
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceM12L24 nanospheres as supramolecular templates for the controlled synthesis of Ir-nanoclusters and their use in the chemo-selective hydrogenation of nitro styrene†
Lotte L.
Metz
 a,
Rens
Ham
a,
Eduard O.
Bobylev
a,
Kelly J. H.
Brouwer
b,
Alfons
van Blaaderen
b,
Rim C. J.
van de Poll
c,
Victor R.
Drozhzhin
c,
Emiel J. M.
Hensen
c and
Joost N. H.
Reek
*a
a,
Rens
Ham
a,
Eduard O.
Bobylev
a,
Kelly J. H.
Brouwer
b,
Alfons
van Blaaderen
b,
Rim C. J.
van de Poll
c,
Victor R.
Drozhzhin
c,
Emiel J. M.
Hensen
c and
Joost N. H.
Reek
*a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, 1098 XH Amsterdam, The Netherlands. E-mail: j.n.h.reek@uva.nl
bSoft Condensed Matter, Debye Institute for Nanomaterials Science, Utrecht University, Princetonplein 1, 3584 CC, Utrecht, The Netherlands
cDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
First published on 9th November 2024
Abstract
Controlled preparation of ultrafine metal nanoclusters (<2 nm) is challenging, yet important as the properties of these clusters are inherently linked to their size and local microenvironment. In the present work, we report the utilization of supramolecular pre-organization of organometallic complexes within well-defined M12L24 coordination spheres for the controlled synthesis of ultrafine Ir nanoclusters by reduction with molecular hydrogen. For this purpose, 24 sulfonate functionalized N-heterocyclic carbene (NHC) Ir complexes (Ir-s) were bound within a well-defined M12L24 nanosphere that is equipped with 24 guanidinium binding sites (G-sphere). Reduction of these pre-organized metal complexes by hydrogenation led to the templated formation of nanoclusters with a narrow size distribution (1.8 ± 0.4 nm in diameter). It was demonstrated through 1H-DOSY-NMR and HAADF-STEM-EDX experiments that the resulting nanoclusters reside within the nanospheres. The reduction of similar non-encapsulated metal complexes in the presence of nanosphere systems (Ir-s + M-sphere or Ir-p + G-sphere) resulted in larger particles with a broader size distribution (2.3 ± 2.1 nm and 6.6 ± 3.2 nm for Ir-s + M-sphere and Ir-p + G-sphere respectively). The encapsulated nanoclusters were used as a homogeneous catalyst in the selective hydrogenation of 4-nitrostyrene to 4-ethylnitrobenzene and display absolute selectivity, which is even maintained at full conversions, whereas the larger non-encapsulated clusters were less selective as these also showed reduction of the nitro functionality.
Introduction
The preparation and study of ultrafine nanoclusters (NCs) is a topic of increasing interest in various fields. NCs are defined as nanoparticles with a size of ≤2 nm.1,2 As a result of their high surface-to-volume atom ratio, particles of such minute dimensions exhibit unique physical and chemical properties compared to their larger analogues and bulk materials. Due to their extraordinary properties, these materials have made their way into various applications such as solid-state memory,3 sensing/imaging4–7 and catalysis.7–9 The size, dispersion and surface environment of nanoparticles and nanoclusters are key to their unique physical properties and catalytic features. Subtle changes can result in substantial differences in their electronic and geometric properties. Therefore, it is of considerable interest to develop methodologies to make nanoclusters with precise control over these properties.1,2In the past decades, various strategies have been reported to gain synthetic control over the size, shape and distribution including the use of biphasic systems10,11 and dendrimers.12,13 More recently, templated strategies involving porous materials such as covalent organic frameworks (COFs),14–17 porous organic cages (POCs)18–32 and discrete coordination-spheres33–42 have been emerging.43–46 Such porous templates can provide a protective cavity for nanoparticle synthesis by sterically impeding certain growth modes, limiting particle aggregation and enhancing particle stability, solubility and size control.47
Recently, our group has developed a method for organometallic catalyst encapsulation within well-defined M12L24 coordination spheres (M = Pt2+ or Pd2+),48–53 internally functionalized with guanidinium anchors (Fig. 1, G-sphere).54–56 These guanidinium functional groups were demonstrated to bind strongly to sulfonate-functionalized molecular catalysts and carboxylate-functionalized substrates through hydrogen-bonding. This strategy was proven to be a powerful and versatile tool for substrate and catalyst pre-organization and was demonstrated to enhance the catalytic activity and/or selectivity for several homogeneously catalysed transformations.54–56
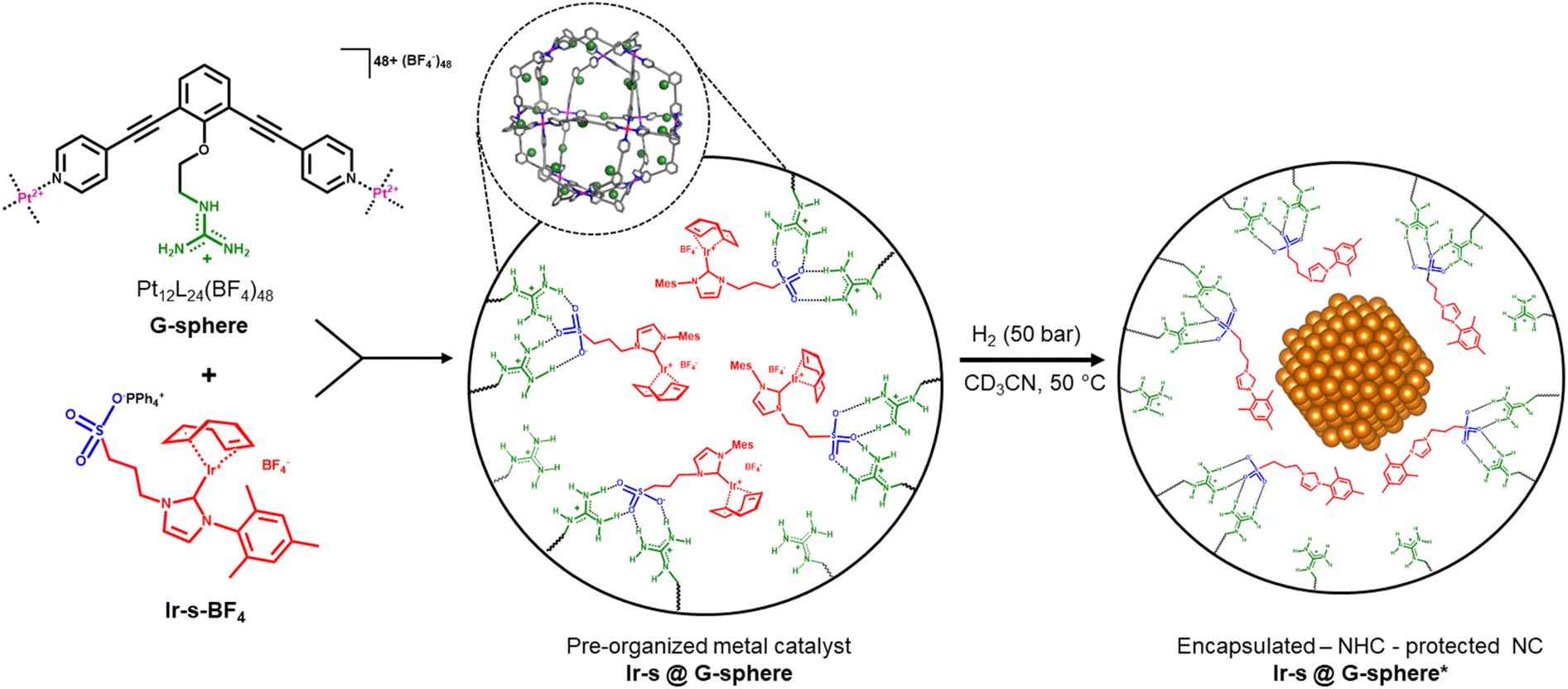 | ||
| Fig. 1 Schematic representation of the supramolecular pre-organization strategy for controlled nanocluster formation. | ||
