bond. Upon the treatment with strong Lewis base PMe3, compound 3 undergoes nucleophilic attack on boron to produce a boronium cation 4. This finding provides some hints for the activation of enthalpically strong bonds by using diboranes.
Data availability
All data are available in the ESI† of this article.Author contributions
Z. X. directed and conceived this project. M. Z. conducted the experiments. J. Z. did the theoretical work. All authors discussed the results and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by grants from Shenzhen Science and Technology Program (Project No. QTD20221101093558015 to ZX), the National Natural Science Foundation of China (Project No. 22331005 to ZX) and Research Grants Council of HKSAR (Project No. 14307421 to ZJ).Notes and references
- R. A. Moss, Acc. Chem. Res., 2006, 39, 267–272 CrossRef CAS PubMed.
- M. T. Liu, Chem. Soc. Rev., 1982, 11, 127–140 RSC.
- A. Blencowe and W. Hayes, Soft Matter, 2005, 1, 178–205 RSC.
- R. A. Moss, J.-M. Fedé and S. Yan, J. Am. Chem. Soc., 2000, 122, 9878–9879 CrossRef CAS.
- N. J. Porter, E. Danelius, T. Gonen and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 8892–8896 CrossRef CAS.
- J. Das, Chem. Rev., 2011, 111, 4405–4417 CrossRef CAS.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS.
- M. Hashimoto and Y. Hatanaka, Eur. J. Org Chem., 2008, 2008, 2513–2523 CrossRef.
- J. R. Hill and A. A. Robertson, J. Med. Chem., 2018, 61, 6945–6963 CrossRef CAS PubMed.
- K. Dey, S. R. Chowdhury, E. Dykstra, A. Koronatov, H. P. Lu, R. Shinar, J. Shinar and P. Anzenbacher, J. Mater. Chem. C, 2020, 8, 11988–11996 RSC.
- S. F. Musolino, M. Mahbod, R. Nazir, L. Bi, H. A. Graham, A. S. Milani and J. E. Wulff, Polym. Chem., 2022, 13, 3833–3839 RSC.
- C. Wu, C. Li, X. Yu, L. Chen, C. Gao, X. Zhang, G. Zhang and D. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21521–21528 CrossRef CAS.
- G. Avar, W. Rüsseler and H. Kisch, Z. Naturforsch. B Chem. Sci., 1987, 42b, 1441–1446 CrossRef.
- P. Kolářová, V. Čmolík, I. Linhart, I. Á. Martínez and T. Martinů, Tetrahedron Lett., 2013, 54, 6764–6767 CrossRef.
- R. Navrátil, J. Tarábek, I. Linhart and T. Martinů, Org. Lett., 2016, 18, 3734–3737 CrossRef PubMed.
- G. Alcaraz, A. Baceiredo, M. Nieger and G. Bertrand, J. Am. Chem. Soc., 1994, 116, 2159–2160 CrossRef CAS.
- G. Alcaraz, V. Piquet, A. Baceiredo, F. Dahan, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 1996, 118, 1060–1065 CrossRef CAS.
- S. Bennaamane, B. Rialland, L. Khrouz, M. Fustier-Boutignon, C. Bucher, E. Clot and N. Mézailles, Angew. Chem., Int. Ed., 2023, 62, e202209102 CrossRef CAS PubMed.
- (a) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed; (b) L. C. Haufe, L. Endres, M. Arrowsmith, R. Bertermann, M. Dietz, F. Fantuzzi, M. Finze and H. Braunschweig, J. Am. Chem. Soc., 2023, 145, 23986–23993 CrossRef CAS PubMed.
- M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef.
- B. Xu, H. Beckers, H. Ye, Y. Lu, J. Cheng, X. Wang and S. Riedel, Angew. Chem., Int. Ed., 2021, 60, 17205–17210 CrossRef CAS PubMed.
- D. Mandal, T. Chen, Z. W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2022, 61, e202209241 CrossRef CAS.
- L. Zhu and R. Kinjo, Angew. Chem., Int. Ed., 2023, 62, e202306519 CrossRef CAS.
- M. Zhong, J. Zhang, Z. Lu and Z. Xie, Dalton Trans., 2021, 50, 17150–17155 RSC.
- H. Lebel, H. Piras and J. Bartholoméüs, Angew. Chem., Int. Ed., 2014, 53, 7300–7304 CrossRef CAS.
- F. Pichierri, Chem. Phys. Lett., 2010, 487, 315–319 CrossRef CAS.
- X. Xiao, S. Huang, S. Tang, G. Jia, G. Ou and Y. Li, J. Org. Chem., 2019, 84, 7618–7629 CrossRef CAS.
- M. A. Dureen, A. Lough, T. M. Gilbert and D. W. Stephan, Chem. Commun., 2008, 4303–4305 RSC.
- A. J. V. Marwitz, J. T. Jenkins, L. N. Zakharov and S.-Y. Liu, Organometallics, 2011, 30, 52–54 CrossRef CAS.
- S. R. Wang, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, V. Paprocki and L. Winner, Chem. Commun., 2017, 53, 11945–11947 RSC.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS.
- F. Dielmann and G. Bertrand, Chem.–Eur. J., 2015, 21, 191–198 CrossRef CAS.
- M. Janssen, T. Frederichs, M. Olaru, E. Lork, E. Hupf and J. Beck-mann, Science, 2024, 385, 318–321 CrossRef CAS.
- Y. Ding, S. K. Sarkar, M. Nazish, S. Muhammed, D. Luert, P. N. Ruth, C. M. Legendre, R. Herbst-Irmer, P. Parameswaran, D. Stalke, Z. Yang and H. W. Roesky, Angew. Chem., Int. Ed., 2021, 60, 27206–27211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2324682 and 2324683. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04980b |
| This journal is © The Royal Society of Chemistry 2024 |