
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous generation of hydroxyl and hydrogen radicals from H+/OH− pairs caused by water–solid contact electrification†
Fengjie
Chen‡
a,
Jingde
Wu‡
b,
Dou
Wang
c,
Yu
Xia
a,
Qingyuan
Song
d,
Ying
Liang
a,
Pu
Wang
a,
Bolei
Chen
 *ad,
Yong
Liang
a,
Yongguang
Yin
d,
Yawei
Wang
ad,
Maoyong
Song
d and
Guibin
Jiang
d
*ad,
Yong
Liang
a,
Yongguang
Yin
d,
Yawei
Wang
ad,
Maoyong
Song
d and
Guibin
Jiang
d
aHubei Key Laboratory of Environmental and Health Effects of Persistent Toxic Substances, School of Environment and Health, Jianghan University, Wuhan, 430056, China
bSchool of Environment, Hangzhou Institute for Advanced Study, UCAS, Hangzhou, 310000, China
cState Key Laboratory for Managing Biotic and Chemical Treats to the Quality and Safety of Agro-products, Institute of Agro-product Safety and Nutrition, Zhejiang Academy of Agricultural Science, Hangzhou, 310012, China
dState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, 10085, China. E-mail: blchen@rcees.ac.cn
First published on 11th November 2024
Abstract
Water–solid contact electrification is a common physical phenomenon involving interfacial electron and ion transfer, recently discovered to trigger unique redox reactions. Here, we demonstrate the generation of both hydroxyl and hydrogen radicals when water contacts SiO2. The coexistence of hydroxyl and hydrogen radicals is confirmed by simultaneous nitrate reduction and nitrite oxidation during the contact. Increased density of hydroxyl groups on the SiO2 surface enhances its surface electronegativity before the contact, as well as boosting charge transfer and radical generation during the contact. We propose that the simultaneous generation of hydroxyl and hydrogen radicals originates from electron gain and loss between hydroxide anions in water and hydrogen cations adsorbed on the solid surface, which are ion pairs separated by the interfacial electric field. This discovery advances our understanding of redox processes induced by contact electrification.
Introduction
Water–solid contact electrification is relevant to a variety of mechanochemical, environmental, and catalytic processes, as well as playing a crucial role in chemical synthesis technology and energy devices.1–6 There is growing evidence suggesting that this physical stimulus would result in transfer of both electrons and ions between water and a solid surface,7,8 influencing oxidation and reduction reactions at the aqueous interface.9–11 Our previous work demonstrated that the contact between aqueous microdroplets and the SiO2 substrate causes the spontaneous generation of reactive oxygen species (ROS) in the microfluidic channel.12 The presence of hydroxyl groups on the solid surface and their hydrogen bonding network play key roles in the oxidation chemical processes.10,13 Other researchers reported that the contact between water and poly-tetrafluoroethylene (PTFE) particles in bulk suspension catalyses ROS generation from hydroxide anions (OH−) under ultrasonication.3,6 Moreover, recent work found that the water–PTFE contact electrification can also lead to continuous reduction of nitrogen (N2) absorbed on a solid surface by bubbling the gas into the bulk aqueous suspension.14 These findings imply that complex reaction pathways exist during water–solid contact. Notably, similar spontaneous oxidation and reduction processes, or even simultaneous redox reactions, can be caused by the strong electric field at the surface of the sprayed water microdroplets.15–20 However, it is not possible to apply the reaction mechanism in microdroplets to explain the observation during water–solid contact, since the electric field and charge transfer behaviour at the water–solid interface are completely different compared to the water–gas interface. In what follows, we present evidence for the simultaneous oxidation and reduction reactions that occurred during water–SiO2 contact electrification. We show that the hydrogen radical and hydroxyl radical act as the reductant and oxidant, respectively, triggering opposite but simultaneous redox reactions. Considering that water loses electrons and solids gain electrons during contact electrification, OH− ions in water and H+ absorbed on SiO2 may work as electron donors and acceptors, respectively, and thus become sources of the radicals during the contact.Results and discussion
An ideal water–solid contact model was constructed based on SiO2 microspheres with a diameter of 60 μm sealed in a homemade chamber as shown in Fig. 1A. The SiO2 microspheres were chosen as the solid phase material in order to create a large contact surface area to initiate a strong enough chemical process to facilitate subsequent detection.10 We applied water-carrying argon gas contact with SiO2 to minimize the effect of bulk water on the reaction process at the contact interface. The use of argon excluded the influence of reactive gases such as oxygen and ozone in our reaction system. Notably, the water-carrying argon gas was obtained by bubbling the gas through DI water, which was deionized reagent-grade with a resistivity larger than 18 MΩ cm. A ROS-sensitive probe (2′,7′-dichlorodihydrofluorescein diacetate, DCFH-DA) and a reductive species-sensitive water-soluble probe (resazurin) were used to detect the generation of ROS and reductive species at the water–solid interface, respectively (shown in Fig. S1†). The fluorescence probes dissolved in the source water were carried by argon gas to the SiO2 surface to react with possible ROS and reductive species. After 2 hours of continuous contact between H2O and SiO2 microspheres, the SiO2 microspheres were placed on a glass slide for fluorescence microscopy measurement. Typically, red fluorescence emission was observed on the SiO2 surface under 570 nm laser excitation (as shown in Fig. 1B), while green fluorescence emission was observed on the surface of the SiO2 micro-sphere under 488 nm laser excitation (as shown in Fig. 1C). To rule out the artificial error caused by the fluorescence measurement, we compared the fluorescence intensity on the SiO2 surface before and after the contact. As shown in Fig. S2,† the fluorescence emission from the SiO2 microsphere surface can hardly be observed before the water–solid contact occurred. These results indicated that both ROS and reductive species can be generated during contact electrification. Moreover, we carried out electron spin resonance spectroscopy (ESR) to identify the radicals generated during the contact. As shown in Fig. 1D, the signal from hydrogen radicals, highlighted by the red “*” symbol, was observed on the surface of SiO2 microspheres after the contact. However, four peaks with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 intensity ratio (highlighted by the green “•” symbol) appeared, which can be attributed to hydroxyl radicals.21,22 This result reveals that the response signals of fluorescence probes observed in Fig. 1B and C can be attributed to the generation of hydrogen and hydroxyl radicals, respectively.
