
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactivated hydride therapy under hypoxia beyond ROS†
Xia
Wang‡
a,
Yijian
Gao‡
b,
Ting
Wang
c,
Zhaobin
Wang
a,
He
Hang
a,
Shengliang
Li
*b and
Fude
Feng
 *a
*a
aMOE Key Laboratory of High Performance Polymer Material and Technology of Ministry of Education, Department of Polymer Science & Engineering, School of Chemistry & Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: fengfd@nju.edu.cn
bCollege of Pharmaceutical Sciences, Soochow University, Suzhou 215123, China
cState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China
First published on 12th November 2024
Abstract
As compared to oxidative phototherapy, studies on reactive reductive species-participating photodynamic therapy (PDT) are rare. Porphyrins are typical photosensitizers restricted by the oxygen level, but efficacy and selectivity are always incompatible in PDT. Herein, we report that phlorins are ideal hydride (H−) donors and explore a water-soluble triphenylphosphonium-modified zinc-coordinated porphyrin (mitoZnPor) for in situ photogeneration of zinc-cored phlorin (mitoZnPhl). Driven by 1,4-dihydronicotinamide adenine dinucleotide (NADH), the mitoZnPor/mitoZnPhl couple can reduce electron acceptors like iron heme and ubiquinone that play key roles in the mitochondrial electron transport chain (Mito-ETC). Under hypoxia, mitoZnPor showed excellent cancer-selectivity and a highly efficient in vitro PDT effect with IC50 at nanomolar levels and potent tumor growth inhibition in a 4T1 tumor-xenografted mouse model with good biosafety, which underlines the great potential of Mito-ETC targeted non-classical PDT via a H−-transfer mechanism beyond reactive oxygen species (ROS) in precision cancer phototherapy using NADH as a biomarker and original electron donor.
Introduction
Generally, photodynamic therapy (PDT) relies on light, photosensitizers (PSs) and molecular oxygen (O2) and has special merits of non-invasiveness, spatiotemporal control and minimal drug resistance in cancer therapy.1 A few 1st generation PSs are generally in the form of a mixture and have been clinically approved for treating cancers.2 2nd generation PSs with accurate structures (e.g., 5-aminolevulinic acid (5-ALA), phthalocyanines, and phenothiazines) are developed, but they suffer from poor solubility and selectivity.3 3rd generation PSs have emerged in the recent decade and the primary contributions lie in the improvement of targeting ability to increase cancer-selective accumulation of PSs. Alternatively, overexpressed or abnormally activated enzymes such as alkaline phosphatase,4 carbonic anhydrase,5 cathepsin B,6 and NAD(P)H quinone oxidoreductase 1 (NQO1)7 are chosen to induce the activation of PSs at the surface or interior of cancer cells. However, cancers with different types and differentiation grades greatly vary in the expression level of these proteins.8 Therefore, lacking sufficient efficacy and selectivity, merely targeting a single enzyme or pathway results in scarce success.Hypoxic cancers suppress the photochemical production of short-lived cytotoxic reactive oxygen species (ROS) such as hydroxyl radicals (·OH) and nonradical singlet oxygen (1O2). Recognized as a hallmark of solid tumors, hypoxia alters a series of biological functions (e.g., gene expression, signal transduction, and metabolic homeostasis) and raises the threshold of ROS resistance.9 As compared to normal cells, hypoxic cancers produce abundant antioxidant species such as reduced glutathione (GSH) and 1,4-dihydronicotinamide adenine dinucleotide (NADH), which counteract the efficacy of type I and II PDT. Photoexcitation of typical PDT agents induces apoptosis or necrosis of cancer cells with poor cell selectivity, as both cancer and normal cells are sensitive to excessive ROS. Most recently, we proposed a new concept of reactive reductive species (RRS)-participating PDT in which the hydrogen radical (H·) plays a key role in the reduction of biotargets or cascade reactions.10,11 In the small family of reactive hydrogen species (RHS), the negatively charged hydride ion (H−) possesses coincident redox potential with H· (−2.3 V).12 In the cells, NADH is an organic hydride compound and participates in a variety of metabolic processes. The mitochondrial free NADH concentration is approximately 20–230 μM in normal cells and 168–870 μM in cancer cells, respectively,13,14 which makes NADH a valuable biomarker for cancer diagnosis. Various fluorescent probes have been reported for distinguishing cancer cells from normal cells through specific recognition of NADH.15–17 Moreover, the recent bioimaging research conducted by Wu and co-workers revealed that the changes in the NADH level can be used for tumor differentiation grade recognition.18 NADH and H− donating systems can potentially be combined for selective hypoxia-tolerant cancer therapy. Hence, the exploration of photo-controllable H− donors and the investigation of H− transfer to bio-relevant oxidants (Bio-Ox) under physiological conditions are urgently demanded.
Since the 1st generation of PSs, molecular porphyrins have received the most interest in clinical PDT, but few of them are effective against hypoxic cancers on various differentiation grades. To date, successful redox-enhanced PDT under hypoxia using porphyrins or porphyrin analogues has been scarcely available. An impressive approach was introduced by Xu and co-workers. They observed that the reduction of 5,10,15,20-tetrakis-(4-N-methylpyridyl)-porphyrin (TMPyP) by facultative anaerobes afforded the phlorin form and applied this reaction to photothermal antimicrobial treatment (Scheme 1a).19 Although phlorin could be prepared by chemical, electrochemical, and photochemical methods,20,21 so far, no additional bio-related studies with phlorin derivatives have been reported, which inspires efforts in phlorin chemistry for cancer therapy (Scheme 1b).
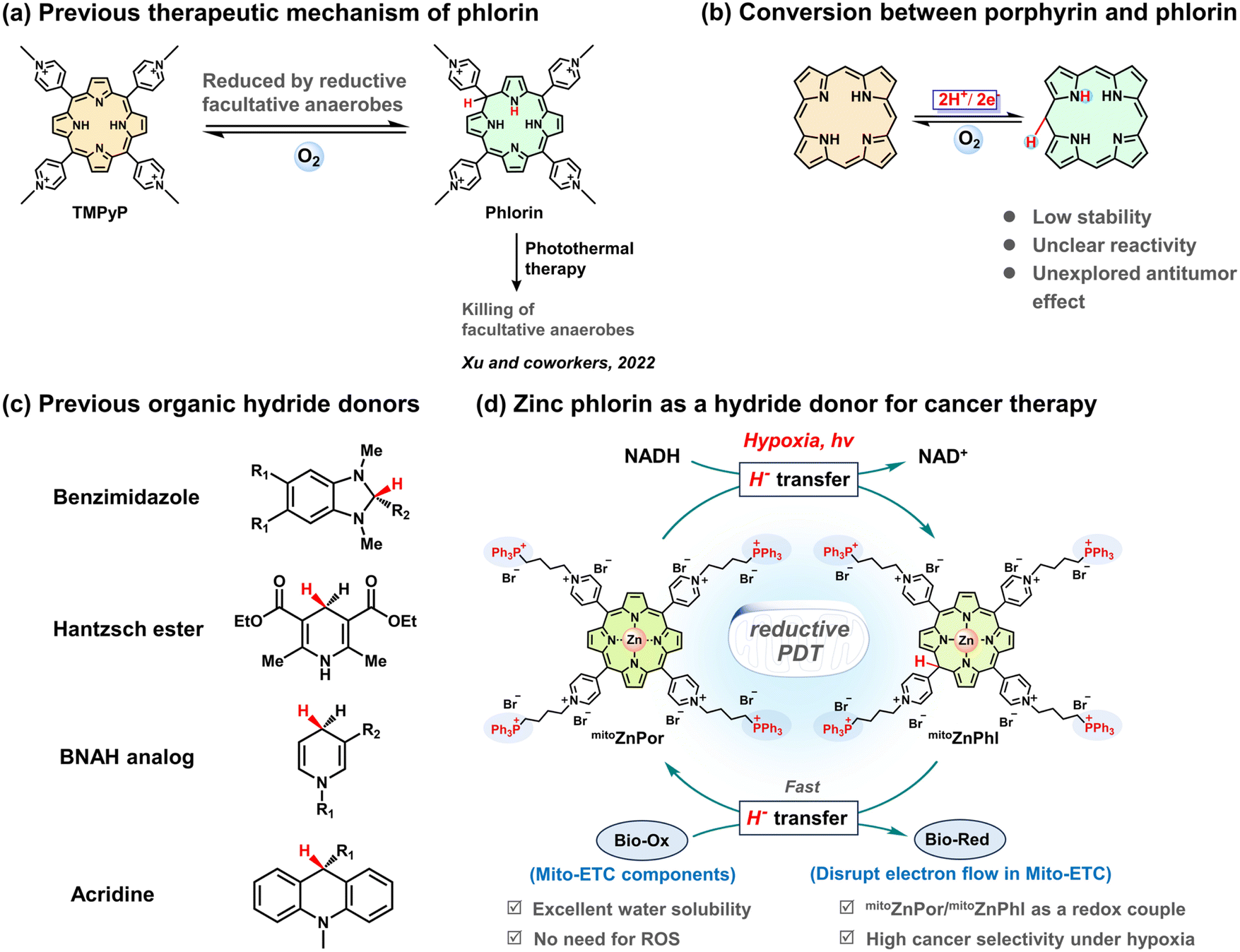 | ||
| Scheme 1 Schematic representation of porphyrin/phlorin chemistry and the use of zinc-cored phlorin as a hydride donor for cancer therapy in the present work. (a) Previous work by Xu et al. using phlorin to kill facultative anaerobes through the photothermal effect. (b) Reversible reaction between porphyrin and phlorin. (c) Representative organic hydride donors. (d) Schematic illustration of in situ NADH-driven photoconversion of mitoZnPor into mitoZnPhl, which disrupts Mito-ETC, leading to cancer cell-selective killing under hypoxia. | ||
NADH analogues (e.g., benzimidazoles, Hantzsch esters, 1-benzyl-1,4-dihydronicotinamide derivatives, and acridines) (Scheme 1c) have been reported with reducing capability (e.g., for CO2 reduction),22–24 although under harsh conditions. In the present study, we investigated the reducing properties of metal-free and zinc-cored phlorins and demonstrated that phlorin is a strong H− donor with the capability to reduce bio-relevant redox molecules like heme and ubiquinone. Using mitochondrial NADH as a hallmark of cancer and mitoZnPor as a mitochondria-targeting precursor of mitoZnPhl, we established a strategy to in situ form a mitoZnPor/mitoZnPhl redox couple in the mitochondria and proposed a reductive PDT mechanism based on cascade H− transfer processes including NADH to mitoZnPor and mitoZnPhl to Bio-Ox (Scheme 1d). Especially, the chemical understanding of the relationship between zinc phlorin and endogenous hydride or electron acceptors such as iron heme and ubiquinone, which are essential redox-active components of the mitochondrial electron transport chain (Mito-ETC), provides a foundation for the H−-participating PDT concept. The NADH-driven disruption of the electron flow in Mito-ETC leads to cell death with high therapeutic indices and selectivity indices against diverse hypoxic tumor cells. We also applied mitoZnPor in a 4T1 tumor-xenografted mouse model to demonstrate the great potential of reductive PDT in fighting hypoxic cancers.
