
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic dopant cyclization and significantly improved RTP properties†
Shiguo
Zhang
,
Guanyu
Liu
,
Zhichao
Mao
,
Shanfeng
Xue
 ,
Qikun
Sun
* and
Wenjun
Yang
*
,
Qikun
Sun
* and
Wenjun
Yang
*
Key Laboratory of Rubber-plastics of Ministry of Education/Shandong Provincial, Key Laboratory of Rubber-plastics, School of Polymer Science & Engineering, Qingdao University of Science & Technology, Qingdao, China. E-mail: qksun@qust.edu.cn; ywjph2004@qust.edu.cn
First published on 30th October 2024
Abstract
The internal rotation of triplet-generating molecules is detrimental to room temperature phosphorescence (RTP) radiation, and this rotation is usually mitigated by doping into rigid microenvironments. The chemical locking of internal rotation units in advance should be an effective strategy but is rarely studied in comparison. Herein, a triplet-generating molecule with two rotatable phenyls (DIA) was designed, synthesized, and then cyclized using two types of bonding bridges. We found that DIA/PMMA film shows little observable RTP afterglow despite a 148 ms lifetime, whereas carbon bridge cyclized DIA (CDIA) and oxygen bridge cyclized DIA (ODIA) emitted green and blue ultralong RTP in PMMA film, with lifetimes of 2146 ms and 2656 ms, respectively, demonstrating the potent role of pre-locking of internal rotation units in promoting RTP. Benefiting from the good spectral overlap between the RTP emissions of dopants and the absorption of perylene red (PR) in PMMA film, the almost complete triplet-to-singlet Förster resonance energy transfer was achieved under trace doping (0.1%), providing red room temperature afterglow materials with lifetimes of 1567–1800 ms. Preliminary applications of blue, green, and red afterglow materials in optical encryption and anti-counterfeiting are demonstrated. This work not only develops new triplet-generating and -radiating molecules but also introduces an effective molecular strategy for achieving ultralong RTP polymers.
Introduction
Physically doping trace amounts of conjugated organic molecules into rigid and polar polymers is the simplest and most economical method to produce room temperature phosphorescence (RTP) polymers. In this method, organic triplet-generating and -radiating molecules are the key components, and their chemical composition and structure require rational design for realizing bright and ultralong RTP afterglow.1–14 The introduction of heavy halogens and heteroatoms can usually enhance molecular spin–orbit coupling (SOC) to facilitate singlet-to-triplet inter-system crossing (ISC), for triplet generation (population).2–8 Triplet population is the precondition of RTP; however, the triplet radiation at room temperature is usually affected seriously by molecular thermal motions.14–18Molecular thermal motions, especially the presence of molecular internal rotation, may completely disable triplet RTP radiation.19,20 The most typical example is rhodamine derivatives that emit long-lived thermally activated delayed fluorescence rather than RTP even in a rigid and polar polyvinyl alcohol matrix; however, they have very high SOC values and multiple ISC channels.20 Therefore, RTP production is the cooperative result of triplet population and radiation, and the internal rotation of conjugated organic molecules is rather detrimental to triplet radiation.
Molecular internal rotation can be mitigated by low-temperature freezing and/or embedding into rigid matrices. After being frozen by liquid nitrogen, many conjugated organic molecules in a solvent glassy state, amorphous state, or crystalline state emit phosphorescence, but the phosphorescence behavior ceases at room temperature, even in a crystalline state. This indicates that crystallization itself often cannot effectively inhibit triplet thermal motion deactivation unless a proper guest–host system exists.21–23
Interestingly, some molecules without crystalline RTP can emit molecular RTP in rigid and polar polymer matrices as molecular thermal motion deactivation is greatly suppressed by polymeric rigidity and cohesion.21,22 However, it can be imagined that, although rigid polymeric chain segments are frozen at room temperature, the thermal motions of local chain units and side groups (secondary relaxations) continue to stimulate dopant molecular motions, especially the most detrimental dopant internal rotation. Therefore, chemically locking internal rotation units in advance should be a radical strategy to prevent the adverse effect of dopant molecular internal rotation and is beneficial for inducing and increasing the molecular RTP radiation of organic-doped polymers.
To validate the above rational idea, herein, three conjugated organic molecules (DIA, CDIA, and ODIA) were designed, synthesized, and doped into poly(methyl methacrylate) (PMMA) film to examine the effect of locking internal rotation units on RTP properties (Fig. 1). DIA contains two rotatable phenyls, and CDIA and ODIA can be regarded as the products of carbon–carbon linking and oxygen bridging DIA between the two phenyls, respectively. From molecular structures, it was determined that cyclization and hetero-cyclization were responsible for locking the internal rotation of the two phenyls.
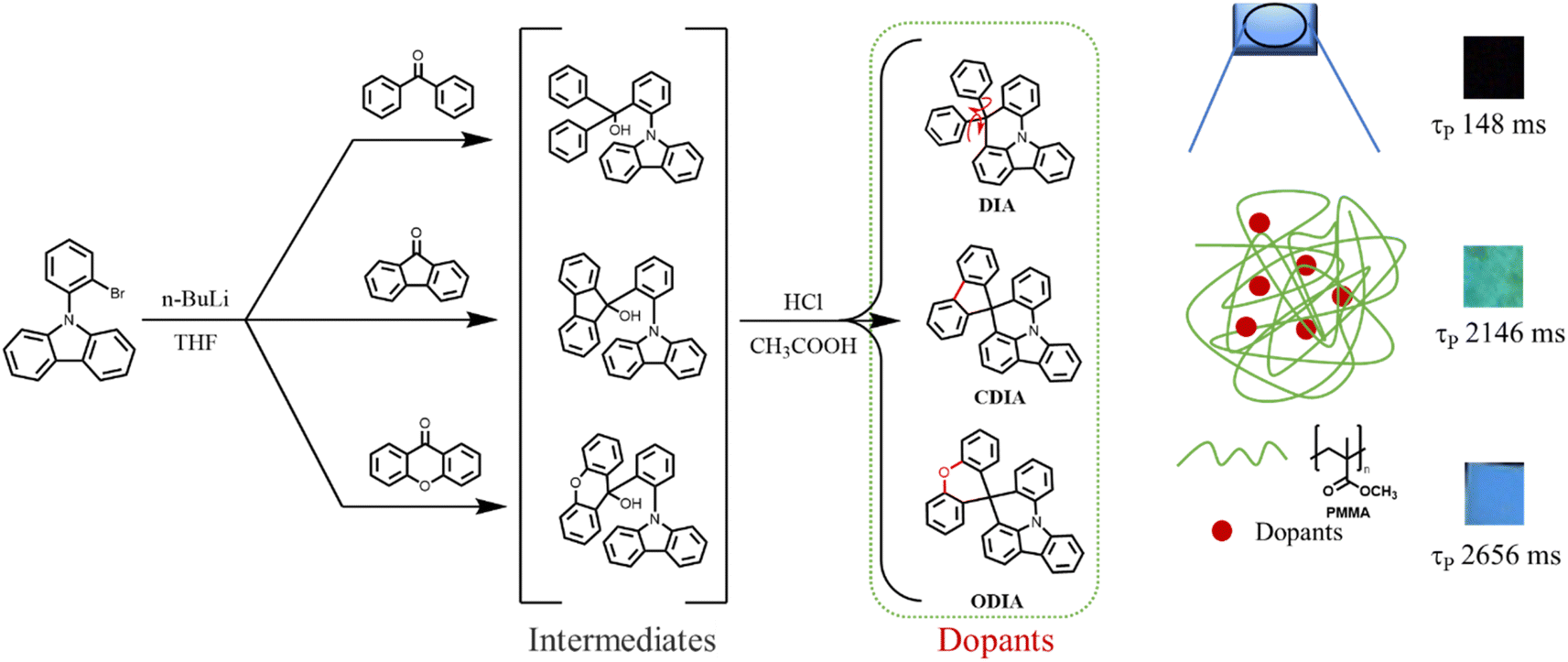 | ||
| Fig. 1 Chemical strategies to prepare polymer-based RTP materials. | ||
Herein, we report three compounds that exhibit little crystal RTP. DIA@PMMA film also emits little observable RTP afterglow despite a detectable 148 ms of lifetime, whereas CDIA and ODIA in PMMA film emit green and blue ultralong RTP afterglow with lifetimes of 2146 ms and 2656 ms, respectively. This fully demonstrates the potent role of pre-locking of the internal rotation unit in promoting RTP. In view of the satisfactory spectral overlaps between the RTP emissions of dopants and the absorption of perylene red (PR) in PMMA film, PR/CDIA@PMMA and PR/ODIA@PMMA films were prepared to enrich afterglow colors via almost complete triplet-to-singlet Förster resonance energy transfer under trace doping (0.1%). The obtained red afterglow materials have the room temperature lifetimes of up to 1567 ms and 1800 ms, respectively. Preliminary applications of blue, green, and red afterglow materials in optical encryption and anti-counterfeiting are demonstrated. Our experimental results reveal that the cyclization of organic dopant molecules is an effective strategy for developing new triplet-generating and -radiating molecules for ultralong RTP polymers.
Results and discussion
DIA, CDIA, and ODIA were synthesized through the lithium substitution of N-(2-bromophenyl)carbazole (BPCZ) and then the addition of benzophenone, 9-fluorenone, and xanthone at 78 °C in a nitrogen atmosphere, respectively (Fig. 1). The intermediates without separation were subjected to aqueous HCl treatment in acetic acid to afford the target products in moderate yields of 52–58%. The details of synthesis and characterization are depicted in the ESI.† The absorption bands appeared before 360 nm, and the emission peaks were approximately 365 nm in THF solution (Fig. S1†). The emission of these compounds was very weak and short, with even a total lack of RTP afterglow in crystals (Fig. S2†). However, DIA, CDIA, and ODIA emitted a few seconds of low temperature phosphorescence (LTP) afterglow in liquid nitrogen, wherein the LTP of DIA crystal was the strongest and lasted the longest (Fig. S3†).DIA, CDIA, and ODIA (5 mg) were doped into PMMA (5 g) by co-dissolving in dichloromethane and evaporating the solvent, respectively, and the solution-cast thermoplastic films were processed into 1 mm thick sheets for the following measurements (see the ESI†). After 100 s of illumination by a 365 nm lamp, the DIA@PMMA sheet exhibited little visible afterglow (Fig. 2b), but in the steady-state PL spectra, there were highly-efficient (52.1%) prompt and lowly-efficient (0.8%) delayed emissions with peak wavelengths at 370 nm and 449 nm, respectively (Fig. 2a and Table 1). Additionally, the time-resolved RTP (at 449 nm) decay curve revealed a lifetime of approximately 148 ms (Fig. 2c). However, the CDIA@PMMA and ODIA@PMMA sheets emitted clearly visible and recordable afterglows (Fig. 2b), and their peak emission wavelengths were at 537 nm (green emission) and 454 nm (blue emission) (Fig. 2a and Table 1), respectively. Significantly, the measured RTP efficiencies (ΦP) for CDIA@PMMA and ODIA@PMMA were 10-fold higher than that for DIA@PMMA, and the fitted corresponding long-component and average RTP lifetimes (2146 and 2656 ms, and 1619 and 2133 ms, respectively) from the time-resolved RTP decay curves were fairly longer than that for DIA@PMMA (77 and 148 ms, increased by approximately 20 times) (Fig. 2c and Table 1).
 | ||
| Fig. 2 (a) The prompt and delayed PL spectra of DIA@PMMA, CDIA@PMMA, and ODIA@PMMA under 280 nm light excitation. Due to the overlap between the 365 nm excitation peak and the fluorescence peak, the test was conducted using 320 nm excitation. (b) Room temperature PL photographs of DIA, CDIA, and ODIA in PMMA after removing 365 nm UV light (3 mW cm−2). (c) The time-resolved RTP decay curves and fitted long-component RTP lifetimes under 365 nm light excitation. | ||
| Dopant | λ FL (nm) | λ P (nm) | τ F (ns) | τ P (ms) | Φ F (%) | Φ P (%) | k ISC (s−1) | k Pnr (s−1) |
|---|---|---|---|---|---|---|---|---|
| a The detection wavelength of average fluorescent and average phosphorescent lifetime was according to the maximum emission. b The phosphorescent quantum efficiency integrated ranges were 350–700 nm for DIA@PMMA, 350–750 nm for CDIA@PMMA, and 350–700 nm for ODIA@PMMA. c k ISC = ΦP/τF, kPnr = (1 − ΦP)/τP. | ||||||||
| DIA | 370 | 449 | 6.1 | 77 | 51.3 | 0.8 | 1.3 × 106 | 13.0 |
| CDIA | 376 | 537 | 6.5 | 1619 | 10.4 | 8.3 | 1.3 × 107 | 0.60 |
| ODIA | 371 | 454 | 5.7 | 2133 | 17.3 | 10.3 | 1.8 × 107 | 0.42 |
The photophysical data for the organic doped PMMA sheets are summarized in Table 1. The calculated rate constants for ISC and RTP radiation are remarkably higher for CDIA@PMMA and ODIA@PMMA as compared to DIA@PMMA, and the rate constants for triplet non-radiation for CDIA@PMMA and ODIA@PMMA are much lower than that for DIA@PMMA. Therefore, cyclization has importantly improved ISC and stabilized triplet exciton radiation. However, freezing these sheets in liquid nitrogen hardly changes the PL emission positions, but the LTP intensity and duration were greatly increased (30–40 s). The LTP spectra also show finer vibrational structures (Fig. S4†), implying that the internal rotation rather than bond vibration thermal deactivation was effectively restricted at room temperature.
To gain insight into the effect of internal rotation on molecular RTP, quantum theoretical calculations were conducted. Fig. 3a shows the calculated Huang–Rhys (HR) factors, where the total HR factors (41.39) for T1 → S0 for DIA are much greater than those for CDIA (17.32) and ODIA (21.47). Moreover, the HR factors for the three molecules in high-frequency vibrational regions are similar; however, in the middle and low frequency regions, CDIA and ODIA show obviously lower HR factors as compared to DIA, which can be referred to as the inhibition effect of cyclization on internal rotation and non-radiation transition.15,24,25 Conversely, as the total reorganization energy (Λ) upon photoexcitation-emission is positively correlated with the non-radiative transition rate, the Λ was calculated for the three molecules (Fig. 3b), and the results revealed reduced Λ values (i.e., suppressed structural relaxation) for CDIA and ODIA as compared to DIA, which supports the experimentally calculated non-radiative transition rates (Table 1).
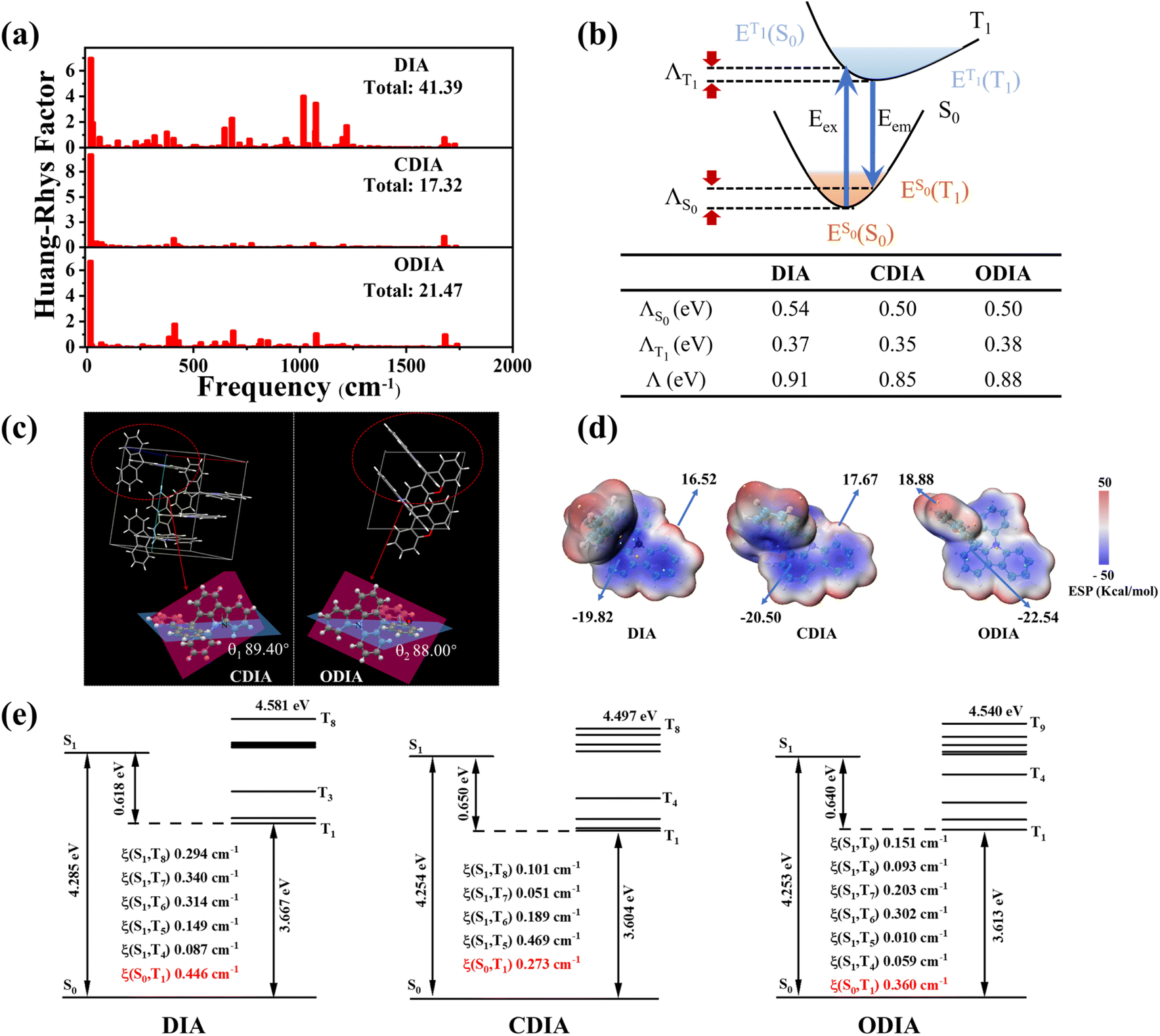 | ||
| Fig. 3 (a) The Huang–Rhys factors versus frequencies of DIA, CDIA, and ODIA. (b) Theoretical estimation of the total reorganization energies (Λ). (c) The molecular geometries of CDIA and ODIA single crystals. (d) Calculated ESP distribution of DIA, CDIA, and ODIA. (e) Calculated energy diagram and spin–orbit coupling (SOC) matrix elements (ξ). | ||
The crystal structures of the CDIA and ODIA molecules were successfully analyzed, and the results show that the planes of fluorene and xanthene are almost perpendicular to the plane of indolo[3,2,1-de]acridine (Fig. 3c), which are consistent with theoretically optimized single molecular structures.24 We calculated the energy levels and distributions of excited states, as well as the SOC values of energy-allowed ISC channels between S1 → Tn (S1 ± 0.3 eV) (Fig. 3e). The ISC (or ability to increase the triplet population) of DIA is not theoretically second to CDIA and ODIA. However, despite non-inferior LTP, the lack of observable RTP radiation by DIA@PMMA indicates that the presence of internal rotation moieties is indeed very detrimental to triplet exciton stabilization and RTP radiation.
Regarding the more excellent RTP properties of ODIA@PMMA compared to CDIA@PMMA, the enhancement of dopant–matrix interactions may be due to the introduction of electronegative oxygen. It has been widely reported that improving dopant–matrix interactions is conducive to RTP radiation. Fig. 3d shows the calculated electrostatic potential (ESP) distribution on the surfaces of the DIA, CDIA, and ODIA molecules. The positive and negative ESP values in ODIA are increased by the introduction of oxygen atoms, signifying the increased compatibility and stronger interactions with the polar PMMA matrix.26,27
To verify the mechanism, we also preliminarily tested the acrylonitrile butadiene styrene (ABS) matrix. As shown in Fig. S5,† the afterglow time for ODIA@ABS is obviously longer, which verifies the role of cyclization. Among the current RTP polymers, intrinsic red afterglow emissions are rare. To achieve red afterglow, co-doping red fluorescent dyes into long-lived RTP polymers is a common method as the triplet-to-singlet Förster resonance energy transfer (FRET) is not limited by spin multiplicity.28–30 However, the satisfactory spectral overlap between the RTP emission of the organic dopant (donor) and the absorption of the fluorescent dye (acceptor) is conducive to FRET efficiency. Otherwise, it is difficult to obtain high FRET efficiency by doping a small amount of acceptor molecules, and high FRET efficiency achieved by doping a large amount of acceptor molecules will severely impair the brightness and lifetime of the red afterglow.
Perylene red (PR) is a highly efficient red dye with an absorption band ranging from 400 to 620 nm, which overlaps well with the RTP emissions of CDIA and ODIA in PMMA (Fig. 4a). Thus, 0.1% PR and 0.1% CDIA or 0.1% ODIA (mass percent in PMMA) were co-doped into the PMMA matrix, respectively, and the same preparation procedure was used for CDIA@PMMA and ODIA@PMMA. Fig. 4b shows the afterglow photographs of PR/CDIA@PMMA and PR/ODIA@PMMA sheets after excitation by 365 nm light for 100 s, and bright and ultralong red afterglow is observed for both sheets, with duration of over 10 s and 15 s, respectively, to the naked eye in the dark.
 | ||
| Fig. 4 (a) The ultraviolet-visible absorption spectra (Abs.) of PR@PMMA and delayed PL spectra of CDIA@PMMA and ODIA@PMMA. (b) Room temperature PL photographs of PR/CDIA and PR/ODIA in PMMA after removing 365 nm UV light (3 mW cm−2). (c) The prompt and delayed PL spectra of PR/CDIA@PMMA and PR/ODIA@PMMA under 280 nm light excitation. (d) Schematic diagram of light excitation and afterglow emission of RTP polymer. (e) The time-resolved RTP decay curves and fitted long-component RTP lifetimes under 365 nm light excitation. (f) Commission Internationale de I'Eclairage (CIE) coordinates of afterglow emission for CDIA@PMMA, ODIA@PMMA, and PR/ODIA@PMMA. (g) The preliminary applications of afterglow materials in optical encryption are demonstrated. | ||
The prompt PL spectra do not show RTP bands but only red emission bands, and the delayed PL spectra are also dominated by a red afterglow emission (Fig. 4c), indicating the complete and the almost complete FRET of triplet excitons of dopant donor to the excited singlet state of the red fluorescent acceptor before and after removing UV light excitation according to the mechanisms depicted in Fig. 4d, respectively. The red afterglow lifetimes fitted from the time-resolved PL decay curves (monitored at 629 nm) are 1567 and 1800 ms for the PR/CDIA@PMMA and PR/ODIA@PMMA sheets, respectively (Fig. 4e). Thus, ultralong blue, green, and red afterglow hydrophobic polymer films were achieved, and the corresponding Commission International de I'Eclairage (CIE) chromaticity coordinates are (0.26, 0.37), (0.35, 0.44), and (0.57, 0.32), respectively (Fig. 4f).
The above results indicate that long-lived red afterglow materials were obtained by doping PR. Taking advantage of the distinct time-resolved luminescent feature of the RTP polymer after doping with different luminophores, we proceeded to demonstrate an information encryption and decryption technology. We chose CDIA/PR@PMMA, ODIA/PR@PMMA, CDIA@PMMA, and ODIA@PMMA films as materials for information storage because they exhibit different afterglow durations when excited by a 365 nm UV lamp.
Fig. 4g shows how we used four materials to construct a “711” pattern. The pattern is clearly visible under daylight and 365 nm excitation. After removing the excitation source, within 4 s, the “711” pattern remains. Depending on the duration of the afterglow, the pattern becomes “111” in 4 to 5 s, “11” in 6 to 8 s, and “1” in 9 to 14 s. If the correct pattern is “11”, then we need to read the information between 6 and 8 s after the light excitation. When the signal is immediately read, another false “711, 111” message appears (within 6 s) after the lights are turned off. As a result, the accuracy of these time-resolved RTP polymers is satisfactory for information encryption applications for dates.
Conclusions
We produced ultralong organic RTP polymer materials through cyclization and heterocyclization. To explore the effect of heterocyclization on pure organic RTP, we systematically studied the RTP properties and corresponding mechanisms of three carbazole-derived polymers. In addition to cyclization by molecular bonds to promote radiative transitions, the introduction of oxygen atoms promoted SOC, enhanced noncovalent interactions, and stabilized the triplet population. As a result, an afterglow of up to 16 s and a lifetime of 2656 ms were obtained, reaching the forefront level (Table S4†). By doping PR, three colors of red, green, and blue afterglow polymer were obtained, and the display application of encryption was demonstrated. There are vast applications for polymer materials due to their properties such as processability, biocompatibility, potential applications in encryption, display, and storage, and they are also easy to synthesize with low cost. Through these works, we can design phosphorescent molecules with improved performance.Data availability
The authors confirm that the data supporting the findings of this study are available within the article or its ESI.†Author contributions
S. Zhang carried out all experiments and wrote the first draft of the manuscript with the assistance of K. Sun and W. Yang. G. Liu was involved in the synthesis and characterization of the compounds. Z. Mao was involved in the measurements of optical properties. S. Xue was involved in the design of experiments and characterization. Q. Sun and W. Yang co-supervised the project, conceptualized the study, analysed the data and completed the editing and revision of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided by the National Natural Science Foundation of China (No. 52273183), the Natural Science Foundation of Shandong Province (No. ZR2020QE-083), and the Taishan Scholar Constructive Engineering Foundation of Shandong Province of China (No. tsqn202211164). We thank the open project of The State Key Laboratory of Supramolecular Structure and Materials of Jilin University (No. sklssm2024032).References
- X. Yang, G. I. N. Waterhouse, S. Lu and J. Yu, Chem. Soc. Rev., 2023, 52, 8005–8058 RSC.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- H. Sun, S. Shen and L. Zhu, ACS Mater. Lett., 2022, 4, 1599–1615 CrossRef.
