DOI:
10.1039/D4SC05375C
(Edge Article)
Chem. Sci., 2024,
15, 20013-20021
Pd(II)-catalyzed enantioselective C–H olefination and photoregulation of sterically hindered diarylethenes†
Received
10th August 2024
, Accepted 1st November 2024
First published on 7th November 2024
Abstract
Sterically hindered diarylethenes with intrinsic chirality have shown great potential in chiral signal regulation, light-controlled liquid crystals (LCs), etc. Their unique enantiospecific phototransformation between axial chirality of ring-open isomers and central chirality of ring-closed isomers can break through the bottleneck of interference between multiple chiral centers in traditional chiral diarylethenes. However, these intrinsic chiral diarylethenes require necessary chiral resolution through preparative chiral HPLC, typically resulting in limited separation efficiency and production scale. Here, we present an enantioselective olefination strategy to directly construct intrinsic chiral diarylethenes from a prochiral sterically hindered diarylethene, achieving high yields and enantioselectivity. The resulting isomers can be further decorated by incorporating mesogenic units, and the derivatives enable the successful reversible photoregulation of blue, green, and red reflection colors of LCs with excellent thermal stability, fatigue resistance, and little texture disorderliness, demonstrating the practical application potential of direct enantioselective olefination in photoregulation with intrinsic chiral diarylethenes.
Introduction
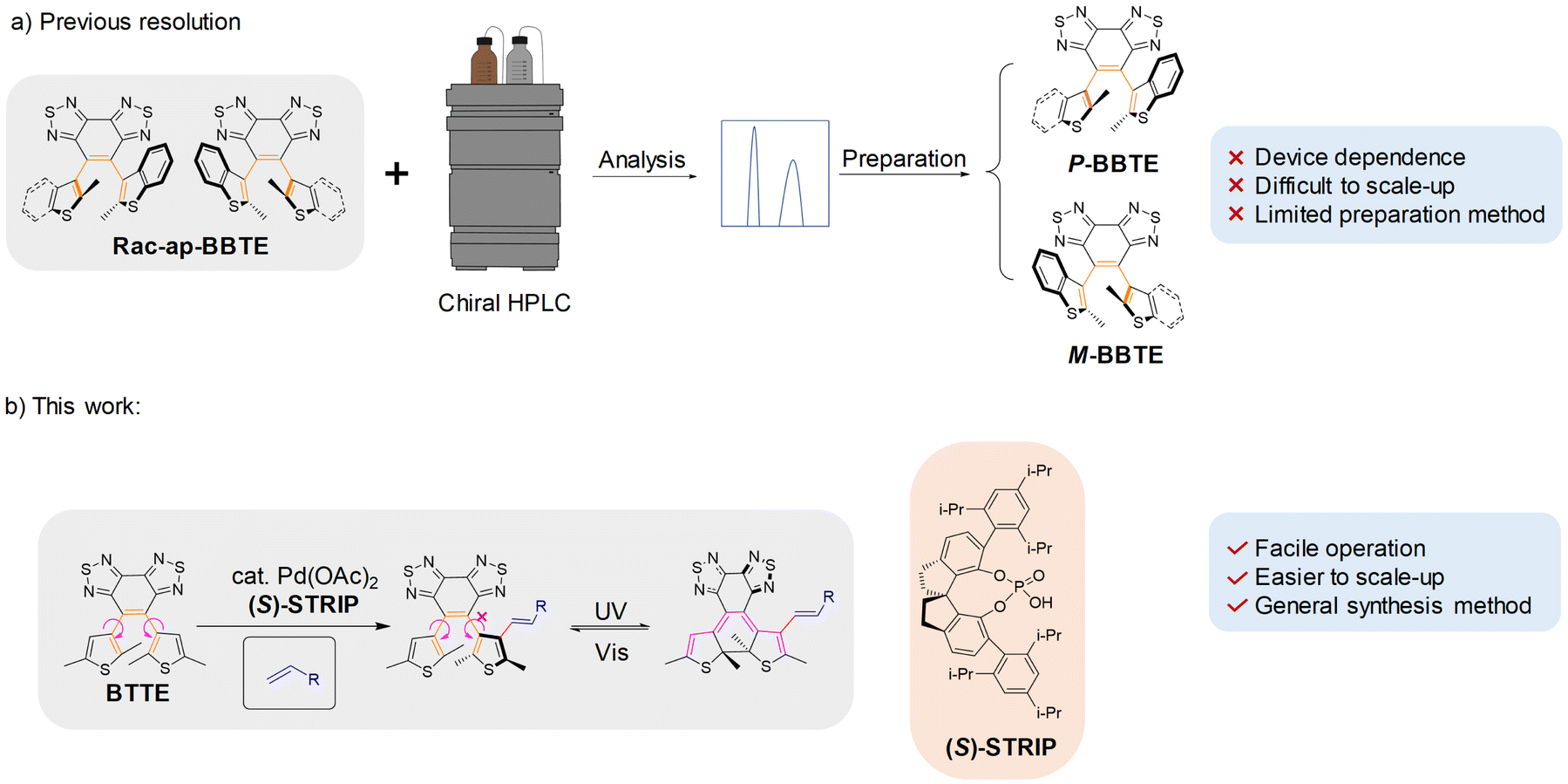
Diarylethene (DAE) is a kind of photoswitch that exhibits remarkable advantages including high thermal stability,1 rapid response,2 robust fatigue resistance,3etc., attracting increasing attention in data storage,4 light-controlled self-assembly,5 super-resolution imaging,6 and so on. Besides, chiral diarylethenes emerge as a compelling choice for chiral signal regulation,7 light-controlled liquid crystal modulation,8etc. Traditionally, due to the small steric hindrance of ethene bridges, most diarylethenes exhibit racemic behavior resulting from the rapid rotation of side aryl groups, and extrinsic chiral groups are inevitable to induce a chiral preference for these diarylethenes, which will decrease the efficiency of chiral photoregulation because of the multiple chiral centers.9 In recent years, we have developed an intrinsic chiral diarylethene by incorporating sterically hindered group benzobisthiadiazole as an ethene bridge and large bulky benzothiophene as side aryl groups. Complete inhibition of racemization and successful isolation of intrinsic chiral enantiomers were achieved.10 The unique enantiospecific phototransformation between the axial chirality of the ring-open isomer and the central chirality of the ring-closed isomer can effectively avoid interference between multiple chiral centers in traditional chiral diarylethenes. However, this kind of diarylethene requires necessary chiral resolution through preparative chiral HPLC for application, typically accompanied by limited separation efficiency and production scale (Fig. 1a).
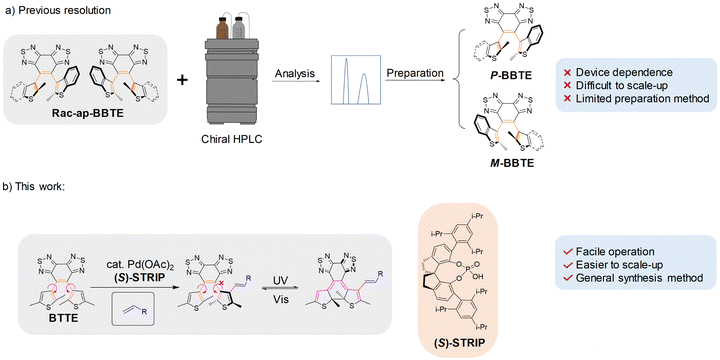 |
| | Fig. 1 Comparison between previous chiral resolution and direct enantioselective olefination. (a) Traditional chiral resolution process of the sterically hindered diarylethene 4,5-bis(2-methylbenzo[b]thien-3-yl)benzo[1,2-c:3,4-c′]bis([1,2,5]thiadiazole) (BBTE) by preparative chiral HPLC. (b) A novel strategy for the direct enantioselective olefination of sterically hindered diarylethenes with several advantages, such as facile operation, being easier to scale up, and a general synthesis method. Note: enantioselective olefination can increase the rotation barrier of side aryl groups of sterically hindered diarylethenes, resulting in the steady existence of their chiral enantiomers and achieving specific enantioselectivity at the same time, which can efficiently reduce the dependence on preparative chiral HPLC. | |
Asymmetric catalysis has been proven to be a promising strategy for efficiently synthesizing chiral molecules.11 Besides, enantioselective C–H activation has emerged as a powerful strategy to construct biaryl atropisomers12 in recent years, simply introducing chirality by one step. Motivated by this, we propose the asymmetrical synthesis of intrinsic chiral diarylethenes by enantioselective C–H activation, starting from prochiral diarylethene 4,5-bis(2,5-dimethylthien-3-yl)benzo[1,2-c:3,4-c′]bis([1,2,5]thiadiazole) (BTTE, 1a)13 with a sterically hindered ethene bridge (Fig. 1b). A series of sterically hindered intrinsic chiral diarylethenes have been constructed by enantioselective olefination with high yields and enantioselectivity, which can efficiently reduce the dependence on preparative chiral HPLC. Moreover, this strategy could achieve better accommodation and tolerance of the structures of diarylethenes than the original chiral resolution. The resulting intrinsic chiral diarylethenes are further modified by incorporating mesogenic units, and subsequently, we embed these derived diarylethenes into a LC to photoregulate its helical superstructure. Precise photoregulation of blue, green, and red reflection colors of the LC has been achieved with good thermal stability, fatigue resistance, and little texture disorderliness, illustrating the practical application potential of direct enantioselective olefination in photoregulation with intrinsic chiral diarylethenes.