Results and discussion
Preparation of porphyrins and photochemical generation of phlorins
The cationic porphyrins (cPor) flanked with four pyridinium units were prepared by the reaction of 5,10,15,20-tetra(4-pyridyl)porphyrin (TPyP) with different organic halides in heated N,N-dimethyl formamide (DMF) (Fig. 1a) and structurally characterized by NMR and high-resolution mass spectrometry (HRMS) (Fig. S1–S4†). Por-2 was frequently termed TMPyP in quite a few previous reports.19,25 The compounds Por-1–Por-4 showed similar spectra in ultraviolet-visible (UV-vis) absorption and fluorescence emission (Fig. S5†) in phosphate buffered saline (PBS). Por-1 was chosen as a representative for metalation to obtain ZnPor (Fig. 1b) and for comparison with ZnPor through parallel experiments including NADH binding and photophysical and photoredox processes.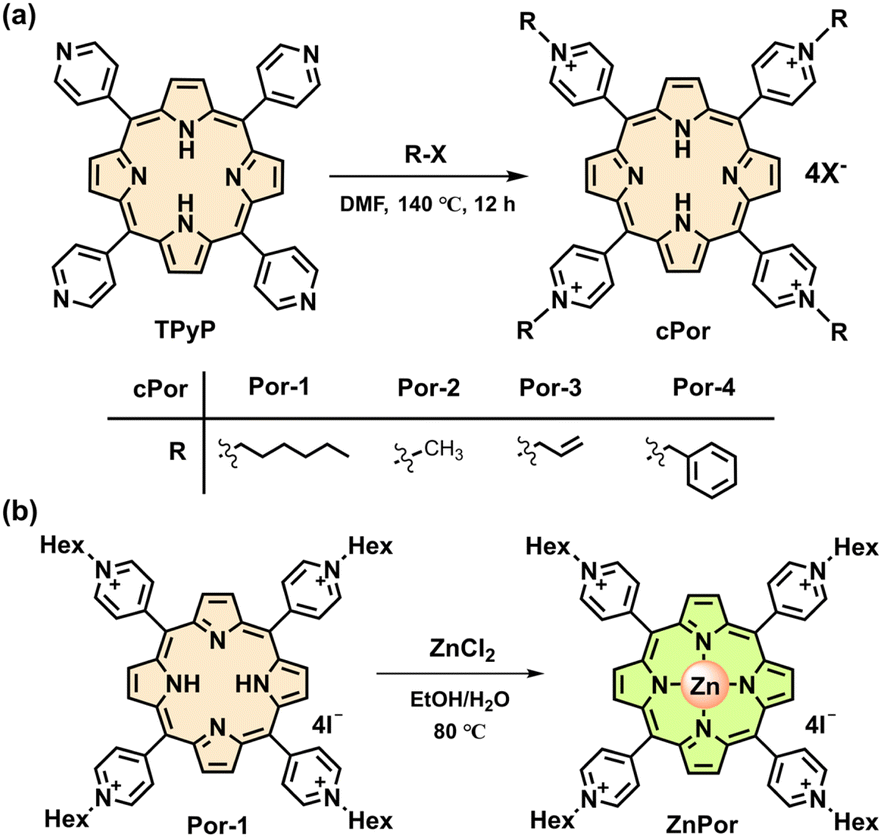 | ||
| Fig. 1 (a) Synthetic route of cPor molecules. (b) Synthetic route of ZnPor. | ||
Negatively charged NADH is expected to electrostatically interact with Por-1. The addition of NADH to Por-1 caused a slight decrease and redshift of both S- and Q-bands in the absorption spectra (Fig. S6a†) as well as a concentration-dependent decline of Por-1 fluorescence with the maximum at 720 nm (Fig. S6b†), although the fluorescence lifetime was unchanged (τ = 4.72 ns) (Fig. S6c†). Moreover, the result of isothermal titration calorimetry (ITC) measurement in PBS (Fig. 2a) revealed that the interaction between NADH and Por-1 was an exothermic process with the binding constant (Ka) estimated to be 5.81 × 103 M−1. The binding ratio between NADH and Por-1 was calculated to be 2.04, in good agreement with the charge ratio of Por-1 to NADH. Therefore, the binding mode driven by electrostatic interaction is proposed in Fig. 2b. The close contact of NADH under physiological conditions would facilitate photochemical processes.
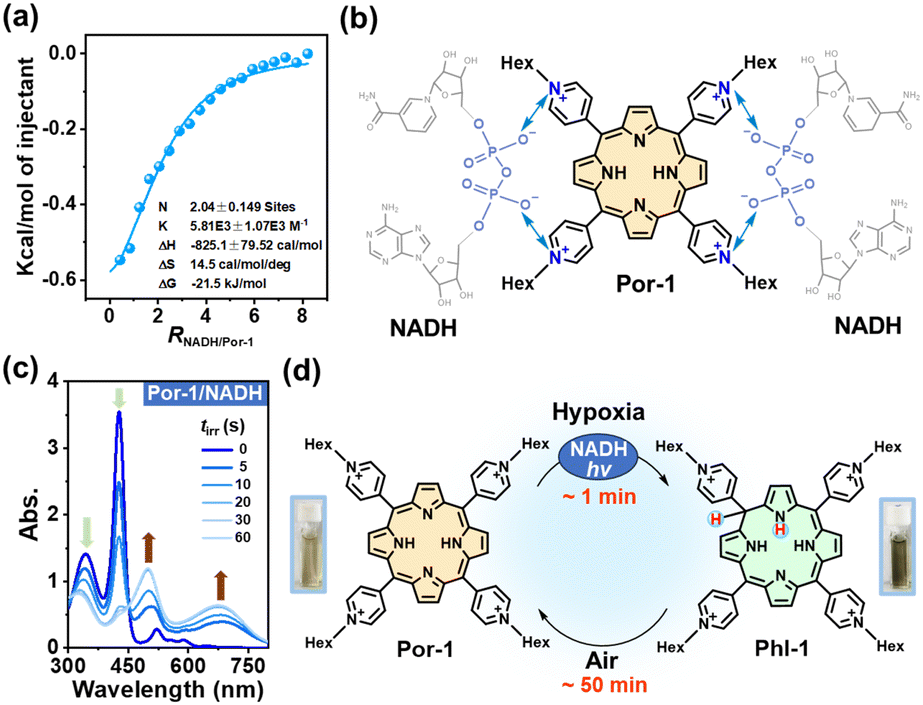 | ||
| Fig. 2 (a) ITC data measured by titrating 8 mM NADH into 0.2 mM Por-1 in 10% DMSO/PBS. (b) Schematic illustration of electrostatic interactions between Por-1 and NADH. (c) Time-dependent UV-vis spectra of Por-1 (20 μM) and NADH (200 μM) in deoxygenated PBS upon LED light irradiation (λ 405 nm and 25 mW cm−2). (d) The illustration of the conversion process between Por-1 and Phl-1 and the photographs of the solution containing Por-1 (left) or Phl-1 (right). | ||
The anaerobic photochemical reaction was monitored using UV-vis absorption spectra under LED light irradiation. The S-band disappeared at a short irradiation time (tirr) of 60 s, accompanied by the emergence of broad phlorin absorptions at around 500 and 680 nm (Fig. 2c) and a decrease in NADH absorption at 340 nm. Simultaneously, the red fluorescence of Por-1 disappeared (Fig. S7†) and the reaction mixture changed color from yellow to tawny (Fig. 2d), in good agreement with a previous study.19 Direct evidence of Phl-1 generation came from the HRMS analysis, by which Phl-1 was discriminated from Por-1 (Fig. S8†). These data demonstrated successful NADH-driven photoconversion of porphyrin into phlorin. In the open air, Phl-1 was unstable and slowly oxidized back to fluorescent Por-1 within 50 min (Fig. 2d and S9†). In a similar way, the phlorins (Table S1†) were detected from photoreactions with other cPor compounds (Fig. S10†), showing that NADH is an efficient electron donor or hydride donor for the photocatalytic cPor reduction. Oxygen concentration affected the photoreaction. As an electron acceptor,26,27 oxygen at high levels caused a decrease in photoconversion efficiency (Fig. S11†).
To elucidate whether one-electron transfer was involved in the photochemical process, we introduced 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger into the reaction. Phl-1 was still generated upon irradiation (Fig. S12a†), which ruled out a one-electron transfer mechanism. Therefore, a two-electron transfer process was more likely to occur. NADH transferred H− to photoexcited porphyrin yielding the hydrogenated product Phl-1 and the oxidized product NAD+ (Fig. S12b†), analogous to the typical role of NADH involved in many metabolic processes.28,29
The coordination of metal ions at the porphyrin center impacts the photophysical and photochemical properties of porphyrins. We chose zinc-cored porphyrin for the present study, as zinc is a biologically essential element with excellent biocompatibility as well as the second most abundant trace metal in the human body.30 ZnPor was synthesized from Por-1 (Fig. 1b) and characterized by 1H NMR analysis (Fig. S13†). As compared to Por-1, ZnPor exhibited features as follows: (1) redshifted absorption and blue shifted fluorescence (Fig. S14†); (2) unaffected fluorescence lifetime by NADH in PBS (Fig. S15†); (3) similar binding mode with NADH (Fig. 3a and S16†).
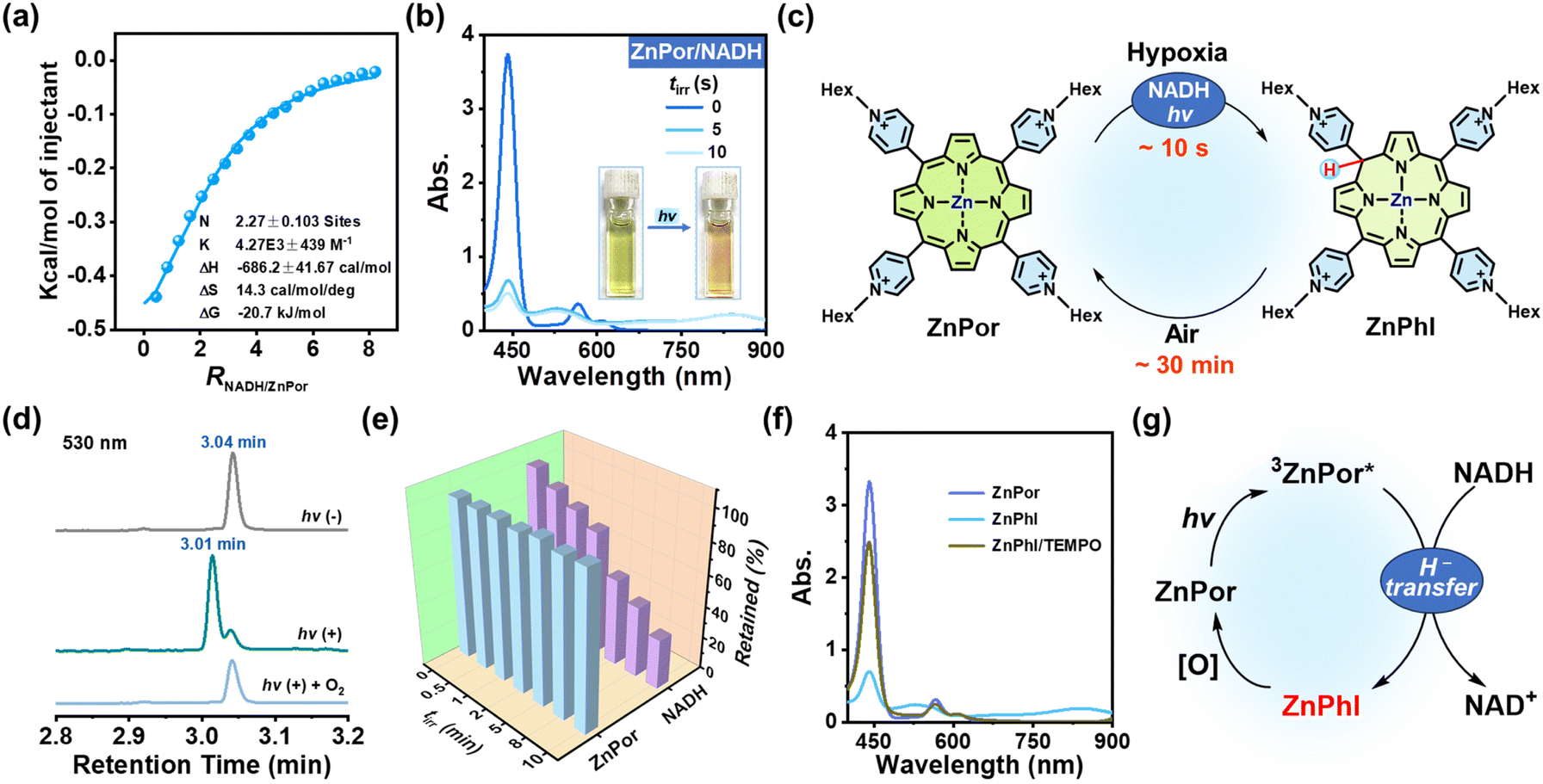 | ||
| Fig. 3 (a) ITC data measured by titrating 8 mM NADH into 0.2 mM ZnPor in 10% DMSO/PBS. (b) Time-dependent UV-vis spectra of ZnPor (20 μM) and NADH (200 μM) in deoxygenated PBS upon LED light irradiation (λ 405 nm and 25 mW cm−2). Inset: photographs of the solution containing ZnPor and NADH in deoxygenated PBS before and after light irradiation. (c) The illustration of the conversion process between ZnPor and ZnPhl. (d) UPLC analysis of the solution containing ZnPor (20 μM) and NADH (200 μM) in deoxygenated pH 7.4 PBS before and after light irradiation (λ 405 nm, 25 mW cm−2, and tirr = 5 s), and after exposure to oxygen following irradiation. (e) Time-dependent percentage of remaining ZnPor and NADH in the solution containing ZnPor (20 μM), NADH (200 μM) and TEMPO (10 mM) in deoxygenated PBS upon LED light irradiation (λ 405 nm and 25 mW cm−2). (f) UV-vis spectra of solution containing ZnPhl before and after the addition of TEMPO (10 mM). (g) Proposed mechanism of the ZnPor/ZnPhl cycle in the presence of NADH and light. | ||
Under irradiation, a photoreaction process between ZnPor and NADH was observed in the deoxygenated PBS (Fig. 3b), manifested as new broad absorption bands at 530 and 840 nm, which indicated the generation of hydrogenated ZnPor in the form of phlorin (termed ZnPhl). The decreased current in cyclic voltammograms further demonstrated the photoconversion of ZnPor (Fig. S17†). Similarly, ZnPhl was air-intolerant and slowly air-oxidized back to ZnPor (Fig. 3c and S18†). The presence of oxygen can affect the photoreduction efficiency from ZnPor to ZnPhl (Fig. S19†). Moreover, the reaction process was confirmed by ultrahigh performance liquid chromatography (UPLC) analysis (Fig. 3d and S20†). The signal peak of ZnPor at a retention time of 3.04 min declined significantly upon irradiation, accompanied by the appearance of a new peak at a retention time of 3.01 min, corresponding to the hydrogenated product ZnPhl. Under exposure to oxygen, the re-appearance of a signal peak at 3.04 min indicated oxidation from ZnPhl to ZnPor, in agreement with the result of UV-vis spectral changes.
Interestingly, in the presence of TEMPO, the photoreaction of deoxygenated solution (tirr 0–10 min) led to continuous consumption of NADH along with negligible loss of ZnPor (Fig. 3e). To check whether a single electron transfer (SET) process was involved, we applied TEMPO to capture the radical intermediate. However, no TEMPO adduct was detected by HRMS, which suggested that the SET process may not account for the ZnPhl generation. To test the hypothesis that the suppression of ZnPhl production was associated with the reducing properties of ZnPhl, we detected the response of ZnPhl upon exposure to TEMPO according to the changes in UV-vis absorption spectra. Surprisingly, the characteristic ZnPhl absorption instantly vanished and ZnPor was regenerated (Fig. 3f). However, the oxidation of TEMPO to Phl-1 proceeded at a relatively lower rate (∼2 min to complete the reaction) (Fig. S21†), which correlated with the phenomenon that TEMPO can inhibit the photoreduction of ZnPor but not that of Por-1. The big difference between Phl-1 and ZnPhl in tolerance to TEMPO may suggest the role of zinc coordination in promoting H− donation and increasing the reducing capability of phlorin. The overall photochemical process is illustrated in Fig. 3g. Of note, typical porphyrins without a phlorin intermediate involved, exemplified by well-documented tetraamine-substituted porphyrin (TAPP) and zinc-coordinated TAPP (ZnTAPP)31 (Scheme S1 and Fig. S22†), were inefficient in photochemical NADH oxidation under hypoxic conditions.
Reactivity of ZnPhl
Organic H− donors, particularly NADH analogues, have attracted considerable attention in energy and green chemistry.22–24 ZnPhl seems akin to an NADH analogue based on H− transfer mediated by the H-bound sp3 hybrid carbon of the phlorin ring, and therefore ZnPhl/ZnPor forms an NADH-dependent photoredox couple under hypoxic physiological conditions. To date, the reactivity of phlorins towards bio-relevant species remains largely unknown, which impedes the biological applications of phlorins.Besides TEMPO, we also checked the response of ZnPhl to methylene blue (MB), a well-known redox dye that was reported to interact with the mitochondrial respiratory chain via acting as an alternative electron carrier.32 The immediate conversion of ZnPhl to ZnPor took place with concomitant bleaching of MB absorption at 665 nm (Fig. 4a). Upon a short period of photochemical reaction (tirr 1 min), MB was reduced by ZnPor/NADH with an efficiency of 91% (Fig. 4b). The absorption of MB was recovered after exposure of the irradiated solution to the open air (Fig. S23†), which supported the reversible transformation between MB and leucoMB and agreed well with the MB activity in accepting H−.33 The photoreaction is illustrated in Fig. 4c. Other H− acceptors such as quinone derivatives like para-quinone and p-chloranil were good substrates of the photosystem as well (Fig. 4d and S24†).
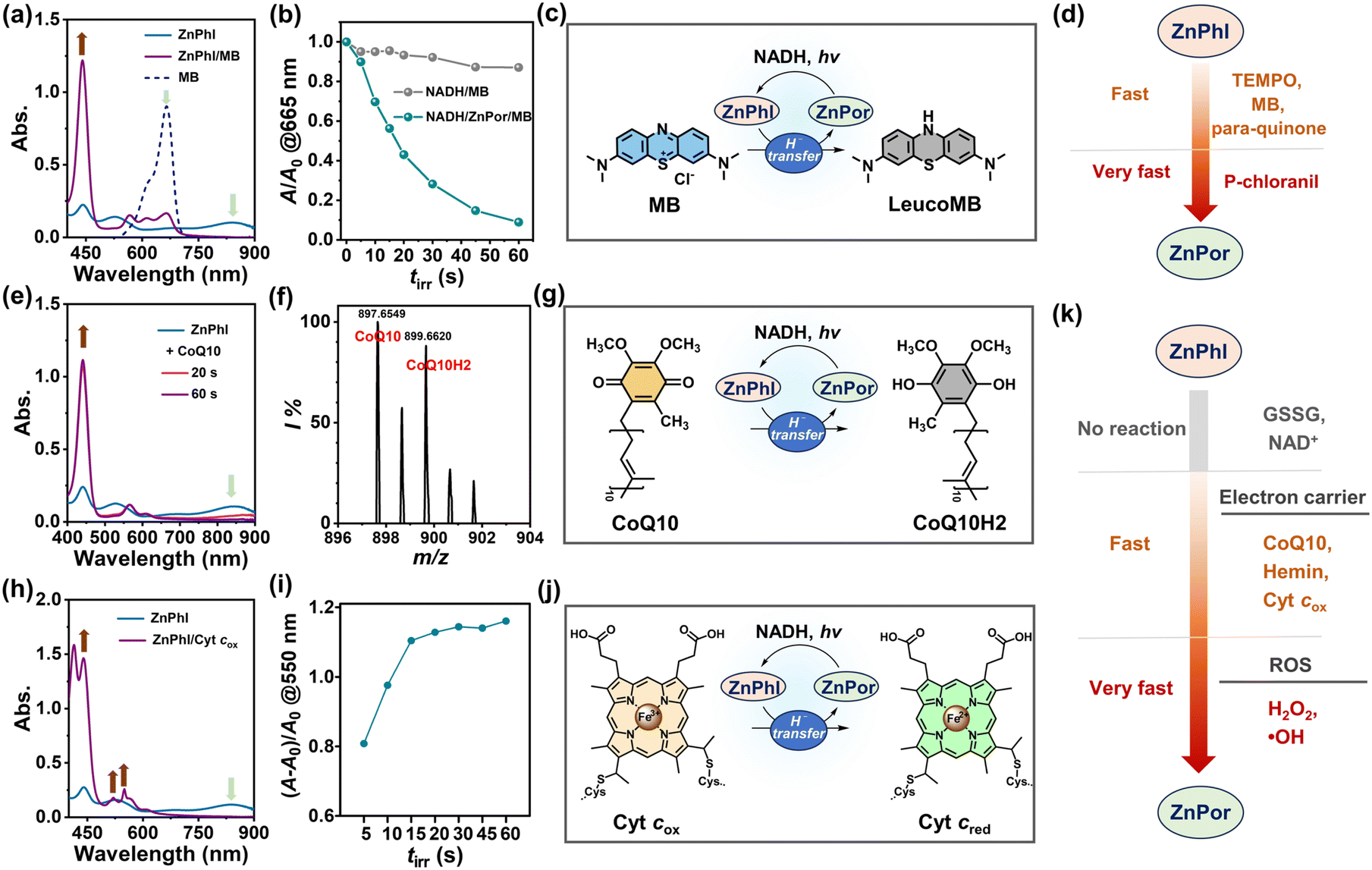 | ||
| Fig. 4 (a) UV-vis spectra of ZnPhl solutions before and after the addition of MB (10 μM). (b) ZnPor (1 μM)-mediated MB (10 μM) photoreduction efficiency in deoxygenated PBS. (c) Schematic diagram of the photochemical ZnPor/ZnPhl cycle in reducing MB. (d) The outlined reactivity of ZnPhl as a hydride donor towards various organic oxidants. (e) UV-vis spectra of ZnPhl solutions before and after the addition of CoQ10 (0.1 mM). (f) HRMS spectrum of the ZnPhl/CoQ10 mixture. (g) Schematic diagram of the photochemical ZnPor/ZnPhl cycle in reducing CoQ10. (h) UV-vis spectra of solutions containing ZnPhl before and after the addition of Cyt cox (0.1 mg mL−1). (i) Changes in absorption at 550 nm in the photochemical reduction of Cyt cox (0.1 mg mL−1) in PBS. (j) Schematic diagram of the photochemical ZnPor/ZnPhl cycle in reducing Cyt cox. (k) The outlined reactivity of ZnPhl towards various endogenous electron acceptors. | ||
The reactivity of ZnPhl to a wide range of endogenous oxidative species was investigated in the subsequent experiments. Firstly, ZnPhl was unreactive to glutathione (GSSG) and NAD+, avoiding undesired scavenging of ZnPhl by the most abundant oxidative species in the living cells. Secondly, ZnPhl efficiently scavenged ROS such as OH· and H2O2 (Fig. S25†). Lastly, redox reversible coenzyme quinone and cytochrome (Cyt) are preferred targets of ZnPhl and thus are discussed in more detail as they play key roles in living systems.34–38
Ubiquinone CoQ10 is an important electron carrier in Mito-ETC and essential for tumor growth.34–36 The hydrophobic CoQ10 oxidized ZnPhl to ZnPor at a fast rate (Fig. 4e). The reduced form of CoQ10, ubiquinol CoQ10H2, was detected with an m/z of 899.6620 ([CoQ10H2 + Cl]−) by HRMS that clearly distinguished ubiquinol from ubiquinone (Fig. 4f and S26†). In the mitochondrial respiratory chain, ubiquinone accepts electrons from NADH and is reduced to ubiquinol by complex I (CI).36,39 Considering the rapid regeneration of ZnPhl from ZnPor, the results suggested that the ZnPor/ZnPhl-based photochemical system is a potential photoenzyme mimicking CI with oxidoreductase-like activity (Fig. 4g). In contrast, the redox reaction between Phl-1 and CoQ10 proceeded at a notably lower rate (Fig. S27†).
Heme is a redox component of Cyt cox, CII, CIII and CIV in Mito-ETC, transferring electrons to quinone or other acceptors.40 With a high oxidation potential (−0.36 V versus SCE),41 the hemin centered with FeIII was efficient in oxidizing ZnPhl (Fig. S28†). As an electron carrier, Cyt cox plays crucial roles in Mito-ETC, regulating ETC activity, oxygen consumption, ATP synthesis and initiating apoptosis.34 The reduction from Cyt cox to Cyt cred took place upon exposing Cyt cox to ZnPhl, as seen by the increased absorption intensity at the β band (520 nm) and α band (550 nm) (Fig. 4h). In the photochemical experiment, the enhancement of absorption intensity at 550 nm reached a plateau after 15 s of light irradiation (Fig. 4i and S29†), with a turnover frequency (TOF) of 20.6 min−1, which was even greater than that of a previously reported photoredox system using an Ir(III) photocatalyst (TOF 16.7 h−1).27 Hence, it was reasonable to infer that the photocatalytic Cyt cox reduction was feasible using the ZnPor/ZnPhl redox couple in biological systems (Fig. 4j).
Taken together, the reactivity of ZnPhl toward various Bio-Ox is outlined in Fig. 4k, which provided a basis for developing metal phlorin-based novel PDT against hypoxic cancers.
Selective in vitro PDT against hypoxic tumors
We applied the above principle of H− donor-participating photoreaction in cancer therapy to improve both therapeutic efficacy and selectivity. Four triphenylphosphonium (TPP) substituents were covalently conjugated to the pyridinium groups of ZnPor for the following considerations: (1) the cellular uptake of TPP-modified agents is more selective to tumor cells than normal cells, ascribed to the hyperpolarized tumor cell membrane potential.42 (2) The excellent mitochondria-targeting capability of the TPP moiety guarantees mitochondrial accumulation driven by the negative mitochondrial membrane potential (MMP).43 (3) The mitochondria-penetration capability of TPP carries cargoes into the Mito-ETC sites,44 which is a prerequisite to ensure the location of the photochemical system close to Mito-ETC with abundant availability of NADH. (4) The positive charges of TPP moieties enhance the binding affinity between NADH and cationic PSs. Consequently, mitoZnPor combines the merits of peripheral TPP moieties in selectivity/Mito-ETC-targeting ability and enclosed photoactivatable mitoZnPor in hypoxia-tolerant antitumor efficacy/NADH-dependence (Fig. 5a).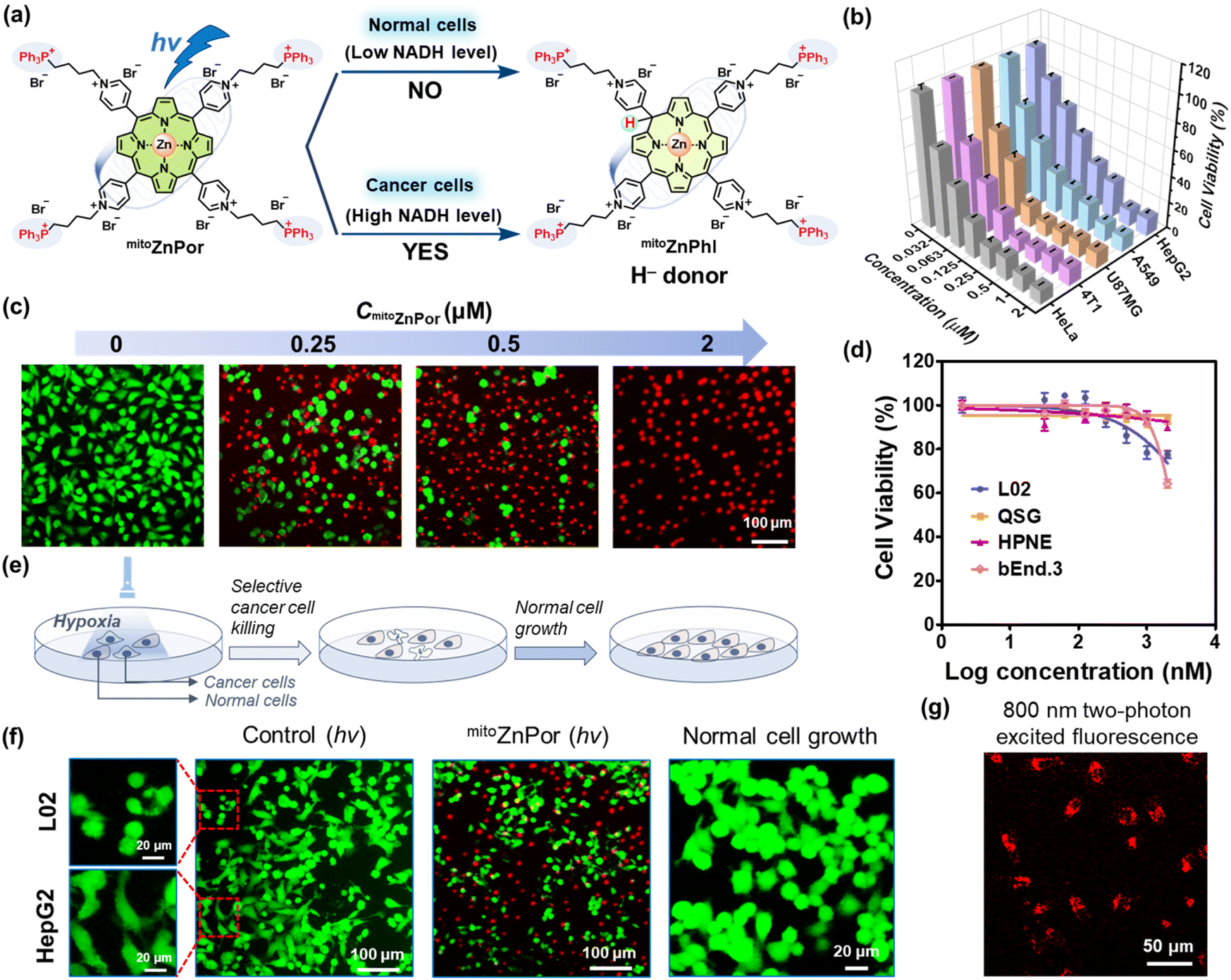 | ||
| Fig. 5 (a) Schematic illustration of mitoZnPor for cancer cells-selective killing under hypoxia with NADH as a biomarker. (b) Viabilities of cancer cells (HeLa, 4T1, U87MG, A549, and HepG2) treated with varied concentrations of mitoZnPor (0–2 μM) after white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia. (c) Fluorescence imaging of A549 cells after treatment with varied concentrations of mitoZnPor and light irradiation under hypoxia. The live and dead cells were stained with Calcein-AM and propidium iodide (PI) before imaging. (d) Viabilities of normal cells (L02, QSG, HPNE, and bEnd.3) treated with varied concentrations of mitoZnPor (0–2 μM) after white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia. (e) Schematic illustration of mitoZnPor for selective cancer cell killing and normal cell growth. (f) The images of mitoZnPor-treated HepG2/L02 cells in the co-cultured model after white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia and normal cell growth after PDT treatment. The cells were stained with Calcein-AM and PI before imaging. (g) 800 nm two-photon CLSM images of mitoZnPor (4 μM)-treated HeLa cells after irradiation with an 800 nm two-photon laser. | ||
mitoZnPor was synthesized through coordination of mitoPor with Zn2+ (Scheme S2†). mitoPor and mitoZnPor were characterized by NMR, HRMS and UV-vis absorption spectroscopy (Fig. S30–S31†). Furthermore, the NADH-driven photogeneration of phlorins in deoxygenated PBS was verified by the emergence of absorption bands in the near infrared region (Fig. S32†). Under light, both mitoPor and mitoZnPor were efficient in reducing Cyt cox (Fig. S33†) and CoQ10 (Fig. S34†).
The mitochondria-targeting properties of mitoZnPor in A549 cells were studied using commercial Mito-Tracker as the mitochondria-specific staining agent. The co-localization imaging data were analyzed with Pearson's correlation coefficient estimated to be 0.87 (Fig. S35†), in good agreement with the ability of TPP moieties in mitochondrial accumulation.44
Next, we harnessed a Mitsubishi™ device to mimic the hypoxic environment (1% O2) for cell studies and the actual O2 content was 0.6% based on three repeated tests (Fig. S36†). To assess the biocompatibility of mitoZnPor, we performed a standard MTT assay after 28 h of treatment of different cells with various concentrations of mitoZnPor in the dark under hypoxia.
The normal cell lines included L02, QSG, HPNE and bEnd.3 cells. The cancer cell lines included HeLa (poorly differentiated),45 4T1 (poorly differentiated),18 U87MG (poorly differentiated),46 A549 (moderately differentiated),47 and HepG2 (well differentiated)48 cells. According to Fig. S37,† the IC50(dark) values were calculated and are summarized in Table 1. All the cells maintained high viability after treatment with 10 μM mitoZnPor. In contrast, strong phototoxicity of mitoZnPor was observed with all the cancer cells at low IC50(hv) values (0.041–0.14 μM) (Fig. 5b), underlining the remarkable PDT effect. MTT assays were performed using white LED light alone (Fig. S38†) or in the presence of sodium azide (NaN3), a known 1O2 scavenger (Fig. S39†), to exclude the possible ROS effect on cell death. The therapeutic index (TI), defined as the ratio of IC50(dark) to IC50(hv), was recorded (Table 1) up to 5.9 × 102, which was a considerable value in comparison to those of previously reported porphyrin-derived PSs under hypoxia.49,50 The in vitro PDT effect of mitoZnPor was also confirmed by the imaging data after fluorescent live/dead staining of A549 cells (Fig. 5c).
| Cell | Typeb | IC50(dark) (μM) | IC50(hv) (μM) | TI | SIc |
|---|---|---|---|---|---|
| a Data are presented as the means ± standard deviations of three repeated measurements. b diff., differentiated. c Selectivity index (SI), calculated with the L02 cell line as a reference (marked as L02*). | |||||
| L02* | Normal | 18.9 ± 1.4 | 20.4 ± 2.6 | 0.92 | |
| QSG | Normal | 18.6 ± 1.0 | 14.8 ± 6.9 | 1.26 | |
| HPNE | Normal | 20.7 ± 1.7 | 15.9 ± 0.2 | 1.30 | |
| bEnd.3 | Normal | 11.2 ± 0.5 | 9.3 ± 2.9 | 1.20 | |
| RAW264.7 | Immune | 12.8 ± 3.7 | 15.8 ± 0.2 | 0.81 | |
| HeLa | Poorly diff. | 29.8 ± 2.8 | 0.067 ± 0.011 | 444 | 305 |
| 4T1 | Poorly diff. | 24.1 ± 0.9 | 0.041 ± 0.003 | 588 | 498 |
| U87MG | Poorly diff. | 17.4 ± 1.5 | 0.045 ± 0.002 | 387 | 453 |
| A549 | Moderately diff. | 45.7 ± 4.8 | 0.079 ± 0.014 | 578 | 259 |
| HepG2 | Well diff. | 23.6 ± 1.7 | 0.14 ± 0.01 | 168 | 146 |
As compared to cancer cells, the normal cells were insensitive to the light treatment with mitoZnPor under hypoxia (Fig. 5d) and the TI values were obtained in a very low range (0.92–1.30) (Fig. S40† and Table 1). The selectivity index (SI), defined as the ratio of IC50(hv) between normal cells and cancer cells, was calculated using L02 cells as a reference (Table 1). The SI is an important indicator for therapeutic selectivity. However, PSs with a high SI against hypoxic tumors are rare because of nonspecific photodamage. mitoZnPor exhibited high SI values (>102) for all the tested cancer cells, despite the differentiation grades.
We assessed the efficacy and selectivity of PDT in a co-cultured model of cancer cells and normal cells (Fig. 5e). HepG2 cells were co-cultured with L02 cells and clearly distinguishable by the cell shape. After PDT treatment, HepG2 cells were selectively killed while L02 cells survived (Fig. 5f), in good agreement with the results of trypan blue staining (Fig. S41†). The co-cultured L02 cells after photo-irradiation proliferated well without the growth of HepG2 cells. These data indicated that mitoZnPor is an exceptional PDT agent with the ability to selectively kill cancer cells and spare normal cells in the same growth environment.
Concerning the special role of macrophages that are known as a major population of immune cells in solid cancers, we also applied the co-cultured model to check the safety of PDT. Like normal cells, macrophage RAW264.7 cells tolerated mitoZnPor treatment under dark and light conditions (Table 1) and survived under the co-cultured conditions after PDT treatment that led to selective killing of A549 cancer cells (Fig. S42†).
For comparison, we tested chlorin e6 (Ce6) using the same co-cultured protocol. However, Ce6 was phototoxic at 2 μM concentration to both cancer cells (HepG2 or A549 cells) and normal cells (or macrophages) (Fig. S43†), particularly normal cells, which was consistent with the results of MTT assays on separately cultured cells (Fig. S44†). The results underline the changes in therapeutic outcomes if the PDT mechanism differs. The strong dependence on ROS for oxidative cell damage is a double-edged sword for traditional PDT, because of the limitation in selectivity.
The limited light penetration depth has been a concern for PDT, which can be improved using NIR two-photon lasers.51 Two-photon absorption only occurs under high illumination intensity, and thus the PDT effect is confined to a small volume with high spatial resolution that circumvents damage to normal cells.51,52 Fortunately, mitoZnPor is photoactivatable in living cells using an 800 nm two-photon laser. The bright fluorescence of mitoZnPor observed in HeLa cells (Fig. 5g) suggested the potential application of mitoZnPor in NIR two-photon PDT.
The investigation of mitochondrial dysfunction
According to the chemical reactivity of mitoZnPhl with Mito-ETC components, we proposed a possible mechanism for the disruption of Mito-ETC by mitoZnPhl photogenerated from mitoZnPor in the presence of abundant mitochondrial NADH (Fig. 6a). In short, complexes CI, CII and CIII require the ubiquinone (CoQ) pool to transport electrons starting from NADH and pump protons into intermembrane space to drive ATP synthesis by CV. Cyt cox accepts electrons and is restored by CIV, with oxygen serving as the ultimate electron acceptor. As a stronger H− donor than NADH, mitoZnPhl can “scavenge” CoQ10 and Cyt cox into ubiquinol CoQ10H2 and Cyt cred like a “photoenzyme” to block electron flow in Mito-ETC after exceeding the repair capability of the intrinsic regulatory system. The PDT process depletes oxygen under hypoxia, which inhibits the function of CIV in restoring Cyt cox and pumping protons.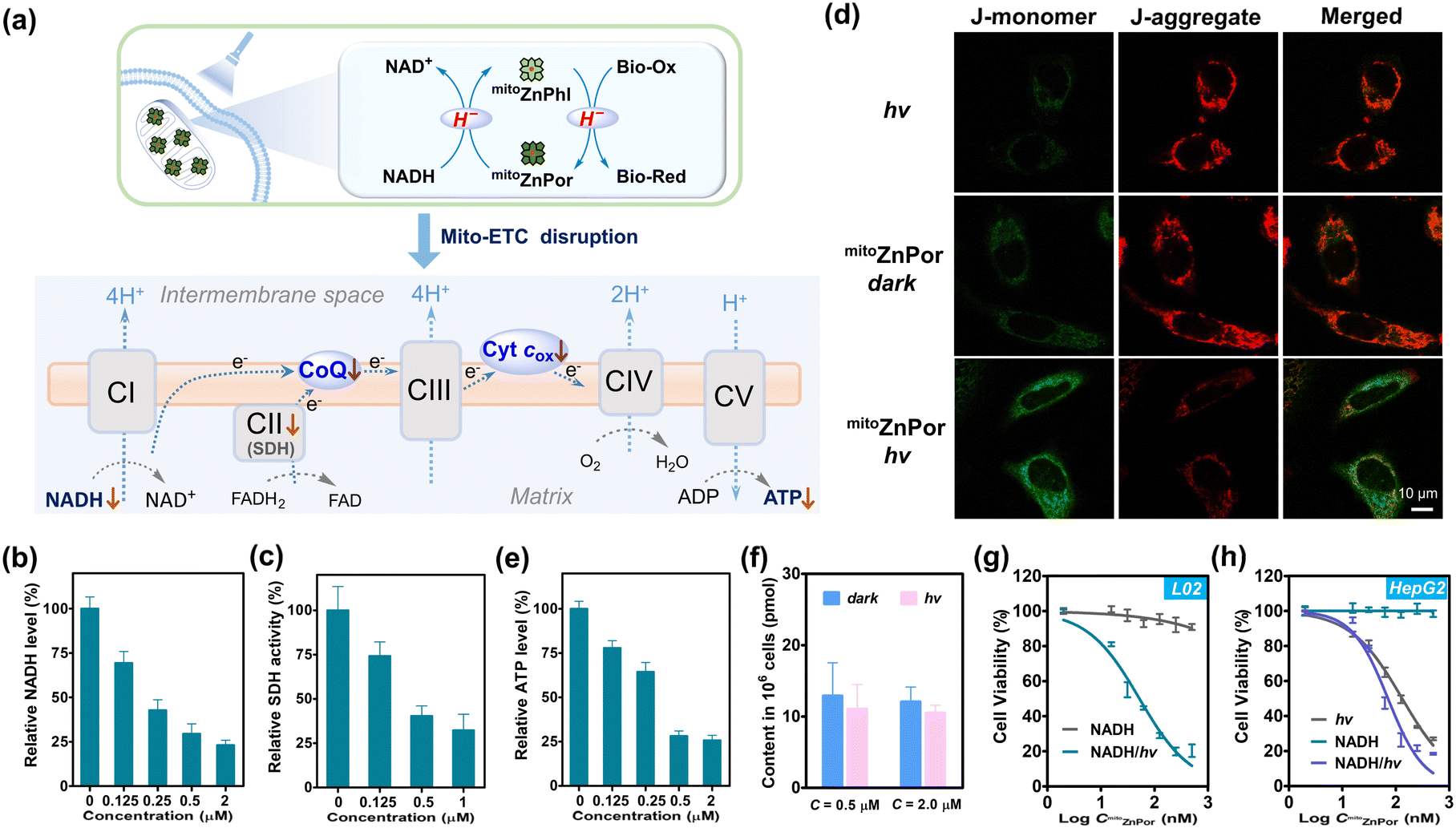 | ||
| Fig. 6 (a) Plausible mechanism of mitochondrial ETC disruption by photoactivation of mitoZnPor under hypoxia. (b) The changes in the intracellular NADH level in HeLa cells after treatment with mitoZnPor and light irradiation under hypoxia. (c) Intracellular SDH activity of HeLa cells after treatment with mitoZnPor and light irradiation under hypoxia. (d) CLSM images of HeLa cells after treatment with mitoZnPor (1 μM) under hypoxia. The cells were stained with JC-1 before imaging. (e) The changes in the intracellular ATP level in HeLa cells after treatment with mitoZnPor and light irradiation under hypoxia. (f) The changes in the intracellular NADH level in L02 cells after treatment with mitoZnPor (0.5 or 2.0 μM) in the absence and presence of white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia. (g) Viabilities of L02 cells treated with NADH (1 mM) and varied concentrations of mitoZnPor (0–0.5 μM) in the absence and presence of white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia. (h) Viabilities of HepG2 cells treated with NADH (0 or 1 mM) and varied concentrations of mitoZnPor (0–0.5 μM) in the absence and presence of white LED light irradiation (30 mW cm−2 and tirr = 10 min) under hypoxia. | ||
The intracellular NADH level was attenuated in a mitoZnPor concentration-dependent manner after PDT treatment of HeLa cells (Fig. 6b). In addition, the formation of formazan in mitochondria, a downstream redox enzyme reaction that needs NADH as the coenzyme,53 was significantly prevented (Fig. S45†). Succinate dehydrogenase (SDH), known as mitochondrial CII, was inactivated (Fig. 6c). SDH not only acts as a pivotal enzyme to generate NADH in the tricarboxylic acid (TCA) cycle, but also serves as CII to catalyze the reduction of ubiquinone to ubiquinol,54 in which two proposed ubiquinone binding sites have been reported.55 These observations demonstrated the requirement of NADH for driving photoconversion cycles that in turn suppress the activity of mitochondrial enzymes.
The Mito-ETC dysfunction was characterized by the collapse of MMP using a commercial JC-1 probe (Fig. 6d). The ATP level was notably reduced (Fig. 6e), attributable to the inhibition of oxidative phosphorylation (OXPHOS), thereby inevitably resulting in cell death. Recently, increasing studies have clarified that in cancer cells, mitochondria are still active and functional, and OXPHOS is crucial in producing ATP and regulating tumor proliferation.56,57 At present, targeting OXPHOS has emerged as a promising strategy for cancer therapy.39,58,59
PDT treatment induced weak ROS generation in the HeLa cells (Fig. S46†), due to the low level of oxygen. To evaluate the effect of ROS on cell death, we treated cells with various ROS scavengers including sodium ascorbate (NaAA) and N-acetyl-L-cysteine (NAC). The remarkable cell killing effect of mitoZnPor was not affected (Fig. S47†), which ruled out the possibility of the ROS effect on cell death. In fact, no oxidative damage of lipid peroxide (LPO) could be detected after normal hydride-PDT treatment (Fig. S48†). Consequently, we concluded that instead of ROS, the reductive effect of mitoZnPhl on mitochondrial function accounts for the killing of hypoxic cancer cells.
In contrast to HeLa cells, L02 cells were insensitive to PDT treatment with negligible fluctuation in the NADH content and MMP (Fig. 6f and S49†). Dehydrogenase activity was not impaired as well, according to the result of the formazan formation assay (Fig. S50†). These data confirmed that normal cells may survive hydride-PDT.
The superb phototherapeutic selectivity with mitoZnPor against hypoxic cancer cells was correlated with the significant difference in the mitochondrial microenvironment of cancer cells and normal cells. Cancer cells are widely known to possess high MMP relative to normal cells (−220 mV versus −140 mV),60 which increased mitochondrial accumulation of mitoZnPor. In addition, cancer cells overexpress mitochondrial NADH to drive H+ across the inner mitochondrial membrane for MMP maintenance and ATP synthesis.56,61 To evaluate the role of NADH in phototoxicity, we re-measured mitoZnPor concentration-dependent phototoxicity on L02 cells with the exception that the mitochondrial NADH level was elevated by supplementing exogenous NADH.54 As a result, L02 cells were induced to be sensitive to PDT treatment due to the perturbance of Mito-ETC and cellular metabolism (Fig. 6g and S51†). Likewise, supplementing exogenous NADH could amplify the phototoxicity of mitoZnPor on HepG2 cells (Fig. 6h). These findings verified that the intracellular NADH level is a reliable driving force for hydride-PDT. Obviously, the low level of mitochondrial NADH in the normal cells is inadequate to drive the photoredox reaction, thereby protecting cells from lethal damage.
It should be noted that other zinc-cored porphyrins such as ZnPor and ZnTAPP exhibited minimal photocytotoxicity (Fig. S52†). At the same time, the parent porphyrin mitoPor was dwarfed by mitoZnPor (Fig. S53†), in agreement with the fact that the zinc-free phlorin reacted with ubiquinone at a low rate (Fig. S54†). The superiority of mitoZnPor over these PS counterparts highlighted that key factors including the mitochondria-targeting ligand, reductive mechanism and phlorin reactivity are indispensable for the efficacy of PDT under hypoxia.
In vivo PDT against hypoxic tumors
The in vivo anti-cancer potency of mitoZnPor was evaluated in 4T1 tumor-xenografted mice. When the initial tumor volume reached ∼80 mm3, the mice were randomly divided into four groups and subjected to mitoZnPor or PBS treatment two times. The irradiation groups were then exposed to white light (100 mW cm−2 and 30 min) at 0.5 h after injection (Fig. 7a). Immunofluorescence imaging of the hypoxia inducible factor (HIF-1α, green signal) revealed that tumor tissues were in a severely hypoxic state (Fig. 7b). During the 14 days following treatment, the body weight of the mice had no obvious variations in different groups (Fig. 7c), but compared to the three groups of PBS, PBS + light, and mitoZnPor, the tumors in the mitoZnPor and light-treated mice were completely eliminated, without evidence of any recurrence (Fig. 7d), exhibiting remarkable PDT efficacy of mitoZnPor. The tumor size and weight further verified the efficient tumor growth inhibition (TGI) for the mitoZnPor + light group after 14 days of treatment (Fig. 7e and f). Moreover, the histological examination of tumor tissues using hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining showed that mitoZnPor and light-treated tumors exhibited sparser nuclei and increased DNA fragmentation, further revealing the potent photocatalytic tumor cell apoptosis of mitoZnPor (Fig. 7g). Fortunately, no obvious damage in the main organs (Fig. S55†) and no effect on the expression levels of blood biochemistry indices (Fig. S56†) were observed at the end of the therapeutic course, underlining the excellent biocompatibility and safety of mitoZnPor in living hosts. These results indicated that mitoZnPor can exert significant anti-cancer activity in vivo and is expected to be a good candidate for clinical trials in PDT.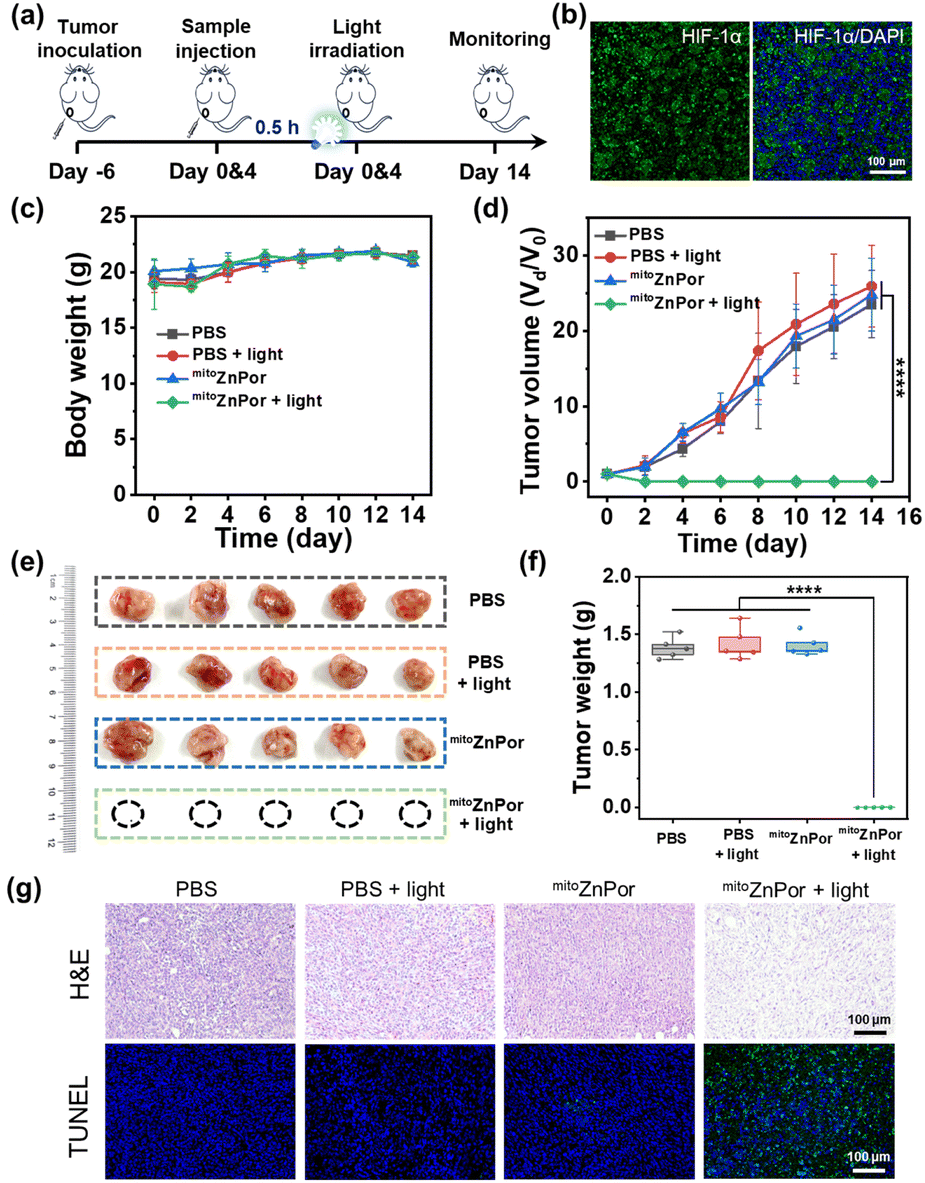 | ||
| Fig. 7 (a) Schematic illustration of the mitoZnPor-based PDT treatment schedule. (b) Immunofluorescence imaging (HIF-1α) of tumors collected 6 days after tumor inoculation for hypoxia confirmation. The nucleus was stained with DAPI (in blue). (c) Body weight change curves of 4T1 tumor-bearing BALB/c nude mice after various treatments. (d) Relative tumor volume changes in different groups as a function of treated time. (e) Photographs of tumors taken from different groups of mice after 14 days. (f) Average tumor weights for each group of mice on day 14 after various treatments. (g) H&E and TUNEL staining of tumor tissues separated from the mice with various treatments on the fifth day after treatment. Mean ± SD and n = 5. ****p < 0.0001. | ||
Conclusions
Inhibiting mitochondrial metabolism in biosynthesis and bioenergetic processes has emerged as a new cancer treatment strategy. Although multiple mitochondrial inhibitors targeting distinctive complexes of Mito-ETC to interrupt more than one enzymatic pathway can improve therapeutic efficacy through synergistic effects in inducing apoptosis, a major challenge remains in achieving selective cancer therapy with these inhibitors. Current PDT against tumor hypoxia is widely limited by PSs in terms of sub-organelle targeting, cancer-selective killing and therapeutic efficacy. In most cases, cytotoxic highly reactive ROS produced from the PDT process are neutralized by the endogenous antioxidant defense system. Metabolic microenvironment-responsive and RRS-participating PDT provides an option to escape from the ubiquitous antioxidant defense system that is inferior in scavenging RRS.We discovered that phlorins are a kind of H− donor with stronger reducing capability than NADH. The coupling of NADH to the porphyrin/phlorin redox cycle bridges NADH and electron acceptors in a photochemically controlled manner, rather than through the oxidoreductase-catalyzed pathway. Further, we demonstrated that in H−-based PDT using mitoZnPor as a precursor of mitoZnPhl, the level of mitochondrial NADH is a key factor in selective killing of hypoxic cancer cells from normal cells. The merits of H−-based phototherapy include: (1) photoactivatable H− transfer under hypoxic conditions circumvents the problems of ROS-participating PDT associated with low efficacy due to the counteraction of antioxidant species; (2) relative to short-lived ROS at a nanosecond scale, zinc phlorin derivatives have improved stability in the hypoxic environment, which allows for efficient redox reactions via H− transfer along Mito-ETC; (3) as compared to traditional PDT, the need for oxidative stress in cancer cells is alleviated. Hence, as a precursor of H−, mitoZnPor exhibited satisfactory in vitro PDT performances. mitoZnPor also possesses outstanding antitumor potency and excellent biocompatibility and safety in vivo, and thus is promising in extensive precision cancer phototherapy.
Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
F. Feng designed and guided the overall study. S. Li guided the in vivo experiments. X. Wang and Y. Gao performed the experiments. F. Feng and X. Wang wrote and revised the manuscript. T. Wang, Z. Wang, and H. Hang helped with data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22077065, 22277054, and 52173135), the Natural Science Foundation of Jiangsu Province (No. BK20231523), and Jiangsu Specially Appointed Professorship for financial support.Notes and references
- X. Wang, J. Peng, C. Meng and F. Feng, Chem. Sci., 2024, 15, 12234–12257 RSC.
- J. Tian, B. Huang, M. H. Nawaz and W. Zhang, Coord. Chem. Rev., 2020, 420, 213410 CrossRef CAS.
- Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC.
- Y. Li, C. Zhang, Q. Wu, Y. Peng, Y. Ding, Z. Zhang, X. Xu and H. Xie, Angew. Chem., Int. Ed., 2023, 63, e202317773 CrossRef.
- X. Su, W.-J. Wang, Q. Cao, H. Zhang, B. Liu, Y. Ling, X. Zhou and Z.-W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115800 CrossRef CAS PubMed.
- L. K. B. Tam, J. C. H. Chu, L. He, C. Yang, K.-C. Han, P. C. K. Cheung, D. K. P. Ng and P.-C. Lo, J. Am. Chem. Soc., 2023, 145, 7361–7375 CrossRef CAS.
- C. Yao, Y. Li, Z. Wang, C. Song, X. Hu and S. Liu, ACS Nano, 2020, 14, 1919–1935 CrossRef CAS.
- Z. Gu, L. Wang, Q. Dong, K. Xu, J. Ye, X. Shao, S. Yang, C. Lu, C. Chang, Y. Hou, Y. Zhai, X. Wang, F. He and A. Sun, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215744120 CrossRef CAS.
- Y. Wan, L.-H. Fu, C. Li, J. Lin and P. Huang, Adv. Mater., 2021, 33, 2103978 CrossRef CAS.
- J. Peng and F. Feng, Chem.–Eur. J., 2024, 30, e202302842 CrossRef CAS.
- J. Peng, K. Du, J. Sun, X. Yang, X. Wang, X. Zhang, G. Song and F. Feng, Angew. Chem., Int. Ed., 2023, 62, e202214991 CrossRef CAS.
- K. Fukui, S. Iimura, T. Tada, S. Fujitsu, M. Sasase, H. Tamatsukuri, T. Honda, K. Ikeda, T. Otomo and H. Hosono, Nat. Commun., 2019, 10, 2578 CrossRef PubMed.
- S. Shibata, S. Sogabe, M. Miwa, T. Fujimoto, N. Takakura, A. Naotsuka, S. Kitamura, T. Kawamoto and T. Soga, Sci. Rep., 2021, 11, 21353 CrossRef CAS PubMed.
- T. Li, Y. Zou, S. Liu, Y. Yang, Z. Zhang and Y. Zhao, Cell Regener., 2022, 11, 5 CrossRef CAS.
- K. Ma, H. Yang, X. Wu, F. Huo, F. Cheng and C. Yin, Angew. Chem., Int. Ed., 2023, 62, e202301518 CrossRef CAS PubMed.
- S. Shamjith, M. M. Joseph, V. P. Murali, G. S. Remya, J. B. Nair, C. H. Suresh and K. K. Maiti, Biosens. Bioelectron., 2022, 204, 114087 CrossRef CAS PubMed.
- A. Podder, V. P. Murali, S. Deepika, A. Dhamija, S. Biswas, K. K. Maiti and S. Bhuniya, Anal. Chem., 2020, 92, 12356–12362 CrossRef CAS PubMed.
- C. Yang, C. Jiang, M. Yang, Q. Bai, Y. Zhen, Y. Zhang, W. Yin, J. Wang, X. Zhou, G. Li, M. Wu, Y. Qin, Q. Wang, H. Ji and L. Wu, Chem. Biomed. Imaging, 2023, 1, 738–749 CrossRef CAS PubMed.
- H. Hu, H. Wang, Y. Yang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200799 CrossRef CAS PubMed.
- H. M. Castro-Cruz, L. R. Arias-Aranda, N. Farfán, E. Xochitiotzi-Flores and N. A. Macías-Ruvalcaba, J. Electrochem. Soc., 2020, 167, 155507 CrossRef CAS.
- K. T. Oppelt, E. Wöβ, M. Stiftinger, W. Schöfberger, W. Buchberger and G. Knör, Inorg. Chem., 2013, 52, 11910–11922 CrossRef CAS PubMed.
- S. Ilic, J. L. Gesiorski, R. B. Weerasooriya and K. D. Glusac, Acc. Chem. Res., 2022, 55, 844–856 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- C.-H. Lim, S. Ilic, A. Alherz, B. T. Worrell, S. S. Bacon, J. T. Hynes, K. D. Glusac and C. B. Musgrave, J. Am. Chem. Soc., 2019, 141, 272–280 CrossRef CAS PubMed.
- H. Shinoda, R. Higano, T. Oizumi, A. J. Nakamura, T. Kamijo, M. Takahashi, M. Nagaoka, Y. Sato and A. Yamaguchi, ACS Appl. Bio Mater., 2024, 7, 1204–1213 CrossRef CAS.
- M. Li, K. H. Gebremedhin, D. Ma, Z. Pu, T. Xiong, Y. Xu, J. S. Kim and X. Peng, J. Am. Chem. Soc., 2022, 144, 163–173 CrossRef CAS.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS.
- S. Chornyi, C. F. Costa, L. Ijlst, M. Fransen, R. J. A. Wanders, C. W. T. van Roermund and H. R. Waterham, Free Radical Biol. Med., 2023, 206, 22–32 CrossRef CAS PubMed.
- T. Nomura, T. Kimura, Y. Kanematsu, D. Yamada, K. Yamashita, K. Hirata, G. Ueno, H. Murakami, T. Hisano, R. Yamagiwa, H. Takeda, C. Gopalasingam, R. Kousaka, S. Yanagisawa, O. Shoji, T. Kumasaka, M. Yamamoto, Y. Takano, H. Sugimoto, T. Tosha, M. Kubo and Y. Shiro, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101481118 CrossRef CAS.
- M. Pellei, F. Del Bello, M. Porchia and C. Santini, Coord. Chem. Rev., 2021, 445, 214088 CrossRef CAS.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS.
- J. C. Rojas, A. K. Bruchey and F. Gonzalez-Lima, Prog. Neurobiol., 2012, 96, 32–45 CrossRef CAS.
- S. Ogo, K. Ichikawa, T. Kishima, T. Matsumoto, H. Nakai, K. Kusaka and T. Ohhara, Science, 2013, 339, 682–684 CrossRef CAS PubMed.
- H. Li, S. K. Kolluri, J. Gu, M. I. Dawson, X. Cao, P. D. Hobbs, B. Lin, G.-Q. Chen, J.-S. Lu, F. Lin, Z. Xie, J. A. Fontana, J. C. Reed and X.-K. Zhang, Science, 2000, 289, 1159–1164 CrossRef CAS.
- E. Lapuente-Brun, R. Moreno-Loshuertos, R. Acín-Pérez, A. Latorre-Pellicer, C. Colás, E. Balsa, E. Perales-Clemente, P. M. Quirós, E. Calvo, M. A. Rodríguez-Hernández, P. Navas, R. Cruz, Á. Carracedo, C. López-Otín, A. Pérez-Martos, P. Fernández-Silva, E. Fernández-Vizarra and J. A. Enríquez, Science, 2013, 340, 1567–1570 CrossRef CAS PubMed.
- I. Martínez-Reyes, L. R. Cardona, H. Kong, K. Vasan, G. S. McElroy, M. Werner, H. Kihshen, C. R. Reczek, S. E. Weinberg, P. Gao, E. M. Steinert, R. Piseaux, G. R. S. Budinger and N. S. Chandel, Nature, 2020, 585, 288–292 CrossRef.
- W. Chen, X. Hu, A. Hu, L. Ji, Y. Huang, H. Heng, F. Feng and S. Wang, CCS Chem., 2023, 6, 812–823 CrossRef.
- W. Zheng, P. Chai, J. Zhu and K. Zhang, Nature, 2024, 631, 232–239 CrossRef CAS.
- Y. Yang, Y. An, M. Ren, H. Wang, J. Bai, W. Du and D. Kong, Front. Pharmacol, 2023, 14, 1243613 CrossRef CAS PubMed.
- H. J. Kim, O. Khalimonchuk, P. M. Smith and D. R. Winge, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 1604–1616 CrossRef CAS.
- Y. Zhao, X. Chu and B. Yang, Bioelectrochemistry, 2017, 117, 15–22 CrossRef CAS PubMed.
- S. Kim, L. Palanikumar, H. Choi, M. T. Jeena, C. Kim and J.-H. Ryu, Chem. Sci., 2018, 9, 2474–2479 RSC.
- X. Lin, L. Li, S. Li, Q. Li, D. Xie, M. Zhou and Y. Huang, Adv. Sci., 2021, 8, 2002834 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS.
- S. Kina, T. Kinjo, F. Liang, T. Nakasone, H. Yamamoto and A. Arasaki, Eur. J. Pharmacol., 2018, 840, 70–78 CrossRef CAS.
- Y. Zou, Y. Wang, S. Xu, Y. Liu, J. Yin, D. B. Lovejoy, M. Zheng, X.-J. Liang, J. B. Park, Y. M. Efremov, I. Ulasov and B. Shi, Adv. Mater., 2022, 34, 2203958 CrossRef CAS.
- S. Pasquale, P. Aparamita, F. Eileen, H. Abbie, Y. Jane, F. R. Erika, R. G. Tristan, G. Giorgio, N. Giovanni, S. Annamaria, W. Alessandro, M. D. Steven and S. Claudio, J. Thorac. Oncol., 2023, 18, 190 CrossRef.
- M.-L. Kung, S.-L. Hsieh, C.-C. Wu, T.-H. Chu, Y.-C. Lin, B.-W. Yeh and S. Hsieh, Nanoscale, 2015, 7, 1820–1829 RSC.
- Y. Ding, W. Yu, R. Shen, X. Zheng, H. Zheng, Y. Yao, Y. Zhang, C. Du and H. Yi, Adv. Healthcare Mater., 2024, 13, 2303308 CrossRef CAS.
- S. Kuang, L. Sun, X. Zhang, X. Liao, T. W. Rees, L. Zeng, Y. Chen, X. Zhang, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 20697–20703 CrossRef CAS PubMed.
- Z. Sun, L.-P. Zhang, F. Wu and Y. Zhao, Adv. Funct. Mater., 2017, 27, 1704079 CrossRef.
- W. Pang, C. Wang, W. Wu, X. Wei and B. Gu, Laser Photonics Rev., 2024, 2400753 CrossRef CAS.
- M. V. Berridge, P. M. Herst and A. S. Tan, Biotechnol. Annu. Rev., 2005, 11, 127–152 CAS.
- J. Liu, W. Zhang, X. Wang, Q. Ding, C. Wu, W. Zhang, L. Wu, T. D. James, P. Li and B. Tang, J. Am. Chem. Soc., 2023, 145, 19662–19675 CrossRef CAS PubMed.
- F. Sun, X. Huo, Y. Zhai, A. Wang, J. Xu, D. Su, M. Bartlam and Z. Rao, Cell, 2005, 121, 1043–1057 CrossRef CAS PubMed.
- K. Vasan, M. Werner and N. S. Chandel, Cell Metab., 2020, 32, 341–352 CrossRef CAS.
- R. J. DeBerardinis and N. S. Chandel, Sci. Adv., 2016, 2, e1600200 CrossRef PubMed.
- W. Zhou, S. Chen, Y. Ouyang, B. Huang, H. Zhang, W. Zhang and J. Tian, Chem. Sci., 2023, 14, 11481–11489 RSC.
- M. R. Schreier, B. Pfund, X. Guo and O. S. Wenger, Chem. Sci., 2020, 11, 8582–8594 RSC.
- X. Cheng, D. Feng, J. Lv, X. Cui, Y. Wang, Q. Wang and L. Zhang, Cancers, 2023, 15, 666 CrossRef CAS PubMed.
- N. N. Pavlova, J. Zhu and C. B. Thompson, Cell Metab., 2022, 34, 355–377 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06576j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |