
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBINAP-CuH-catalysed enantioselective allylation using alkoxyallenes to access 1,2-syn-tert,sec-diols†
N.
Navaneetha
ad,
Sundaram
Maurya
ad,
Prativa
Behera
b,
Sandip B.
Jadhav
a,
Lakshmi Revati
Magham
ad,
Jagadeesh Babu
Nanubolu
 cd,
Lisa
Roy
*be and
Rambabu
Chegondi
*ad
cd,
Lisa
Roy
*be and
Rambabu
Chegondi
*ad
aDepartment of Organic Synthesis and Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad 500007, India. E-mail: cramhcu@gmail.com; rchegondi@iict.res.in; Web: https://cramhcu.wixsite.com/rambabu-chegondi
bInstitute of Chemical Technology Mumbai, IOC Odisha Campus Bhubaneswar, Bhubaneswar 751013, India
cDepartment of Analytical and Structural Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad 500007, India
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201 002, India
eDepartment of Education, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: L.Roy@edu.iitkgp.ac.in
First published on 21st November 2024
Abstract
Herein, we present an economical method for highly enantioselective and diastereoselective Cu-BINAP-catalysed reductive coupling of alkoxyallenes with a range of electronically and structurally diverse ketones to afford 1,2-syn-tert,sec-diols, using PMHS as the hydride source. This reductive coupling has also been efficiently employed in the enantioselective desymmetrization of prochiral cyclic ketones harboring quaternary centres, in high yields with exclusive diastereoselectivity. Density Functional Theory (DFT) calculations are used to elucidate that the reaction is facilitated by a kinetically favourable “open” Z-enolate copper–alkoxyallene conformer, occurring at a lower Gibbs free energy barrier (by 3.9 kcal mol−1) than its E-enolate counterpart, dictating the stereoselectivity. Subsequently, this Z-enolate conformer synchronizes with appropriate nucleophilic faces to achieve the targeted syn-diastereoselectivity in the product through six-membered chair-like transition states during ketone addition.
Introduction
Enantiomerically enriched complex alcohols are ubiquitously found in a wide range of small molecule therapeutics and biologically active natural polyketides.1 Therefore, exploring general methods for the enantioselective synthesis of substituted alcohols is an attractive target in organic synthesis. In this regard, several asymmetric methods have been developed based on nucleophilic addition of various organometallic reagents2 to prochiral carbonyls. Particularly, several elegant enantioselective approaches have been developed to access homoallylic alcohols using allylmetal reagents.1b,2b,2d,2e,3 However, the use of stoichiometric amounts of an organometallic reagent in either the reaction or substrate preparation and chiral auxiliaries are major limitations for these reactions.4–9 In addition, the stereoselectivity of the allylation step depends on the geometry of the corresponding substrates and it is highly challenging to synthesize those precursors having sensitive functional groups to achieve the desired geometry. Alternatively, catalytic hydrometalation of easily accessible unsaturated compounds such as dienes, allenes or alkynes as common (pro)nucleophiles could generate allyl organometallic intermediates in situ to undergo nucleophilic additions.10 This approach is highly convenient and has garnered significant attention owing to its utilization of cost-effective reagents, while also minimizing the generation of a stoichiometric amount of waste.Recently, Krische and other research groups have conducted elegant studies on allylations using Rh, Ru, and Ir-catalysed hydrofunctionalization.11 In particular, Krische and coworkers first reported allene-mediated carbonyl allylations using a hydrogen atmosphere or transfer hydrogenation.12 Over the last few decades, a variety of C–C and C–X (X = heteroatom) bond formation reactions have been developed via CuH-catalysed enantioselective hydrofunctionalization of unsaturated pronucleophiles.13,14 In particular, Buchwald et al. have recently demonstrated asymmetric CuH-catalysed allylation with alkyl-substituted terminal allenes or simple allenes as allyl–metal surrogates using complex chiral ligands (Scheme 1a).15,16 Excellent regio- and enantioselectivity was observed for the 1,2-addition of (E)-allyl-Cu species on carbonyls via a six-membered chair-like transition state. Later, the same research group demonstrated the CuH-catalysed asymmetric allylation of ketones with 1,3-dienes to access substituted homoallyl alcohols.17 In 2021, the Krische group reported catalytic reductive coupling between carbonyls and 1,1-disubstituted benzhydryloxyallene with excellent enantioselectivity.11d Here, the internal chelation caused by the oxophilicity of Ru leads to a (Z)-selective allyl-Ru intermediate, which directs the formation of syn-selective sec,tert-alcohols (Scheme 1b). Inspired by Buchwald's and Krische's pioneering studies, here, we have demonstrated the Cu(I)-BINAP catalysed highly enantioselective synthesis of complex 1,2-syn-tert,sec-diols from the reductive coupling of alkoxyallenes with ketones (Scheme 1c). The hydrometallation of a mono-substituted alkoxyallene with a phosphine ligated CuH-complex could generate the chelation-controlled (Z)-selective allylcopper species Acis.11d,18 The nucleophilic addition of intermediate Acis on carbonyl through a six-membered chair-like transition state (TS) allows syn-diastereoselectivity.19 However, DFT calculations suggest the formation of kinetically favourable “open” Z-enolate of copper–alkoxyallene rather than a chelation assisted “closed” intermediate. Additionally, the “open” (Z)-selective π-allyl Cu-complex has higher affinity to accommodate carbonyl binding to undergo faster nucleophilic addition than the “closed” intermediate. Notably, the enantioselective synthesis of these valuable 1,2-syn-sec,tert-diols is limited to only two approaches using geometrically defined chiral allylboron reagents by the Roush group20 and Ru-catalysed enantioselective reductive coupling using alkoxyallene pronucleophiles by the Krische group.11d Herein, we have achieved excellent enantioselectivity as well as syn-selectivity using a simple and inexpensive BINAP ligand and air-stable hydride source, PMHS. In addition, this method allows the enantioselective desymmetrization of prochiral 4-substituted cyclohexanones with high diastereoselectivity.
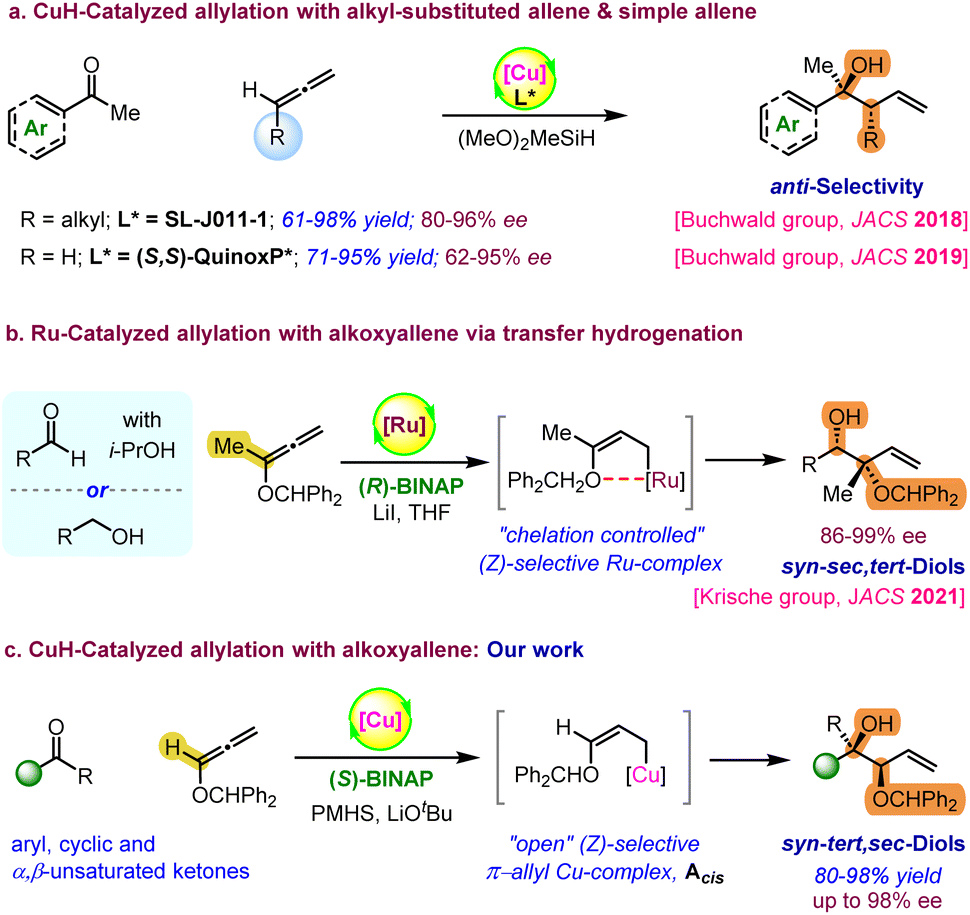 | ||
| Scheme 1 Previous and present approaches for enantioselective allylation using an allene. | ||
Results and discussion
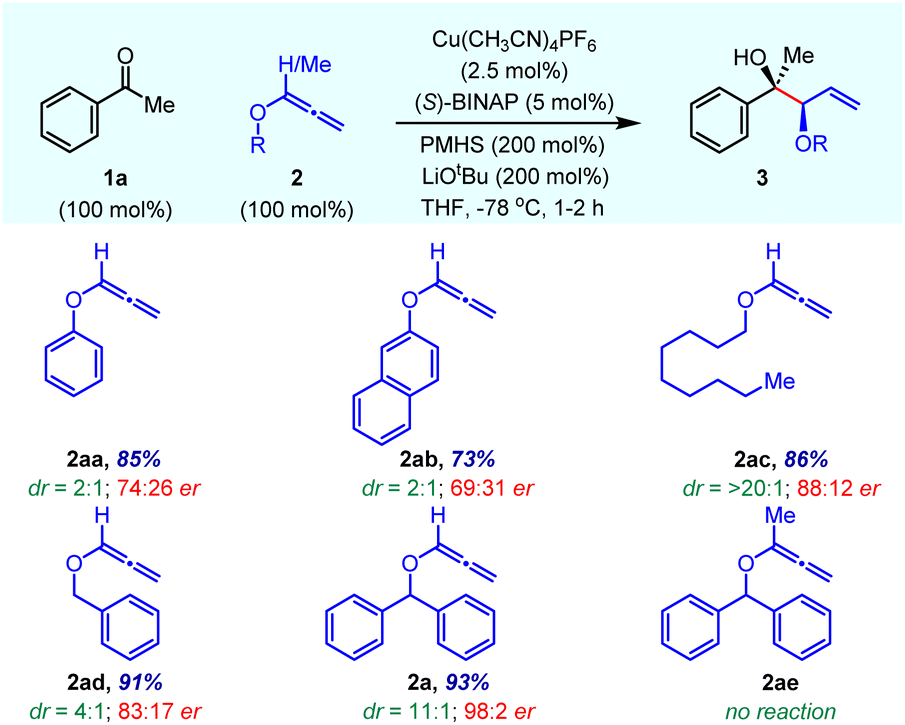
At the outset, instead of screening a variety of chiral ligands, we focused on the effect of various O-substituents on alkoxyallene 2 on reaction yield, enantioselectivity and diastereoselectivity (Table 1). Using Cu(CH3CN)4PF6 as the precatalyst and the commercially inexpensive, easily accessible chiral ligand (S)-BINAP, the allylation reaction on acetophenone 1a was evaluated in the presence of PMHS/LiOtBu at −78 °C in THF. Aryloxyallenes 2aa and 2ab provided the desired product 3 in good yield with poor stereoselectivities in favour of the syn-isomer. However, alkoxyallene 2ac gave an allylation product with high dr, albeit with moderate enantioselectivity. Notably, benzyloxyallene 2ad furnished good enantioselectivity in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers. On increasing the steric crowding by the installation of another phenyl ring at the benzylic position, the enantioselectivity drastically increased up to 98:2 with excellent yield and good diastereoselectivity (entry 2a). This clearly indicates that the O-benzhydryl group plays a crucial role in stereoselectivity probably due to better π–π stacking interactions with the chiral ligand of the Cu complex. The methyl substituted O-benzhydryl 3-alkoxyallene 2ae failed to give the desired product due to the cis-orientation of both substrates' methyl groups in the six-membered cyclic transition state (see TS2acis).
:1 mixture of diastereomers. On increasing the steric crowding by the installation of another phenyl ring at the benzylic position, the enantioselectivity drastically increased up to 98:2 with excellent yield and good diastereoselectivity (entry 2a). This clearly indicates that the O-benzhydryl group plays a crucial role in stereoselectivity probably due to better π–π stacking interactions with the chiral ligand of the Cu complex. The methyl substituted O-benzhydryl 3-alkoxyallene 2ae failed to give the desired product due to the cis-orientation of both substrates' methyl groups in the six-membered cyclic transition state (see TS2acis).
| a Reaction conditions: 1a (80 mg, 0.67 mmol), 2 (0.67 mmol), Cu(CH3CN)4PF6 (6.2 mg, 2.5 mol%), (S)-BINAP (21.0 mg, 5.0 mol%), LiOtBu (1.3 mL, 1.33 mmol, 1.0 M THF solution), and PMHS (178 μL, 1.33 mmol) in THF (3 mL, 0.3 M). Combined yields of both isomers after column chromatography. Enantiomeric ratio (er) was determined by HPLC analysis using a chiral stationary phase. The minor isomer enantiomeric ratio was reported in the ESI. The dr was determined from 1H NMR analysis of crude products 3. |
|---|
|
Later, acetophenone 1a was treated with O-benzhydryl 3-alkoxy-1,2-butadiene 2a in the presence of PMHS, LiOtBu and the Cu(CH3CN)4PF6/(S)-BINAP complex in THF solvent at various temperatures (Table 2). Both enantioselectivity and diastereoselectivity were highly influenced by the reaction temperature from rt to −90 °C without substantial changes in the reaction yield (entries 1–6). As mentioned previously, an excellent er (98:2) and high dr (11:1) were achieved at −78 °C with simple (S)-BINAP ligand L1 in 93% overall yield (entry 5). The effect of various bidentate ligands was also screened at optimized reaction temperature. Other BINAP, SEGPHOS, and Josiphos ligands L2–L7 gave either moderate yield or moderate diastereoselectivity with up to 98:2 er (entries 7–12). In addition, DIOP (L8) and Ph-BPE (L9) ligands led to different levels of erosion in yield and enantioselectivity (entries 13–14). Interestingly, only a trace amount of desired product was observed with the oxazoline ligand L10 (entry 15). From the above reaction optimization, the chiral (S)-BINAP ligand (L1) was the best choice at −78 °C for Cu(I)-catalysed reductive allylation (entry 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | T °C | Yield [%] | 3a (dr) | 3a (er) |
| a Reaction conditions: 1a (80 mg, 0.67 mmol), 2a (148 mg, 0.67 mmol), Cu(CH3CN)4PF6 (6.2 mg, 2.5 mol%), ligand (5.0 mol%), LiOtBu (1.3 mL, 1.33 mmol, 1.0 M THF solution), and PMHS (178 μL, 1.33 mmol) in THF solvent (3 mL, 0.3 M). b Isolated yields. c Enantiomeric ratio (er) was determined by HPLC analysis using a chiral stationary phase. d Diastereoselectivity was observed from 1H NMR analysis of crude product 3. | |||||
| 1 | (S)-BINAP, L1 | r.t. | 87 | 4:1 |
88:12 |
| 2 | (S)-BINAP, L1 | 0 | 90 | 6:1 |
90:10 |
| 3 | (S)-BINAP, L1 | −20 | 94 | 7:1 |
92:8 |
| 4 | (S)-BINAP, L1 | −40 | 93 | 9:1 |
94:6 |
| 5 | (S)-BINAP, L1 | −78 | 93 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
98:2
|
| 6 | (S)-BINAP, L1 | −90 | 91 | 11:1 |
98:2 |
| 7 | (R)-Tol-BINAP, L2 | −78 | 81 | 9:1 |
5:95 |
| 8 | (S)-SEGPHOS, L3 | −78 | 67 | 10:1 |
98:02 |
| 9 | (R)-DM-SEGPHOS, L4 | −78 | 72 | 4:1 |
3:97 |
| 10 | (S)-DTBM-SEGPHOS, L5 | −78 | 66 | 8:1 |
88:12 |
| 11 | Josiphos SL-J505-1, L6 | −78 | 41 | 3:1 |
83:17 |
| 12 | Josiphos SL-J011-1, L7 | −78 | 37 | 4:1 |
80:20 |
| 13 | (R,R)-DIOP, L8 | −78 | 53 | 6:1 |
63:37 |
| 14 | (S,S)-Ph-BPE, L9 | −78 | 76 | 7:1 |
35:65 |
| 15 | (S)-Oxazoline, L10 | −78 | >10 | — | — |

With the optimal reaction conditions, the substrate scope of the enantioselective reduction coupling was investigated with various activated ketones using O-benzhydryl allene 2a (Table 3). The allylation reaction efficiently proceeded with aryl methyl ketones having electron-donating or electron-withdrawing substituents at the para-position and gave high yields with good diastereocontrol and excellent enantioselectivity (entries 3a–3j). The use of meta-substituted aryl methyl ketones also furnished syn-alcohols in high yields with similar enantiomeric and diastereomeric ratios (entries 3k–3m). Sterically demanding ortho-substituted aryl ketones and disubstituted aryl ketones were successfully converted into the corresponding chiral homoallylic tertiary alcohols with high yields and high enantiomeric purity (entries 3n–3p). Interestingly, ethyl and n-hexyl aryl ketones provided the corresponding products 3q–3s with excellent yield and enantioselectivity. However, the sterically hindered i-propyl aryl ketone failed to give the desired product 3t. Moreover, heteroaryl methyl ketones having pyridine, furan and thiophene rings were also suitable substrates under optimal reaction conditions to afford desired products in an efficient and selective manner (entries 3u–3w). Generally, heterocycles are among the most prevalent structural motifs in pharmaceuticals; however, they are challenging substrates in transition-metal-catalysed enantioselective transformations. The scope of reductive coupling reactions was further highlighted with α,β-unsaturated ketones. Both electron donating and withdrawing substituents on methyl styryl ketone are well-tolerated to afford good yield with high diastereoselectivity (entries 3x and 3y). The α-substituted enone also gave desired product 3z in 86% yield with a 5:2 diastereoselectivity ratio. It is important to mention that high enantioselectivity was observed for the minor isomer (96:4 er), whereas the major isomer gave moderate enantioselectivity. Interestingly, we did not observe the reduction of α,β-unsaturated ketones alone via 1,4-addition due to the superior reactivity of alkoxyallene 2a under standard reaction conditions. Next, we set out to investigate allylation reactions with a variety of cyclic ketones. The symmetrical 4- to 12-membered cyclic ketones were well-tolerated under standard reaction conditions to afford the corresponding products 3aa–3ae in good yields and good enantioselectivity. Particularly, 5- and 7-membered ketones gave the highest enantiomeric ratio. Additionally, α-tetralones, β-tetralone and indanone also underwent the current nucleophilic allylation with high yield and excellent enantiocontrol (entries 3af–3aj).
| a Reaction conditions: 1a (0.36 mmol), 2a (80 mg, 0.36 mmol), Cu(CH3CN)4PF6 (3.4 mg, 2.5 mol%), (S)-BINAP (11.2 mg, 5.0 mol%), LiOtBu (0.7 mL, 0.72 mmol, 1.0 M THF solution), and PMHS (97 μL, 0.72 mmol) in THF solvent (2 mL, 0.2 M). b Isolated yields after column chromatography. c Enantiomeric ratio (er) was determined by HPLC analysis using a chiral stationary phase. d The dr was obtained from 1H NMR analysis of crude products 3. |
|---|
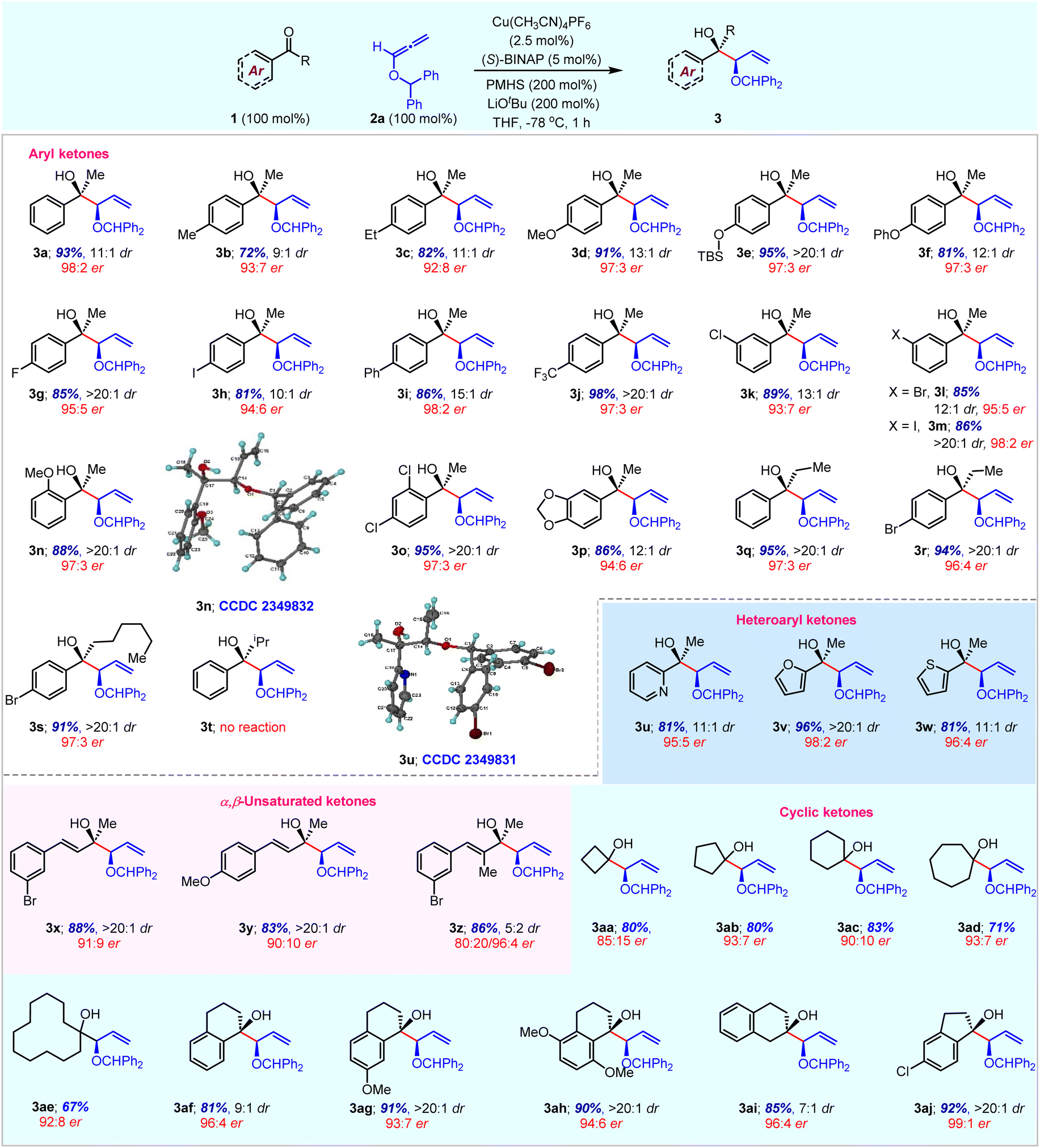
|
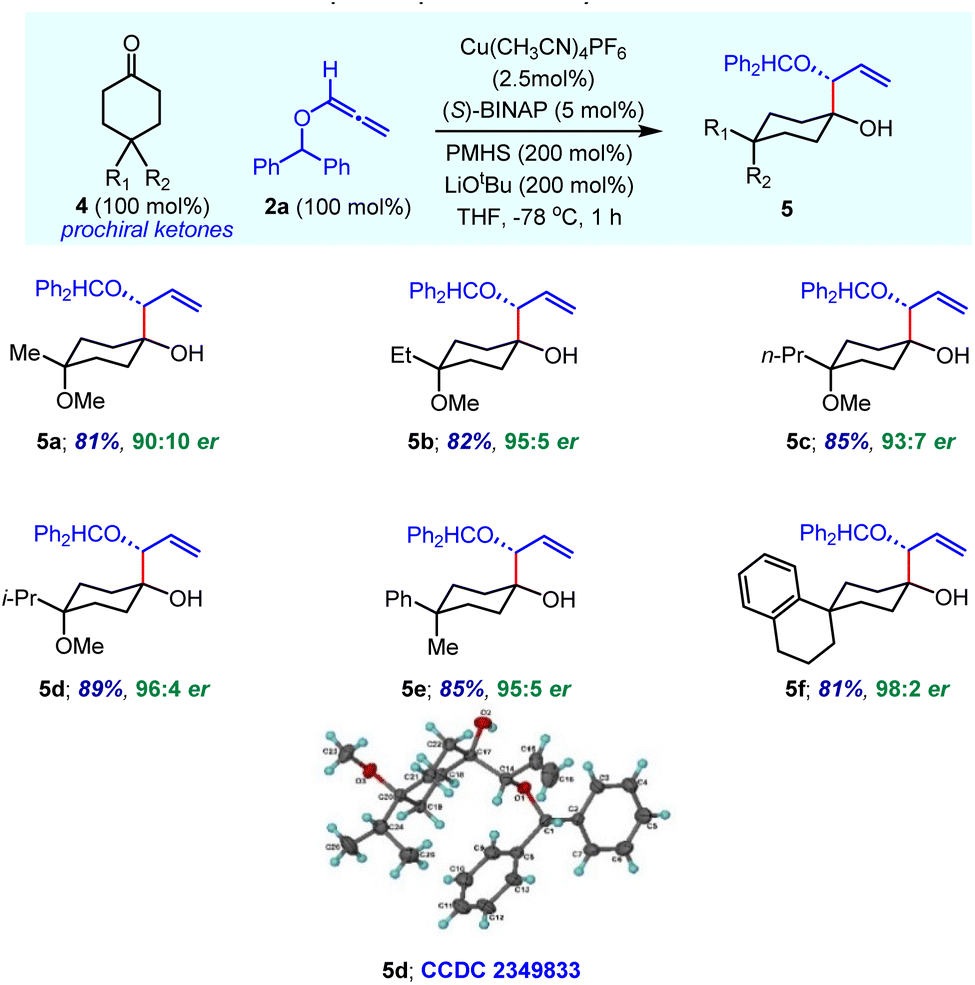
We next explored the use of reductive allylation for the enantioselective desymmetrization of prochiral 4,4-disubstituted cyclohexanones 4 having a plane of symmetry (Table 4). Simple methyl- and methoxy-substituted cyclohexanone was readily functionalized to produce desired product 5a with high enantioselectivity. The assignment of product stereochemistry is non-trivial. There are three stereogenic centres, two of which are chirotopic (R/S) and one is achirotopic (r/s). In the same vein, products 5b–5d bearing various alkyl substituents in combination with the OMe group were obtained with an excellent enantiomeric ratio under optimized reaction conditions. Overall, enantioselectivity increases with the steric bulk of alkyl substituents. The relative stereochemistry was confirmed by single crystal X-ray analysis of compound 5d. In the case of γ,γ-disubstituted cyclohexanones having aryl and alkyl groups, the corresponding products 5e and 5f containing an all-carbon quaternary centre were obtained with excellent yield and enantioselectivity. We believe that the bulky aryl ring occupies the equatorial position in these products, which dictates the high diastereoselectivity.
The Cu(I)-catalysed enantioselective reductive coupling reaction was demonstrated to be scalable even at reduced catalyst loading (Scheme 2). We were able to execute a gram-scale (4.5 mmol) reaction to synthesize 3a, using 1.5 mol% Cu(CH3CN)4PF6 and 3.0 mol% (S)-BINAP without lowering the yield (1.43 g, 93%) or the stereoselectivity (97:3 er, 10:1 dr) of the reaction. The synthetic utility of this reaction is highlighted by the conversion of adduct 3a into highly functionalized scaffolds with additional transformations (Scheme 2b). The selective removal of the diphenylmethylene group on 3a in under Li/naphthalene conditions in the presence of a double bond was achieved with excellent yield (6, 96% yield). The cross-metathesis of 6 with methyl acrylate afforded α,β-unsaturated ester 8 and subsequent hydrogenation followed by acid-mediated cyclization gave γ-lactone 9 in good yield. The ozonolysis of the terminal olefin furnished aldehyde 10, which was then subjected to Zn-mediated Barbier-type allylation leading to the homoallyl alcohol 11 with excellent diastereoselectivity in decent yields. The absolute configuration of 3a was identified to be 2R,4S by Mosher ester analysis of its diastereomeric α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) esters 7a and 7b (see the ESI†). Furthermore, the robustness and scalability of this enantioselective allylation reaction and the synthetic versatility of 1,2-syn-tert,sec-diol scaffolds can be illustrated by its potential application in the synthesis of highly enantioenriched molecular architectures and bioactive natural products. We envisioned that reductive coupling of alkoxyallenes could be used to construct the C3–C11 fragment of fostriecin, an antitumor and antibiotic natural product isolated from Streptomyces pulveraceus. To this end, known ester 12 was converted to α,β-unsaturated ketone 14 to set the stage for the key reaction through DIBAL-H reduction followed by Wittig olefination of 13 in two steps. The CuH-catalysed enantioselective allylation of 14 under our optimized conditions using the (S)-BINAP ligand gave 0.56 grams of fostriecin C3–C11 fragment 15 with excellent yield and diastereoselectivity (Scheme 2c).
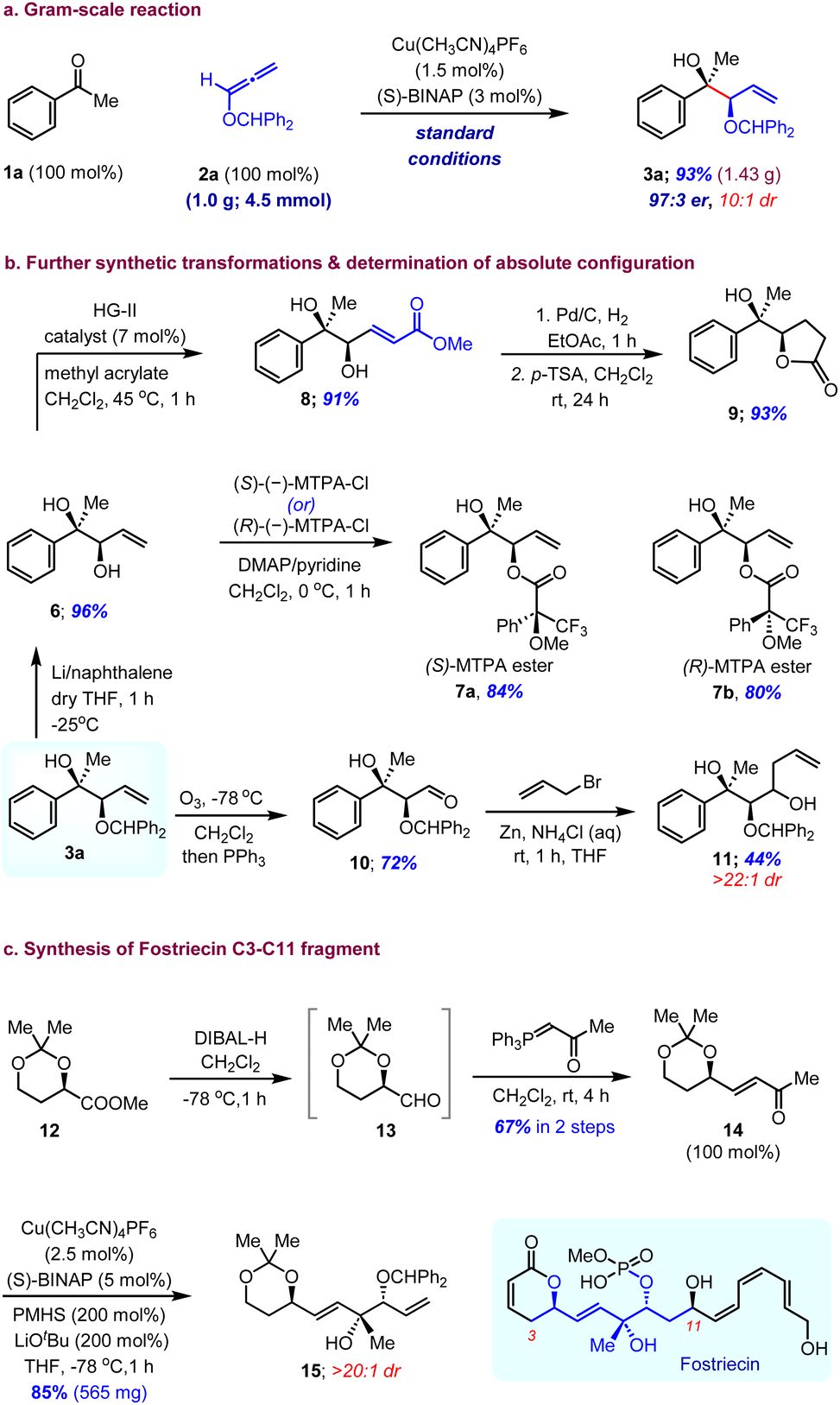 | ||
| Scheme 2 Gram-scale reaction, synthetic utility and confirmation of absolute stereochemistry. | ||
We have conducted density functional investigation at B3LYP-D3BJ/def2-TZVPP/SMD(THF) to understand the reaction mechanism of copper catalysed allylation of acetophenone and to rationalize the origin of 1,2-syn-diol enantioselectivity. Our proposed catalytic mechanism for the hydrocupration of alkoxyallene 2a by [(S)-BINAP]CuH followed by nucleophilic attack on ketone 1a to generate the product 3a is illustrated in Fig. 1a. Our computational studies reveal that the hydrocupration of the allene through the cis configuration viaTS1cis (15.9 kcal mol−1) is kinetically favored over the trans orientation viaTS1trans (19.8 kcal mol−1). As shown in Fig. 1, the rate-determining strained four-membered transition state (TS1cis) features concerted hydride transfer and Cu–C bond formation.21 This is followed by the formation of a (Z)-selective allyl-copper intermediate (Acis) in a highly exergonic irreversible fashion. Unlike analogous high-valent octahedral Ru(II)-BINAP complexes reported earlier to involve a chelated (Z)-selective π-allyl-metal complex,11d low-valent Cu(I) species feature triple coordination around the metal in the allyl-copper intermediate (see ESI, Fig. S8†).22
 | ||
| Fig. 1 (a) Computed free energy profile at 298.15 K for copper catalysed allylation of acetophenone at the B3LYP-D3BJ/def2-TZVPP/SMD(THF) level of theory. In parentheses, relative free energies computed at 195.15 K DFT are shown. Calculated prominent transition state structures of the 1,2-hydrocupration step and allylation step consisting of quadrant diagrams in (b) and (d) and steric maps in (c) and (e). In (b) and (d), roman numerals denote the four quadrants of the coordination space with corresponding %Vfree; green indicates the alkoxyallene substrate and purple indicates the ketone; LnCuH has the following color codes: O (red), Cu (brown), P (orange), C (grey), and H (white). In (c), steric mapping (contours within a range of ±3.0 Å in 1 (c) and ±4.0 Å in 1 (e)) along with total %Vfree is shown within a radius of 3.5 Å and 4.5 Å from the copper centre for hydrocupration and allylation steps, respectively. PMHS was modelled as Me(OMe)2SiH to reduce computational cost. | ||
Further, to shed light on the origin of cis-induction during the hydrocupration step, we have illustrated the steric profile of the LnCuH-substrate complex in TS1cis and TS1trans through quadrant diagrams and steric mapping of the buried volume on the xy-plane with a radius of 3.5 Å from the copper centre (Fig. 1b and c). Our analysis reveals that in the preferred transition state (TS1cis), the incoming alkoxyallene substrate is positioned in a sterically less-hindered fashion at the interface of quadrants II and III, as compared to TS1trans where the alkoxy group of the allene is in an out-of-plane fashion (Fig. 1b). This leads to a greater total %Vfree (which accounts for the percentage of total free volume within the pocket of the substrate–catalyst complex) of 15.4% in TS1cis as compared to 14.1% in TS1trans (Fig. 1c), resulting in a better catalyst–substrate interaction at the active site of the hydrocupration event. Indeed, the orientation of the substrate leads to lowered intramolecular steric repulsion in TS1cis as observed from the steric map here (red contours in Fig. 1c) and energy decomposition analysis (see ESI, Table S3†). Our study therefore reveals that the larger structural reorganization for both LnCuH (ΔΔEreorg = 1.5 kcal mol−1) and the alkoxyallene (ΔΔEreorg = 2.4 kcal mol−1) in TS1trans (ΔΔG‡ = 3.9 kcal mol−1) accounts for the observed Z-selectivity through TS1cis. In fact, the kinetically controlled Z-enolate copper–alkoxyallene conformer Acis synchronizes with appropriate nucleophilic faces on carbonyl engagement to ensure observed stereoselectivity during ketone addition (see Fig. 1a, d, 1e, and S1 in the ESI†).
The attack of the cis-allyl–Cu complex could occur on the carbonyl group via Re/Si, Re/Re, Si/Re and Si/Si faces, through six-membered chair-like transition states, to obtain four possible stereoisomers (Fig. 1a, Fig. S1, and S9†). Hence, the allylation step is our enantioselective and diastereoselective transition state. Careful inspection of the quadrant diagrams (Fig. 1d) and steric mapping data (Fig. 1e) suggests that Si-face attack of the ketone from the Si-face of the allyl may be the most feasible approach due to lower steric interference with the bulky phosphine ligands. The alkoxy group of the allene also engages cooperatively in strong non-covalent forces such as C–H–π, π–π and C–H–O interactions, aiding in lowering of TS2acis energy (see ESI, Fig. S10†).23TS2acis requires the lowest free energy barrier (8 kcal mol−1) and should rapidly promote the generation of intermediate B2acis. Energy decomposition analysis further shows that the total reorganization energy in TS2acis is least among the four allylation pathways, which highlights that its stereo-electronic environment is most conducive (Table S4†). Thereby, we envision the involvement of LiOtBu and PMHS to release syn-selective lithiated diol product 3a′ and the regeneration of the catalyst. Finally, the diol product 3a can be produced with acidic work-up.
Based on our experimental results and computational studies, the overall plausible mechanism for enantioselective CuH-catalysed reductive coupling of alkoxyallenes was proposed, as depicted in Scheme 3. Initially, a hydride copper complex LnCuH forms in situ with a copper catalyst, phosphine ligand, base, and silane reductant. The hydrocupration of a phosphine-ligated Cu–H complex across the alkoxyallene 2a furnishes a transient, kinetically favourable Z-selective π-allyl-copper complex Acis, which readily undergoes nucleophilic addition to ketone 1a through a closed six-centred, cyclic transition state TS2acis, resulting in the formation of the homoallylic Cu–alkoxide complex B. Subsequent lithium exchange and hydrolysis release the 1,2-syn-diol 3a, followed by metathesis with the PMHS regenerating the LnCuH complex.
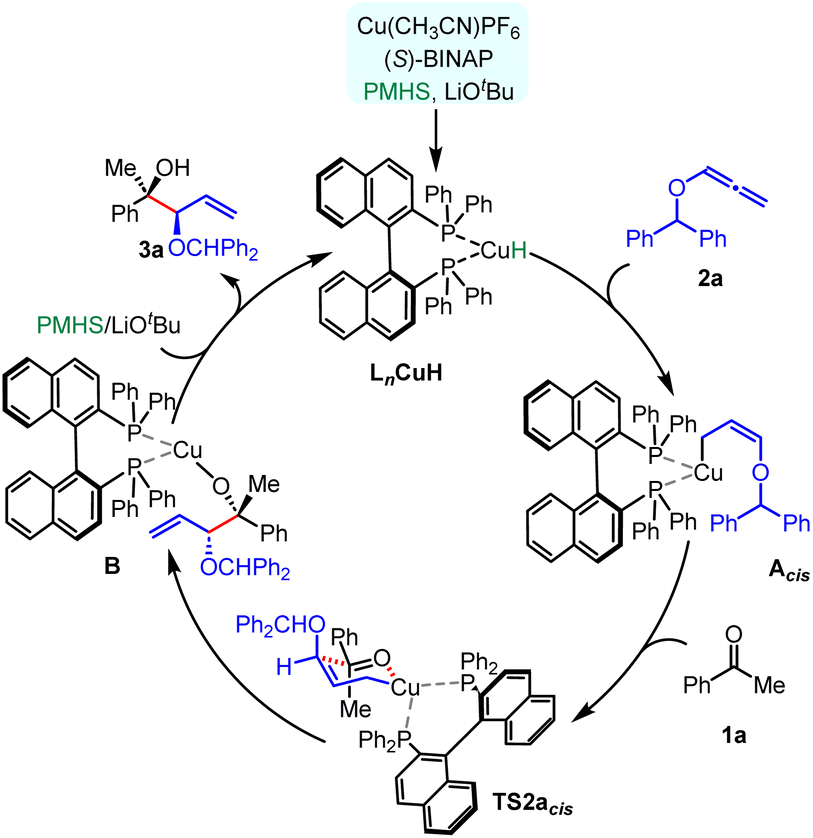 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a highly enantioselective and diastereoselective copper-hydride-catalysed synthesis of 1,2-syn-tert,sec-diols via the reductive coupling of electronically and structurally diverse ketones with easily accessible O-benzhydryl 3-alkoxyallene. The reaction proceeds via the formation of a Z-enolate copper–alkoxyallene complex, which smoothly undergoes carbonyl binding to furnish the syn-selective product. The reaction utilizes an economical chiral Cu–BINAP complex, which provides 1,2-syn-tert,sec-diol products in high yields, in the presence of PMHS as a commercially inexpensive and air-stable hydride source. Furthermore, this reductive coupling with alkoxyallenes proves effective in the desymmetrization of prochiral cyclic ketones containing quaternary centres, affording products with excellent enantioselectivity. The synthetic utility of this approach is exemplified by the enantioselective synthesis of a key fragment of fostriecin. Density Functional Theory (DFT) computations suggest that the reaction proceeds through a kinetically favorable “open” Z-enolate copper-alkoxyallene conformer, occurring at ΔΔG‡ = 3.9 kcal mol−1 lower as compared to its E-enolate counterpart, which determines the syn-diastereoselectivity of the nucleophilic addition.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
N. N., S. R. M., L. R. M. and S. B. J. conducted the experiments and HPLC analysis; P. B. and L. R. conducted the computational studies; J. B. N. conducted the single X-ray crystallographic analysis; R. C. designed and supervised the project and wrote the manuscript with the assistance of co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the SERB Core Research Grant (CRG/2022/001419) and SERB-STAR Award for a research grant (STR/2022/000007) by ANRF, New Delhi, for financial support. L. R. acknowledges SERB (SPG/2020/000754) and CSIR (01WS(001)/2023-24/EMR-II/ASPIRE) for funding. N. N and L. R. M thank DST-INSPIRE, New Delhi, India; P. B. thanks ICT-IOCB; S. M and S. B. J thank UGC, New Delhi, for a research fellowship. The IICT Communication Number for this manuscript is IICT/pubs./2024/140.Notes and references
- For selected reviews, see: (a) J. F. Bower, I. S. Kim, R. L. Patman and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 34–46 CrossRef CAS PubMed; (b) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595–5698 CrossRef CAS PubMed; (c) D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893–907 RSC.
- For selected reviews, see: (a) L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757–824 CrossRef CAS PubMed; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2793 CrossRef CAS PubMed; (c) O. Riant and J. Hannedouche, Org. Biomol. Chem., 2007, 5, 873–888 RSC; (d) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853–2873 CrossRef CAS PubMed; (e) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774–7854 CrossRef CAS; (f) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed.
- For selected reviews on allylation, see: (a) H. Yamamoto and M. Wadamoto, Chem.–Asian J., 2007, 2, 692–698 CrossRef CAS PubMed; (b) M. Hatano and K. Ishihara, Synthesis, 2008, 11, 1647–1675 Search PubMed; (c) L. F. Tietze, T. Kinzel and C. C. Brazel, Acc. Chem. Res., 2009, 42, 367–378 CrossRef CAS PubMed; (d) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303–320 RSC.
- For selected references for asymmetric allylation using allylboron reagents, see: (a) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2006, 128, 12660–12661 CrossRef CAS; (b) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262–11265 CrossRef CAS; (c) D. W. Robbins, K. Lee, D. L. Silverio, A. Volkov, S. Torker and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 9610–9614 CrossRef CAS; (d) K. Lee, D. L. Silverio, S. Torker, F. Haeffner, D. W. Robbins, F. W. van der Mei and A. H. Hoveyda, Nat. Chem., 2016, 8, 768–777 CrossRef CAS PubMed; (e) R. J. Morrison and A. H. Hoveyda, Angew. Chem., Int. Ed., 2018, 57, 11654–11661 CrossRef CAS.
- For selected references for asymmetric allylboration using copper catalysts, see: (a) R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8910–8911 CrossRef CAS; (b) M. Kanai, R. Wada, T. Shibuguchi and M. Shibasaki, Pure Appl. Chem., 2008, 80, 1055–1062 CrossRef CAS; (c) S.-L. Shi, L.-W. Xu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 6638–6639 CrossRef CAS PubMed.
- For selected references for asymmetric allylation using titanium catalysts, see: (a) S. Kii and K. Maruoka, Chirality, 2003, 15, 68–70 CrossRef CAS PubMed; (b) D. S. Barnett, P. N. Moquist and S. E. Schaus, Angew. Chem., Int. Ed., 2009, 48, 8679–8682 CrossRef PubMed; (c) J. Yadav, G. R. Stanton, X. Fan, J. R. Robinson, E. J. Schelter, P. J. Walsh and M. A. Pericas, Chem.–Eur. J., 2014, 20, 7122–7127 CrossRef CAS PubMed.
- For selected references for asymmetric allylation using indium, scandium and silver catalysts, see: (a) M. Wadamoto and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 14556–14557 CrossRef CAS PubMed; (b) Y. C. Teo, J. D. Goh and T. P. Loh, Org. Lett., 2005, 7, 2743–2745 CrossRef CAS; (c) X. Zhang, D. Chen, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS; (d) N. V. Hanhan, Y. C. Tang, N. T. Tran and A. K. Franz, Org. Lett., 2012, 14, 2218–2221 CrossRef CAS PubMed.
- For selected references for asymmetric allylation using stoichiometric reductant, see: (a) R.-Y. Chen, A. P. Dhondge, G.-H. Lee and C. A. Chen, Adv. Synth. Catal., 2015, 357, 961–966 CrossRef CAS; (b) X.-R. Huang, C. Chen, G.-H. Lee and S.-M. Peng, Adv. Synth. Catal., 2009, 351, 3089–3095 CrossRef CAS; (c) J. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 2752–2753 CrossRef CAS PubMed; (d) Z.-L. Shen, S.-Y. Wang, Y.-K. Chok, Y.-H. Xu and T.-P. Loh, Chem. Rev., 2013, 113, 271–401 CrossRef CAS.
- For selected references for asymmetric allylation using chiral auxiliaries, see: (a) H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS; (b) W. R. Roush, A. E. Walts and L. K. Hoong, J. Am. Chem. Soc., 1985, 107, 8186–8190 CrossRef CAS; (c) R. O. Duthaler, A. Hafner and M. Riediker, Pure Appl. Chem., 1990, 62, 631–642 CrossRef CAS; (d) J. W. A. Kinnaird, P. Y. Ng, K. Kubota, X. Wang and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7920–7921 CrossRef CAS PubMed; (e) J. D. Huber, N. R. Perl and J. L. Leighton, Angew. Chem., Int. Ed., 2008, 47, 3037–3039 CrossRef CAS PubMed; (f) W. A. Chalifoux, S. K. Reznik and J. L. Leighton, Nature, 2012, 487, 86–89 CrossRef CAS; (g) D. Kumar, S. R. Vemula, N. Balasubramanian and G. R. Cook, Acc. Chem. Res., 2016, 49, 2169–2178 CrossRef CAS PubMed.
- For recent reviews, see: (a) K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, 5133 CrossRef PubMed; (b) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (c) R. S. Doerksen, C. C. Meyer and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 14055–14064 CrossRef CAS PubMed; (d) C. C. Meyer, E. Ortiz and M. J. Krische, Chem. Rev., 2020, 120, 3721–3748 CrossRef CAS PubMed; (e) M. Xiang, D. E. Pfaffinger and M. J. Krische, Chem.–Eur. J., 2021, 27, 13107–13116 CrossRef CAS PubMed.
- For recent reviews, see: (a) C. G. Santana and M. J. Krische, ACS Catal., 2021, 11, 5572–5585 CrossRef CAS PubMed , for selected examples, see:; (b) K. Spielmann, M. Xiang, L. A. Schwartz and M. J. Krische, J. Am. Chem. Soc., 2019, 141, 14136–14141 CrossRef CAS PubMed; (c) C. Cooze, R. Dada and R. J. Lundgren, Angew. Chem., Int. Ed., 2019, 58, 12246–12251 CrossRef CAS PubMed; (d) M. Xiang, D. E. Pfaffinger, E. Ortiz, G. A. Brito and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 8849–8854 CrossRef CAS PubMed; (e) M. Xiang, A. Ghosh and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 2838–2845 CrossRef CAS PubMed , for anti-alkoxyallylation employing allylic gem-dicarboxylates, see:; (f) S. B. Han, H. Han and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 1760–1761 CrossRef CAS PubMed.
- (a) E. Skucas, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 12678–12679 CrossRef CAS PubMed; (b) J. F. Bower, E. Skucas, R. L. Patman and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 15134–15135 CrossRef CAS PubMed; (c) M.-Y. Ngai, E. Skucas and M. J. Krische, Org. Lett., 2008, 10, 2705–2708 CrossRef CAS PubMed; (d) E. Skucas, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 5054–5055 CrossRef CAS PubMed ; for enantioselective examples, see:; (e) S. B. Han, I. S. Kim, H. Han and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 6916–6917 CrossRef CAS PubMed.
- For reviews, see: (a) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed; (b) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (c) R. Y. Liu and S. L. Buchwald, Acc. Chem. Res., 2020, 53, 1229–1243 CrossRef CAS PubMed; (d) J. Mohr and M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 12148–12149 CrossRef CAS PubMed; (e) Z. Sorádová and R. Šebesta, ChemCatChem, 2016, 8, 2581–2588 CrossRef; (f) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (g) M. T. Pirnot, Y.-M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed; (h) G. L. Larson and R. J. Liberatore, Org. Process Res. Dev., 2021, 25, 1719–1787 CrossRef CAS.
- For recent publications, see: (a) Y. Dong, K. Shin, B. K. Mai, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 16303–16309 CrossRef CAS PubMed; (b) S. Feng, Y. Dong and S. L. Buchwald, Angew. Chem., Int. Ed., 2022, 61, e202206692 CrossRef CAS PubMed; (c) Y. Yuan, Y. Zhang, W. Li, Y. Zhao and X.-F. Wu, Angew. Chem., Int. Ed., 2023, 62, e202309993 CrossRef CAS PubMed; (d) L. T. Brechmann, B. Kaewmee and J. F. Teichert, ACS Catal., 2023, 13, 12634–12642 CrossRef CAS.
- (a) E. Y. Tsai, R. Y. Liu, Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 2007–2011 CrossRef CAS PubMed; (b) R. Y. Liu, Y. Zhou, Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 2251–2256 CrossRef CAS PubMed.
- For selected reviews on allene-mediated allylations, see: (a) A. P. Pulis, K. Yeung and D. J. Procter, Chem. Sci., 2017, 8, 5240–5247 RSC; (b) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (c) G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC; (d) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed.
- (a) C. Li, R. Y. Liu, L. T. Jesikiewicz, Y. Yang, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 5062–5070 CrossRef CAS PubMed; (b) C. Li, K. Shin, R. Y. Liu and S. L. Buchwald, Angew. Chem., Int. Ed., 2019, 58, 17074–17080 CrossRef CAS PubMed; (c) Y. Dong, A. W. Schuppe, B. K. Mai, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 5985–5995 CrossRef CAS PubMed.
- For internal chelation in alkoxy-substituted allylmetal intermediates, see: (a) P. Almendros and E. J. Thomas, J. Chem. Soc., Perkin Trans. 1, 1997, 2561–2568 RSC; (b) B. K. Zambron, Synthesis, 2020, 52, 1147–1180 CrossRef CAS.
- During the preparation of this manuscript, a Cu(I)-catalyzed enantioselective allylation with siloxypropadienes using the TANIAPHOS ligand was reported. For reference, see: N. Jiang, P. Liu, Z. Pan, S. Q. Wang, Q. Peng and L. Yin, Angew. Chem., Int. Ed., 2024, 63, e202402195 CrossRef CAS PubMed.
- M. Chen, M. Handa and W. R. Roush, J. Am. Chem. Soc., 2009, 131, 14602–14603 CrossRef CAS PubMed.
- V. Arun, L. Roy and S. De Sarkar, Chem.–Eur. J., 2020, 26, 16649–16654 CrossRef CAS PubMed.
- M. Piñeiro-Suárez, A. M. Álvarez-Constantino and M. Fañanás-Mastral, ACS Catal., 2023, 13, 5578–5583 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2349832, 2349831 and 2349833. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07002j |
| This journal is © The Royal Society of Chemistry 2024 |