
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriple-function porphyrin in glycopolymeric photosensitizers: from photoATRP to targeted PDT†
Jiahui
Lin
,
Zhiyuan
Ma
 *,
Weiwei
Zuo
and
Meifang
Zhu
*,
Weiwei
Zuo
and
Meifang
Zhu
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, 2999 North Renmin Road, Shanghai 201620, China. E-mail: maz@dhu.edu.cn
First published on 12th November 2024
Abstract
Porphyrin derivatives serve as photocatalysts in reversible-deactivation radical polymerization and as photosensitizers in photodynamic therapy (PDT). Herein, a triple function porphyrin, ZnTPPC6Br, was synthesized as a photocatalyst and initiator for photoATRP. Oxygen-tolerant photoATRP produced fructose-based star-shaped glycopolymers as targeted photosensitizers for PDT. ZnTPPC6Br/CuII/PMDETA could synthesize polymer photosensitizers with predictable Mn and low Đ. Mechanistic studies unveiled the transition of ZnTPPC6Br from a singlet excited state (1PC*) to a triplet excited state (3PC*), enabling the activator CuI/L generation and initiating photoATRP. The excess ligands facilitate return of the active species to the ground state, while the presence of DMSO assists in oxygen depletion. Three fructose-based monomers with different polymerizable groups (acrylated, methacrylated, and p-vinylbenzoated) were employed to scale up polymerization, yielding glycopolymeric photosensitizers post-deprotection. In vitro cellular studies showed enhanced PDT efficacy of glycopolymeric photosensitizers against MCF-7 cells, attributed to specific GLUT5 binding for targeted endocytosis, highlighting their potential for precise cancer treatment compared to L929 cells. The multifunctional capabilities of ZnTPPC6Br are anticipated to serve as a strategic avenue for the advancement of polymer photosensitizers with potential PDT applications.
Introduction
Polysaccharides are fundamental and widespread natural polymers across both plant and animal kingdoms,1,2 serving crucial functions as energy sources (e.g., starch and glycogen) and structural components (e.g., cellulose, chitin, and collagen).3,4 A multitude of complex biomolecular recognition processes, such as cancer metastasis, cell migration, microbial and viral infections, and inflammatory responses, are mediated by carbohydrate-binding proteins (lectins) located on cell surfaces that possess the ability to selectively bind to these polymers.5–10 Mimicking nature is a significant objective in the advancement of functional polymers.11,12 Glycopolymers, synthetic macromolecules with attached sugar moieties, have been developed as manageable alternatives.13–15 Glycopolymers may be synthesized by polymerizing glycomonomers containing vinyl groups, strained olefin rings, lactones, or N-carboxyanhydride rings using appropriate polymerization techniques.16–19 The advancement of (meth)acrylated and (meth)acrylamided glycomonomers, particularly via reversible-deactivation radical polymerization like atom transfer radical polymerization (ATRP), has opened a wealth of opportunities for the development and application of well-defined glycopolymers.20,21 The initial glycopolymer, synthesized in 1998 via ATRP using methacrylated isopropylidene-protected glucose as the monomer, allowed generation of polymers with molar mass up to 2 × 105 Da and Đ from 1.27 to 1.82 by adjusting the monomer-to-initiator ratio.22 Miktoarm ABA and star amphiphilic block copolymers of poly(vinyl sugars) and poly(ε-caprolactone) segments have been synthesized through the ring-opening polymerization of ε-caprolactone and ATRP of methacrylated isopropylidene-protected galactose.23 Poly(2-aminoethyl methacrylate hydrochloride)-co-poly(2-lactobionamidoethyl methacrylate) cationic glycopolymers were synthesized using activators regenerated by electron transfer ATRP, facilitating the development of a high-efficiency gene delivery nanosystem with optimal physicochemical properties, ASGPR specificity, and transfection efficiency.24 AB and ABA block copolymers, composed of acetyl-mannopyranoside methacrylate, were synthesized using a PEG macroinitiator through ATRP, creating a water-soluble, biocompatible glycopolymer that enhanced cell proliferation, adhesion, and osteointegration efficiency.25 The utilization of ATRP facilitated the synthesis of comb-shaped glycopolymers, leading to a range of serum-tolerant and low-toxicity complexes with polyethyleneimine that enhance gene transfection efficiency, notably in serum conditions, for in vitro gene delivery.26 The versatility and flexibility of ATRP, coupled with the ability for modulation and control of glycomoieties, has fostered potential applications of glycopolymers in cell-targeted vectors and therapeutics, promoting integration with therapeutic modalities to enhance diagnostic and treatment efficacy.27–29Photodynamic therapy (PDT) has emerged as an effective modality for treating superficial tumors and diseased tissues, contributing to an improved understanding of its biophysical mechanisms through significant advancements in preclinical and clinical research over the past two decades.30,31 The photosensitizer (PS) is a crucial element activated by light, leading to the production of reactive oxygen species (ROS) responsible for cell destruction.32–35 Numerous synthetic photosensitizers, such as porphyrin, phthalocyanine, and phenothiazine derivatives, have been meticulously designed and extensively investigated for transitioning from theoretical studies to clinical applications.36–38 The Boyer group successfully utilized ZnTPP monomers containing polymerizable groups to catalyse PET-RAFT polymerization, while simultaneously achieving photo-enhanced antimicrobial activity of the resulting porphyrin-containing polymers under light irradiation.39 Recent studies have interestingly discovered the pivotal role of porphyrin in establishing a photocatalytic system that facilitates photoATRP under light irradiation.40–42 This system integrates a copper (Cu) catalyst that effectively modulates polymerization through the ATRP equilibrium, subsequently generating the activator CuI species. Notably, this system displays oxygen tolerance, attributed to the consumption of oxygen during the photoredox reactions, an interaction leading to favorably regulated polymerizations. The versatile use of porphyrins, both in photoinduced ATRP and as PSs, exemplifies the multifunctional strategy, providing a promising approach for synthesizing robust glycopolymeric photosensitizers. In addition, combining sugar-containing polymers with PSs can mitigate aggregation-caused quenching (ACQ), enhancing ROS production for enhanced PDT and improving cell-specific endocytosis through customized polymer design and glycomoiety modulation.43,44
In this study, zinc(II) tetra(p-hydroxyphenyl)porphine was transformed into corresponding alkyl (pseudo)halide, ZnTPPC6Br, serving dually as a photocatalyst and an initiator for photoATRP of fructose-based glycomonomers, yielding star-shaped glycopolymeric photosensitizers. This strategy, employing a triple functional approach, facilitates photoATRP under dual photoredox/copper catalysis systems and photosensitizers for PDT application. The ZnTPPC6Br/CuII/PMDETA photoATRP system effectively controls the polymerization of fructose functionalized with acrylate, methacrylate, and p-vinylbenzoate groups, illustrating proficient “on–off” light responsiveness. Water-soluble glycopolymeric photosensitizers were successfully synthesized with satisfactory Mn and relatively narrow Đ. The star-shaped polymers, characterized by the presence of a porphyrin core and four fructose-containing arms, displayed exceptional photophysical and photochemical properties, leading to effective targeted PDT against MCF-7 cells. The versatile integration of porphyrins significantly contributes to the efficacious triple functional strategy, presenting an innovative and promising methodology for developing and applying glycopolymeric photosensitizers (Scheme 1).
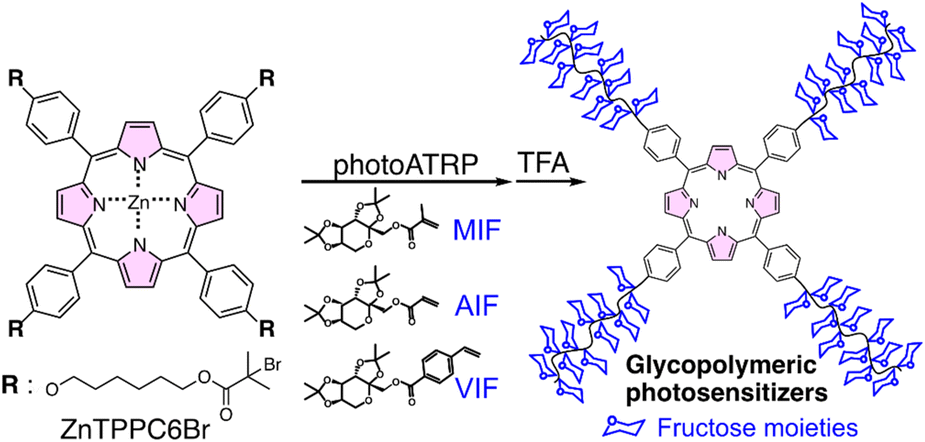 | ||
| Scheme 1 Synthesis of a star-shaped glycopolymeric photosensitizer using ZnTPPC6Br via photoATRP. | ||
Results and discussion
Polymerization
The synthesis of ZnTPPC6Br, demonstrating dual roles in photoATRP, was accomplished and its structure was confirmed through 1H and 13C NMR spectra (Fig. S1 and S2†). Its UV-visible spectrum (Fig. S3†) in DMF was characterized by a strong Soret band at 431 nm and two weaker Q bands at 563 and 605 nm, respectively. The initial absorption Q band ranging from 540–583 nm aligns with the wavelength range of yellow LED lights (560–580 nm), strategically chosen for efficiently exciting the photocatalyst and initiating the photoATRP. Glycomonomers, including AIF, MIF and VIF, were successfully prepared and confirmed via1H and 13C NMR spectra (Fig. S4–S9†). Under degassed conditions, photoATRP of AIF was initially conducted using CuBr2/diverse ligands as the ATRP catalysts in the presence of ZnTPPC6Br as the photocatalyst and initiator under yellow light irradiation (560–580 nm, 15 mW cm−2, Table 1). Considering the solubility of ZnTPPC6Br, which is soluble in DMF but not in DMSO, preliminary testing was conducted with DMF as the solvent. As expected, in the absence of CuBr2, ligands, photosensitizers, or initiators, no monomer conversion or polymer formation was observed (Table 1, entries 1–4). Although it has been reported that Cu(II) can be reduced in the presence of amines,45,46 polymerization using 4 equivalents of EIBB under the same PMDETA to Cu(II) bromide ratio (Table 1, entry 5) resulted in minimal monomer conversion, suggesting that amine-based photoreduction is insignificant. Despite employing an equimolar ratio of PMDETA to Cu(II) bromide (Table 1, entry 6), minimal monomer conversion was observed, indicating a limited polymerization process. Interestingly, increasing the ligand ratio in the photoATRP resulted in enhanced monomer conversion, reduced Đ, and satisfactory molecular weights (Table 1, entries 7–10, Đ < 1.2), highlighting the crucial role of an excess of ligand in initiating polymerization. It was found that a notable difference was observed between the measured and theoretically calculated molar masses of the polymers, likely due to exclusion volume disparities between the synthesized star-shaped polymers and the linear polystyrene standards utilized for SEC calibration. Maintaining a constant ratio of CuBr2 to ligand at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and concurrently increasing the quantity of catalyst compared to the photosensitizer (Table 1, entries 10–13) yielded no substantial alterations in AIF conversion, Mn, and Đ. Substituting the ligand with Me6TREN resulted in an increased Đ (Table 1, entry 14, Đ = 1.31), while a reduction in monomer conversion was noted with TPMA and BPY (Table 1, entries 15–16), potentially attributable to differing ligand activities.47
:10 and concurrently increasing the quantity of catalyst compared to the photosensitizer (Table 1, entries 10–13) yielded no substantial alterations in AIF conversion, Mn, and Đ. Substituting the ligand with Me6TREN resulted in an increased Đ (Table 1, entry 14, Đ = 1.31), while a reduction in monomer conversion was noted with TPMA and BPY (Table 1, entries 15–16), potentially attributable to differing ligand activities.47
| Entry | Ligand | CuBr2:L |
Conv.b (%) | M n,th (g mol−1) | M n (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| a Reaction conditions: [AIF]/[ZnTPPC6Br]/[CuBr2]/[L] = 200/1/x/y (L = PMDETA, Me6TREN, TPMA or BPY; x = 0, 1, 2, 4, or 6; y = 0, 3, 5, 7, 10, 20, 40 or 60) in DMF irradiated for 10 h under yellow LEDs (560–580 nm, 15 mW cm−2). b Calculated by 1H NMR spectra. c Measured by SEC. d Without ZnTPPC6Br. e Replacing ZnTPPC6Br with ZnTPPC6OH (without initiation sites). f Represents no ZnTPPC6Br, with 4 equivalents of EBIB as the initiator. | ||||||
| 1 | PMDETA | 0:10 |
<5 | — | — | — |
| 2 | PMDETA | 1:0 |
<5 | — | — | — |
| 3d | PMDETA | 1:10 |
<5 | — | — | — |
| 4e | PMDETA | 1:10 |
<5 | — | — | — |
| 5f | PMDETA | 1:10 |
<5 | — | — | — |
| 6 | PMDETA | 1:1 |
<5 | — | — | — |
| 7 | PMDETA | 1:3 |
13 | 9900 | 4700 | 1.02 |
| 8 | PMDETA | 1:5 |
27 | 18700 |
3900 | 1.03 |
| 9 | PMDETA | 1:7 |
27 | 18700 |
4100 | 1.04 |
| 10 | PMDETA | 1:10 |
57 | 38800 |
11600 |
1.15 |
| 11 | PMDETA | 2:20 |
51 | 33800 |
9800 | 1.11 |
| 12 | PMDETA | 4:40 |
58 | 38200 |
9900 | 1.13 |
| 13 | PMDETA | 6:60 |
57 | 37600 |
7000 | 1.21 |
| 14 | Me6TREN | 1:10 |
55 | 37000 |
15900 |
1.31 |
| 15 | TPMA | 1:10 |
34 | 23000 |
6000 | 1.11 |
| 16 | BPY | 1:10 |
<5 | — | — | — |
By significantly enhancing efficiency and safety, oxygen-tolerant ATRP plays a crucial role in free radical polymerization processes and finds broad applications in fields including pharmaceuticals, advanced materials manufacturing, and bioengineering.48,49 Upon reviewing the conditions delineated in Table 1 (entry 10), it was observed that non-deoxygenated vials accounted for an 18% monomer conversion of AIF, highlighting the weak oxygen-tolerance polymerization (Table 2, entry 1). The influence of various factors such as cosolvents, reducing agents, and monomers on photoATRP has been reported and systematically examined.50–52 Accounting for the potential role of DMSO in the deoxidation process,53,54 the effects of the DMSO/DMF mixture were initially investigated based on the optimized polymerization conditions. Introducing DMSO in sealed vials facilitated the photoATRP of AIF, forming glycopolymers without requisite deoxygenation. Increased DMSO proportions led to favorable adjustments in molecular weight and Đ (Table 2, entries 2–5). By utilizing a DMF to DMSO ratio of 4:6, a glycopolymer was synthesized with a Mn of 11700 g mol−1 and a relatively narrow Đ of 1.30 (Table 2, entry 4), meriting subsequent investigative study.
| Entry | Monomer | Solvent | Conv.b (%) | M n,th (g mol−1) | M n (g mol−1) | Đ |
|---|---|---|---|---|---|---|
|
a Reaction conditions: [M]/[ZnTPPC6Br]/[CuBr2]/[L] = 200/1/1/10, irradiated for 10 h under yellow LEDs (560–580 nm, 15 mW cm−2) in 4 mL vials sealed with a stopper without deoxygenation, using a 2 mL 4:6 DMF:DMSO mixture as the solvent.
b Calculated by 1H NMR spectra.
c Measured by SEC.
d Reaction vials without stoppers and deoxygenation.
e Carried out for 4 h.
f Initiated with EBIB.
g Initiated with PETB.
h Represents no stoppers conditions.
i Represents reaction with stoppers. The numbers enclosed in parentheses indicate varying quantities of solvent.
|
||||||
| 1 | AIF | DMF | 18 | 13000 |
2900 | 1.28 |
| 2 | AIF | DMF:DMSO = 8:2 |
22 | 15600 |
3000 | 1.27 |
| 3 | AIF | DMF:DMSO = 6:4 |
36 | 24400 |
7600 | 1.35 |
| 4 | AIF | DMF:DMSO = 4:6 |
55 | 36300 |
11700 |
1.30 |
| 5 | AIF | DMF:DMSO = 2:8 |
57 | 37600 |
11000 |
1.31 |
| 6 | AIFd | DMF:DMSO = 4:6 |
88 | 57000 |
20700 |
2.10 |
| 7 | AIF | DMF:DMSO = 4:6 (1 mL) |
49 | 32500 |
12300 |
1.30 |
| 8 | AIF | DMF:DMSO = 4:6 (4 mL) |
52 | 34400 |
21800 |
1.20 |
| 9 | MIFe | DMF:DMSO = 4:6 (4 mL) |
94 | 63500 |
19200 |
1.21 |
| 10 | VIF | DMF:DMSO = 4:6 (4 mL) |
73 | 58700 |
22400 |
1.06 |
| 11 | MIFf | DMF:DMSO = 4:6 (4 mL) |
55 | 36300 |
10200 |
1.45 |
| 12 | MIFg | DMF:DMSO = 4:6 (4 mL) |
66 | 43200 |
7400 | 1.37 |
| 13 | MIFh | DMF:DMSO = 4:6 (4 mL) |
33 | 22500 |
3200 | 1.33 |
| 14 | MIFi | DMF:DMSO = 4:6 (4 mL) |
29 | 19900 |
3300 | 1.32 |
Despite facilitating the synthesis of a high-molecular-weight glycopolymer, the open flask conditions led to an uncontrollable ATRP, as underscored by a broad Đ of 2.10 (Table 2, entry 6). The increased polymerization rate observed in the presence of oxygen can be attributed to the enhanced Cu(I)/Cu(II) redox cycle, where the rapid oxidation of Cu(I) by oxygen accelerates the catalytic cycle, thereby increasing the availability of active Cu(I) species for chain activation and growth.55,56 Triethylamine (TEA) and triethanolamine (TEOA) play essential roles in controlling the polymerization process and safeguarding the catalyst during free radical polymerization,57,58 prompting further investigation into their functions. The addition of TEA and TEOA expedited polymerization, resulting in higher molecular weight polymers with increased Đ, reaching up to 1.47, in setups with and without stoppers under non-degassing conditions (Table S1†). It was reported that regulating the solution volume in the vial can influence the oxygen content of the mixture, ultimately controlling the polymerization process.59 Compared to the conditions of smaller solution volumes (Table 2, entries 4 and 7 for 2 and 1 mL, respectively), conducting a photoATRP of AIF with a nearly full vial (4 mL) yielded a Đ of 1.20 and a Mn of 21800 g mol−1 (Table 2, entry 8). By replacing the AIF monomer with MIF and VIF, significant discrepancies in polymerization were noted. The MIF exhibited a 94% conversion and a Đ of 1.21 following 4 h of yellow light irradiation (Table 2, entry 9). The VIF demonstrated a 73% conversion with a Đ of 1.03 after 10 h of light exposure (Table 2, entry 10). To demonstrate the benefits of the ZnTPPC6Br, linear and star-shaped glycopolymers were synthesized using the photocatalyst and initiator concentrations specified in Table 2, entry 9, with EIBB and PETB serving as initiators and ZnTPPC6OH as the photocatalyst (Table 2, entries 11 and 12). The decreased conversion of monomers, lower than the expected Mns, and wider Đ of the both resulting linear and star-shaped glycopolymers suggest that the porphyrin conjugated with the ATRP initiator may facilitate the photoATRP process. To compare ZnTPPC6Br with the related TPPC6Br in polymerization, additional photoATRP was conducted in both open and sealed vials (Table 2, entries 13 and 14). The results showed a reduction in both monomer conversion and the molecular weight of the resulting polymers. This finding underscores the superior performance of ZnTPPC6Br over TPPC6Br in improving polymerization efficiency, underscoring the crucial role of zinc in the system.
Kinetics and temporal control of polymerization
The ZnTPPC6Br/CuII/PMDETA photoATRP was performed in a 4 mL vial under the conditions specified in Table 2 (entry 10), with the mixture rapidly cooled using liquid nitrogen at the designated time for subsequent analysis by 1H NMR and SEC. Linear semilogarithmic kinetic plots were observed for various glycomonomers. The kinetics of the photoATRP of MIF indicated that the polymerization proceeded with the highest apparent rate constant of kp = 0.64 h−1, followed by VIF at kp = 0.22 h−1, and AIF exhibiting the slowest rate at kp = 0.06 h−1 (Fig. 1A). The polymer molecular weight increased proportionally with monomer conversion, as evidenced by the SEC traces shifting towards shorter elution times as polymerization progressed (Fig. S11†), demonstrating high initiator efficiency and well-controlled polymerizations with relatively low Đ (<1.20) (Fig. 1B).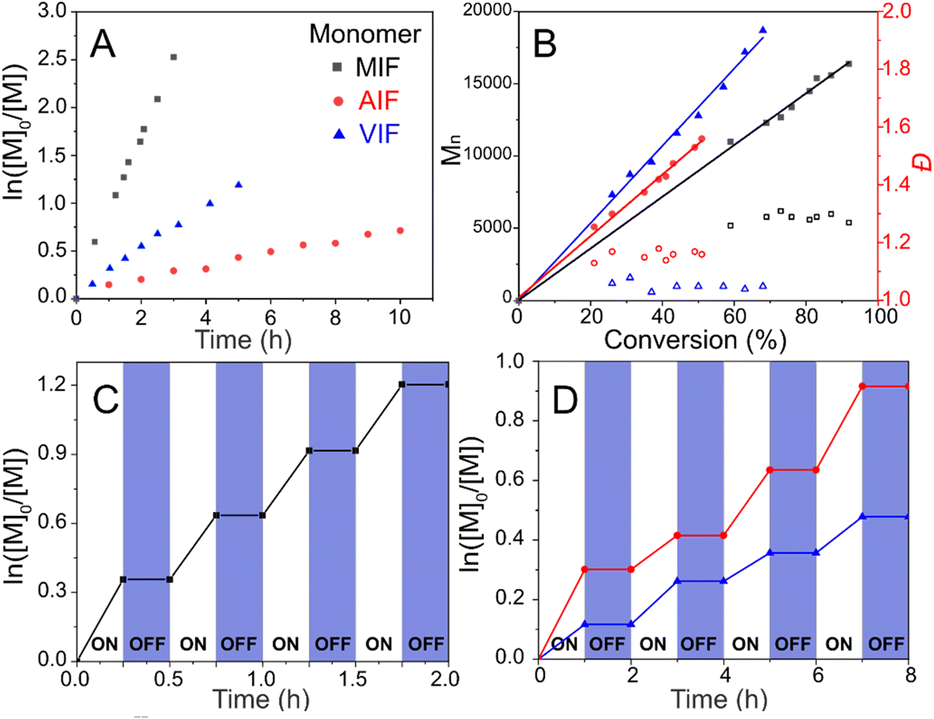 | ||
| Fig. 1 ZnTPPC6Br/CuII/PMDETA photoATRP: (A) first-order kinetic plots, (B) evolution of molar masses and Đ with monomer conversion, and temporal control of (C) MIF, (D) AIF (red) and VIF (blue). Polymerization conditions: [M]/[ZnTPPC6Br]/[CuBr2]/[PMDETA] = 200/1/1/10, [ZnTPPC6Br] = 8.66 mM, irradiated under yellow LEDs (560–580 nm, 15 mW cm−2) in a sealed 4 mL vial containing 4 mL of DMF/DMSO solvent (4/6, v/v). | ||
Moreover, the photoATRP of three glycomonomers showed temporal control through light activation and deactivation, as shown in Fig. 1C and D. Polymerizations advanced under light irradiation, with minimal monomer conversion observed when the light was turned off. The CuI/L activator can be consumed via radical termination to transform into the deactivator CuII/L-Br, halting polymerization in the absence of light. Upon reirradiating the mixture with light, polymerization resumed, triggered by the photoexcitation of ZnTPPC6Br. Multiple cycles of alternating yellow light on and off showcased exceptional temporal control in the polymerization process.
Varying targeted degree of polymerization and chain extension
The degree of polymerization of glycomonomers varied from 10 to 50 monomer units per arm to produce star-shaped glycopolymers with diverse molecular weights (Table S2†). Twelve glycopolymers were synthesized by varying the glycomonomer concentration while maintaining the constant concentration of other reagents, using three corresponding glycomonomers at feed ratios ranging from 40 to 200. The resulting star-shaped glycopolymers displayed predictable Mns and low Đ values, indicating that the ZnTPPC6Br/CuII/PMDETA photoATRP system offers precise control in synthesizing glycopolymers with diverse molecular weights from different monomers. The length, composition, and versatility of glycoarms in polymer play a significant role in targeted therapy,60 and the established design and synthesis approach have the potential to expedite further research in this area.To determine the chain terminal fidelity of polymers synthesized via ZnTPPC6Br/CuII/PMDETA photoATRP, ZnTPP-P(MIF20)4 (Mn = 12900, Đ = 1.22, Table S2,† entry 6) served as a macroinitiator for extending the chain of additional MIF. The resulting glycopolymer had a Mn of 37400 and a low Đ of 1.23. The SEC traces displayed a distinct shift towards the high molecular weight range, devoid of tailing and shoulder peaks in the low molecular weight region (Fig. S12†). A similar trend was observed in the chain extension of ZnTPP-P(MIF20)4 with OEGMA, resulting in ZnTPP-P(MIF20)4-b-P(OEGMA20)4 (Mn = 25000, Đ = 1.24) (Fig. S12†).
PhotoATRP mechanism initiated by ZnTPPC6Br
In the presence of light, PCs can undergo a transition to a singlet excited state (1PC*) and subsequently to a triplet excited state (3PC*) through intersystem crossing, which may be utilized for the activation of the CuII/L catalyst during photoATRP. Transient fluorescence spectroscopy was utilized to investigate the interaction between the 1PC* and CuII/L catalyst. The fluorescence decay kinetics of ZnTPPC6Br remained unchanged upon the addition of CuBr2/PMDETA or PMDETA only, suggesting that these compounds did not quench the 1PC* of ZnTPPC6Br (Fig. 2A). Steady-state fluorescence experiments indicated that the 3PC* was quenched upon addition of CuBr2/PMDETA (1:1) (Fig. 2B and C). Conversely, the presence of solely PMDETA (Fig. 2C and D) did not lead to a reduction in fluorescence intensity. These findings validate that the 3PC* reacts with CuII/L to produce the PC radical cation (PC˙+)61 and the activator CuI/L, which are utilized to initiate photoATRP. The employment of an excess ligand is essential, as it can react with PC˙+ to facilitate PC return to the ground state, enabling the continuous polymerization process, as supported by the findings outlined in Table 1, entries 6–10.
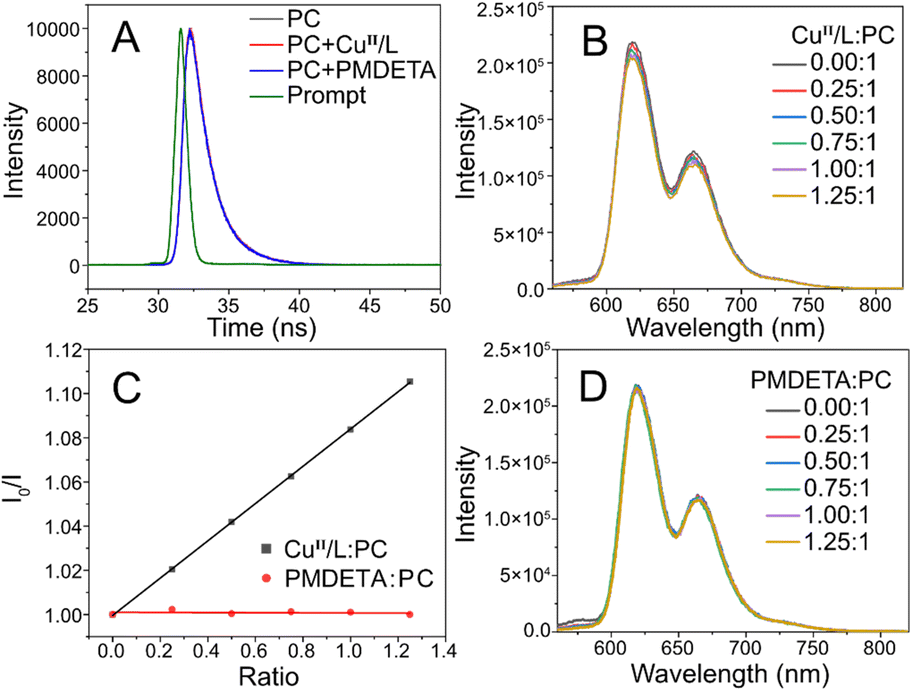 | ||
| Fig. 2 (A) Transient absorption decay profiles at 630 nm of ZnTPPC6Br (8.9 μM) recorded in the absence and presence of CuII/L (8.9 μM) or PMDETA (8.9 μM) after excitation at 407 nm; emission spectra of ZnTPPC6Br excited at 420 nm upon addition of different ratios of (B) CuII/L and (D) PMDETA as a quencher of 3PC*; (C) the variation in fluorescence intensity of ZnTPPC6Br in the presence of CuII/L or PMDETA as a quencher, with varying ratios to PC. | ||
Upon light irradiation, porphyrin can undergo photochemical reactions with oxygen, generating various reactive oxygen species (ROS) and depleting the dissolved oxygen present in the solution.62 To investigate the ROS species generated by ZnTPPC6Br in photoATRP, 1,3-diphenylisobenzofuran (DPBF), known for its high sensitivity and rapid degradation in response to singlet oxygen (1O2) attributed to its low β value.63 Additionally, 3,3′,5,5′-tetramethyl benzidine (TMB) can react with hydroxyl radicals (OH˙) at a rate constant of 11.8 × 10−12 cm3 per molecule per s at 298 K,64 while nitro blue tetrazolium chloride (NBT) was selected for selective detection of superoxide anions (O2˙−).65 During light irradiation, DPBF exhibited substantial degradation in the presence of all polymerization components except the monomer, reaching complete degradation within 60 s (Fig. S13A†). In contrast, there were minimal UV absorbance changes for NBT and TMB (Fig. S13B and C†). These observations suggest that oxygen is mainly consumed in the polymerization process through the conversion of O2 to 1O2. The addition of DMSO can enhance the monomer conversion and boost the apparent polymerization rate of photoATRP by facilitating its reaction with 1O2 and aiding in oxygen depletion in the system, as evidenced by the results presented in Table 2 entries 1–5.
Taking into account these findings and the quenching results of active substances, a photoATRP mechanism initiated by ZnTPPC6Br is proposed (Scheme 2). Under light irradiation, the ZnTPPC6Br transitions form a singlet excited state (1PC*) to a triplet excited state (3PC*), allowing it to react with the CuII/L to generate the CuI/L activator and subsequently initiate photoATRP. The excess ligand reacts with the active photocatalyst species to facilitate its return to the ground state, enabling the continuous photoATRP. Furthermore, the DMSO can react with 1O2 produced by the photochemical reaction between PC and O2 under light irradiation, assisting in depleting oxygen during the polymerization process.
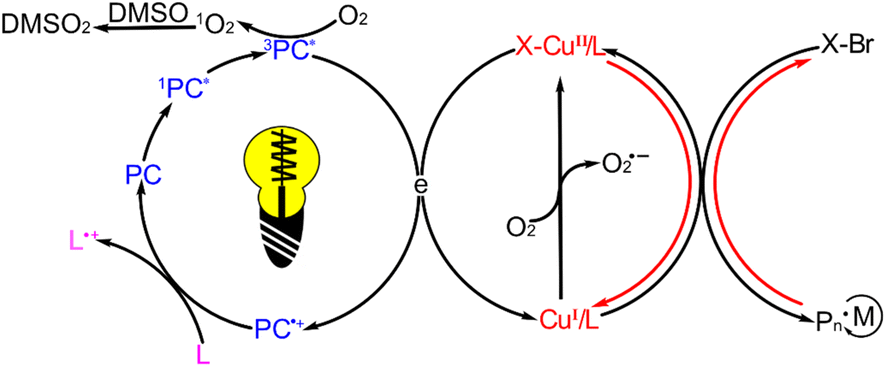 | ||
| Scheme 2 Proposed photoATRP mechanism under light irradiation using ZnTPPC6Br. | ||
Scaling up PhotoATRP
Scale-up polymerizations of three glycomonomers were conducted to assess the reproducibility and efficiency of the ZnTPPC6Br/CuII/PMDETA photoATRP system. The mixture volumes were scaled up to nearly fill a 100 mL flask, with consistent reactant ratios and conditions maintained under yellow light irradiation (Fig. S14†). Subsequently, the mixtures were poured into cold methanol to precipitate the resulting glycopolymers and remove any remaining unreacted monomers and CuII/L catalyst. The 1H NMR spectra of the obtained star-shaped polymers with protective isopropylidene groups are presented in Fig. S15.† Proton signals at δ 1.2–1.6 correspond to the protons adjacent to the isopropyl groups, while the signal at δ 4.62 is assigned to protons on the anomeric carbon of the sugar ring. All polymers displayed proton signals at δ 8.87 and 8.16, which were characteristic of ZnTPPC6Br, confirming the incorporation of porphyrin into the star-shaped polymers. The polymer compositions were calculated by integrating the characteristic peaks of porphyrin and glycomonomers, demonstrating a close alignment with the original monomer feed ratio (Table 3). SEC traces confirmed the successful synthesis of star-shaped polymer precursors with predictable molecular weights and relatively narrow Đ values (Fig. S16†). The prepolymers were deprotected in TFA/H2O (8:2, v/v) for 3 h, followed by dialysis against deionized water for 3 days to obtain the final products after freeze-drying. The 1H NMR spectra of three deprotected star-shaped polymers showed the absence of protective groups and the presence of the NH proton from the porphyrin center (Fig. S17†), indicating both the formation of the resulting glycopolymer and the removal of Zn coordination within the porphyrin structure.
| Glycopolymer | Conv.a (%) | M n,th , (g mol−1) | M n (g mol−1) | Đ | TPP wt% (theo./obs.) | Φ F | Φ Δ |
|---|---|---|---|---|---|---|---|
| a Calculated by 1H NMR spectra. b Evaluated via copolymers featuring isopropylidene groups. c Assessed through the examination of UV-vis and fluorescence spectra utilizing glycopolymers after deprotection. | |||||||
| ZnTPP-P(MIF20)4 | 95 | 26700 |
15200 |
1.25 | 2.42/1.54 | 0.274 | 0.445 |
| ZnTPP-P(AIF20)4 | 85 | 23100 |
12600 |
1.21 | 2.53/2.44 | 0.291 | 0.427 |
| ZnTPP-P(VIF20)4 | 93 | 29800 |
18000 |
1.02 | 2.06/2.77 | 0.299 | 0.436 |
Photophysical properties and ROS generation of star-shaped glycopolymers
The resulting glycopolymer solutions in DMF and water are presented in Fig. S18.† Compared to TPP-P(MF20)4 and TPP-P(AF20)4, TPP-P(VF20)4 demonstrated insolubility in water, possibly attributed to the hydrophobic benzene ring in the modified fructose moiety. The porphyrin content within the polymers significantly influences their PDT performance, which was quantified by comparison with a calibration curve (Fig. S19†) outlined in Table 3. The results suggested that the porphyrin content closely aligned with the theoretical values, with any minor deviations potentially attributed to experimental errors or the elimination of porphyrin interactions by the glycoarms. The UV-visible spectra of these polymer solutions were recorded, as shown in Fig. S20A and B.† In DMF, all polymer spectra displayed a Soret band at 422 nm along with four weaker Q-bands at 518, 556, 596, and 650 nm, respectively. In water, the spectra of the soluble polymers also exhibited a Soret band at 423 nm and Q-bands at 521, 557, 595, and 652 nm, while the TPP-P(VF20)4 polymer, insoluble in water, showed no absorbance in the upper aqueous layer. The shifts in the positions of the Soret and Q-bands may be attributed to the solvent effects.66 Upon excitation at 420 nm, all polymeric photosensitizers exhibited prominent fluorescence emission peaks at 660 and 725 nm (Fig. S21A and B†), which were evident in both DMF and water, except for the water-insoluble TPP-P(VF20)4. These bands were assigned to the Q (0–0) and Q (0–1) transitions of the porphyrin core present in the star-shaped glycopolymers. The fluorescence quantum yield (ΦF), indicative of photon conversion efficiency, was determined using tetraphenylporphyrin as a standard, as shown in Table 3. The ΦF values for the three polymers ranged between 0.27 and 0.30, reflecting their strong luminescence efficiency.The ability of the polymeric photosensitizers to generate 1O2 was assessed using DPBF, which is known for its susceptibility to photobleaching by 1O2 (Scheme S1†). The DPBF solution showed 20% self-degradation under light irradiation within 30 s (Fig. S22†). Upon the introduction of the polymer photosensitizers, the absorbance of DPBF significantly decreased during the irradiation, becoming nearly undetectable after 30 s. This underscores the impressive 1O2 generation capability of the star-shaped glycopolymeric photosensitizers, highlighting their potential for PDT applications. The singlet oxygen quantum yield (ΦΔ) is another crucial parameter for assessing photosensitizers, where a higher ΦΔ value signifies a more effective generation of 1O2 upon light exposure.67 The ΦΔ values of the resulting polymers were tested using DPBF as a probe, as presented in Table 3. It is noteworthy that all polymers exhibited ΦΔ values above 0.4, indicating a satisfactory ability to generate 1O2. In addition, the solution properties of the glycopolymers were investigated using Dynamic Light Scattering (DLS), as shown in Fig. S23.† The findings revealed that all water-soluble polymers exhibited small hydrodynamic diameters (all under 30 nm), and possessed a narrow size distribution.
Cellular uptake of star-shaped glycopolymeric photosensitizers
Cells can take up sugar-containing polymers either through nonspecific endocytosis or through specific uptake facilitated by sugar transporters.68 In our previous work, it was found that a high density of fructose with longer glycoarms on star-shaped photosensitizers was crucial for achieving optimal uptake by MCF-7 cells through GLUT5 receptors.15 In this work, we aim to evaluate the efficacy of star-shaped glycopolymeric photosensitizers synthesized using photoATRP for targeted PDT. The variances in endocytosis of two glycopolymers by L929 and MCF-7 cells, sourced from the National Collection of Authenticated Cell Cultures in Shanghai, China, were investigated by conducting uptake experiments with the water-soluble TPP-P(MF20)4 and TPP-P(AF20)4. To explore the specific uptake facilitated by sugar transporters, free fructose (F), a fructose-based homopolymer (F100, synthesized via RAFT polymerization), and (−)-epicatechin gallate (ECG), a designated inhibitor of the glucose transporter 5 (GLUT5), were employed.In the L929 group (Fig. 3A), both polymers exhibited similar mean fluorescence intensity (MFI) due to the lack of fructose-binding sites, and the impact on cellular uptake remained consistent even after the introduction of the three inhibitors. Conversely, the MCF-7 group showed an elevated MFI following an 18 h co-incubation with both polymers, suggesting an enhanced endocytosis of the fructose-containing polymer photosensitizers (Fig. 3B). However, pre-incubating MCF-7 cells with the inhibitors for 6 h prior to exposure to the polymers resulted in a significant reduction in polymer uptake. These results suggest that the three endocytosis inhibitors likely occupied the GLUT5, hindering the binding of fructose-containing polymer photosensitizers to MCF-7, consequently impeding endocytosis.
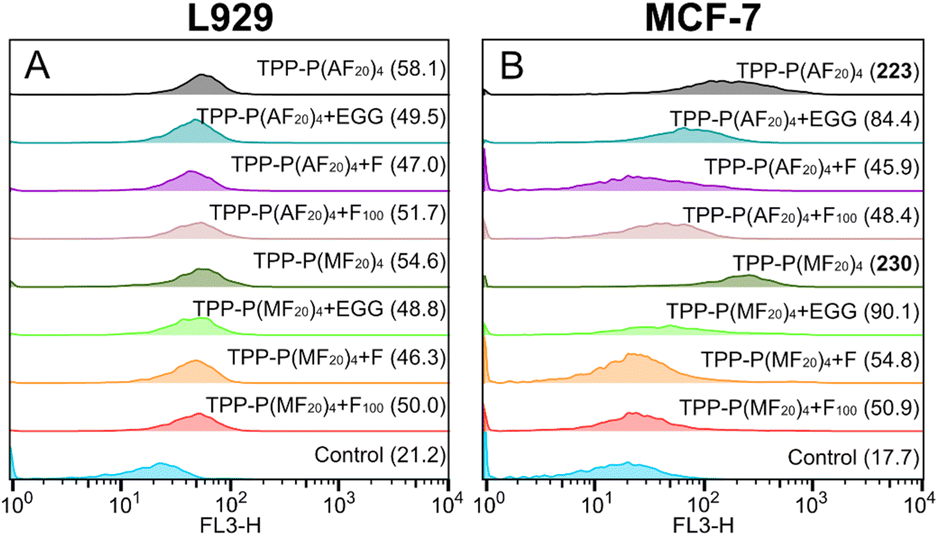 | ||
| Fig. 3 (A) L929 and (B) MCF-7 pretreated with F, F100 or EGG were assayed for endocytosis by flowcytometry after 18 h of incubation with polymer photosensitizers, where the values in brackets indicate the MFI. | ||
In vitro PDT of star-shaped glycopolymeric photosensitizers
In our previous study, a porphyrin concentration of 442.5 μM was sufficient to completely eradicate cancer cells after 1 h of light exposure.15 Consequently, the same porphyrin concentration was utilized for evaluating both dark toxicity and phototoxicity, while control experiments included the complete culture medium and polymer solutions with porphyrin concentrations of 147.5 μM and 295 μM.The cytotoxicity of both polymers against L929 and MCF-7 was assessed using the CCK-8 assay under both dark and light conditions (Fig. 4A). The exceptional biocompatibility of the fructose moieties ensured that both polymers did not show noteworthy cytotoxicity in the dark, maintaining cell viability at approximately 100% even at porphyrin concentrations as high as 442.5 μM. Nevertheless, upon light irradiation, a notable and concentration-dependent decrease in cell viability was observed, indicating that the polymer photosensitizers effectively generated 1O2, leading to cancer cell damage. It is noteworthy that L929 treated with the polymeric photosensitizers displayed a viability of about 16%, whereas MCF-7 showed nearly 0% viability. This difference may be attributed to the enhanced cellular uptake of MCF-7 towards fructose-containing polymeric photosensitizers. Live-dead staining results using calcein-AM (green) and propidium iodide (PI, red) were in agreement with the CCK-8 assay findings (Fig. 4B and S24†). Under dark conditions, minimal red spots were observed, suggesting limited internalization of the porphyrin-containing polymers by the cells. Conversely, under light exposure, escalating concentration of the polymeric photosensitizer led to a decrease in green fluorescence intensity alongside an increase in red fluorescence intensity. These findings demonstrate the favorable biocompatibility of star-shaped glycopolymers in the dark and their robust 1O2 generation ability under light irradiation, highlighting the potential of glycopolymeric photosensitizers as promising assets in PDT.
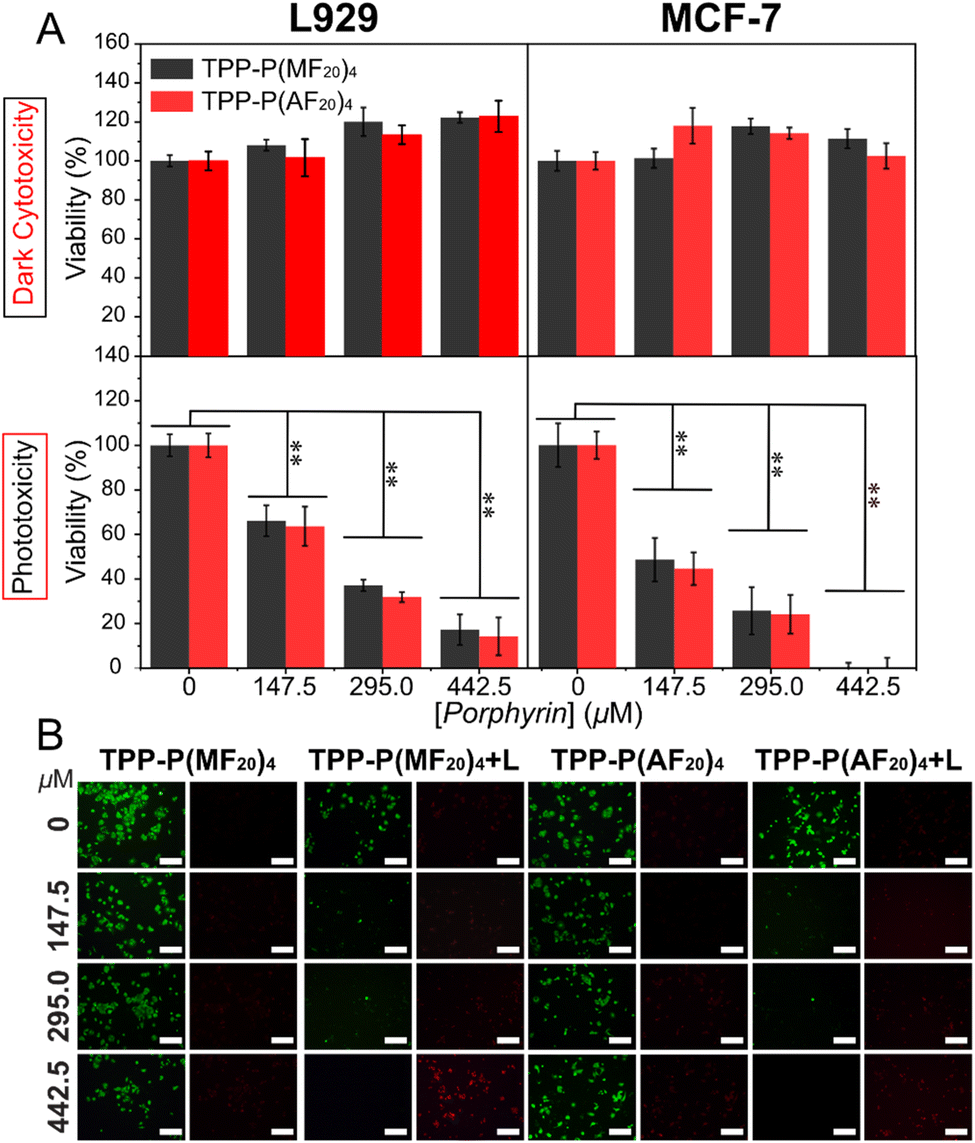 | ||
| Fig. 4 (A) Evaluation of dark cytotoxicity and phototoxicity (white LED light, 20 mW cm−2) in L929 and MCF-7 cells treated with star-shaped glycopolymeric photosensitizers. Values are mean ± standard deviation analysis (n = 3) with significance level **p < 0.01. (B) Live/dead cell staining of MCF-7 cells under various treatments (green for live cells, red for dead cells; scale bar: 200 μm). | ||
The intracellular ROS generation induced by glycopolymeric photosensitizers was further investigated using DFCH-DA as a probe. Under dark conditions, cells incubated with the polymer photosensitizers exhibited minimal green fluorescence, but upon 10 min of light irradiation, pronounced green fluorescent signals indicative of ROS generation within the cells were observed (Fig. S25†). Under light irradiation, MCF-7 cells exhibited higher green fluorescence intensity compared to L929 cells treated with the same glycopolymeric photosensitizer. The difference in intracellular ROS production between the two cell lines is likely due to the enhanced endocytosis of the fructose-containing polymeric photosensitizer by MCF-7 cells, leading to increased ROS generation and stronger fluorescence intensity under light conditions. These findings were consistent with the results of targeted cell uptake. Flow cytometry was utilized to investigate the MFI changes induced by intracellular ROS generation, aligning with the fluorescence staining observations (Fig. S26†).
Conclusions
In this study, triple functional ZnTPPC6Br was synthesized for serving as both the photocatalyst and initiator in photoATRP, as well as acting as a photosensitizer in PDT. Polymerization condition scanning revealed that ZnTPPC6Br/CuII/ligands could synthesize star-shaped fructose-containing polymers with predictable Mn and low Đ, even without degassing the reaction. Mechanistic studies unveiled the transition of ZnTPPC6Br from a singlet excited state to a triplet excited state, enabling it to react with CuII to generate the activator CuI/L and initiate photoATRP. The excess ligand addition facilitates the return of active photocatalyst species to the ground state, enabling continuous photoATRP, while DMSO reacts with 1O2 produced during the photochemical reaction between the photocatalyst and oxygen under light irradiation, assisting in oxygen depletion throughout the polymerization process. The ZnTPPC6Br/CuII/PMDETA system exhibited excellent temporal control over polymerization for three fructose-based monomers, allowing the synthesis of polymers with varying degrees of polymerization. This resulted in the generation of two glycopolymers with high solubility in both water and DMF. The presence of characteristic porphyrin peaks in UV-visible, 1H NMR and fluorescence spectra confirmed the integrity of the porphyrin structure within the star-shaped polymers, which acts as a photosensitizer for the robust 1O2 and fluorescence generation capabilities of the resulting glycopolymers. In vitro cellular experiments showed that star-shaped glycopolymers were more effectively endocytosed by MCF-7 cells, thereby enhancing the PDT efficacy against MCF-7 cells and showcasing their potential for precise cancer treatment in comparison to L929 cells. The versatile attributes of ZnTPPC6Br are expected to provide a versatile pathway for the development of cell-specific targeted glycopolymers, leveraging its potential as a photosensitizer to advance PDT.Data availability
The data supporting this article have been included in the ESI.† Additional data are available upon request from the corresponding author.Author contributions
Jiahui Lin: methodology, formal analysis, investigation, writing – original draft. Zhiyuan Ma: methodology, writing – review & editing, supervision, project administration, funding acquisition. Weiwei Zuo and Meifang Zhu: formal analysis, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52003047).Notes and references
- M. Kurzyna-Szklarek, J. Cybulska and A. Zdunek, Food Chem., 2022, 394, 133466 CrossRef CAS PubMed.
- G. A. Valencia, E. N. Zare, P. Makvandi and T. J. Gutiérrez, Compr. Rev. Food Sci. Food Saf., 2019, 18, 2009–2024 CrossRef CAS PubMed.
- T. Pelras and K. Loos, Prog. Polym. Sci., 2021, 117, 101393 CrossRef CAS.
- S. Mizrahy and D. Peer, Chem. Soc. Rev., 2012, 41, 2623–2640 RSC.
- S. Li, N. Wang, B. Yu, W. Sun and L. Wang, Nat. Chem., 2023, 15, 33–42 CrossRef CAS PubMed.
- N. Sharon, Trends Biochem. Sci., 1993, 18, 221–226 CrossRef CAS PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–4503 RSC.
- E. C. Woods, N. A. Yee, J. Shen and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 15782–15788 CrossRef CAS PubMed.
- J. S. Basuki, L. Esser, H. T. T. Duong, Q. Zhang, P. Wilson, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Chem. Sci., 2014, 5, 715–726 RSC.
- D. A. Mitchell, Q. Zhang, L. Voorhaar, D. M. Haddleton, S. Herath, A. S. Gleinich, H. S. Randeva, M. Crispin, H. Lehnert, R. Wallis, S. Patterson and C. R. Becer, Chem. Sci., 2017, 8, 6974–6980 RSC.
- S. Liese and R. R. Netz, ACS Nano, 2018, 12, 4140–4147 CrossRef CAS PubMed.
- J. Gu, Y. Li, G. Lu, Y. Ma, Y. Zhang and J. Chen, Int. J. Biol. Macromol., 2023, 253, 126975 CrossRef CAS PubMed.
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- J. Lin, Z. Ma, W. Zuo and M. Zhu, Biomacromolecules, 2024, 25, 1950–1958 CrossRef CAS PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- I. Pramudya and H. Chung, Biomater. Sci., 2019, 7, 4848–4872 RSC.
- L. Su, Y. Feng, K. Wei, X. Xu, R. Liu and G. Chen, Chem. Rev., 2021, 121, 10950–11029 CrossRef CAS PubMed.
- S. C. Purcell, M. H. Zhang, D. J. Honigfort, H. J. C. Ng, A. L. Michalak and K. Godula, Chem. Sci., 2022, 13, 6626–6635 RSC.
- A. Ghadban and L. Albertin, Polymers, 2013, 5, 431–526 CrossRef.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- K. Ohno, Y. Tsujii and T. Fukuda, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2473–2481 CrossRef CAS.
- Y.-M. Chen and G. Wulff, Macromol. Rapid Commun., 2002, 23, 59–63 CrossRef CAS.
- D. Santo, R. A. Cordeiro, P. V. Mendonca, A. C. Serra, J. F. J. Coelho and H. Faneca, Biomacromolecules, 2023, 24, 1274–1286 CrossRef CAS PubMed.
- N. N. M. Rao, K. K. Palodkar, T. S. Kumar, V. Sadhu, T. M. Aminabhavi, R. R. Kakarla and A. V. S. Sainath, Int. J. Biol. Macromol., 2023, 237, 124119 CrossRef CAS PubMed.
- L. Zhao, Y. Li, D. Pei, Q. Huang, H. Zhang, Z. Yang, F. Li and T. Shi, Carbohydr. Polym., 2019, 205, 167–175 CrossRef CAS PubMed.
- M. H. Stenzel, Macromolecules, 2022, 55, 4867–4890 CrossRef CAS.
- S. R. S. Ting, G. J. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- C. Zhang, X. Hu, L. Jin, L. Lin, H. Lin, Z. Yang and W. Huang, Adv. Healthcare Mater., 2023, 12, 2300530 CrossRef CAS PubMed.
- C. Ji, Q. Gao, X. Dong, W. Yin, Z. Gu, Z. Gan, Y. Zhao and M. Yin, Angew. Chem., Int. Ed., 2018, 57, 11384–11388 CrossRef CAS PubMed.
- M. Kolarikova, B. Hosikova, H. Dilenko, K. Barton-Tomankova, L. Valkova, R. Bajgar, L. Malina and H. Kolarova, Med. Res. Rev., 2023, 43, 717–774 CrossRef CAS PubMed.
- M. Warszyńska, P. Repetowski and J. M. Dąbrowski, Coord. Chem. Rev., 2023, 495, 215350 CrossRef.
- X. Zhou, C. Shi, S. Long, Q. Yao, H. Ma, K. Chen, J. Du, W. Sun, J. Fan, B. Liu, L. Wang, X. Chen, L. Sui, K. Yuan and X. Peng, ACS Cent. Sci., 2023, 9, 1679–1691 CrossRef CAS PubMed.
- N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248–7268 RSC.
- H. Zhou, D. Tang, Y. Yu, L. Zhang, B. Wang, J. Karges and H. Xiao, Nat. Commun., 2023, 14, 5350 CrossRef CAS PubMed.
- C. Kütahya, C. Schmitz, V. Strehmel, Y. Yagci and B. Strehmel, Angew. Chem., Int. Ed., 2018, 57, 7898–7902 CrossRef PubMed.
- X. Wang, J. Peng, C. Meng and F. Feng, Chem. Sci., 2024, 15, 12234–12257 RSC.
- P. R. Judzewitsch, N. Corrigan, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 24248–24256 CrossRef CAS PubMed.
- S. Dadashi-Silab, K. Kim, F. Lorandi, G. Szczepaniak, S. Kramer, L. Peteanu and K. Matyjaszewski, ACS Macro Lett., 2022, 11, 376–381 CrossRef CAS PubMed.
- X. Luo, J. Wan, N. Meckbach, V. Strehmel, S. Li, Z. Chen and B. Strehmel, Angew. Chem., Int. Ed., 2022, 61, e202208180 CrossRef CAS PubMed.
- Y. Zhang, D. S. Chen, Z. F. Guo, Z. H. Wei, X. C. Zhang and H. Z. Xing, New J. Chem., 2020, 44, 5235–5242 RSC.
- F. Hu, S. Xu and B. Liu, Adv. Mater., 2018, 30, 1801350 CrossRef PubMed.
- K. Wei, Y. Wu, X. Zheng, G. Ma, C. Ji and M. Yin, Adv. Funct. Mater., 2023, 33, 2305187 CrossRef CAS.
- T. G. Ribelli, M. Fantin, J.-C. Daran, K. F. Augustine, R. Poli and K. Matyjaszewski, J. Am. Chem. Soc., 2018, 140, 1525–1534 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953–4959 CrossRef CAS.
- K. Kapil, A. M. Jazani, G. Szczepaniak, H. Murata, M. Olszewski and K. Matyjaszewski, Macromolecules, 2023, 56, 2017–2026 CrossRef CAS PubMed.
- W. Yan, S. Dadashi-Silab, K. Matyjaszewski, N. D. Spencer and E. M. Benetti, Macromolecules, 2020, 53, 2801–2810 CrossRef CAS.
- S. A. Mountaki, R. Whitfield, E. Liarou, N. P. Truong and A. Anastasaki, J. Am. Chem. Soc., 2024, 146, 18848–18854 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- S. Dadashi-Silab, F. Lorandi, M. J. DiTucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef CAS PubMed.
- X. Hu, G. Szczepaniak, A. Lewandowska-Andralojc, J. Jeong, B. Li, H. Murata, R. Yin, A. M. Jazani, S. R. Das and K. Matyjaszewski, J. Am. Chem. Soc., 2023, 145, 24315–24327 CrossRef CAS PubMed.
- N. Corrigan, D. Rosli, J. W. J. Jones, J. Xu and C. Boyer, Macromolecules, 2016, 49, 6779–6789 CrossRef CAS.
- G. Szczepaniak, J. Jeong, K. Kapil, S. Dadashi-Silab, S. S. Yerneni, P. Ratajczyk, S. Lathwal, D. J. Schild, S. R. Das and K. Matyjaszewski, Chem. Sci., 2022, 13, 11540–11550 RSC.
- K. Parkatzidis, N. P. Truong, R. Whitfield, C. E. Campi, B. Grimm-Lebsanft, S. Buchenau, M. A. Rübhausen, S. Harrisson, D. Konkolewicz, S. Schindler and A. Anastasaki, J. Am. Chem. Soc., 2023, 145, 1906–1915 CrossRef CAS PubMed.
- X. Hu, R. Yin, J. Jeong and K. Matyjaszewski, J. Am. Chem. Soc., 2024, 146, 13417–13426 CrossRef CAS PubMed.
- M. Rolland, R. Whitfield, D. Messmer, K. Parkatzidis, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2019, 8, 1546–1551 CrossRef CAS PubMed.
- H. Cao, G. Wang, Y. Xue, G. Yang, J. Tian, F. Liu and W. Zhang, ACS Macro Lett., 2019, 8, 616–622 CrossRef CAS PubMed.
- Y. Abdouni, G. Yilmaz, A. Monaco, R. Aksakal and C. R. Becer, Biomacromolecules, 2020, 21, 3756–3764 CrossRef CAS PubMed.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef PubMed.
- T. Zhang, Z. Wu, G. Ng and C. Boyer, Angew. Chem., Int. Ed., 2023, 62, e202309582 CrossRef CAS PubMed.
- R. H. Young, K. Wehrly and R. L. Martin, J. Am. Chem. Soc., 1971, 93, 5774–5779 CrossRef CAS.
- S. Ponnusamy, L. Sandhiya and K. Senthilkumar, New J. Chem., 2017, 41, 10259–10271 RSC.
- R.-h. Liu, S.-y. Fu, H.-y. Zhan and L. A. Lucia, Ind. Eng. Chem. Res., 2009, 48, 9331–9334 CrossRef CAS.
- G. Lu, X. Jiang, Z. Ou, S. Yan and K. M. Kadish, J. Porphyrins Phthalocyanines, 2017, 21, 465–475 CrossRef CAS.
- E. Pang, S. Zhao, B. Wang, G. Niu, X. Song and M. Lan, Coord. Chem. Rev., 2022, 472, 214780 CrossRef CAS.
- A. Lacroix, H. H. Fakih and H. F. Sleiman, J. Controlled Release, 2020, 324, 34–46 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06466f |
| This journal is © The Royal Society of Chemistry 2024 |