Motivated by the challenges involved in nanocluster synthesis and based on our previous results, we were curious if we could use these nanospheres as templates for confined nanocluster synthesis. It was previously demonstrated that porous microenvironments such as dendrimers,12,13 COFs14–17 and POCs18–32 can limit the growth of nanoparticles prepared from metal salts through steric confinement and by using coordinating functional groups such as amines or sulfides to preorganize metal salts for the initiation of nanoparticle formation. The pre-organization of organometallic complexes in confined space has not been demonstrated and subsequent reduction should lead to the controlled formation of encapsulated nanoclusters that reside in a unique microenvironment in which the ligand of the metal complex precursor may still be present. We herein report the supramolecular pre-organization and reduction of a sulfonate functionalized Ir–NHC complex (Ir-s-BF4) within a guanidinium functionalized M12L24 sphere (G-sphere, Fig. 1). The hydrogenation of encapsulated Ir metal complexes resulted in ultrafine encapsulated nanoclusters (1.8 ± 0.4 nm), which display excellent selectivity in the hydrogenation of 4-nitrostyrene (1).
Results and discussion
Strategy/design of system
The strategy is illustrated in Fig. 1. To start, 24 sulfonated metal complexes (Ir-s) should be bound and pre-organized in a well-defined M12L24 nanosphere as the sulfonate groups form hydrogen bonds with the 24 guanidinium binding sites located at the inside of the sphere (G-sphere). Then, upon subsequent reduction by hydrogenation, these pre-organized metal complexes should lead to the templated formation of nanoclusters. The metal precursor (Ir-s-BF4) was chosen based on three specific requirements: the metal complex should be (1) stable and remain strongly bound to the sulfonate-bearing ligand during encapsulation, (2) readily and cleanly be converted upon reduction by hydrogenation and (3) function as a stabilizing ligand for the resulting nanocluster to retain the cluster within the nanosphere. We anticipated that the N-heterocyclic carbene-based (NHC) metal complex Ir-s-BF4 would embody these requirements.57–60Synthesis of the host (G-sphere) and guest (Ir-s)
The guanidinium functionalized building block (GuaniBB) and corresponding coordination sphere (G-sphere) were prepared via a modified literature procedure (pages S15–S25†).54,61 The formation of the M12L24 spheres (M = Pt2+ or Pd2+) was confirmed with 1H-NMR, DOSY NMR and cold-spray-ionization time-of-flight mass spectrometry CSI-TOF-MS (Fig. S19–S29†), which correspond to previously reported values.54,55 The sulfonate functionalized iridium complex (Ir-s-Cl) was synthesized via a modified literature procedure through transmetallation of the corresponding silver carbene (Ag-s, Scheme 1).62 The chloride counter ion of the Ir-s-Cl complex was exchanged with BF4−, to prevent possible compatibility issues of the chloride ions with the Pt12L24 coordination sphere (Ir-s-BF4, Scheme 1). The sulfonate functionalized metal complexes (Ag-s, Ir-s-Cl and Ir-s-BF4) were characterized by 1H-NMR, 13C-NMR and CSI-TOF-MS (pages S3–S14†).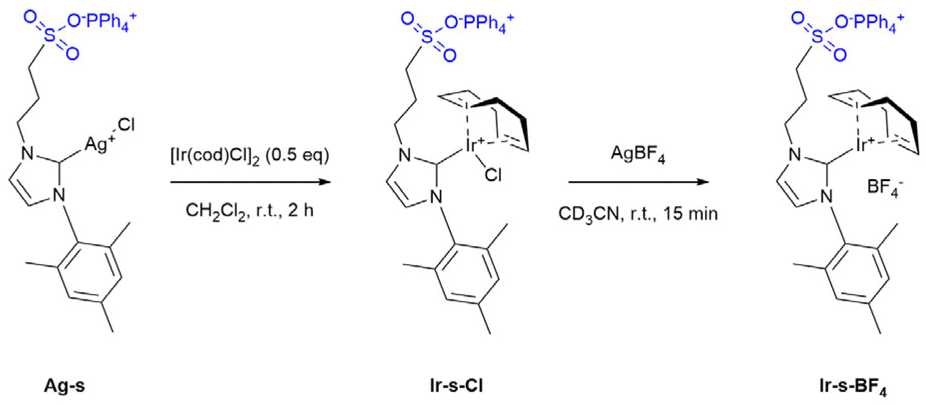 | ||
| Scheme 1 Synthesis of Ir-s-BF4. | ||
Encapsulation of the metal complex
Endohedral binding of Ir-s-BF4 inside the nanosphere (G-sphere) was established by CSI-TOF-MS analysis and various 1H-NMR experiments, which will be discussed in more detail in this section. Direct evidence for the binding of Ir-s-BF4 was provided by CSI-TOF-MS spectra of a solution of nanosphere in the presence of 8 equivalents of Ir-s-BF4, which display multiple charged species that can be assigned to the nanosphere hosting different numbers of Ir-s complexes, ranging from 5–9 equivalents of Ir-s-BF4 per nanosphere (Fig. S44, S45 and Table S2†). It should be noted that under the MS conditions, water coordination and hydrolysis of the carbene ligand to its imidazolium salt is observed, resulting in a variety of encapsulated metal complex/imidazolium salt/H2O combinations (Fig. S46–S49 and Table S3†). As a result, the signals in the MS spectra are substantially broadened obscuring precise interpretation. Consequently, as the number of variations increases with the number of Ir-s complexes bound in the nanosphere, the signals become too broad for detection or meaningful interpretation for mixtures containing more than 9 equivalents of complex with respect to the sphere.Encapsulation was further confirmed by DOSY-NMR (Fig. S36–S39†) of a solution containing 8, 12 or 24 eq. of Ir-s-BF4 complex with respect to the G-sphere, as in such experiment the complex (Ir-s-BF4) has signals at the same diffusion as that of the host, in line with encapsulation (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D = 9.51 m2 s−1 for 8 eq.). In contrast, the free complex in absence of the nanosphere displays a significantly lower diffusion coefficient (LogD = 8.93 m2 s−1). It should be noted that at full capacity of the host, i.e. 24 equivalents of Ir-s-BF4 with respect to the sphere (Fig. S32 and S34†), there is a subtle increase in the diffusion coefficient for some of the signals of the sphere. This suggests that the hydrodynamic radius of the sphere slightly increases as a result of the crowded interior of the host at full capacity.
D = 9.51 m2 s−1 for 8 eq.). In contrast, the free complex in absence of the nanosphere displays a significantly lower diffusion coefficient (LogD = 8.93 m2 s−1). It should be noted that at full capacity of the host, i.e. 24 equivalents of Ir-s-BF4 with respect to the sphere (Fig. S32 and S34†), there is a subtle increase in the diffusion coefficient for some of the signals of the sphere. This suggests that the hydrodynamic radius of the sphere slightly increases as a result of the crowded interior of the host at full capacity.
To gain more insight into the encapsulation capacity of the host, the binding behaviour was investigated by a titration experiment monitored by 1H-NMR spectroscopy (pages S30 and S31†). In the resulting spectra, the N–H-signal of the endohedral guanidinium functionality (G-sphere) most significantly shifts upon introduction of the guest (Ir-s-BF44), whereas the other signals of the sphere display modest to no changes. This result indicates that binding of the guest indeed primarily occurs inside the sphere. A linear correlation between the N–H-signal-shift (G-sphere) and guest (Ir-s-BF4) concentration was observed up to 18 equivalents of guest. Addition of more equivalents resulted in overlap of peaks, complicating the monitoring of the N–H signal by 1H-NMR spectroscopy (Fig. S42†). This indicates that at least 18 equivalents of Ir-s bind to the inside of the G-sphere, while they are still able to exchange. Importantly, these experiments combined clearly illustrate the host–guest complexation and anticipated encapsulation capacity of this system.
As a control, we synthesized variations of the nanosphere and metal complex for which the binding element on each side was removed (M-sphere and Ir-pFig. 3). Naturally, the lack of encapsulation for both combinations was confirmed by DOSY-NMR and 1H-NMR experiments (Fig. S40 and S41†).
Nanocluster formation
To demonstrate that nanoclusters could be prepared within the nanospheres, reduction of the iridium complexes was performed in the presence and absence of nanospheres. The clusters were prepared by hydrogenation of a 24:1 mixture of Ir-s-BF4 and G-sphere to ensure full loading, (1 ml, 8.7 mM, Ir-s@G-sphere), and the CD3CN solution was kept at 50 bar H2 and 50 °C for 18 h to allow full reduction. After 18 h, the solution had changed colour from light orange to amber brown and no precipitation was formed, indicating that all iridium was still in solution and no agglomeration had occurred (Fig. 2). The observed colour corresponds to observations reported in literature for small Ir(0) nanoclusters.63 In contrast, when the Ir-s-BF4 complex was hydrogenated in absence of the nanosphere, under otherwise identical conditions, precipitates were formed and a dark film was visible on the reaction vessel (Fig. S50†).
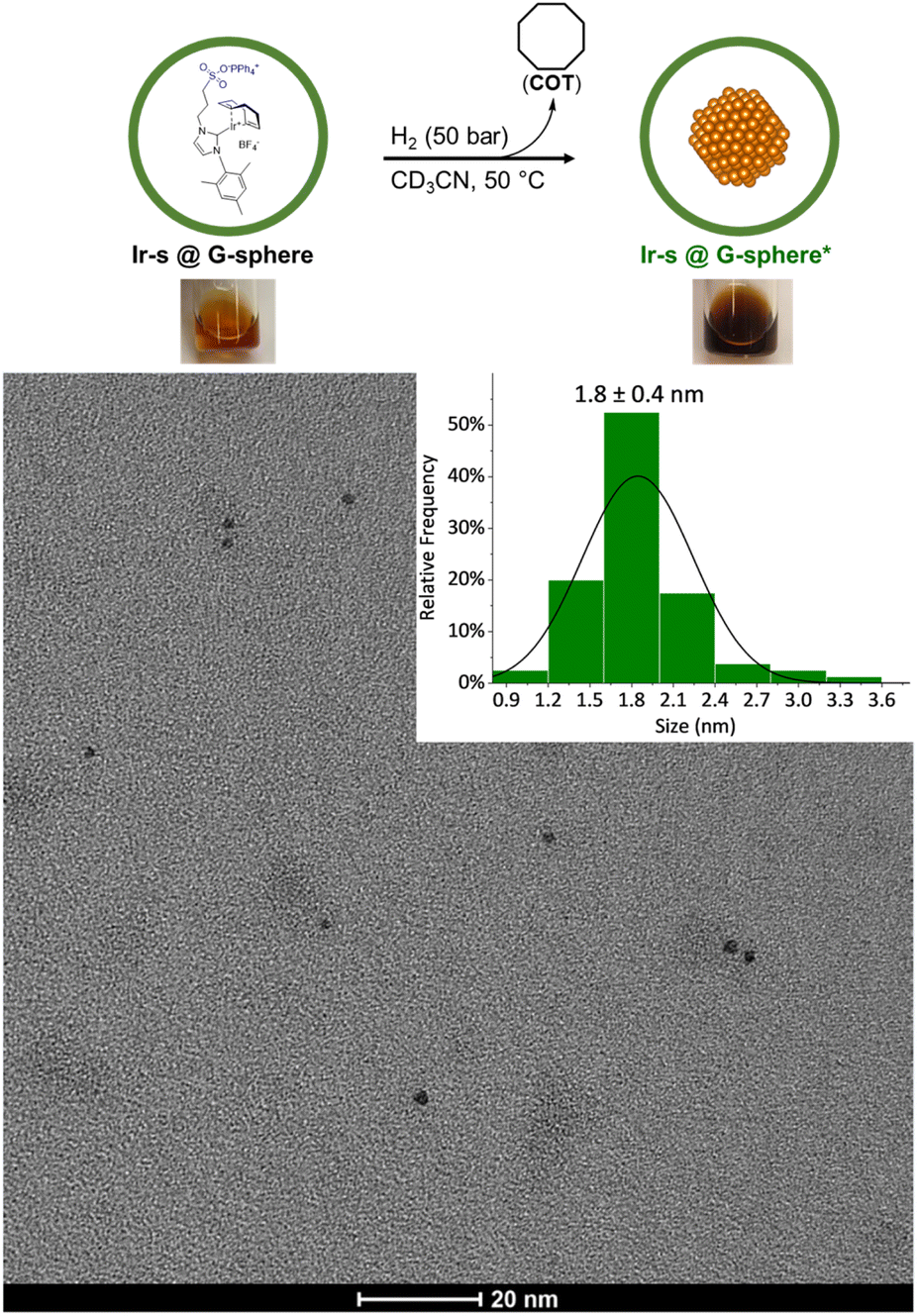 | ||
| Fig. 2 Top: nanocluster synthesis conditions and representative images of the solutions of Ir-s@G-sphere before and after hydrogenation (Ir-s@G-sphere*) respectively. Bottom: representative TEM image of Ir-s@G-sphere* and corresponding size distribution (in diameter). | ||