:2:2:1 intensity ratio (highlighted by the green “•” symbol) appeared, which can be attributed to hydroxyl radicals.21,22 This result reveals that the response signals of fluorescence probes observed in Fig. 1B and C can be attributed to the generation of hydrogen and hydroxyl radicals, respectively.
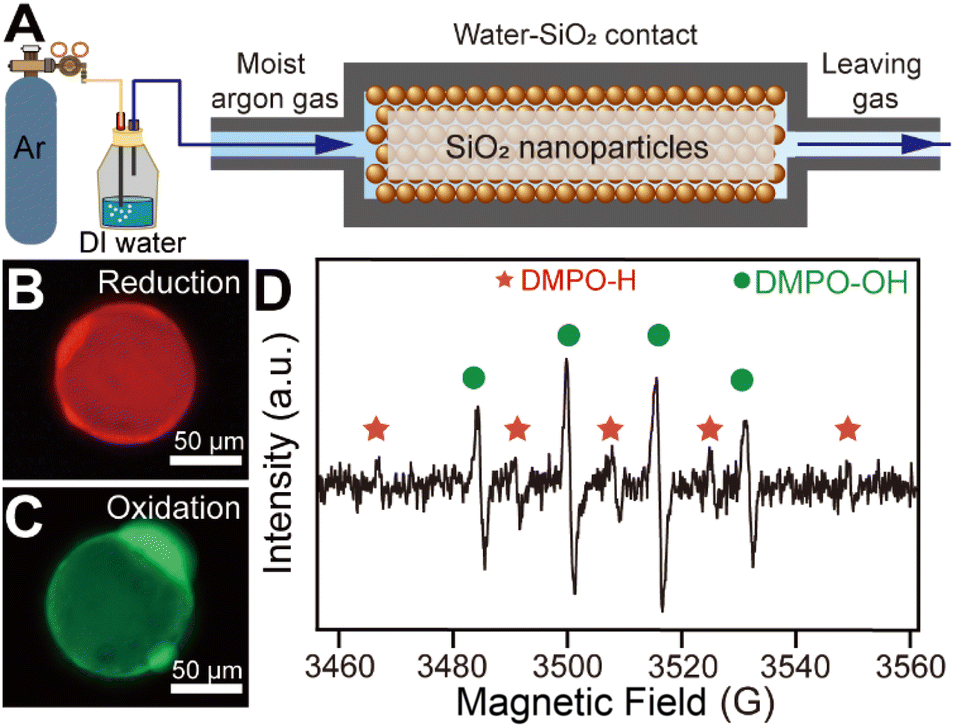 | ||
| Fig. 1 (A) Schematic representation of the experimental setup. (B) A typical fluorescence microscopy image of reductive species generated on the surface of a SiO2 microsphere. (C) A typical fluorescence microscopy image of ROS generated on the surface of a SiO2 microsphere. (D) Corresponding ESR result for SiO2 micro-spheres after contacting with water-carrying argon gas. | ||
In order to elucidate the simultaneous generation of hydroxyl and hydrogen radicals during the contact process, we contacted moist argon gas carrying both sodium nitrate and sodium nitrite with SiO2 microspheres and analyzed the reaction products. The ion chromatography measurements were carried out immediately after the contact occurred between water and SiO2. By comparing the NO3−/NO2− ratio before and after the contact shown in Fig. 2A, we found that the reduction of nitrate or oxidation of nitrite may happen.23 And the nitrite and nitrate generation during water–solid contact increased with the increase of the flow rate of argon gas (Fig. S3†). Notably, we employed 15N isotopically labeled sodium nitrate and normal sodium nitrite as starting reactants to distinguish their products during the contact.24 Their mass spectra are displayed in Fig. S4.† After the contact between water and SiO2 occurred, the 15N isotopically labeled nitrite cation (15NO2−) was observed at m/z at 47, while the generation of the nitrate cation (NO3−) was confirmed by the peak at m/z 62 (shown in Fig. 2B). The peaks at m/z 63 and m/z 46 can be attributed to incompletely reacted starting reactants. By comparing the changes in the mass spectra before and after the water–solid contact, it is clear that 15N isotope-labeled nitrite and normal nitrite are generated spontaneously and simultaneously during the contact process.
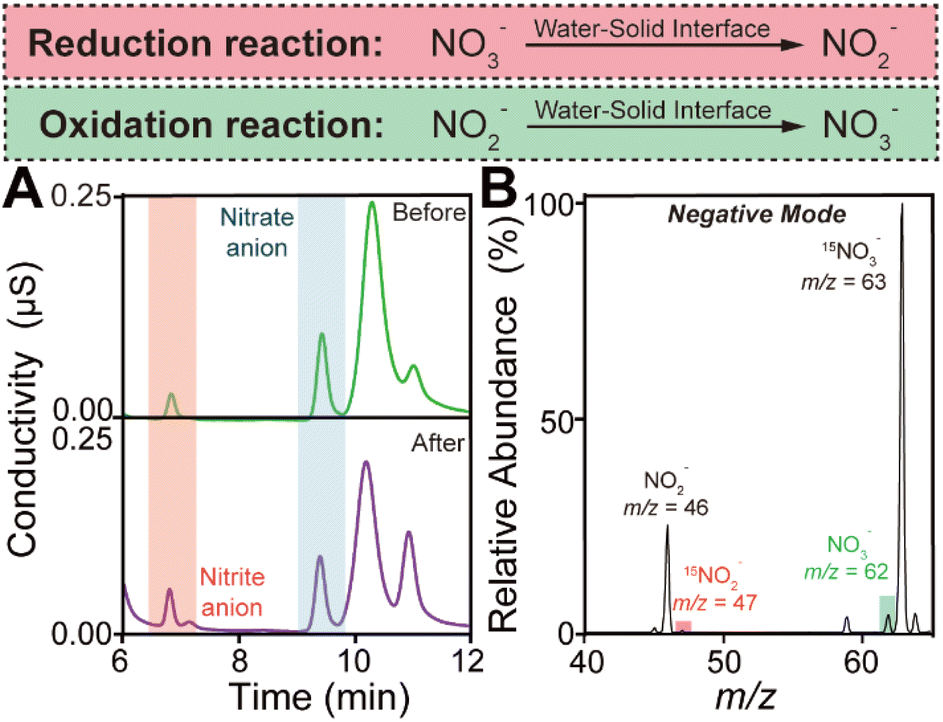 | ||
| Fig. 2 (A) Ion chromatography showing the transformation of NO2− and NO3− before and after the water–solid contact. (B) Mass spectrum in negative mode showing the oxidation of NO2− and the reduction of 15NO3−. | ||
To further understand the above redox reactions, we analyze the oxidation and reduction during the contact process separately. When moist argon gas containing NaNO3 was contacted with SiO2, ion chromatography measurements indicated that nitrate was converted to nitrite as shown in Fig. S5.† The corresponding mass spectra obtained in the negative and positive modes are displayed in Fig. S6A and B.† The peak observed in negative mode at m/z 46 further confirms the reduction of NaNO3 during the contact. Observation of the peak at m/z 63 could be attributed to the intermediate product produced in the reduction of NO3− by hydrogen radicals (NO3− + ˙H → NO3H˙−).25 Then the NO3H˙− further reacted with hydrogen radicals to generate NO3− (NO3H˙− + ˙H → NO2˙− + H2O). Furthermore, in the positive mode, the peak at m/z 36 suggests that the hydroxyl radical is generated from an ion in the water during the contact process, regardless of whether the ion is OH− or the water radical cation.20,26 Replacing water with heavy water (D2O) gave the corresponding mass spectrum in the negative mode of deuterated ions as shown in Fig. S7.† Similarly, the oxidation of NO2− was confirmed by ion chromatography measurements and mass spectra shown in Fig. S8A and B.† Notably, in negative mode, the peak at m/z 63 was hardly observed during the oxidation process caused by water–solid contact. We believe that this result provides further evidence for the participation of hydrogen radicals in the reduction reaction caused by contact electrification. Moreover, in positive mode, we calculated the relative peak intensities at m/z 36 representing the hydroxyl radical generation when using the mass spectral peak of the hydrated sodium ion as an internal standard. The relative peak intensity at m/z 36 observed during the oxidation of NO2− is lower than that obtained during the reduction of NO3−. This result further confirmed the generation of the hydroxyl radical caused by the water–solid contact process and induced the NO2− oxidation (NO2− + 2˙OH → NO3− + H2O). These observations are in good agreement with the ESR measurements displayed in Fig. S9.† Considering the coexistence of hydroxyl and hydrogen radicals, we believe that both radicals are generated by the gain and loss of electrons from ion pairs, which are separated by the electric double layer at the water–solid interface. Importantly, based on the common sense that electrons are transferred from water to SiO2 during contact electrification, we conjectured that the generation of hydroxyl radicals originates from the process of electron loss by the OH− in water, while the production of hydrogen radicals is attributed to the process of electron gain by the H+ adsorbed on the surface of the solid.
To confirm our conjecture, we obtained SiO2 microspheres with different surface electronegativity by varying the hydroxyl group density on the solid surface as shown in Fig. 3A and S10.† We believe that microspheres with high surface hydroxyl group density can adsorb more H+ when water is in contact with the solid. Fig. 3B reveals that electron transfer caused by water–solid contact electrification increased with increasing surface hydroxyl density. This result could be attributed to more H+ which acts as an electron acceptor during the contact, while OH− in water is considered to be in excess as an electron donor. Furthermore, the ESR measurements and ion chromatography quantitative analysis, as shown in Fig. 3C and D, indicate that the simultaneous generation of hydroxyl and hydrogen radicals was enhanced by increasing the charge transfer during contact electrification. We believe that these findings provide evidence for our hypothesis that H+ from the stem layer and OH− in the diffusion layer at the water–solid interface are responsible for the simultaneous reduction and oxidation under the physical stimuli of the contact.
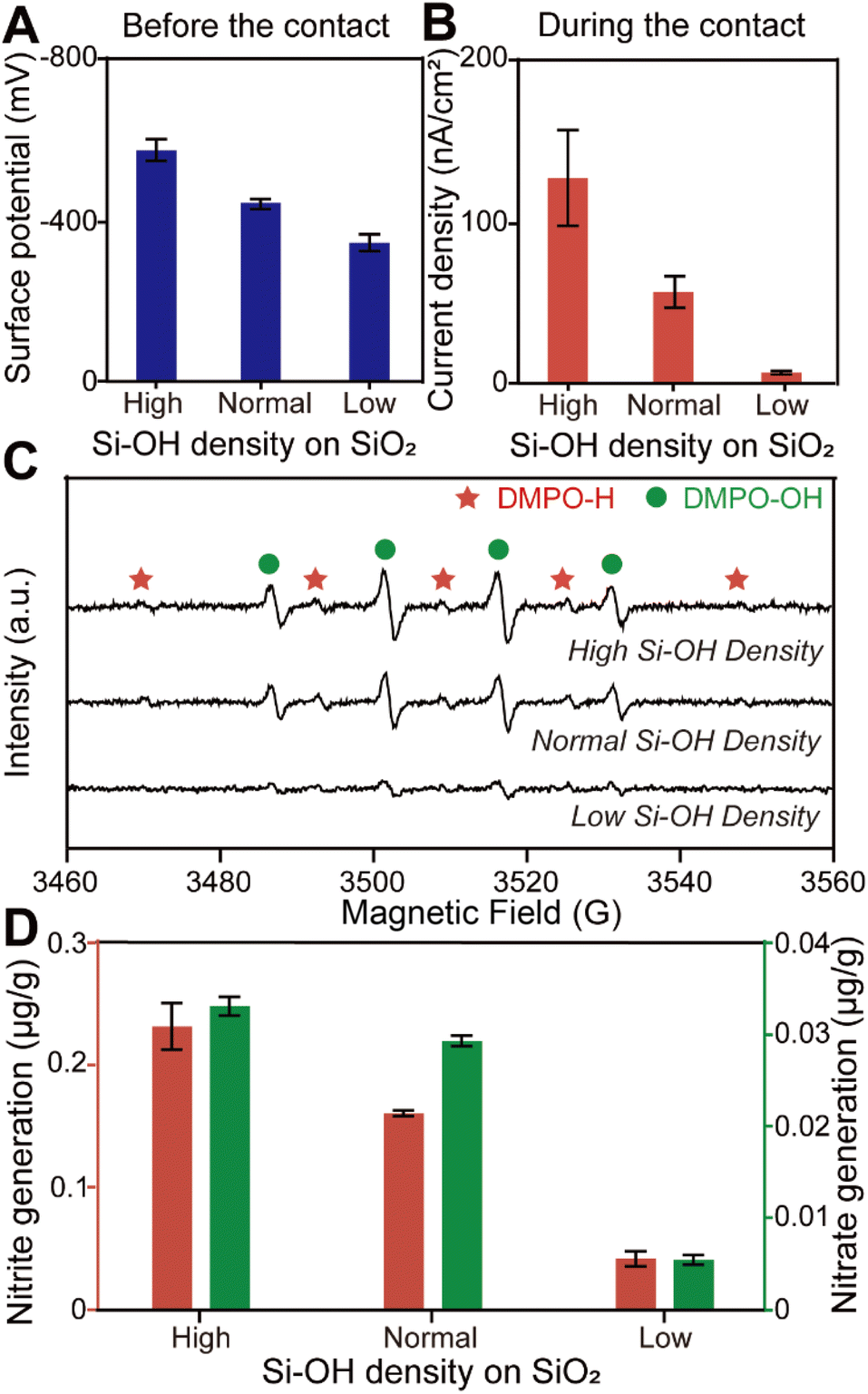 | ||
| Fig. 3 (A) Surface potential of SiO2 samples with different hydroxyl group density before the contact measured by Kelvin probe force microscopy (KPFM). (B) The corresponding current density of SiO2 samples with different hydroxyl group density during the contact. (C) The corresponding ESR spectra of SiO2 samples after the contact process. (D) Nitrite and nitrate generation during water–solid contact depending on the density of hydroxyl groups on the SiO2 surface. | ||
As shown in Fig. 4, a possible reaction pathway was constructed to describe the simultaneous generation of hydrogen and hydroxyl radicals when water–solid contact occurs. Firstly, electrons are transferred from water to the solid surface during the initial stage of the contact (contact electrification), leading to an increase in the electronegativity of the solid surface. Secondly, the solid surface absorbs H+ ionized from the acidic hydroxyl functional groups, which in turn forms a double electric layer. Notably, although each material carries a net charge of either positive or negative polarity, their surface supports a random “mosaic” of oppositely charged regions of nanoscopic dimension.27,28 We speculate that the non-uniform distribution of these charges during water–solid contact can cause enrichment of H+ in some regions of the solid surface, which in turn leads to the formation of a strong electric field directly between H+ in the stem layer and OH− in the diffusion layer, causing electrons to be stripped from OH− and transfer toward the adsorbed H+ on the solid surface. Simultaneously, a small portion of OH− loses their electrons and form hydroxyl radicals, while H+ gains electrons and generates hydrogen radicals. Finally, the hydrogen and hydroxyl radicals together mediate the interfacial redox chemical processes.
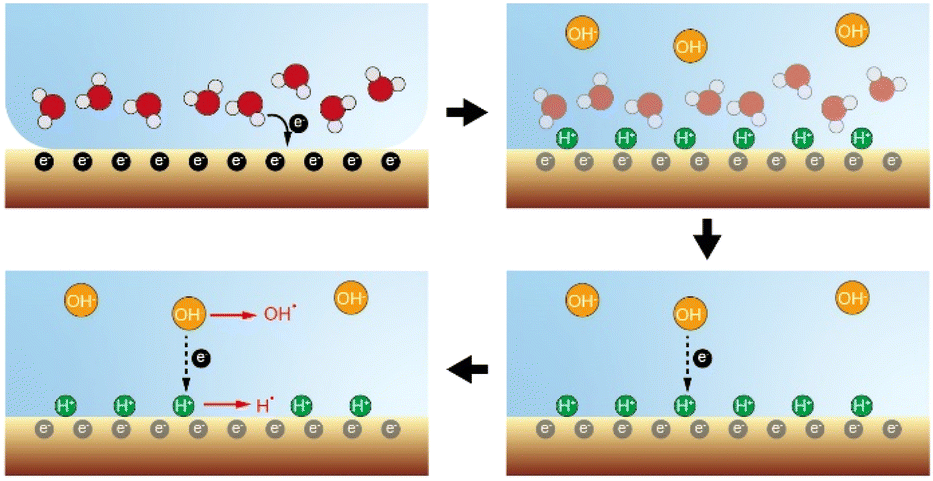 | ||
| Fig. 4 Proposed mechanism for simultaneous generation of hydrogen and hydroxyl radicals caused by water–solid contact electrification. | ||
Conclusions
In conclusion, we report the simultaneous and spontaneous oxidation and reduction reactions when the contact occurred between water and the SiO2 surface. We have provided a series of evidence showing that the redox reactions observed during the water–solid contact are triggered by hydroxyl and hydrogen radicals. Importantly, the coexistence of hydroxyl and hydrogen radicals suggests that their generation can be attributed to electron gain and loss of OH−/H+ ion pairs, which are separated by an electric field at the water–solid interface. We believe that our hypothesis is applicable to explain the redox processes in sprayed microdroplets, as well as the observation at the water–oil interface. Moreover, these findings might provide new insight into understanding the redox chemistry in the atmosphere under dark conditions.Data availability
All data utilized in the manuscript have been uploaded to the Science Data Bank repository and are available at https://www.scidb.cn/s/rQN7Rr.Author contributions
F. C. and J. W. conceptualized the specific research in close consultation with B. C., who conceived the overall project and acquired necessary funding. D. W. and Y. X. developed and implemented the water–solid model. Q. S. and Y. L. conducted fluorescence experiments and image segmentation. F. C. conducted the remaining experiments. P. W., Y. L., Y. Y., Y. W., M. S. and G. J. participated in the discussion of the experiments and related results. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2023YFC3905300), the National Natural Science Foundation of China (22376080, 22306073, 22125606, and 22193051), and the Hubei Provincial Natural Science Foundation of China (2024AFA089).Notes and references
- S. Lin, X. Chen and Z. L. Wang, Chem. Rev., 2022, 122, 55209–55232 Search PubMed.
- C. Cai, B. Luo, Y. Liu, Q. Fu, T. Liu, S. Wang and S. Nie, Mater. Today, 2022, 52, 299–326 CrossRef CAS.