- B. Chang, J. Chen, J. Bao, T. Sun and Z. Cheng, Chem. Rev., 2023, 123, 13966–14037 CrossRef PubMed.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 1, 582–594 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 Search PubMed.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef PubMed.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS.
- Y. Zhao, B. Ding, Z. Huang and X. Ma, Chem. Sci., 2022, 13, 8412–8416 RSC.
- Z. Yang, H. Liu, X. Zhang, Y. Lv, Z. Fu, S. Zhao, M. Liu, S. Zhang and B. Yang, Adv. Mater., 2024, 36, 2306784 CrossRef CAS.
- S. Xiong, Y. Xiong, D. Wang, Y. Pan, K. Chen, Z. Zhao, D. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2301874 CrossRef CAS PubMed.
- Z. Yang, Z. Fu, H. Liu, M. Wu, N. Li, K. Wang, S.-T. Zhang, B. Zou and B. Yang, Chem. Sci., 2023, 14, 2640–2645 RSC.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- Y. Niu, Y. Guan, C. Long, C. Ren, J. Lu, C. Jin, P. Wang, X. Fan and H.-L. Xie, Sci. China Chem., 2023, 66, 1161–1168 CrossRef CAS.
- D. Wang, H. Wu, J. Gong, Y. Xiong, Q. Wu, Z. Zhao, L. Wang, D. Wang and B. Z. Tang, Mater. Horiz., 2022, 9, 1081–1088 RSC.
- Z. Lin, P. Zhang, F. Song, Y. Yang, X. Miao and W. Liu, Chem. Sci., 2024, 15, 8052–8061 RSC.
- C. Qian, X. Zhang, Z. Ma, X. Fu, Z. Li, H. Jin, M. Chen, H. Jiang and Z. Ma, CCS Chem., 2024, 6, 798–811 CrossRef CAS.
- S. Zhang, G. Liu, J. Chen, Y. Zhang, Q. Sun, S. Xue and W. Yang, Adv. Opt. Mater., 2023, 11, 2301147 CrossRef CAS.
- Y. Zhang, S. Zhang, G. Liu, Q. Sun, S. Xue and W. Yang, Chem. Sci., 2023, 14, 5177–5181 RSC.
- J. Chen, Y. Zhang, S. Zhang, G. Liu, Q. Sun, S. Xue and W. Yang, Small Struct., 2023, 4, 2300101 CrossRef CAS.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS.
- X. Yang, S. Wang, K. Sun, H. Liu, M. Ma, S. Zhang and B. Yang, Angew. Chem., Int. Ed., 2023, 62, e202306475 CrossRef CAS PubMed.
- X. Lv, J. Miao, M. Liu, Q. Peng, C. Zhong, Y. Hu, X. Cao, H. Wu, Y. Yang, C. Zhou, J. Ma, Y. Zou and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201588 CrossRef CAS.
- C. Wang, L. Qu, X. Chen, Q. Zhou, Y. Yang, Y. Zheng, X. Zheng, L. Gao, J. Hao, L. Zhu, B. Pi and C. Yang, Adv. Mater., 2022, 34, 2204415 CrossRef CAS.
- Z. Lin, P. Zhang, F. Song, Y. Yang, X. Miao and W. Liu, Chem. Sci., 2024, 15, 8052–8061 RSC.
- J. Wang, Y. Yang, K. Li, L. Zhang and Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202304020 CrossRef CAS PubMed.
- J. Chen, S. Zhang, G. Liu, Y. Zhang, S. Xue, Q. Sun and W. Yang, Adv. Opt. Mater., 2023, 11, 2301163 CrossRef CAS.
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2350554 and 2350558. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06213b |
| This journal is © The Royal Society of Chemistry 2024 |