The dimensions of the Ir clusters formed inside the sphere, were characterized by transmission electron microscopy (TEM). The clusters were determined to be 1.8 ± 0.4 nm in diameter (Fig. 2), which is much smaller than the size of the nanosphere (5 nm).64,65 However, for nanoparticles consisting of 24 Ir-atoms, a particle size of 0.9 nm would be expected. As the metal complexes are pre-organized in a supramolecular fashion, the Ir-s-BF4 complexes can exchange between nanocages, and as this apparently is faster than nanoparticle formation, the final number of Ir-atoms per particle can be larger. Importantly, these results altogether indicate that pre-organization of the Ir-s-BF4 within the G-sphere results in controlled nanocluster formation (Ir-s@G-sphere*).
To compare the effects of sphere interaction and encapsulation on cluster size and distribution, the non-encapsulated metal complexes (Ir-s + M-sphere and Ir-p + G-sphere, Fig. 3) were hydrogenated under otherwise identical conditions. From TEM analysis (Fig. S51 and S52†) it becomes evident that the resulting particle size and distribution for these systems (Fig. 3) are generally larger and significantly broader than the encapsulated clusters (2.3 ± 2.1 nm and 6.6 ± 3.2 nm for Ir-s + M-sphere and Ir-p + G-sphere respectively). The non-encapsulated sulfonate functionalized metal complex (Ir-s + M-sphere) displays a smaller particle size than the non-sulfonated metal complex (Ir-p + G-sphere). Importantly, the superior control over the size and size distribution of the encapsulated clusters (Ir-s@G-sphere) clearly demonstrates the power of pre-organization.
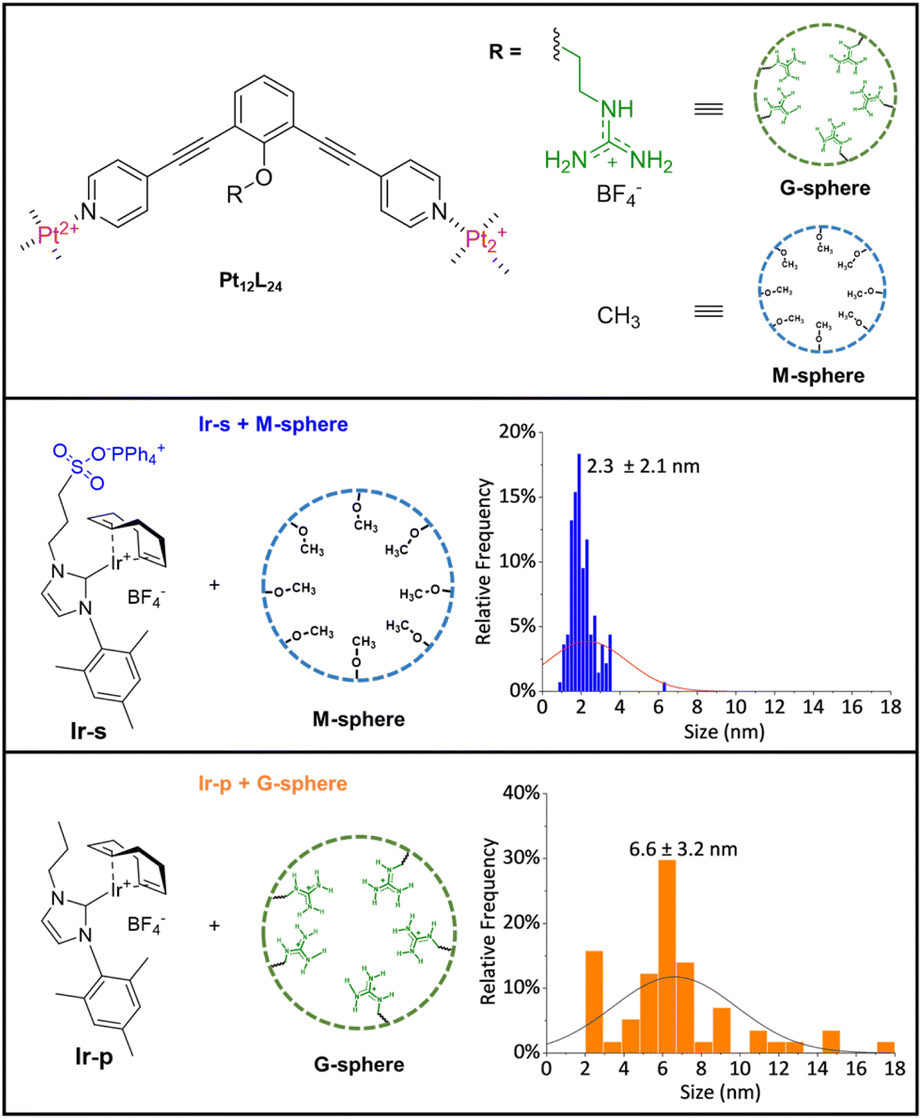 | ||
| Fig. 3 Schematic representations of control groups and resulting particle size distribution (diameter). Top: schematic representation of G-sphere and M-sphere, middle and bottom: Ir-s + M-sphere and Ir-p + G-sphere and corresponding size distribution (in diameter). | ||
To gain further insight into the conversion of the metal precursor and the spatial relationship between the sphere and the encapsulated Ir cluster, the resulting mixture was studied by 1H-NMR and DOSY-NMR. The 1H-NMR spectrum of Ir-s@G-sphere* (Fig. S54†) shows the disappearance of the Ir-s ligand signals and the formation of its corresponding imidazolium salt, which is expected to form upon nanocluster formation. In addition, the formation of cyclooctane (COT at 1.54 ppm) is observed, which is a result of Ir-catalysed hydrogenation of the cyclooctadiene (COD) ligand. Integration of this signal, compared to an internal standard (1,3,5-trimethoxybenzene), confirmed full conversion (Table S4†). Notably, the signals of the nanosphere have become broadened after hydrogenation, which is commonly a result of local restricted motion and the heterogeneous character of the encapsulated Ir clusters.66 Importantly, the DOSY-NMR spectrum (Fig. 4f) shows that the diffusion coefficient of the encapsulated clusters (Ir-s@G-sphere*, 9.49 m2 s−1) remains similar to that of the host–guest system before hydrogenation (Ir-s@G-sphere, 9.48 m2 s−1). These results combined confirm that the nanospheres remain of similar dimensions after nanocluster formation and indicate that the clusters are residing inside the spheres.