- Z. Wang, A. Berbille, Y. Feng, S. Li, L. Zhu, W. Tang and Z. L. Wang, Nat. Commun., 2022, 13, 130 CrossRef CAS PubMed.
- Z. L. Wang, T. Jiang and L. Xu, Nano Energy, 2017, 39, 9–23 CrossRef CAS.
- W. Xu, H. Zheng, Y. Liu, X. Zhou, C. Zhang, Y. Song, X. Deng, M. Leung, Z. Yang, R. X. Xu, Z. L. Wang, X. Zeng and Z. Wang, Nature, 2020, 578, 392–396 CrossRef CAS PubMed.
- Z. Wang, X. Dong, W. Tang and Z. L. Wang, Chem. Soc. Rev., 2024, 53, 4349–4373 RSC.
- J. Nie, Z. Ren, L. Xu, S. Lin, F. Zhan, X. Chen and Z. L. Wang, Adv. Mater., 2020, 32, 1905696 CrossRef CAS.
- S. Lin, L. Xu, A. Wang and Z. L. Wang, Nat. Commun., 2020, 11, 399 CrossRef CAS.
- C. Yun, S. Lee, J. Ryu, K. Park, J. Jang, J. Kwak and S. Hwang, J. Am. Chem. Soc., 2018, 140, 14687–14695 CrossRef CAS.
- Y. Xia, J. Li, Y. Zhang, Y. Yin, B. Chen, Y. Liang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2302014120 CrossRef CAS.
- Y. Li, K. W. Kolasinski and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2304735120 CrossRef CAS PubMed.
- B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209056119 CrossRef CAS.
- S. Shaik, D. Danovich and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 20132–20140 CrossRef CAS PubMed.
- J. Li, Y. Xia, X. Song, B. Chen and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2318408121 CrossRef CAS PubMed.
- S. Jin, H. Chen, X. Yuan, D. Xing, R. Wang, L. Zhao, D. Zhang, C. Gong, C. Zhu, X. Gao, Y. Chen and X. Zhang, JACS Au, 2023, 3, 1563–1571 CrossRef CAS.
- X. Yuan, D. Zhang, C. Liang and X. Zhang, J. Am. Chem. Soc., 2023, 145, 2800–2805 CrossRef CAS PubMed.
- S. Jin, R. Wang, H. Chen, X. Yuan and X. Zhang, J. Phys. Chem. A, 2023, 127, 2805–2809 CrossRef CAS.
- X. Chen, Y. Xia, Z. Zhang, L. Hua, X. Jia, F. Wang and R. N. Zare, J. Am. Chem. Soc., 2023, 145, 21538–21545 CrossRef CAS PubMed.
- X. Chen, Y. Xia, Y. Wu, Y. Xu, X. Jia, R. N. Zare and F. Wang, J. Am. Chem. Soc., 2024, 145, 10868–10874 CrossRef.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CrossRef CAS PubMed.
- J. Peng, K. Du, J. Sun, X. Yang, X. Wang, X. Zhang, G. Song and F. Feng, Angew. Chem., Int. Ed., 2022, 62, e202214991 CrossRef PubMed.
- S. Pei, S. You, J. Ma, X. Chen and N. Ren, Environ. Sci. Technol., 2020, 54, 13333–13343 CrossRef CAS.
- Y. Wu, L. Bu, X. Duan, S. Zhu, M. Kong, N. Zhu and S. Zhou, J. Clean. Prod., 2022, 273, 123065 CrossRef.
- C. Hu, Y. Chang, C. Yen, J. Chen, R. Muthukumaran and M. Chao, Free Radicals Biol. Med., 2019, 143, 193–202 CrossRef CAS.
- M. Han and M. Mohseni, Water Res., 2020, 168, 115169 CrossRef CAS PubMed.
- D. Xing, Y. Meng, X. Yuan, S. Jin, X. Song, R. N. Zare and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207587 CrossRef CAS.
- H. Baytekin, A. Patashinski, M. Branicki, B. Baytekin, S. Soh and B. Grzybowski, Science, 2011, 333, 108–312 CrossRef.
- Y. Sobolev, W. Adamkiewicz, M. Siek and B. Graybowski, Nat. Phys., 2022, 18, 1347–1355 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional data and experimental details. See DOI: https://doi.org/10.1039/d4sc06227b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |