
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-constraint induced increase in Lewis acidity of tris(ortho-carboranyl)borane and selective complexation with Bestmann ylides†
Libo
Xiang
ab,
Junyi
Wang
c,
Alexander
Matler
ab and
Qing
Ye
 *ab
*ab
aInstitute for Inorganic Chemistry Julius-Maximilians-Universität Würzburg Am Hubland, 97074 Würzburg, Germany. E-mail: qing.ye@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron Julius-Maximilians-Universität Würzburg Am Hubland, 97074 Würzburg, Germany
cDepartment of Chemistry, Southern University of Science and Technology, 518055 Shenzhen, P. R. China
First published on 4th October 2024
Abstract
The Lewis acidity of tris(ortho-carboranyl)borane has been slightly increased by mimicking the structural evolution from triarylborane to 9-aryl-9-borafluorene. The o-carborane-based analogue (C2B10H10)2B(C2B10H11), obtained via salt elimination between LiC2B10H11 and (C2B10H10)2BBr, has been fully characterized. Gutmann–Beckett and computational fluoride/hydride ion affinity (FIA/HIA) studies have confirmed the increase in Lewis acidity, which is attributable to structural constraint imposed by the CC-coupling between two carboranyl groups. Selective complexation of (C2B10H10)2B(C2B10H11) with Bestmann ylides R3PCCO (R = Ph, Cy) has been achieved, enabling further conversion into the zwitterionic phospholium salt through NHC-catalyzed proton transfer.
Triarylboranes feature a three-coordinate six-electron boron center. They constitute a highly significant class of Lewis acids. Over the past century, the continuous focus on triarylborane chemistry has led to its substantial development.1 This has resulted in a wide array of applications across diverse fields, such as sensory materials,2 optoelectronic materials,3 small molecule activation,4 and catalysis.5 A significant benefit derived from the triarylborane motif is the feasibility of tuning the steric hindrance and electronic properties of the boron center by introducing appropriate substituents on the aryl groups.
Remarkably, the coupling of one ortho-carbon atom from each of the two aryl groups in triarylborane results in the creation of another significant class of molecules known as 9-borafluorenes.6 In these molecules, the boron atom is positioned within an anti-aromatic borole unit,7 giving rise to distinctive properties,8 including an increase of the Lewis acidity. For instance, triphenylborane (Fig. 1. A) demonstrates a fluoride ion affinity (FIA) of 354.35 kJ mol−1 (Table S2†), while the FIA of 9-phenyl-9-borafluorene (B) is elevated to 366.27 kJ mol−1 (Table S2†).9 Moreover, despite the removal of two ortho-electron-withdrawing fluorine atoms, the FIA of D (456.67 kJ mol−1, Table S2†) is still higher than that of C (452.68 kJ mol−1, Table S2†).9
 | ||
| Fig. 1 The trend in Lewis acidity among triarylboranes, 9-aryl-9-borafluorenes, and their respective carborane-based three-dimensional analogues. | ||
On the other hand, three-dimensional (3D) aromatic ortho-dicarbadodecaborane (o-carborane) can be regarded as an 3D analogue of benzene, and there has been a growing interest in carborane-based 3D analogues of conventional aryl-substituted boron compounds in recent years.10 In fact, the carboranyl substituted boron compounds have displayed numerous properties worthy of further exploration and development, including Lewis superacidity,11 fluorescence,11d,12 the capability of small molecule activation,13 and unique boron radical electronic structures.14 The successful synthesis of the three-dimensional analogue of A, i.e.E as a Lewis superacid (LSA) stands as a recent exemplary case.15 In this context, curiosity arises as to whether a similar structural alteration of E, as illustrated in Fig. 1, could also enhance the Lewis acidity. Herein, we present the synthesis and Lewis acidity assessment of the carborane-based 3D analogue of B, i.e.1. Furthermore, it is believed that the ball-shaped carboranyl substituents create a sterically hindered spatial environment around the central boron atom. To validate this hypothesis, we examined its reaction with Bestmann ylide,16 a Lewis base known for its ditopicity.
Compound 1 was attained by the reaction of LiC2B10H11 with an equimolar amount of (C2B10H10)2BBr11d at −78 °C (Scheme 1). 11B NMR spectrum displays a broad signal at δB 65, which is comparable to E (δB 67.2)15 and falls in the range of three-coordinate boron centers. The C–H proton NMR signal at δH 3.70 is diagnostic while the corresponding C–H displays a singlet at δC 58.7 in the 13C{1H} NMR spectrum. The atom connectivity was confirmed by single-crystal X-ray diffraction analysis (Fig. 2). Remarkably, the three Ccarborane–B bonds (B–C1: 1.558(2) Å, B–C3/B–C6: 1.597(16) Å) are shorter than the Ccaroborane–B bonds (1.614(8)–1.627(7) Å) in BoCb3 (E).15 In addition, the sum of angles around the central boron atom is 359.8°, indicating the trigonal-planar geometry. The C3–B–C6 angles of 110.74(14)° is close to the interior angle of a pentagon (108°). In addition, we exposed the C6D6 solution of 1 to air and conducted hourly NMR monitoring, which revealed complete hydrolysis within 6 h.
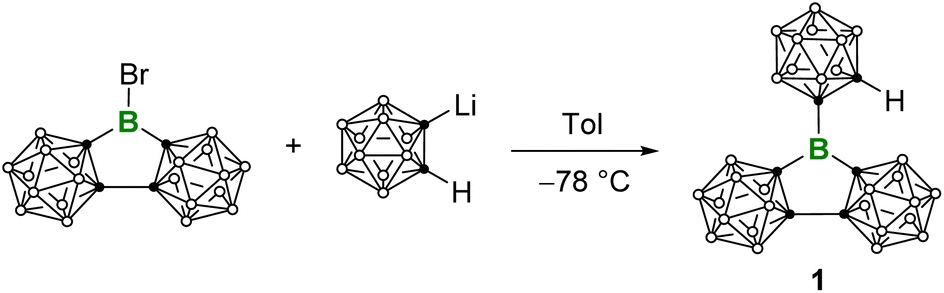 | ||
| Scheme 1 Synthesis of 1. | ||
 | ||
| Fig. 2 Single crystal structure of complex 1 in the solid state. The hydrogen atoms were omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: B–C1 1.558(2), B–C3 1.5966(16), B–C6 1.5966(16), C3–C4 1.6554(16), C4–C5 1.537(2), C5–C6 1.6554(16), C1–B–C3 124.55(7), C1–B–C6 124.55(7), C3–B–C6 110.74(14). | ||
The Gutmann–Beckett method17 was applied to assess the Lewis acidity of 1. With an LA/Et3PO ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the 31P Δδ of 34.9 for 1/Et3PO was slightly larger than that of C/Et3PO (31P Δδ 33.9 in C6D6). The acceptor number (AN) was 76.8 for 1 in C6D6, confirming its greater Lewis acidity than that of B(C6F5)3 (AN 65.6 in C6D6) and C (AN 74.9 in C6D6),15 and comparable to that of the pyramidal borane: 9-boratriptycene (AN 76.2 in CD2Cl2).18d It should be noted that the Gutmann–Beckett values are steric dependent. We calculated the buried volumes (% Vbur) of 1 and E considering the steric bulk of the carboranyl substituents,19 which indicate that both compounds have large % Vbur values (1: 67.7%, E: 73.3%, Table S5†). We also conducted calculations to further determine the FIA and hydride ion affinity (HIA) of 1 alongside A, to E, following the established literature protocols (Fig. 3).9 The calculations indicate that 1 displays the highest FIA and HIA energies, clearly designating it as a Lewis superacid. Notably, 1 exhibits larger FIA and HIA values than E, which is consistent with the results obtained through the Gutmann–Beckett method. One possible primary reason why 1 is slightly more acidic than E should be due to the structural constraint through CC-coupling (marked in red in Fig. 1), which reduces the C3–B–C6 angle by ca. 10°, from 120° to ca. 110° compared to that in E (ca. 120°). This deformation can be regarded as a ring-closure-enforced pre-pyramidalization, lowering the energy required for the structural deformation accompanying the complexation process.18 Indeed, the calculated pyramidization energy of 1 (122.0 kJ mol−1) was lower than that of E (130.1 kJ mol−1). Overall, the results of FIA, HIA and Gutmann–Beckett method are all consistent, indicating the Lewis acidity order of 1 > E > D > C > B > A. Among these, 1 and E are the strongest acids in the sequence, further demonstrating that the inductive effect of o-carborane can effectively enhance the Lewis acidity of boranes. Additionally, the relatively lower pyramidization energy of 1 leads to its higher Lewis acidity compared to E.