Results and discussion
Direct enantioselective olefination of sterically hindered diarylethene BTTE
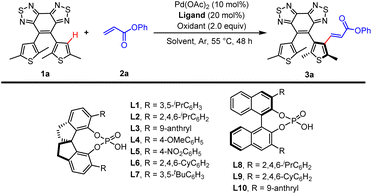
According to our previous research, a large sterically hindered ethene bridge and β-substituted thiophene group with large bulk can significantly increase the rotation barrier and completely inhibit the axis's rotation of diarylethenes.14 To further verify the impact of the benzothiadiazole ethene bridge, we calculated the rotation barrier of the parallel conformers of BTTE and a reference DAE (1,2-bis(2,5-dimethyl-3-thienyl)perfluorocyclopentene, 1b) using M06-2X functional with a def2tzvp basis set (Table S2†).15 The rotation barrier of BTTE (19.03 kcal mol−1) is much higher than that of 1b (5.10 kcal mol−1), demonstrating that the benzothiadiazole ethene bridge can introduce large steric hindrance to the axis rotation in DAE. Based on the discussions above, it is possible to introduce chirality into prochiral sterically hindered diarylethene BTTE by enantioselective substitution of β-proton. In addition, the α-methyl group of thiophene in BTTE can avoid other possible reacting sites. Additionally, previous studies have demonstrated that benzothiadiazole can serve as an effective directing group (DG) for realizing C–H activation.16 Thus, we believe that benzobisthiadiazole will have a similar ability. Hence, we decided to employ asymmetric C–H olefination, which has been proved to have good reactivity, enantioselectivity, and endurance of heteroatoms to introduce chirality into achiral BTTE.17
Optimizing reaction conditions
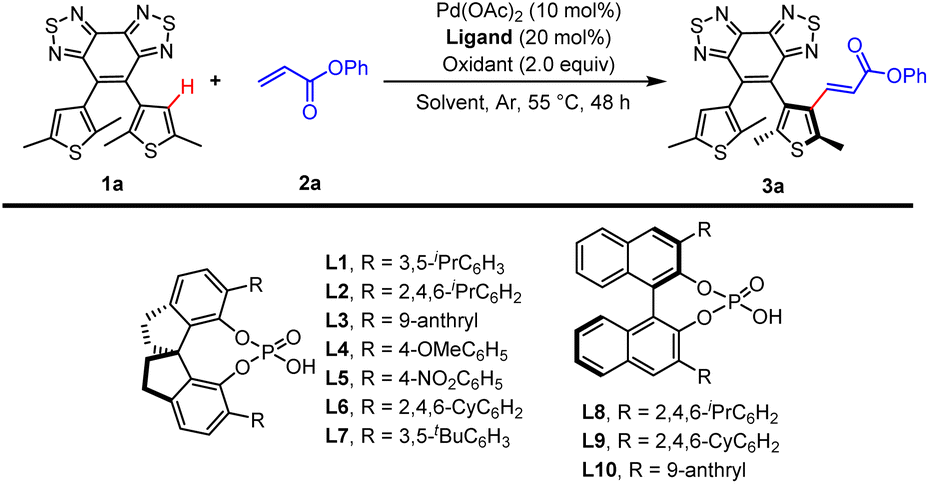
We first investigated the reaction of BTTE bearing a benzobisthiadiazole DG as the model substrate with phenyl acrylate (2a) as the olefination reagent. We have found that spiro phosphoric acids (SPAs) could enhance both the reactivity and enantioselectivity in Pd-catalyzed enantioselective synthesis of atropisomeric biaryl via asymmetry C–H activation.18 Therefore, optimization was first done with a survey of chiral phosphoric acids, including spiro phosphoric acids (Table 1, entries 1–7) and BINOL-derived phosphoric acids (entries 8–10). The reactivity and enantioselectivity were sensitive to the steric environment of SPAs. (S)-STRIP L2 bearing 2,4,6-tri-iso-propylphenyl gave the highest enantioselectivity (86.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13.5 er) in 37% yield (entry 2) possibly because L2 can impose more effective steric repulsion in the C–H activation process.17a The solvent effects were then examined (entries 11–17). The use of dibutyl ether may reduce the background reaction and lead to better enantiocontrol (96:4 er; entry 11), but with dramatically reduced yield (15%). Using trichloromethane as the solvent gave a high yield (80%) and poor enantiocontrol (80:20 er; entry 13) possibly due to better solubility of substrates. Then, further investigation of solvents revealed that a mixture containing trichloromethane/1,2-dichlorobenzene/dibutyl ether (1:1:1, v/v/v) which could enhance the solubility of substrates and potentially inhibit the background reaction at the same time was optimal (78%, 95:5 er; entry 17). The yield could be improved to 85% with a slightly increased er value (95.5:4.5 er; entry 19) when the reaction time was prolonged to 60 h. The screening of oxidants demonstrated that AgOAc was superior for the transformation (entries 21–23 in Table 1; and entries 1–9 in Table S1†). Overall, the optimal reaction conditions were identified as the following: 1a (0.1 mmol), 2a (0.15 mmol), Pd(OAc)2 (10 mol%), (S)-STRIP L2 (20 mol%), AgOAc (2 equiv.) under Ar in trichloromethane/1,2-dichlorobenzene/dibutyl ether (1:1:1, v/v/v), 55 °C, 60 h.
:13.5 er) in 37% yield (entry 2) possibly because L2 can impose more effective steric repulsion in the C–H activation process.17a The solvent effects were then examined (entries 11–17). The use of dibutyl ether may reduce the background reaction and lead to better enantiocontrol (96:4 er; entry 11), but with dramatically reduced yield (15%). Using trichloromethane as the solvent gave a high yield (80%) and poor enantiocontrol (80:20 er; entry 13) possibly due to better solubility of substrates. Then, further investigation of solvents revealed that a mixture containing trichloromethane/1,2-dichlorobenzene/dibutyl ether (1:1:1, v/v/v) which could enhance the solubility of substrates and potentially inhibit the background reaction at the same time was optimal (78%, 95:5 er; entry 17). The yield could be improved to 85% with a slightly increased er value (95.5:4.5 er; entry 19) when the reaction time was prolonged to 60 h. The screening of oxidants demonstrated that AgOAc was superior for the transformation (entries 21–23 in Table 1; and entries 1–9 in Table S1†). Overall, the optimal reaction conditions were identified as the following: 1a (0.1 mmol), 2a (0.15 mmol), Pd(OAc)2 (10 mol%), (S)-STRIP L2 (20 mol%), AgOAc (2 equiv.) under Ar in trichloromethane/1,2-dichlorobenzene/dibutyl ether (1:1:1, v/v/v), 55 °C, 60 h.
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Ligand |
Solvent |
Oxidant |
Yieldd (%) |
ere |
|
Reaction conditions: 1a (0.10 mmol), 2a (0.15 mmol), Pd(OAc)2 (10 mol%), Ligand (20 mol%), oxidant (2.0 equiv.) in solvent (2 mL), 55 °C, 48 h under Ar.
In air.
60 h.
Isolated yield.
The er value was determined by chiral HPLC. o-DCB = 1,2-dichlorobenzene.
|
| 1 |
L1
|
1,4-Dioxane |
AgOAc |
43 |
78:22 |
| 2 |
L2
|
1,4-Dioxane |
AgOAc |
37 |
86.5:13.5 |
| 3 |
L3
|
1,4-Dioxane |
AgOAc |
54 |
51:49 |
| 4 |
L4
|
1,4-Dioxane |
AgOAc |
50 |
51:49 |
| 5 |
L5
|
1,4-Dioxane |
AgOAc |
10 |
52:48 |
| 6 |
L6
|
1,4-Dioxane |
AgOAc |
53 |
68:32 |
| 7 |
L7
|
1,4-Dioxane |
AgOAc |
44 |
65:35 |
| 8 |
L8
|
1,4-Dioxane |
AgOAc |
64 |
68:32 |
| 9 |
L9
|
1,4-Dioxane |
AgOAc |
34 |
67:33 |
| 10 |
L10
|
1,4-Dioxane |
AgOAc |
54 |
51:49 |
| 11 |
L2
|
n
Bu2O |
AgOAc |
15 |
96:4 |
| 12 |
L8
|
n
Bu2O |
AgOAc |
62 |
84.5:15.5 |
| 13 |
L2
|
CHCl3 |
AgOAc |
72 |
80:20 |
| 14 |
L8
|
CHCl3 |
AgOAc |
79 |
85:15 |
| 15 |
L2
|
CHCl3/nBu2O (1:1) |
AgOAc |
66 |
95:5 |
| 16 |
L2
|
o-DCB/nBu2O (1:1) |
AgOAc |
65 |
97:3 |
| 17 |
L2
|
CHCl3/o-DCB/nBu2O (1:1:1) |
AgOAc |
78 |
95:5 |
| 18b |
L2
|
CHCl3/o-DCB/nBu2O (1:1:1) |
AgOAc |
50 |
94:6 |
| 19c |
L2
|
CHCl3/o-DCB/nBu2O (1:1:1) |
AgOAc |
85 |
95.5:4.5 |
| 20 |
L8
|
CHCl3/o-DCB/nBu2O (1:1:1) |
AgOAc |
76 |
84.5:15.5 |
| 21 |
L9
|
CHCl3/o-DCB/nBu2O (1:1:1) |
AgOAc |
14 |
95:5 |
| 22 |
L2
|
CHCl3/o-DCB/nBu2O (1:1:1) |
Cu(OAc)2 |
11 |
94.5:5.5 |
| 23 |
L2
|
CHCl3o-DCB/nBu2O (1:1:1) |
BQ |
22 |
94:6 |
Scope of the substrates
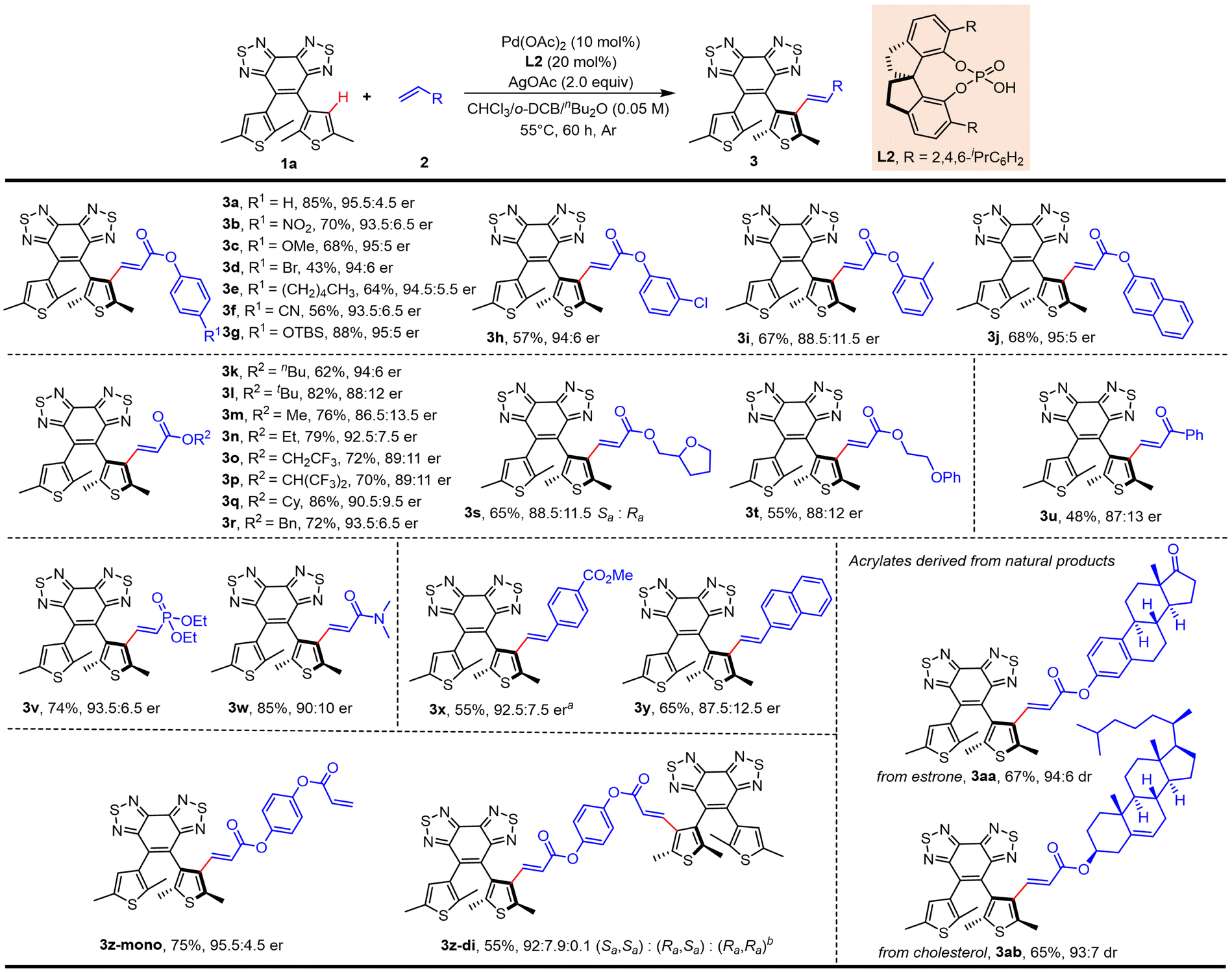
With the optimal reaction conditions in hand, we set out to explore the scope of this methodology concerning the olefin coupling partner (Scheme 1). Phenyl acrylates carrying various electron-withdrawing and -donating substituents at the para-position gave the desired products in moderate to high yields with good enantioselectivity (3b–3g, 43–88%, 93.5:6.5–95:5 er). Phenyl acrylates substituted at the ortho- or meta-position also reacted efficiently (3h, 57%, 94:6 er; 3i, 67%, 88.5:11.5 er). It was worth mentioning that phenyl acrylates substituted with a halogen (3d and 3h) and silicon (3g) were compatible with this reaction. Naphthyl acrylates also participated in the reaction effectively (3j, 68%, 95:5 er). Then we screened several generally available esters, such as n-butyl, t-butyl, methyl, ethyl, cyclohexyl, and benzyl, and all these acrylates proceeded smoothly to afford the desired products (3k–3r, 62–86%, 86.5:13.5–94:6 er; 3s, 65%, Sa:Ra = 88.5:11.5; 3t, 55%, 88:12 er). α,β-Unsaturated ketone was used as the olefination reagent to provide the product in lower yield and enantioselectivity (3u, 48%, 87:13 er). To our delight, diethyl vinyl phosphonate also reacted effectively to give the desired product in good yield and enantioselectivity (3v, 74%, 93.5:6.5 er). Acrylamide also reacted smoothly, giving the alkenylation product in excellent yield and moderate er (3w, 85%, 90:10 er). Notably, aromatic alkenes, such as styrene and vinyl naphthalene, employed as coupling partners were well tolerated (3x, 55%, 92.5:7.5 er; 3y, 65%, 87.5:12.5 er). To further demonstrate the utility of this protocol, we further examined the reaction with olefins derived from the core structures of natural products, affording the desired olefinated products in good yields and diastereoselectivity (estrone, 3aa, 67%, 94:6 er; cholesterol, 3ab, 65%, 93:7 er). To prove the practical potential of this strategy, scale-up synthesis and further transformations were conducted (Scheme 2). The olefination of 1a with phenyl acrylate (2a) on a 2 mmol scale under the standard conditions gave the corresponding product in good yield without the loss of enantioselectivity (3a, 74%, 95.5:4.5 er). To further demonstrate the applicability of this strategy, the obtained diarylethene 3a above was further decorated for LC photomanipulation. To enhance the miscibility and stability of the resulting chiral photoswitches in LC systems, a series of functional molecules were synthesized by introducing mesogenic units (green part in Scheme 2). This was expected to effectively reduce the disorderliness and deformation of LC induced by dopants and increase the photoregulatory ability of corresponding molecules.19 In the presence of DIBAL-H, the ester group of 3a could be selectively reduced to the allyl alcohol 4 (87%, 97:3 er), which could be then condensed with benzoic acid derivatives to obtain axially chiral ester 5 (93%, 96:4 er) with the retention of axial chirality. Treatment of 3a with LiOH resulted in hydrolysis of the ester group to give carboxylic acid 6 (92%, 96:4 er) in a nearly quantitative yield without loss of enantioselectivity. Meanwhile, the oxidative cleavage of the double bond could be performed to aldehyde 7 (86%, 95:5 er) without notable change in enantioselectivity, and it could be readily transformed into the novel axially chiral carboxylic acid 8, which is an essential prerequisite for further introducing mesogenic units. Then, we successfully developed a novel strategy for the effective synthesis of a series of axially chiral esters 9 (81%, 94:6 er), 10 (90%, 94.5:5.5 er), and 11 (78%, 94.5:5.5 er) in good yields and enantioselectivity via condensation of acid 8 with aliphatic or phenol derivatives containing mesogenic units as coupling partners to modify compatibility with the LC. Besides, the absolute configuration of 8 was determined as Sa by single crystal X-ray crystallographic analysis (Fig. S1, and Table S3†). Other molecules were assigned by analogy.
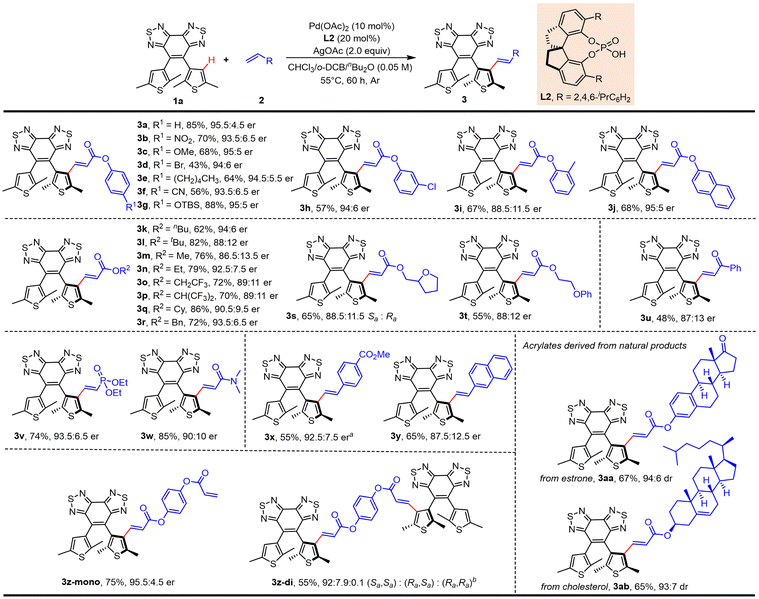 |
| | Scheme 1 Substrate scope. Reaction conditions: 1a (0.10 mmol), 2 (0.15 mmol), Pd(OAc)2 (10 mol%), L2 (20 mol%), AgOAc (2.0 equiv.) in CHCl3/1,2-dichlorobenzene/nBu2O (1:1:1, 2 mL), 55 °C, 60 h under Ar. aIn toluene/nBu2O (1:1, 2 mL). b1a (0.30 mmol), 2a (0.1 mmol), Pd(OAc)2 (20 mol%), L2 (40 mol%), AgOAc (4.0 equiv.) in CHCl3/1,2-dichlorobenzene/nBu2O (1:1:1, 2 mL). | |
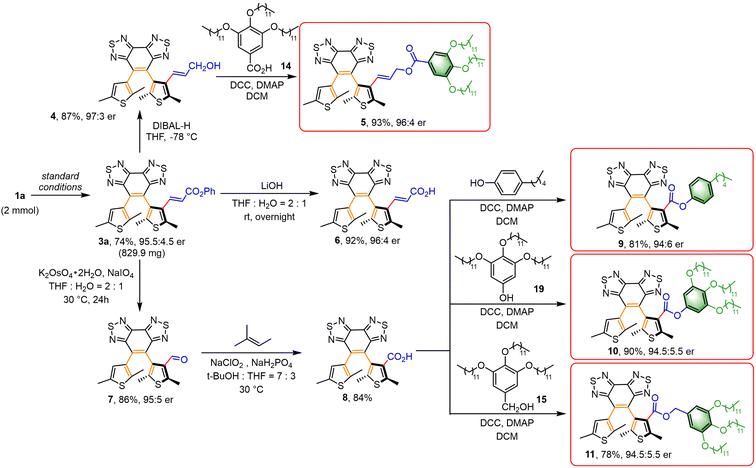 |
| | Scheme 2 Scale-up synthesis and further decoration. | |
Intrinsic chirality and photoswitching behavior
To evaluate the suitability of these derived diarylethenes for photoregulation, we examined their photoswitching characteristics. First, the photoswitching behavior of diarylethene 9 was investigated, which has the simplest molecular structure among the derivatives. Upon exposure to UV light (λ = 313 nm), the ring-open isomer 9o gradually converted into the corresponding ring-closed isomer 9c (Fig. 2a). The solution transformed from colorless into orange, along with an increase in absorption at 335–630 nm and a decrease at 290 nm, and reached the photostationary state (PSS) after 5 min (Fig. 2b). Under irradiation with visible light (λ > 490 nm), the solution at the PSS turned back to colorless as a result of the cycloreversion of 9c. The conversion ratio from 9o to 9c (90% in CD2Cl2, Fig. 2f; 94% in THF-d8, and S16†) was determined by calculating the peak area ratio of protons in the 1H NMR spectrum of the PSS solution. Compared with the reported intrinsic chiral diarylethenes, the photoswitch 9 is mono-substituted by a mesogenic unit with one rotatable thiophene unit and one unrotatable large bulky aryl group. Therefore, the photoactive anti-parallel conformer 9o–ap and photoinert parallel conformer 9o–p (Fig. 2a) can still interconvert into each other in solution. For traditional diarylethenes, this kind of interconversion is typically very fast, thus only average signals appear in their NMR spectra. For 9o, because the increased steric hindrance reduces the rotation rate of thiophene rings, we can observe both signals of anti-parallel and parallel conformers in the NMR spectra to study the spectral changes based on 1D and 2D NMR. For example, in the structure of 9o–ap, due to the shielding effect from the facing aryls, the methyl protons (He) are in a higher field than those in the p-conformer (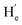 , Fig. 2h). Similarly, in the structure of 9o–p, because
, Fig. 2h). Similarly, in the structure of 9o–p, because 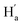 is facing the other thiophene ring, it is also in a higher field compared to Ha of 9o–ap. Considering the complexity of the methyl groups of 9o, we further checked their chemical shifts using the 2D NOESY spectrum (Fig. S4†). In the structure of 9o–ap, Ha may have a possible nuclear Overhauser effect (NOE) with nearby Hd and He, while for 9o–p, only
is facing the other thiophene ring, it is also in a higher field compared to Ha of 9o–ap. Considering the complexity of the methyl groups of 9o, we further checked their chemical shifts using the 2D NOESY spectrum (Fig. S4†). In the structure of 9o–ap, Ha may have a possible nuclear Overhauser effect (NOE) with nearby Hd and He, while for 9o–p, only 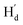 is close to
is close to 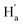 . As illustrated in the 2D NOESY spectrum, we can distinctly find the coupling signals of Ha with Hd and He. Meanwhile, Hc has weak correlations with Hf, which is consistent with the structure of 9o–ap. In contrast, for 9o–p, only signals between
. As illustrated in the 2D NOESY spectrum, we can distinctly find the coupling signals of Ha with Hd and He. Meanwhile, Hc has weak correlations with Hf, which is consistent with the structure of 9o–ap. In contrast, for 9o–p, only signals between 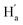 and
and 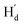 were observed, suggesting that
were observed, suggesting that 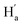 is far from
is far from 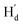 and
and 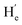 . These results can elaborately describe the interconversion between anti-parallel and parallel conformers in the corresponding 1D and 2D NMR. The ratio of ap/p (43/57 in CD2Cl2, 44/56 in THF-d8, Fig. S17†) can be calculated from the peak area ratio of Ha and
. These results can elaborately describe the interconversion between anti-parallel and parallel conformers in the corresponding 1D and 2D NMR. The ratio of ap/p (43/57 in CD2Cl2, 44/56 in THF-d8, Fig. S17†) can be calculated from the peak area ratio of Ha and 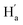 . After UV irradiation, signals of these two conformers in 1H NMR both turned into those of 9c and would recover into two kinds of signals after visible light irradiation. This reversible process can be repeated many times without remarkable degradation (Fig. 2e, and S9†), showing robust fatigue resistance. The thermal stability of 9c was also found to be excellent, which exhibited only a marginal decrease in absorption over 96 hours at 298 K (Fig. 2d) and 24 hours at 333 K (Fig. S15†). Next, the intrinsic chirality of diarylethene 9 was further investigated by circular dichroism (CD) spectroscopy. As depicted in Fig. 2c, the ring-open isomer 9o exhibited a distinct CD peak at 273 nm attributed to Cotton effects, which is uncommon in typical diarylethenes20 but close to overcrowded alkenes21 and binaphthyls22 with axial chirality. Thus, this peak can be attributed to the axial chirality resulting from the substituted thiophene and ethene bridge. Upon irradiation with UV light, distinct signals emerged at 437–612 nm (−) attributed to the formation of ring-closed isomer 9c with the extending π-conjugation. Meanwhile, a strong peak gradually emerged near 250 nm (+), while the initial peak at 273 nm (−) redshifted to 283 nm (−). These peaks can be attributed to the high asymmetry of 9c resulting from the cyclohexadiene center and distorted ethene bridge.10 These observed changes in the overall CD spectrum indicated an enantiospecific transformation of 9 from axial into central chirality. The results of fatigue resistance experiments based on the CD spectrum illustrated that almost no destruction of chirality would occur during the whole transformation (Fig. 2e). As demonstrated, enantioselective olefination can increase the rotation barrier of side aryl groups of sterically hindered diarylethenes, resulting in the steady existence of their chiral enantiomers and achieving specific enantioselectivity at the same time, which can efficiently reduce the dependence on preparative chiral HPLC. Indeed, diarylethene 9 exhibited good photoresponsive properties and chiral regulation ability, which are enough for further photoregulation applications. Additional tests demonstrated that 10 and 11 exhibited comparable performances of photochromism and chiral regulation (Fig. S8–S21, and Table S4†). However, the cycloreversion quantum yields of 10 and 11 exhibited a significant decrease in comparison to 9, which can be likely ascribed to the large hindrance of the trialkoxyphenyl groups and the non-covalent interactions between the three dodecane chains and solvent molecules enhancing the hindrance of photocycloreversion.
. After UV irradiation, signals of these two conformers in 1H NMR both turned into those of 9c and would recover into two kinds of signals after visible light irradiation. This reversible process can be repeated many times without remarkable degradation (Fig. 2e, and S9†), showing robust fatigue resistance. The thermal stability of 9c was also found to be excellent, which exhibited only a marginal decrease in absorption over 96 hours at 298 K (Fig. 2d) and 24 hours at 333 K (Fig. S15†). Next, the intrinsic chirality of diarylethene 9 was further investigated by circular dichroism (CD) spectroscopy. As depicted in Fig. 2c, the ring-open isomer 9o exhibited a distinct CD peak at 273 nm attributed to Cotton effects, which is uncommon in typical diarylethenes20 but close to overcrowded alkenes21 and binaphthyls22 with axial chirality. Thus, this peak can be attributed to the axial chirality resulting from the substituted thiophene and ethene bridge. Upon irradiation with UV light, distinct signals emerged at 437–612 nm (−) attributed to the formation of ring-closed isomer 9c with the extending π-conjugation. Meanwhile, a strong peak gradually emerged near 250 nm (+), while the initial peak at 273 nm (−) redshifted to 283 nm (−). These peaks can be attributed to the high asymmetry of 9c resulting from the cyclohexadiene center and distorted ethene bridge.10 These observed changes in the overall CD spectrum indicated an enantiospecific transformation of 9 from axial into central chirality. The results of fatigue resistance experiments based on the CD spectrum illustrated that almost no destruction of chirality would occur during the whole transformation (Fig. 2e). As demonstrated, enantioselective olefination can increase the rotation barrier of side aryl groups of sterically hindered diarylethenes, resulting in the steady existence of their chiral enantiomers and achieving specific enantioselectivity at the same time, which can efficiently reduce the dependence on preparative chiral HPLC. Indeed, diarylethene 9 exhibited good photoresponsive properties and chiral regulation ability, which are enough for further photoregulation applications. Additional tests demonstrated that 10 and 11 exhibited comparable performances of photochromism and chiral regulation (Fig. S8–S21, and Table S4†). However, the cycloreversion quantum yields of 10 and 11 exhibited a significant decrease in comparison to 9, which can be likely ascribed to the large hindrance of the trialkoxyphenyl groups and the non-covalent interactions between the three dodecane chains and solvent molecules enhancing the hindrance of photocycloreversion.
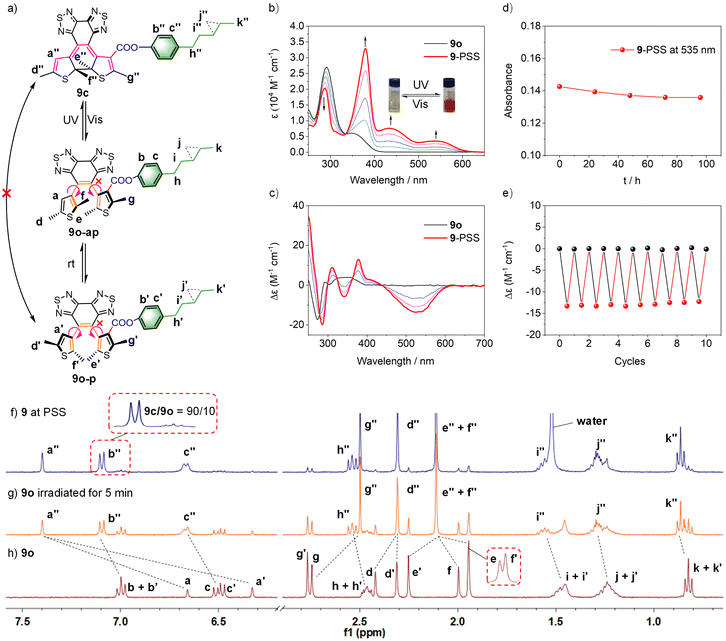 |
| | Fig. 2 Photochromic behavior and chiral regulation of 9. (a) Transformation between the isomers of 9. (b) Normalized UV-vis spectrum changes (c = 4 × 10−5 M, THF) and (c) CD spectrum changes of 9 (c = 4 × 10−5 M, THF) during irradiation with UV light (λ = 313 nm). (d) Absorbance changes of 9 at PSS at 535 nm at room temperature for 96 h (c = 4 × 10−5 M, THF). (e) Changes of CD signals of 9 (c = 4 × 10−5 M, THF) at 535 nm during alternative irradiation with UV light (λ = 313 nm) and visible light (λ > 490 nm). Irradiation with UV light (λ = 313 nm) in CD2Cl2 (c = 5 × 10−3 M): 1H NMR spectra of (f) 9 at the PSS; (g) 9o irradiated for 5 min and (h) 9o. | |
Photomanipulation of the liquid crystal superstructure
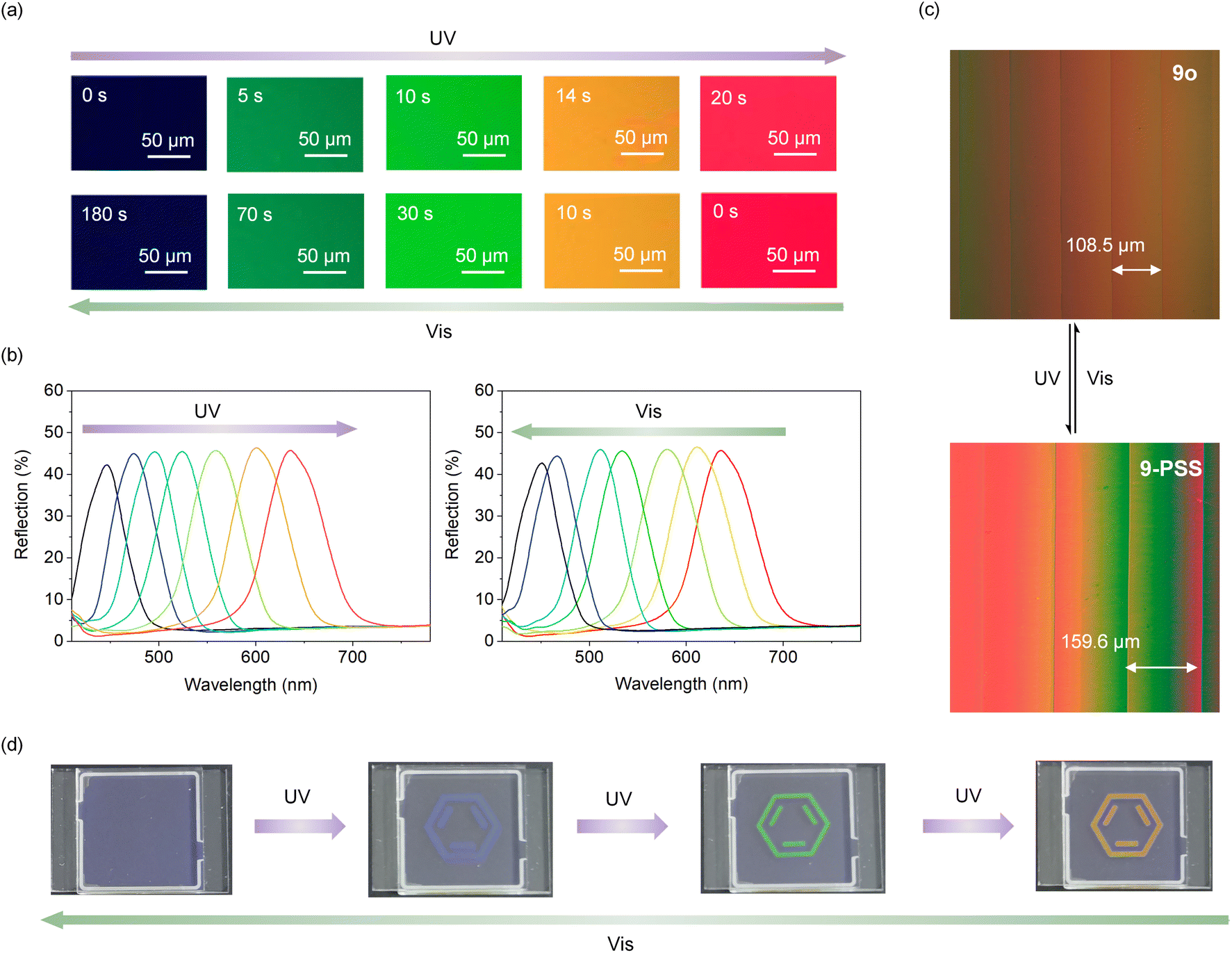
Given the intrinsic chiral photoswitch 9 obtained through enantioselective C–H olefination and decoration with a mesogenic group, we incorporated it into a commercial nematic LC for precise photoregulation of the helical superstructure. The helical twist power (HTP) represents the ability of chiral guest (dopant) compounds to induce a cholesteric liquid crystal (CLC) phase from nematic phases. Chiral photoswitches can effectively utilize their unique optical responsiveness to drive and dynamically adjust the properties of the induced CLCs through the changes of HTP. Thus, we first examined the HTP of 9 by the well-known Cano's method (Fig. S22 and S23†) calculated according to the equation: βM = (P·c)−1, where βM is HTP, P represents the pitch length, and c represents the concentration of the chiral dopant in the LC. Under UV light, the distance between two disclination lines of a mixture comprising 9 and the commercial LC increased from 108.5 to 159.6 μm (Fig. 3c), which is consistent with the decrease of HTP from 69.8 μm−1 to 47.4 μm−1 (Table S5†), indicating that 9 could effectively twist and manipulate the helical superstructure. Similar tests were conducted with 10 and 11 (Fig. S24, and Table S5†). Unfortunately, 10 and 11 exhibited poorer regulation abilities in the LC possibly due to increased steric hindrance of side groups, resulting in diminished conversion rates. Although this effect may not be apparent in solution (Fig. S18 and S19†), it can become evident in LCs where the movements of molecules are more restricted. Subsequently, compound 9 was mixed with TEB300 and chiral dopant S5011 to prepare a photoresponsive LC system. The reflection color of this LC system can continuously transform from blue into green and then into red under UV light (λ = 365 nm, 4.0 mW cm−2) irradiation for 20 s (Fig. 3a) accompanied by a redshift of the reflection wavelength from 430 to 650 nm (Fig. 3b), covering the basic RGB colors. Full recovery was achieved by irradiation under visible light (λ = 530 nm, 1.0 mW cm−2) for 3 min. The reflection bands remained sharp without distinct spectral deformation during the shift process, thus confirming that the photoregulation has negligible destruction on the texture of the LC system attributed to the unique enantiospecific transformation of intrinsic chirality and increased LC compatibility originated from the decorated mesogenic unit. In addition, the color of the LC system remained unchanged after the removal of UV light due to the good thermal stability of 9c. Next, a patterned image was recorded using light passing through a photomask in the photoresponsive LC system. With the irradiation of UV light, a distinct benzene ring gradually emerged, accompanied by a transition in color passing through blue, green, and orange (Fig. 3d). By precisely controlling the duration of irradiation, it is achievable to maintain the pattern at any intermediate state, without any obvious color migration or blurring of the boundary. This photoresponsive LC system exhibits potential application in the field of anti-counterfeiting.
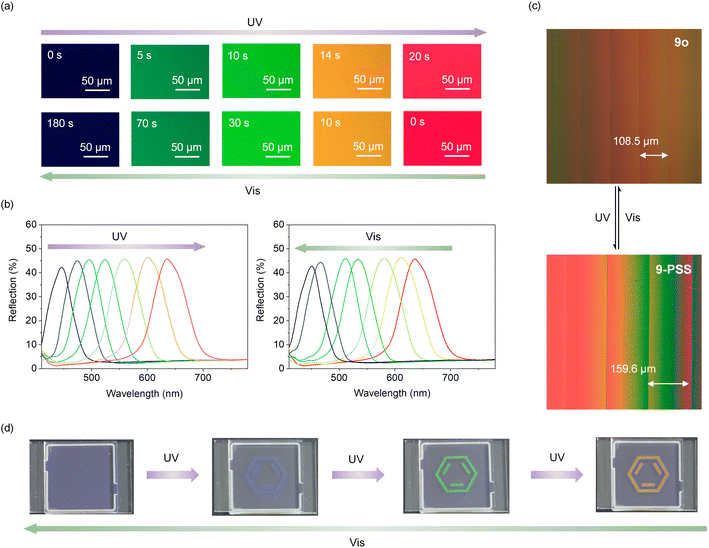 |
| | Fig. 3 LC photomanipulation and anti-counterfeiting application. (a) Texture changes of the LC system during photoregulation. (b) Reflection spectra of the LC system during photoregulation. (c) Distance changes between two disclination lines of a mixture of 9 and TEB300. (d) The color changes of the designed pattern. | |
Conclusions
In conclusion, we proposed a novel strategy to directly construct intrinsic chiral diarylethenes. By employing enantioselective C–H olefination, we successfully synthesized a series of intrinsic chiral diarylethenes with high yields and enantioselectivity. The obtained diarylethenes can be further decorated by incorporating mesogenic units through three-step synthesis for liquid crystal modulation. A photoresponsive liquid crystal system and an anti-counterfeiting method with excellent photoregulatory abilities have been established. Research in the future will primarily concentrate on exploring other applications for these synthesized diarylethenes and further improving their photoregulatory abilities.
Data availability
All data in the manuscript are available in the ESI,† including the experimental details, characterization data, HPLC spectra, and crystallographic data.
Author contributions
G. Z. and X. W. contributed equally to this work. G. Z. and X. W. performed the synthesis and tests and wrote the draft. M. L., W. Z. and B. S. reviewed and edited the manuscript. S. M. and H. L. helped in the experimental process.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (T2488302, 92356301, 22338006, 22108076, 21925109, U22A20388, and 92256302), the National Key R&D Program of China (2022YFA1504302 and 2021YFF0701603), Fundamental Research Funds for the Central Universities (226-2023-00115 and 226-2022-00224), and Zhejiang Provincial NSFC (LD22B030003).
Notes and references
-
(a) M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS;
(b) S. Takami, S. Kobatake, T. Kawai and M. Irie, Chem. Lett., 2003, 32, 892–893 CrossRef CAS;
(c) X. Li, Q. Zou and H. Ågren, J. Phys. Chem. A, 2015, 119, 9140–9147 CrossRef CAS PubMed;
(d) N. M. Wu, H.-L. Wong and V. W.-W. Yam, Chem. Sci., 2017, 8, 1309–1315 RSC.
-
(a) H. Jean-Ruel, R. R. Cooney, M. Gao, C. Lu, M. A. Kochman, C. A. Morrison and R. J. D. Miller, J. Phys. Chem. A, 2011, 115, 13158–13168 CrossRef CAS;
(b) Y. Ishibashi, M. Fujiwara, T. Umesato, H. Saito, S. Kobatake, M. Irie and H. Miyasaka, J. Phys. Chem. C, 2011, 115, 4265–4272 CrossRef CAS;
(c) Y. Ishibashi, T. Umesato, S. Kobatake, M. Irie and H. Miyasaka, J. Phys. Chem. C, 2012, 116, 4862–4869 CrossRef CAS;
(d) P. Hong, J. Liu, K.-X. Qin, R. Tian, L.-Y. Peng, Y.-S. Su, Z. Gan, X.-X. Yu, L. Ye, M.-Q. Zhu and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202316706 CrossRef CAS;
(e) D. Kolarski, P. Steinbach, C. Bannwarth, K. Klaue and S. Hecht, Angew. Chem., Int. Ed., 2024, 63, e202318015 CrossRef CAS.