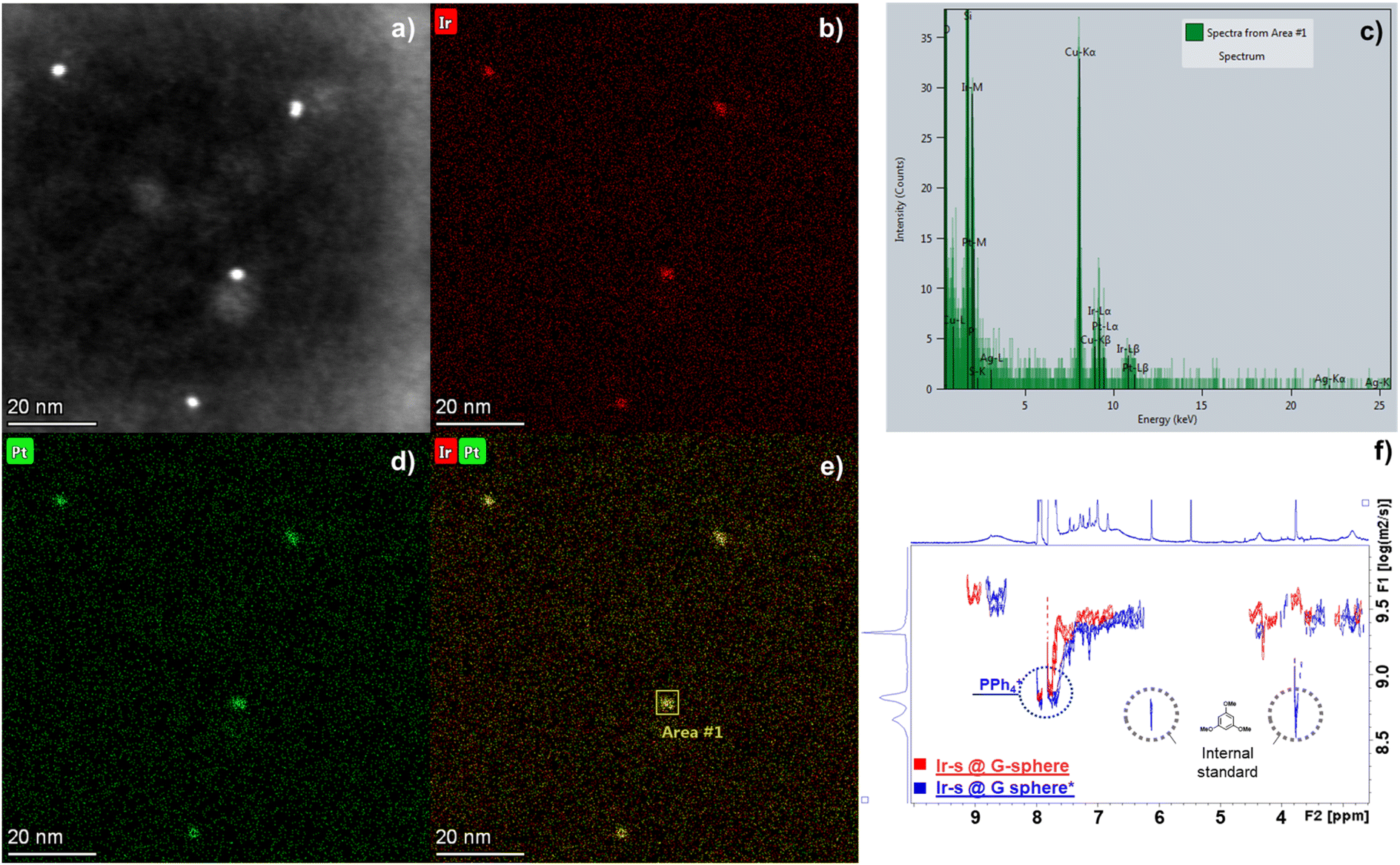 | ||
| Fig. 4 (a) HAADF-STEM image of Ir-s@G-sphere* and corresponding EDX maps of (b) Ir, (d) Pt and (e) an overlay of Ir and Pt, (c) EDX pattern of the selected area in (e), (f) 1H-DOSY-NMR overlay of Ir-s@G-sphere/before (red) and Ir-s@G-sphere*/after hydrogenation. | ||
The formation of an encapsulated cluster was further evidenced by an energy-dispersive X-ray spectroscopy (EDX) study in combination with high-angle annular dark-field scanning transmission microscopy (HAADF-STEM) imaging (Fig. 4). The images reveal that Ir and Pt are clearly located together, affirming the Ir clusters are indeed encapsulated within the sphere. Moreover, quantification of the Pt and Ir atom fraction through area scanning (Fig. 4c and e) confirmed the 12:24 ratio that is expected from the nanosphere – guest system and corresponds to values found with Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) analysis (Table S5†). To further confirm the integrity of the sphere and the formation of metallic Ir nanoclusters in the sphere, the encapsulated systems were studied by XPS analysis before and after hydrogenation conditions that lead to nanoparticle formation (Fig. S59†). The resulting spectra after hydrogenation (Ir-s@G-sphere*) display two characteristic signals centred around 61.3 eV (Ir 4f7/2) and 64.2 eV (Ir 4f5/2). These signals were determined to be composed of Ir0 clusters that are partially oxidized (47% Ir0), confirming the formation of metallic Ir0 species under hydrogenation conditions.67–70 Additionally, the doublets centred around 73.8 eV and 76.8 eV, corresponding to Pt 4f for the PtII cornerstones of the sphere,71 remain unchanged after hydrogenation (Fig. S59†). These results confirm that Pt atoms remain unreduced and further support that the sphere remains intact. Overall, these results, combined with the STEM-EDX and DOSY-NMR analyses, clearly demonstrate that the formed Ir0 nanoclusters are indeed residing within the guanidine containing Pt12L24 nanospheres.
Catalysis
To evaluate the catalytic activity of the newly formed encapsulated nanoclusters, the hydrogenation of 4-nitrostyrene (1) was explored by monitoring the reduction reaction over time (Fig. 5). This reaction was chosen because it can provide insight into both activity and selectivity. For these experiments, the encapsulated and non-encapsulated clusters/NPs (Ir-s@G-sphere* and Ir-s + M-sphere* respectively) were compared and used as prepared. The hydrogenation reactions were carried out in CH3CN under hydrogen pressure (50 bar) at 50 °C, using 2 mol% of Ir vs.1. The product composition was monitored over time by means of GC-MS with an internal standard (Fig. 5). From the observed product compositions, it became evident that both catalytic systems display a preference for alkene reduction, thus favoring the initially selective formation of 4-ethylnitrobenzene (2). Notably, the product compositions over time reveal that the Ir-s@G-sphere* clusters provide fully selective conversion toward 4-ethylnitrobenzene (2) over 17 h (Fig. 5c). In contrast, in the presence of nanoparticles formed by the mixture of Ir-s + M-sphere*, 1 is rapidly converted to 2 and further reduced to form 4-ethylaniline (3) (Fig. 5b). This result highlights a striking difference in catalytic behavior between the encapsulated (Ir-s@G-sphere*) and non-encapsulated particles (Ir-s + M-sphere*). Importantly, in the presence of Ir-s + M-sphere* over-reduction of 2 to 3 already commences before full conversion is attained (@30 min 66% conversion with 1% of 3), thus invariably leading to an impure product mixture and a maximum product yield of 87% after two hours (Fig. 5a). On the contrary, in the presence of Ir-s@G-sphere* complete conversion to 2 occurs entirely selectively without any observable side product formation even at full conversions. Thus, although Ir-s + M-sphere* may be a faster catalyst, Ir-s@G-sphere* eventually provides significantly higher attainable yields. To the best of our knowledge, this is the first iridium nanocatalyst reported to selectively hydrogenate nitrostyrene to ethylnitrobenzene.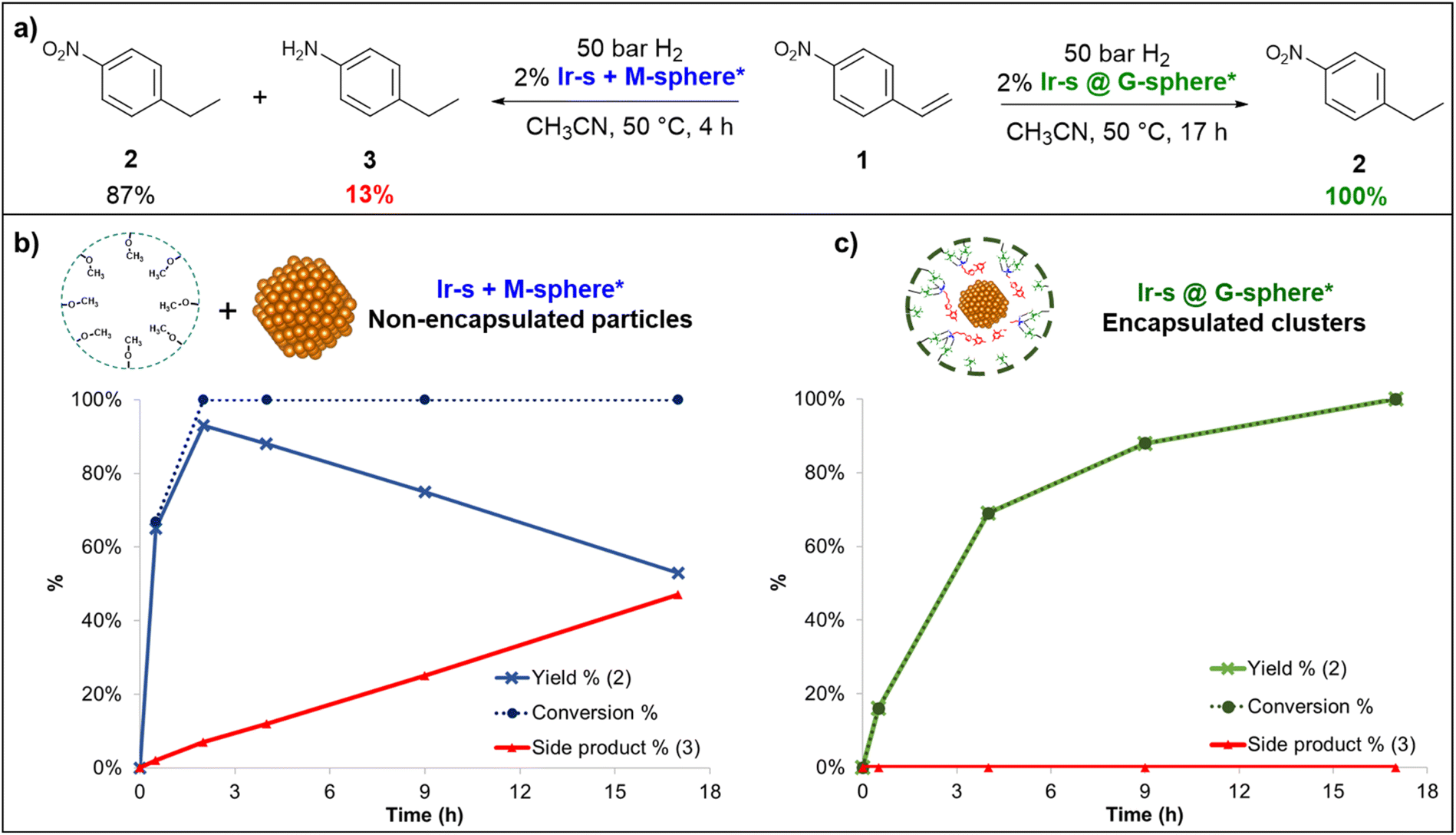 | ||
| Fig. 5 (a) Hydrogenation of 4-nitrostyrene (1): product composition at full conversion in the presence of Ir-s + M-sphere* (left) and Ir-s@G-sphere* (right), (b) hydrogenation of 1 over time in the presence of Ir-s + M-sphere* and (c) in the presence of Ir-s@G-sphere*. ([1] = 60 mM in CH3CN, 2% Ir vs.1). | ||
To compare the selectivity of Ir-s@G-sphere* to nanoclusters with a similar size and size-distribution in a different microenvironment, polyvinylpyrrolidone (PVP) supported Ir-nanoclusters were prepared (Ir@PVP) according to a literature protocol.72 Hydrogenation of 1 under H2 (40 bar) at 50 °C in the presence of Ir@PVP (2 mol%, based on Ir) afforded a mixture of the fully hydrogenated product 3 (4-amino ethylbenzene, 44%) and 4-vinylaniline (<5%). This is in large contrast to the results obtained for Ir-s@G-sphere* which resulted in complete and selective conversion of 1 to 2 (4-nitro ethylbenzene), whereas 2 was not observed at all by 1H-NMR nor GC-MS (Fig. S58†) when Ir@PVP was used at catalyst. This demonstrates that the unique second coordination sphere around the Ir-nanocluster formed by the cage is crucial for the selectivity displayed by the enclosed nanoparticle. To gain more insight into the role of the guanidinium groups of the G-sphere during the catalytic reaction, styrene and 3-vinylbenzoic acid were hydrogenated with Ir-s@G-sphere*. We anticipated that the guanidinium sites should still be hydrogen bonded to the sulfonate groups from the Ir-s ligand. If some of the guanidinium groups would be available for H-bonding, the substrates with carboxylate groups in principle could be pre-organized in close proximity to the nanocluster, and hence should be hydrogenated faster, in line with other previously reported catalytic transformations within the guanidinium functionalized nanosphere.54 Although both substrates undergo selective hydrogenation to their ethylbenzene derivatives, there is no rate acceleration observed for the carboxylate functionalized substrate (Fig. S56†). Apparently, the guanidinium sites are sufficiently engaged in hydrogen bonding to the sulfonate functionalized ligand and thus unavailable for binding additional substrates. Overall, these experiments show that the Ir-s@G-sphere* system contains an iridium cluster with a defined size and a unique second coordination sphere, altogether resulting in a selective catalyst for the hydrogenation of nitrostyrene to ethylnitrobenzene.
Conclusions
To summarize, we have demonstrated supramolecular pre-organization in nanospheres as a effective strategy for controlled nanocluster synthesis. We have shown that hydrogenation of the organo-metallic Ir-s-BF4 complex encapsulated inside a Pt12L24 nano-sphere (G-sphere) led to the formation of fine nanoclusters with a narrow size distribution, whereas non-encapsulated precursors did not. These results together demonstrate that pre-organization within the nanosphere can indeed facilitate the templated formation of ultrafine metal nanoclusters. Moreover, we were able to establish that the nanocluster remains encapsulated inside the nanosphere upon formation, underlining that the cluster is indeed formed and stabilized within the sphere. The encapsulated clusters (Ir-s@G-sphere*) displayed outstanding selectivity in the hydrogenation of 4-nitrostyrene (1). We envision this new system can provide a unique nano environment for nanocluster formation and catalytic conversions, which can be extended to other metals and combinations thereof. Furthermore, we conceive that this preorganization strategy can provide flexibility for subtle ligand modifications and promises to be a versatile tool for controlled synthesis and fine-tuning of small nanoclusters.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: LLM, EOB, JNHR; formal analysis: LLM, RH, EOB, KJHB RCJP, VRD; funding acquisition: JNHR; investigation: LLM, RH; supervision: JNHR, AvB, EJMH; validation: LLM, RH, JNHR; visualization: LLM; writing – original draft: LLM; writing – review & editing: LLM, RH, JNHR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC (project 2018.016.C.UvA.5), which is cofounded and cofinanced by The Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and The Netherlands Ministry of Economic Affairs and Climate. Ed Zuidinga is acknowledged for his assistance with HR-MS analysis. HAAFD-STEM and EDX measurements were made possible in collaboration with the Electron Microscopy Centre of the University of Utrecht. Michel van Son is acknowledged for his assistance with the ICP-OES measurements.Notes and references
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- C. Busche, L. Vilà-Nadal, J. Yan, H. N. Miras, D.-L. Long, V. P. Georgiev, A. Asenov, R. H. Pedersen, N. Gadegaard, M. M. Mirza, D. J. Paul, J. M. Poblet and L. Cronin, Nature, 2014, 515, 545–549 CrossRef CAS PubMed.
- L. Y. Chen, C. W. Wang, Z. Yuan and H. T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS.
- P. Chakraborty, A. Nag, A. Chakraborty and T. Pradeep, Acc. Chem. Res., 2019, 52, 2–11 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Front. Chem., 2014, 2, 63 Search PubMed.
- T. Kawawaki, Y. Negishi and H. Kawasaki, Nanoscale Adv., 2020, 2, 17–36 RSC.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- S. R. K. Perala and S. Kumar, Langmuir, 2013, 29, 9863–9873 CrossRef CAS PubMed.
- V. T. Liveri in Controlled synthesis of nanoparticles in microheterogeneous systems, Springer, New York, NY, United states, 1st edn, 2006, pp. 75–91 Search PubMed.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef CAS PubMed.
- V. S. Myers, M. G. Weir, E. V. Carino, D. F. Yancey, S. Pande and R. M. Crooks, Chem. Sci., 2011, 2, 1632–1646 RSC.
- P. Pachfule, S. Kandambeth, D. Díaz Díaz and R. Banerjee, Chem. Commun., 2014, 50, 3169–3172 RSC.
- M. Bhadra, H. S. Sasmal, A. Basu, S. P. Midya, S. Kandambeth, P. Pachfule, E. Balaraman and R. Banerjee, ACS Appl. Mater. Interfaces, 2017, 9, 13785–13792 CrossRef CAS PubMed.
- J. Wang, Y. Yu, H. Yu, W. Wang, L.-L. Shen, G.-R. Zhang and D. Mei, ACS Catal., 2023, 13, 5135–5146 CrossRef CAS.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS.
- S. Bai, Y. Y. Wang and Y. F. Han, Chin. J. Chem., 2019, 37, 1289–1290 CrossRef CAS.
- G. J. Chen, W. L. Xin, J. S. Wang, J. Y. Cheng and Y. B. Dong, Chem. Commun., 2019, 55, 3586–3589 RSC.
- X. Yang, J. K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- J. K. Sun, W. W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS.
- B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- B. Mondal, P. Bhandari and P. S. Mukherjee, Chem. - Eur. J., 2020, 26, 15007–15015 CrossRef CAS PubMed.
- S. Y. Zhang, Z. Kochovski, H. C. Lee, Y. Lu, H. Zhang, J. Zhang, J. K. Sun and J. Yuan, Chem. Sci., 2019, 10, 1450–1456 RSC.
- V. Sharma, D. De, R. Saha, P. K. Chattaraj and P. K. Bharadwaj, ACS Appl. Mater. Interfaces, 2020, 12, 8539–8546 CrossRef CAS.
- A. Verma, K. Tomar and P. K. Bharadwaj, Inorg. Chem., 2019, 58, 1003–1006 CrossRef CAS PubMed.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- E. H. Peters and M. Mayor, Chem. Commun., 2023, 59, 4895–4898 RSC.
- Y. Ma, X. Kuang, X. Deng, B. Zi, J. Zeng, J. Zhang, Z. Zhu, Y. Zhang and Q. Liu, Microporous Mesoporous Mater., 2022, 335, 111701 CrossRef CAS.
- T. Liu, S. Bai, L. Zhang, F. E. Hahn and Y.-F. Han, Natl. Sci. Rev., 2022, 9, nwac067 CrossRef CAS PubMed.
- S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, W. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef CAS PubMed.
- S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, X. Li, W. Liao and W. Chen, J. Am. Chem. Soc., 2018, 140, 6271–6277 CrossRef CAS PubMed.
- S. L. Hou, J. Dong, Z. H. Zhu, L. C. Geng, Y. Ma and B. Zhao, Chem. Mater., 2020, 32, 7063–7069 CrossRef CAS.
- Y. Fang, Z. Xiao, J. Li, C. Lollar, L. Liu, X. Lian, S. Yuan, S. Banerjee, P. Zhang and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5283–5287 CrossRef CAS PubMed.
- Y. Fang, J. L. Li, T. Togo, F. Y. Jin, Z. F. Xiao, L. J. Liu, H. Drake, X. Z. Lian and H. C. Zhou, Chem, 2018, 4, 555–563 CAS.
- X. Hang, S. Wang, H. Pang and Q. Xu, Chem. Sci., 2022, 13, 461–468 RSC.
- K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS PubMed.
- K. Suzuki, K. Takao, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2011, 50, 4858–4861 CrossRef CAS PubMed.
- T. Ichijo, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 6786–6789 CrossRef CAS PubMed.
- E. Ubasart, I. Mustieles Marin, J. M. Asensio, G. Mencia, Á. M. López-Vinasco, C. García-Simón, I. del Rosal, R. Poteau, B. Chaudret and X. Ribas, Nanoscale Horiz., 2022, 7, 607–615 RSC.
- W. L. Jiang, J. C. Shen, Z. Peng, G. Y. Wu, G. Q. Yin, X. Shi and H. B. Yang, J. Mater. Chem. A, 2020, 8, 12097–12105 RSC.
- C. Wang, F. Sun, G. He, H. Zhao, L. Tian, Y. Cheng and G. Li, Curr. Opin. Colloid Interface Sci., 2023, 63, 101660 CrossRef CAS.
- J.-Y. Li, X.-D. Yang, F.-X. Chen and J.-K. Sun, Mater. Chem. Front., 2023, 7, 5355–5376 RSC.
- L.-M. Cao, J. Zhang, X.-F. Zhang and C.-T. He, Chem. Sci., 2022, 13, 1569–1593 RSC.
- X. Yang and Q. Xu, Trends Chem., 2020, 2, 214–226 CrossRef CAS.
- S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS PubMed.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- K. Harris, Q.-F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS.
- M. Tominaga, K. Suzuki, M. Kawano, T. Kusukawa, T. Ozeki, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5621–5625 CrossRef CAS.
- J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chemie, 2021, 133, 5489–5495 CrossRef.
- X. Yan, P. Wei, Y. Liu, M. Wang, C. Chen, J. Zhao, G. Li, M. L. Saha, Z. Zhou, Z. An, X. Li and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 9673–9679 CrossRef CAS.
- Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanovic-Burmazovic and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- F. Yu, D. Poole, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS PubMed.
- S. Gonell and J. N. H. Reek, ChemCatChem, 2019, 11, 1458–1464 CrossRef CAS PubMed.
- B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441–8449 CrossRef CAS.
- W. Oberhauser, C. Evangelisti, A. Liscio, A. Kovtun, Y. Cao and F. Vizza, J. Catal., 2018, 368, 298–305 CrossRef CAS.
- C. Cerezo-Navarrete, P. Lara and L. M. Martínez-Prieto, Catalysts, 2020, 10, 1144 CrossRef CAS.
- M. Ghosh and S. Khan, ACS Catal., 2023, 13, 9313–9325 CrossRef CAS.
- R. Zaffaroni, E. O. Bobylev, R. Plessius, J. I. Van Der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2020, 142, 8837–8847 CrossRef.
- M. A. N. Virboul, Sulfonate Functionalisation of Transition Metal Complexes: A Versatile Tool Towards Catalyst Recovery, PhD thesis, Utrecht university, Utrecht, 2011, https://dspace.library.uu.nl/handle/1874/203778, accessed 2023-06-01.
- Y. Lin and R. G. Finke, J. Am. Chem. Soc., 1994, 116, 8335–8353 CrossRef CAS.
- S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS PubMed.
- C. Gütz, R. Hovorka, C. Klein, Q. Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef PubMed.
- X. X. Gou, T. Liu, Y. Y. Wang and Y. F. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS.
- W. Oberhauser, C. Evangelisti, A. Liscio, A. Kovtun, Y. Cao and F. Vizza, J. Catal., 2018, 368, 298–305 CrossRef CAS.
- H. Y. Hall and P. M. A. Sherwood, J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases, 1984, 80, 135–152 CAS.
- C. Crotti, E. Farnetti, S. Filipuzzi, M. Stener, E. Zangrando and P. Moras, Dalton Trans., 2007, 133–142 RSC.
- T. Higaki, H. Kitazawa, S. Yamazoe and T. Tsukuda, Nanoscale, 2016, 8, 11371–11374 RSC.
- P. Brant, L. S. Benner and A. L. Balch, Inorg. Chem., 1979, 18, 3422–3427 CrossRef CAS.
- M. J. Sharif, P. Maity, S. Yamazoe and T. Tsukuda, Chem. Lett., 2013, 42, 1023–1025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, preparation and characterization of the catalyst. See DOI: https://doi.org/10.1039/d4sc06324d |
| This journal is © The Royal Society of Chemistry 2024 |