:1, the 31P Δδ of 34.9 for 1/Et3PO was slightly larger than that of C/Et3PO (31P Δδ 33.9 in C6D6). The acceptor number (AN) was 76.8 for 1 in C6D6, confirming its greater Lewis acidity than that of B(C6F5)3 (AN 65.6 in C6D6) and C (AN 74.9 in C6D6),15 and comparable to that of the pyramidal borane: 9-boratriptycene (AN 76.2 in CD2Cl2).18d It should be noted that the Gutmann–Beckett values are steric dependent. We calculated the buried volumes (% Vbur) of 1 and E considering the steric bulk of the carboranyl substituents,19 which indicate that both compounds have large % Vbur values (1: 67.7%, E: 73.3%, Table S5†). We also conducted calculations to further determine the FIA and hydride ion affinity (HIA) of 1 alongside A, to E, following the established literature protocols (Fig. 3).9 The calculations indicate that 1 displays the highest FIA and HIA energies, clearly designating it as a Lewis superacid. Notably, 1 exhibits larger FIA and HIA values than E, which is consistent with the results obtained through the Gutmann–Beckett method. One possible primary reason why 1 is slightly more acidic than E should be due to the structural constraint through CC-coupling (marked in red in Fig. 1), which reduces the C3–B–C6 angle by ca. 10°, from 120° to ca. 110° compared to that in E (ca. 120°). This deformation can be regarded as a ring-closure-enforced pre-pyramidalization, lowering the energy required for the structural deformation accompanying the complexation process.18 Indeed, the calculated pyramidization energy of 1 (122.0 kJ mol−1) was lower than that of E (130.1 kJ mol−1). Overall, the results of FIA, HIA and Gutmann–Beckett method are all consistent, indicating the Lewis acidity order of 1 > E > D > C > B > A. Among these, 1 and E are the strongest acids in the sequence, further demonstrating that the inductive effect of o-carborane can effectively enhance the Lewis acidity of boranes. Additionally, the relatively lower pyramidization energy of 1 leads to its higher Lewis acidity compared to E.
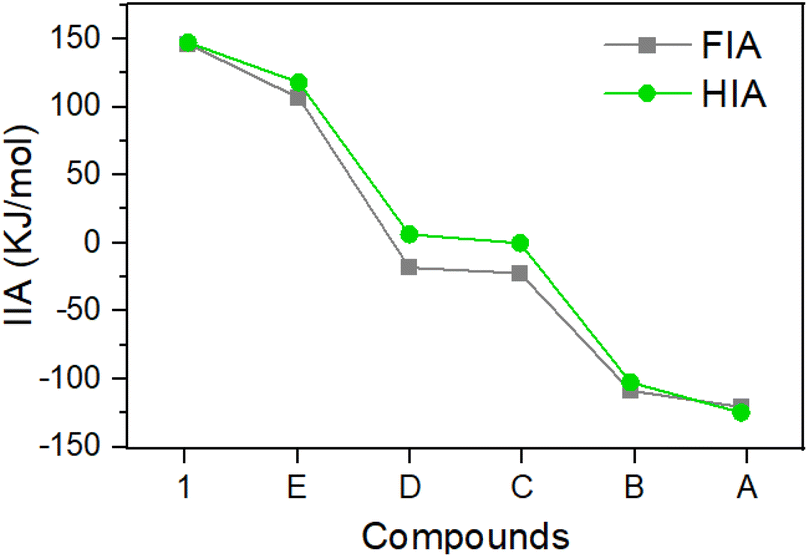 | ||
| Fig. 3 Results of fluoride ion affinity (ref. to SbF5), hydride ion affinity (ref. to B(C6F5)3) analyses and Global Electrophilicity Index (GEI) of compound 1 and A to E. | ||
Density functional theory (DFT) calculations were performed to provide further insight into the electronic properties of 1, E and D. Compound 1 has appreciably lower LUMO and larger HOMO–LUMO gap (Fig. 4) than D, thus indicating the enhanced electron-accepting ability. This can be attributed to the strong electron-withdrawing effect of the carboranyl group. The LUMO of E is 0.03 eV lower in energy than that of 1. Furthermore, Global Electrophilicity Index (GEI)18c,20,21 was calculated for 1 and A to E (Table S4†). The GEI values of 1 (4.17), E (4.20) and D (4.40) are larger than those of C (3.78), B (2.59) and A (2.04). Notably, the GEI results do not correlate well with the GB-method and the FIA results, with D displaying a relatively higher GEI value.
 | ||
| Fig. 4 Frontier orbitals of compound 1, D and E. | ||
With the Lewis superacidic borane 1 in hand, we decided to investigate its complexation reaction with Bestmann ylides. The prototype compound for Bestmann ylide is (triphenylphosphoranylidene)ketene Ph3PCCO.22 Several Bestmann ylide-supported transition metal complexes have been reported, where the ylidic carbon donates a pair of electrons to the transition metal center.23 However, the reactivity of Bestmann ylides with three-coordinate boranes has been less explored. In fact, Bestmann ylides can undergo addition to B(C6F5)3, either through the ylidic carbon or the carbonyl oxygen.24 In contrast, because the central boron of 1 is surrounded by three spherical substituents, this could potentially enable a selective complexation reaction.
To this end, 1 was reacted with an equimolar amount of 2a and 2b in toluene, respectively (Scheme 2). The reactions were monitored by multinuclear NMR spectroscopy. In both cases, the three-coordinate boron 11B signal disappeared within 10 min, accompanied by the formation of a white precipitate. It is worth noting that the phosphonium 31P signal is merely slightly low-field shifted, in the case of 2a from δP 3.1 to 5.2, 2b from δP 19.9 to 23.3, indicating that the electronic environment of the phosphorus center is not much affected upon complexation, and the oxygen-coordinated products 3a and 3b should be formed. Single crystal X-ray diffraction analysis unambiguously confirmed our hypothesis, showing that the terminal oxygen atom is bound to 1 (Fig. 5). While the bond lengths around boron increase from 1 (B–C1 1.558(2), B–C3 1.5966(16), B–C6 1.5966(16)) to 3 (3a: B–C3 1.683(2), B–C4 1.691(2), B–C1 1.653(2); 3b: B–C3 1.691(4), B–C4 1.695(4), B–C1 1.664(4)), the sum of angles decreases from 359.8° in 1 to 339.5° in 3a and 338.6° in 3b, respectively, indicating a geometric change from trigonal planar to tetrahedral. In 3a, the P–C2–C5–O unit features a nearly linear geometry, an elongated C5–O distance (1.264(19) vs. 1.195(2) Å) and a shortened C–C distance ((1.204(2) vs. 1.247(2) Å)), suggesting the oxyethynyl phosphonium type structure. The overall geometry of 3b resembles that of 3a. The infrared data for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching in compound 3 (3a: 2198 cm−1, 3b: 2199 cm−1) show a shift to higher wavenumbers than the uncoordinated Bestmann ylide (2090 cm−1), indicating the strengthening of the carbon–carbon bond upon coordination. In addition, the notably shorter B–O distance in 3b (1.522(4) Å) compared to the corresponding O-coordinated B(C6F5)3-2b adducts (1.573(3) Å)24 further confirms the higher Lewis acidity of 1 than B(C6F5)3.
C stretching in compound 3 (3a: 2198 cm−1, 3b: 2199 cm−1) show a shift to higher wavenumbers than the uncoordinated Bestmann ylide (2090 cm−1), indicating the strengthening of the carbon–carbon bond upon coordination. In addition, the notably shorter B–O distance in 3b (1.522(4) Å) compared to the corresponding O-coordinated B(C6F5)3-2b adducts (1.573(3) Å)24 further confirms the higher Lewis acidity of 1 than B(C6F5)3.
 | ||
| Scheme 2 Synthesis of 3. | ||
 | ||
| Fig. 5 Molecular structure of complex 3a and 3b in the solid state. The hydrogen atoms were omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: 3a, B–O 1.533(2), B–C1 1.653(2), B–C3 1.683(2), B–C4 1.691(2), O–C1 1.264(19), C1–C2 1.204(2), C2–P 1.701(17), O–C1–C2 174.19(17), C1–C2–P 179.96(17), C3–B–C4 104.18(12); 3b, B–O 1.522(4), B–C1 1.664(4), B–C3 1.691(4), B–C4 1.695(4), O–C1 1. 265(4), C1–C2 1.206(4), C2–P 1.713(3), O–C1–C2 172.60(3), C1–C2–P 166.5(3), C3–B–C4 104.1(2). | ||
The zwitterionic phospholium salt 4a was synthesized by treating 3a with 10 mol% MeIiPr (1,3-diisopropyl-4,5-dimethyl-imidazole-2-ylidene) at room temperature (Scheme 3). The 31P NMR spectrum showed the complete consumption of 3a (δ31P 5.17) within 10 min and the formation of a new species showing a downfield-shifted resonance at δ31P 12.64. After workup, colorless single crystals of 4a suitable for X-ray crystallography were obtained in a high yield (82%). X-ray analysis revealed a formal 1,2-addition of the CH of the exocyclic carborane cage to the alkynyl group of the coordinated Bestmann ylide, leading to a cyclization and the formation of 4a featuring a planar C3BO five-membered heterocycle with an internal angle sum of 540.0° (Fig. 6). This reaction can also be evidenced by infrared spectroscopy, which indicated the disappearance of the characteristic CC stretching and a new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1610 cm−1 (Fig. S27 and S28†). A plausible mechanism for the formation of 4a was proposed in Scheme 4. Following the deprotonation of 3a by MeIiPr, the resulting ortho-carborane anion undergoes intramolecular nucleophilic attack on the alkynyl group, leading to cyclization into a carbone intermediate. Due to the generally higher Brønsted basicity of carbone than NHCs,25 the carbone intermediate reclaims the proton from [MeIiPr-H]+, yielding the zwitterionic phospholium product 4a and regenerating the free carbene for the next catalytic cycle. The strong Brønsted basicity of the carbone intermediate can also be reflected by the fact that all attempts using the common strong bases such as NaHMDS and NaH to deprotonate 4a have failed: no reaction was observed.
C stretching at 1610 cm−1 (Fig. S27 and S28†). A plausible mechanism for the formation of 4a was proposed in Scheme 4. Following the deprotonation of 3a by MeIiPr, the resulting ortho-carborane anion undergoes intramolecular nucleophilic attack on the alkynyl group, leading to cyclization into a carbone intermediate. Due to the generally higher Brønsted basicity of carbone than NHCs,25 the carbone intermediate reclaims the proton from [MeIiPr-H]+, yielding the zwitterionic phospholium product 4a and regenerating the free carbene for the next catalytic cycle. The strong Brønsted basicity of the carbone intermediate can also be reflected by the fact that all attempts using the common strong bases such as NaHMDS and NaH to deprotonate 4a have failed: no reaction was observed.
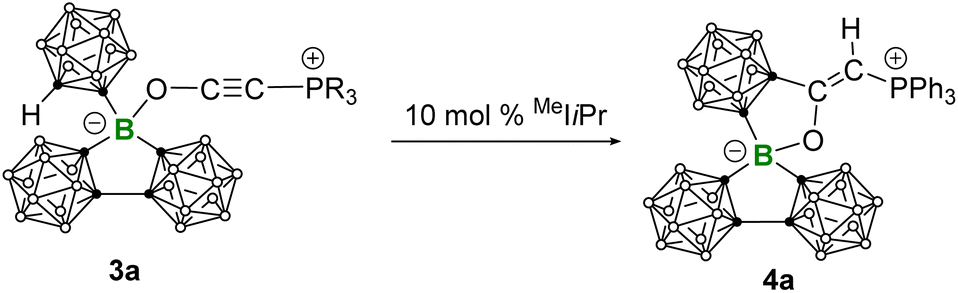 | ||
| Scheme 3 Synthesis of 4a. | ||
 | ||
| Fig. 6 Molecular structure of complex 4a in the solid state. The hydrogen atoms were omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]:4a, B–O 1.5221(14), O–C1 1.3265(14), C1–C2 1.3469(17), C2–P 1.7633(14), C1–C2–P 130.04(10). | ||
 | ||
| Scheme 4 Proposed reaction mechanism for the NHC-catalyzed proton transfer reaction. | ||
In summary, drawing inspiration from the structural evolution from triarylboranes to 9-aryl-9-borafluorenes, we envisioned that the title compound 1 may exhibit even higher Lewis acidity than the Lewis superacidic tris(o-carboranyl)borane. Compound 1 was synthesized via salt elimination between monolithiated o-carborane and the carborane-based analogue of 9-Br-9-borafluorene. The increase in its Lewis acidity with respect to tris(o-carboranyl)borane was confirmed experimentally by Gutmann–Beckett method and computationally through FIA and HIA values. It is noteworthy that unlike the 9-borafluorene system, the exceptional Lewis acidity of 1 primarily stems from the strong inductive effect of o-carborane, with negligible (hyper)conjugation effects, as well as the structural constraint imposed by the CC-coupling between two carboranyl groups. These factors combined render it the strongest Lewis acid among triarylboranes, 9-borafluorenes, and their carborane-based analogues. Furthermore, the unique steric environment of 1 allows for its selective addition to the O-site of Bestmann ylides, representing a rare example of selective coordination of Bestmann ylides with a borane. Moreover, benefiting from this, a unique reactivity, namely the NHC-catalyzed proton transfer has been disclosed. As demonstrated by this reaction, the combination of selective O-complexation and the presence of an acidic proton from the exocyclic carboranyl cage enables the catalytic process.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1, 3a, 3b, 4a have been deposited at the Cambridge Crystallographic Data Centre (CCDC) CODE under 2355581–2355583, 2382930 and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
L. X. carried out all the experiments. J. W. performed the DFT calculations. A. M. performed single crystal X-ray diffraction analysis. Q. Y. conceived and supervised the project. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Q. Y. thanks DFG (Grant No. 517941121, 520987585) and Julius-Maximilians-Universität Würzburg (JMU) for financial support. The Center for Computational Science and Engineering at SUSTech is acknowledged for providing computational resources. We thank Christoph Mahler and Liselotte Michels for their help with testing high-resolution mass spectrometry (HRMS) and elemental analysis (EA), respectively.Notes and references
- (a) M. Hatano and K. Ishihara, Lewis Acids, in Boron Reagents in Synthesis, American Chemical Society, 2016, vol. 1236, pp. 27–66 Search PubMed; (b) D. S. Matteson, in Stereodirected Synthesis with Organoboranes, ed. D. S. Matteson, Springer, Berlin, 1995, pp. 1–20 CrossRef; (c) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS; (d) S. M. Berger, M. Ferger and T. B. Marder, Chem. - Eur. J., 2021, 27, 7043–7058 CrossRef CAS PubMed; (e) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC; (f) A. G. Massey and A. J. Park, Organomet. Chem., 1964, 2, 245–250 CrossRef CAS; (g) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- (a) K. C. Song, K. M. Lee, N. V. Nghia, W. Y. Sung, Y. Do and M. H. Lee, Organometallics, 2013, 32, 817–823 CrossRef CAS; (b) G. Turkoglu, M. E. Cinar and T. Ozturk, Eur. J. Org Chem., 2017, 4552–4561 CrossRef CAS; (c) H. R. Bhat, P. S. S. Gupta, S. Biswal and M. K. Rana, ACS Omega, 2019, 4, 4505–4518 CrossRef CAS PubMed; (d) S. M. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 RSC; (e) H. Zhao, L. A. Leamera and F. P. Gabbaï, Dalton Trans., 2013, 42, 8164–8178 RSC; (f) K. Ishihara and H. Yamamoto, Eur. J. Org Chem., 1999, 527–538 CrossRef CAS; (g) A. Chardon, A. D. Mahaut, A. B. Saida and G. Berionni, Synlett, 2020, 31(17), 1639–1648 CrossRef CAS.
- (a) Z. M. Hudson and S. Wang, Dalton Trans., 2011, 40, 7805–7816 RSC; (b) Y. Yu, C. Dong, A. F. Alahmadi, B. Meng, J. Liu, F. Jäkle and L. Wang, J. Mater. Chem. C, 2019, 7, 7427–7432 RSC; (c) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42(10), 1584–1596 CrossRef CAS; (d) S. Mukherjeea and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
- (a) Y. Su and R. Kinj, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC; (b) M. Pramanik and R. L. Melen, Synthesis, 2023, 55, 3906–3918 CrossRef CAS; (c) M. Ghar, M. G. H. Mondal, R. Pal and P. K. Chattaraj, J. Phys. Chem. A, 2023, 127, 4561–4582 CrossRef PubMed; (d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS; (e) N. Zwettler and N. C. Mösch-Zanetti, Chem. - Eur. J., 2019, 25, 6064–6076 CrossRef CAS.
- (a) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC; (b) J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed; (c) E. Y. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS; (d) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC; (e) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS.
- (a) A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295–1298 RSC; (b) P. A. Chase, W. E. Piers and B. O. Patrick, J. Am. Chem. Soc., 2000, 122, 12911–12912 CrossRef CAS; (c) C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955–2958 CrossRef CAS; (d) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed.
- (a) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS; (b) J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC; (c) G. Varvounis, Comprehensive Heterocyclic Chemistry II, 1996, vol. 2, pp. 919–932 Search PubMed; (d) C. Hong, J. Baltazar and J. D. Tovar, Eur. J. Org Chem., 2022, e202101343 CrossRef CAS; (e) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270, 75–88 CrossRef.
- (a) M. Yamashita, Angew. Chem., Int. Ed., 2010, 49, 2474–2475 CrossRef CAS; (b) J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC; (c) K. Huynh, J. Vignolle and T. D. Tilley, Angew. Chem., Int. Ed., 2009, 48, 2835–2837 CrossRef CAS; (d) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, G. Frenking, S. Pan, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2020, 59, 3850–3852 CrossRef CAS.
- All FIA values are obtained from the same level of calculations: (a) L. Greb, Chem. - Eur. J., 2018, 24, 17881–17896 CrossRef CAS; (b) M. O. Keeffe, J. Am. Chem. Soc., 1986, 108, 4341–4344 CrossRef; (c) C. R. Wade, A. E. Broomsgrove, S. Aldridge and P. F. Gabbai, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed; (d) L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed.
- (a) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (b) M. Scholz and E. H. Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (c) D. Zhao and Z. Xie, Coord. Chem. Rev., 2016, 314, 14–33 CrossRef CAS; (d) Q. Zao and Z. Xie, Acc. Chem. Res., 2021, 54, 4065–4079 CrossRef PubMed; (e) J. Wang and Q. Ye, Chem. - Eur. J., 2023, 30, e202303695 CrossRef; (f) J. Wang, L. Xiang, X. Liu, A. Matler, Z. Lin and Q. Ye, Chem. Sci., 2024, 15, 4839–4845 RSC; (g) H. Zhang, J. Wang, W. Yang, L. Xiang, W. Sun, W. Ming, Y. Li, Z. Lin and Q. Ye, J. Am. Chem. Soc., 2020, 142, 17243–17249 CrossRef CAS PubMed; (h) J. Wang, P. Jia, W. Sun, Y. Wei, Z. Lin and Q. Ye, Inorg. Chem., 2022, 61, 8879–8886 CrossRef CAS PubMed.
- (a) Y. Wei, J. Wang, Y. Li, Z. Lin and Q. Ye, Chem. - Eur. J., 2023, 29, e202203265 CrossRef CAS; (b) C. Zhang, X. Liu, J. Wang and Q. Ye, Angew. Chem., Int. Ed., 2022, 61, e202205506 CrossRef CAS; (c) C. Zhang, J. Wang, W. Su, Z. Lin and Q. Ye, J. Am. Chem. Soc., 2021, 143, 8552–8558 CrossRef CAS PubMed; (d) C. Zhang, J. Wang, Z. Lin and Q. Ye, Inorg. Chem., 2022, 61, 18275–18284 CrossRef CAS; (e) M. Diab, K. Jaiswal, D. Bawari and R. Dobrovetsky, Isr. J. Chem., 2023, e202300010 CrossRef CAS; (f) M. O. Akram, J. R. Tidwell, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2023, 62, e202307040 CrossRef CAS PubMed; (g) M. O. Akram, C. D. Martin and J. L. Dutton, Inorg. Chem., 2023, 62, 13495–13504 CrossRef CAS.
- (a) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed; (b) J. Li, J. Xu, L. Yan, C. Lu and H. Yan, Dalton Trans., 2021, 50, 8029–8035 RSC; (c) M. Chen, J. Xu, D. Zhao, F. Sun, S. Tian, D. Tu, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2022, e202205672 CAS.
- (a) Y. Liu, W. Dong, Z. Li and H. Wang, Chem, 2021, 7, 1843–1851 CrossRef CAS; (b) Y. Liu, B. Su, W. Dong, Z. Li and H. wang, J. Am. Chem. Soc., 2019, 141, 8358–8363 CrossRef CAS; (c) Y. Xu, Y. Yang, Y. Liu, Z. Li and H. Wang, Nat. Catal., 2023, 6, 16–22 CrossRef CAS; (d) K. Vashisth, S. Dutta, M. O. Akrama and C. D. Martin, Dalton Trans., 2023, 52, 9639–9645 RSC.
- L. Xiang, J. Wang, I. Krummenacher, K. Radacki, H. Braunschweig, Z. Lin and Q. Ye, Chem.–Eur. J., 2023, 29, e202301270 CrossRef CAS PubMed.
- M. O. Akram, J. R. Tidwell, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2022, 61, e202212073 CrossRef CAS PubMed.
- (a) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3260–3272 CrossRef CAS PubMed; (b) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3273–3289 CrossRef CAS PubMed; (c) L. Zhao, M. Hermann, N. Holzmann and G. Frenking, Coord. Chem. Rev., 2017, 344, 163–204 CrossRef CAS; (d) H. J. Bestmann and D. Sandmeier, Chem. Ber., 1980, 113, 274–277 CrossRef CAS.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS; (b) M. A. Beckett, G. C. Strickland, J. R. Holland and K. A. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- (a) A. V. Pomogaeva and A. Y. Timoshkin, ACS Omega, 2022, 7, 48493–48505 CrossRef CAS PubMed; (b) M. El-Hamdi, M. Sola, J. Poater and A. Y. Timoshkin, J. Comput. Chem., 2016, 37, 1355–1362 CrossRef CAS PubMed; (c) S. B. H. Karnbrock, C. Golz, R. A. Mata and M. Alcarazo, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef PubMed; (d) A. Chardon, A. Osi, D. Mahaut, T.-H. Doan, N. Tumanov, J. Wouters, L. Fusaro, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2020, 59, 12402–12406 CrossRef CAS PubMed.
- (a) L. Zapf, M. Riethmann, S. A. Föhrenbacher, M. Finze and U. Radius, Chem. Sci., 2023, 14, 2275–2288 RSC; (b) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS; (c) S. D. González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef; (d) M. O. Akram, C. D. Martin and J. L. Dutton, Inorg. Chem., 2023, 62(33), 13495–13504 CrossRef CAS PubMed.
- P. Pérez, L. R. Domingo, A. Aizman and R. Contreras, Theor. Comput. Chem., 2007, 19, 139–201 Search PubMed.
- (a) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC; (b) A. R. Jupp, T. C. Johnstone and D. W. Stephane, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed.
- J. J. Dayl and P. J. Wheatley, J. Chem. Soc. A, 1966, 1703–1706 RSC.
- (a) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fuerstner, Nat. Chem., 2009, 1, 295–301 CrossRef CAS PubMed; (b) R. Bertani, M. Casarin and L. Pandolfo, Coord. Chem. Rev., 2003, 236, 15–33 CrossRef CAS; (c) L. Pandolfo, G. Paiaro, L. K. Dragani, C. Maccato, R. Bertani, G. Facchin, L. Zanotto, P. Ganis and G. Valle, Organometallics, 1996, 15, 3250–3252 CrossRef CAS.
- A. Brar, D. K. Unruh, A. J. Aquino and C. Krempner, Chem. Commun., 2019, 55, 3513–3516 RSC.
- (a) G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC; (b) W. Petz, Coord. Chem. Rev., 2015, 291, 1–27 CrossRef CAS; (c) A. L. Liberman-Martin, Cell Rep. Phys. Sci., 2023, 4, 101519 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray crystallographic and characterisation data. CCDC 2355581–2355583 and 2382930. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06144f |
| This journal is © The Royal Society of Chemistry 2024 |