-
(a) D. Mendive-Tapia, A. Perrier, M. J. Bearpark, M. A. Robb, B. Lasorne and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 18463–18471 RSC;
(b) J. C. H. Chan, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 16994–16997 CrossRef CAS PubMed;
(c) T. Kolmar, S. M. Büllmann, C. Sarter, K. Höfer and A. Jäschke, Angew. Chem., Int. Ed., 2021, 60, 8164–8173 CrossRef CAS PubMed;
(d) M.-Q. Li, H.-L. Hu, B.-H. Liu, X. Liu, Z.-G. Zheng, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2022, 144, 20773–20784 CrossRef CAS;
(e) M. Bovoloni, J. Filo, I. Sigmundová, P. Magdolen, Š. Budzák, E. Procházková, M. Tommasini, M. Cigáň and A. Bianco, Phys. Chem. Chem. Phys., 2022, 24, 23758–23768 RSC.
-
(a) J.-W. Jiang, P.-S. Zhang, L. Liu, Y.-Q. Li, Y.-B. Zhang, T.-C. Wu, H.-L. Xie, C. H. Zhang, J. X. Cui and J. Chen, Chem. Eng. J., 2021, 425, 131557 CrossRef CAS;
(b) Y.-Y. Tang, Y.-L. Zeng and R.-G. Xiong, J. Am. Chem. Soc., 2022, 144, 8633–8640 CrossRef CAS PubMed;
(c) M. Karmakar, S. K. Bag, B. Mondal and A. Thakur, J. Mater. Chem. C, 2022, 10, 8860–8873 RSC.
-
(a) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC;
(b) T. Fukushima, K. Tamaki, A. Isobe, T. Hirose, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi, M. J. Hollamby and S. Yagai, J. Am. Chem. Soc., 2021, 143, 5845–5854 CrossRef CAS;
(c) B. H. Tang, M. Pauls, C. Bannwarth and S. Hecht, J. Am. Chem. Soc., 2024, 146, 45–50 CrossRef CAS;
(d) M. Li, L.-J. Chen, Z. P. Zhang, Q. F. Luo, H.-B. Yang, H. Tian and W.-H. Zhu, Chem. Sci., 2019, 10, 4896–4904 RSC;
(e) W.-L. Zhou, X.-Y. Dai, W.-J. Lin, Y. Chen and Y. Liu, Chem. Sci., 2023, 14, 6457–6466 RSC.
-
(a) L. Gu, L.-J. Zhang, X. Luo, Y. Zheng, Z.-W. Ye, M. Lv, J.-Q. Chen, C.-L. Chen, Y. Xiao, W.-H. Zhu, X.-H. Qian and Y.-J. Yang, Sci. China: Chem., 2021, 64, 253–262 CrossRef CAS;
(b) C. Li, K. Xiong, Y. Chen, C. Fan, Y.-L. Wang, H. Ye and M.-Q. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 27651–27662 CrossRef CAS;
(c) H. Yang, M. Li, C. Li, Q. Luo, M.-Q. Zhu, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2020, 59, 8560–8570 CrossRef CAS.
-
(a) J. X. Hou, J. H. Wang, A. Ryabchun and B. L. Feringa, Adv. Funct. Mater., 2024, 2312831 CrossRef CAS;
(b) Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS;
(c) M. Li, L.-J. Chen, Y. Cai, Q. Luo, W. Li, H.-B. Yang, H. Tian and W.-H. Zhu, Chem, 2019, 5, 634–648 CrossRef CAS;
(d) Y. Wu, Y. R. Ren, X.-X. Zeng, H.-L. Hu, M.-Q. Li, J.-Z. Li, T.-C. He, X.-S. Li, Z.-Q. Yu and W.-H. Zhu, Smart Mol., 2023, 1, e20230003 CrossRef.
-
(a) T. Yamaguchi, T. Inagawa, H. Nakazumi, S. Irie and M. Irie, Chem. Mater., 2000, 12, 869–871 CrossRef CAS;
(b) Y.-N. Li and Q. Li, Org. Lett., 2012, 14, 4362–4365 CrossRef CAS;
(c) Y. Li, M. Wang, A. Urbas and L. Quan, J. Mater. Chem. C, 2013, 1, 3917–3923 RSC;
(d) Z.-G. Zheng, H.-L. Hu, Z.-P. Zhang, B.-H. Liu, M.-Q. Li, D.-H. Qu, H. Tian, W.-H. Zhu and B. L. Feringa, Nat. Photonics, 2022, 16, 226–234 CrossRef CAS;
(e) H.-L. Hu, M. He, X.-S. Liang, M.-Q. Li, C. L. Yuan, B.-H. Liu, X. Liu, Z. G. Zheng and W.-H. Zhu, Matter, 2023, 6, 3927–3939 CrossRef CAS.
-
(a) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS;
(b) S.-M. Guo, M.-Q. Li, H.-L. Hu, T. Xu, H.-C. Xi and W.-H. Zhu, Chem. Sci., 2023, 14, 6237–6243 RSC.
-
(a) W.-L. Li, C.-H. Jiao, Y.-S. Xie, K. Nakatani, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS;
(b) W.-L. Li, X. Li, Y.-S. Xie, Y. Wu, M. Q. Li, X. Y. Wu, W. H. Zhu and H. Tian, Sci. Rep., 2015, 5, 9186 CrossRef CAS PubMed.
-
(a) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pamies and M. Dieguez, Chem. Rev., 2011, 111, 2077–2118 CrossRef CAS;
(b) R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS PubMed;
(c) C. X. Liu, S. Y. Yin, F. N. Zhao, H. Yang, Z. L. J. Feng, Q. Gu and S. L. You, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS;
(d) D.-B. Zhao and K.-L. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS;
(e) G.-Q. Yang and W.-B. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
-
(a) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS;
(b) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC;
(c) J. A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC;
(d) J. K. Cheng, S.-H. Xiang, S.-Y. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS;
(e) C.-X. Liu, W. W. Zhang, S. Y. Yin and S. L. You, J. Am. Chem. Soc., 2021, 143(35), 14025–14040 CrossRef CAS PubMed.
- W. H. Zhu, Y. H. Yang, R. Métivier, Q. Zhang, R. Guillot, Y.-S. Xie, H. Tian and K. Nakatani, Angew. Chem., Int. Ed., 2011, 50, 10986–10990 CrossRef CAS PubMed.
- W.-L. Li, Y.-S. Cai, X. Li, H. Ågren, H. Tian and W.-H. Zhu, J. Mater. Chem. C, 2015, 3, 8665–8674 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
-
(a) J. Guo, H. He, Z. Ye, K. Zhu, Y. Wu and F. Zhang, Org. Lett., 2018, 20, 5692–5695 CrossRef CAS;
(b) C. Y. He, C. Z. Wu, F. L. Qing and X. Zhang, J. Org. Chem., 2014, 79, 1712–1718 CrossRef CAS PubMed;
(c) H. He, J. Guo, W. Sun, B. Yang, F. Zhang and G. Liang, J. Org. Chem., 2020, 85, 3788–3798 CrossRef CAS PubMed;
(d) J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, J. Am. Chem. Soc., 2013, 135, 16376–16379 CrossRef CAS.
-
(a) J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708–6712 CrossRef CAS;
(b) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS PubMed;
(c) L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B. B. Zhan, Y. Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497–511 CrossRef CAS;
(d) Q.-J. Yao, P.-P. Xie, Y.-J. Wu, Y.-L. Feng, M.-Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS PubMed;
(e) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed;
(f) Y. J. Li, Y.-C. Liou, X.-R. Chen and L. Ackermann, Chem. Sci., 2022, 13, 4088–4094 RSC;
(g) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735–2759 RSC;
(h) Z. Dong, J. Li, T. Yao and C. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202315603 CrossRef CAS.
-
(a) B.-B. Zhan, L. Wang, J. Luo, X. F. Lin and B. F. Shi, Angew. Chem., Int. Ed., 2020, 59, 3568–3572 CrossRef CAS;
(b) L. Jin, P. Zhang, Y. Li, X. Yu and B. F. Shi, J. Am. Chem. Soc., 2021, 143, 12335–12344 CrossRef CAS;
(c) G. Liao, T. Zhang, L. Jin, B. J. Wang, C. K. Xu, Y. Lan, Y. Zhao and B. F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202115221 CrossRef CAS.
-
(a) A. Ryabchun, Q. Li, F. Lancia, I. Aprahamian and N. Katsonis, J. Am. Chem. Soc., 2019, 141, 1196–1200 CrossRef CAS PubMed;
(b) Y. Zhan, S. Calierno, J. Peixoto, L. Mitzer, D. J. Broer and D. Liu, Angew. Chem., Int. Ed., 2022, 61, e202207468 CrossRef CAS;
(c) W. Kang, Y. Tang, X. Meng, S. Lin, X. Zhang, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202311486 CrossRef CAS;
(d) J. Hou, R. Toyoda, S. C. J. Meskers and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202206310 CrossRef CAS PubMed.
-
(a) T. Nakashima, K. Yamamoto, Y. Kimura and T. Kawai, Chem.–Eur. J., 2013, 19, 16972–16980 CrossRef CAS PubMed;
(b) H. Jin-nouchi and M. Takeshita, Chem.–Eur. J., 2012, 18, 9638–9644 CrossRef CAS.
-
(a) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS;
(b) D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652–6658 CrossRef CAS PubMed.
-
(a) C. Gao, S. Silvi, X. Ma, H. Tian, A. Credi and M. Venturi, Chem.–Eur. J., 2012, 18, 16911–16921 CrossRef CAS PubMed;
(b) C. Gütz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2359686 (8). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05375c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Wei-Hong
Zhu
*b and
Wei-Hong
Zhu

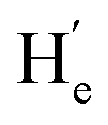 , Fig. 2h). Similarly, in the structure of 9o–p, because
, Fig. 2h). Similarly, in the structure of 9o–p, because 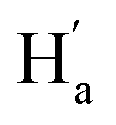 is facing the other thiophene ring, it is also in a higher field compared to Ha of 9o–ap. Considering the complexity of the methyl groups of 9o, we further checked their chemical shifts using the 2D NOESY spectrum (Fig. S4†). In the structure of 9o–ap, Ha may have a possible nuclear Overhauser effect (NOE) with nearby Hd and He, while for 9o–p, only
is facing the other thiophene ring, it is also in a higher field compared to Ha of 9o–ap. Considering the complexity of the methyl groups of 9o, we further checked their chemical shifts using the 2D NOESY spectrum (Fig. S4†). In the structure of 9o–ap, Ha may have a possible nuclear Overhauser effect (NOE) with nearby Hd and He, while for 9o–p, only 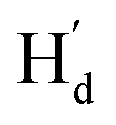 is close to
is close to 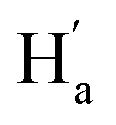 . As illustrated in the 2D NOESY spectrum, we can distinctly find the coupling signals of Ha with Hd and He. Meanwhile, Hc has weak correlations with Hf, which is consistent with the structure of 9o–ap. In contrast, for 9o–p, only signals between
. As illustrated in the 2D NOESY spectrum, we can distinctly find the coupling signals of Ha with Hd and He. Meanwhile, Hc has weak correlations with Hf, which is consistent with the structure of 9o–ap. In contrast, for 9o–p, only signals between 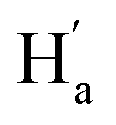 and
and 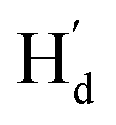 were observed, suggesting that
were observed, suggesting that 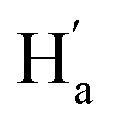 is far from
is far from 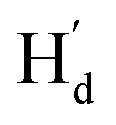 and
and  . These results can elaborately describe the interconversion between anti-parallel and parallel conformers in the corresponding 1D and 2D NMR. The ratio of ap/p (43/57 in CD2Cl2, 44/56 in THF-d8, Fig. S17†) can be calculated from the peak area ratio of Ha and
. These results can elaborately describe the interconversion between anti-parallel and parallel conformers in the corresponding 1D and 2D NMR. The ratio of ap/p (43/57 in CD2Cl2, 44/56 in THF-d8, Fig. S17†) can be calculated from the peak area ratio of Ha and 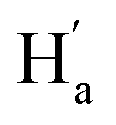 . After UV irradiation, signals of these two conformers in 1H NMR both turned into those of 9c and would recover into two kinds of signals after visible light irradiation. This reversible process can be repeated many times without remarkable degradation (Fig. 2e, and S9†), showing robust fatigue resistance. The thermal stability of 9c was also found to be excellent, which exhibited only a marginal decrease in absorption over 96 hours at 298 K (Fig. 2d) and 24 hours at 333 K (Fig. S15†). Next, the intrinsic chirality of diarylethene 9 was further investigated by circular dichroism (CD) spectroscopy. As depicted in Fig. 2c, the ring-open isomer 9o exhibited a distinct CD peak at 273 nm attributed to Cotton effects, which is uncommon in typical diarylethenes20 but close to overcrowded alkenes21 and binaphthyls22 with axial chirality. Thus, this peak can be attributed to the axial chirality resulting from the substituted thiophene and ethene bridge. Upon irradiation with UV light, distinct signals emerged at 437–612 nm (−) attributed to the formation of ring-closed isomer 9c with the extending π-conjugation. Meanwhile, a strong peak gradually emerged near 250 nm (+), while the initial peak at 273 nm (−) redshifted to 283 nm (−). These peaks can be attributed to the high asymmetry of 9c resulting from the cyclohexadiene center and distorted ethene bridge.10 These observed changes in the overall CD spectrum indicated an enantiospecific transformation of 9 from axial into central chirality. The results of fatigue resistance experiments based on the CD spectrum illustrated that almost no destruction of chirality would occur during the whole transformation (Fig. 2e). As demonstrated, enantioselective olefination can increase the rotation barrier of side aryl groups of sterically hindered diarylethenes, resulting in the steady existence of their chiral enantiomers and achieving specific enantioselectivity at the same time, which can efficiently reduce the dependence on preparative chiral HPLC. Indeed, diarylethene 9 exhibited good photoresponsive properties and chiral regulation ability, which are enough for further photoregulation applications. Additional tests demonstrated that 10 and 11 exhibited comparable performances of photochromism and chiral regulation (Fig. S8–S21, and Table S4†). However, the cycloreversion quantum yields of 10 and 11 exhibited a significant decrease in comparison to 9, which can be likely ascribed to the large hindrance of the trialkoxyphenyl groups and the non-covalent interactions between the three dodecane chains and solvent molecules enhancing the hindrance of photocycloreversion.
. After UV irradiation, signals of these two conformers in 1H NMR both turned into those of 9c and would recover into two kinds of signals after visible light irradiation. This reversible process can be repeated many times without remarkable degradation (Fig. 2e, and S9†), showing robust fatigue resistance. The thermal stability of 9c was also found to be excellent, which exhibited only a marginal decrease in absorption over 96 hours at 298 K (Fig. 2d) and 24 hours at 333 K (Fig. S15†). Next, the intrinsic chirality of diarylethene 9 was further investigated by circular dichroism (CD) spectroscopy. As depicted in Fig. 2c, the ring-open isomer 9o exhibited a distinct CD peak at 273 nm attributed to Cotton effects, which is uncommon in typical diarylethenes20 but close to overcrowded alkenes21 and binaphthyls22 with axial chirality. Thus, this peak can be attributed to the axial chirality resulting from the substituted thiophene and ethene bridge. Upon irradiation with UV light, distinct signals emerged at 437–612 nm (−) attributed to the formation of ring-closed isomer 9c with the extending π-conjugation. Meanwhile, a strong peak gradually emerged near 250 nm (+), while the initial peak at 273 nm (−) redshifted to 283 nm (−). These peaks can be attributed to the high asymmetry of 9c resulting from the cyclohexadiene center and distorted ethene bridge.10 These observed changes in the overall CD spectrum indicated an enantiospecific transformation of 9 from axial into central chirality. The results of fatigue resistance experiments based on the CD spectrum illustrated that almost no destruction of chirality would occur during the whole transformation (Fig. 2e). As demonstrated, enantioselective olefination can increase the rotation barrier of side aryl groups of sterically hindered diarylethenes, resulting in the steady existence of their chiral enantiomers and achieving specific enantioselectivity at the same time, which can efficiently reduce the dependence on preparative chiral HPLC. Indeed, diarylethene 9 exhibited good photoresponsive properties and chiral regulation ability, which are enough for further photoregulation applications. Additional tests demonstrated that 10 and 11 exhibited comparable performances of photochromism and chiral regulation (Fig. S8–S21, and Table S4†). However, the cycloreversion quantum yields of 10 and 11 exhibited a significant decrease in comparison to 9, which can be likely ascribed to the large hindrance of the trialkoxyphenyl groups and the non-covalent interactions between the three dodecane chains and solvent molecules enhancing the hindrance of photocycloreversion.