
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble enhancement of protonation and conjugation in donor–imine–donor covalent organic frameworks for photocatalytic hydrogen evolution†
Huan
He‡
a,
Rongchen
Shen‡
a,
Yuhao
Yan‡
a,
Dejun
Chen
a,
Zhixiong
Liu
a,
Lei
Hao
a,
Xin
Zhang
 *b,
Peng
Zhang
*c and
Xin
Li
*a
*b,
Peng
Zhang
*c and
Xin
Li
*a
aInstitute of Biomass Engineering, Key Laboratory of Energy Plants Resource and Utilization of Ministry of Agriculture and Rural Affairs, Key Laboratory for Biobased Materials and Energy of Ministry of Education, College of Materials and Energy, South China Agricultural University, Guangzhou 510642, China. E-mail: Xinli@scau.edu.cn
bHubei Key Lab Low Dimens Optoelect Mat & Devices, Hubei University of Arts and Science, Xiangyang 441053, People's Republic of China. E-mail: xinzhang@hbuas.edu.cn
cState Centre for International Cooperation on Designer Low-Carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Zhengzhou, Henan 450001, P. R. China. E-mail: zhangp@zzu.edu.cn
First published on 18th November 2024
Abstract
Covalent organic frameworks (COFs) have emerged as highly promising platforms for photocatalytic water splitting. However, exploring the structure–activity relationships in different COF systems remains challenging. In this study, three donor–imine–donor (D–I–D) COFs as relatively pure model materials were carefully selected to investigate the effect of protonation and conjugation on the mechanism of photocatalytic H2 evolution. Unlike widely reported donor–acceptor (D–A) COF systems, these three ideal COFs have short electronic channels and lack chemical bond isomerism and heteroatoms in building blocks. These aspects are beneficial for a comprehensive investigation of the underlying mechanisms at the active sites of the imine bond. Both the calculation and experimental results indicate that increasing the conjugation intensity can enhance the efficiency of exciton dissociation and charge transfer rates. Protonation can also dominantly enhance the light absorption capacity and electron transport efficiency of D–I–D COFs. After protonation, the Py-hCOF with optimal conjugation intensity exhibits a remarkable H2 evolution rate of 44.2 mmol g−1 h−1 under visible light, which is 88.4 times higher than that of Tpe-hCOF. This result highlights the crucial roles of simultaneous enhancement of the protonation and conjugation in improving photocatalytic hydrogen evolution of COFs, providing valuable insights for the design of COF materials to achieve the superior electronic functions in photocatalysis.
Introduction
Photocatalytic hydrogen evolution offers a promising and sustainable approach for converting solar energy into clean chemicals.1–5 Covalent organic frameworks (COFs) have recently garnered significant interest in photocatalysis research due to their tunable semiconducting properties.6–9 Combining different building monomers enables precise control of their porosity, modular functionality, chemical structure, and structural periodicity.10,11 For example, donor–acceptor (D–A) construction represents a typical charge separation strategy, with COFs consisting of two different structural units arranged alternately.12–14 In addition, Yang et al. have designed and synthesized donor–π–acceptor (D–π–A) COFs, which feature two nodes and one linker.15 Although D–A and D–π–A improve the efficiency of intermolecular charge transfer, they are limited by the longer intermolecular charge transfer channel, which seriously impacts free carrier generation. Therefore, it is critical to develop COFs that inhibit charge recombination while promoting charge transfer. This necessitates the rational optimization of the electronic structure of COFs to regulate the local charge distribution and thus improve the catalytic performance.COFs with a donor–imine–donor (D–I–D) structure have recently been synthesized using two building units with the same central module.16,17 Unlike D–A and D–π–A COFs, D–I–D COFs are unaffected by the imine bond orientation and also have shorter intermolecular electron transfer channels.18,19 However, COFs based on D–I–D structures have not been extensively studied in photocatalysis. It is challenging to improve the photocatalytic performance by adjusting the multiple structural driving forces of D–I–D COFs to manipulate the horizontal charge transfer ability.
Changes in the conjugated structure introduce new semiconducting characteristics, offering promising potential in various applications, including chemiresistive sensing,20–22 optoelectronics,13,23 photo-/electro-catalysis,24–29 and magnetism.30,31 The diverse conjugation systems influence the band gaps and contribute to structural stabilization, directly affecting the stability of charged states.32 Additionally, the conjugated structure is crucial in facilitating efficient delocalization and electron transport within the COF backbone, thereby influencing both photovoltaic performance and photocatalytic activity.33 It is important to deeply study how the conjugated structure affects the electronic structure and photocatalytic performance of COFs in the real environment. Protonation has garnered significant attention as a favorable transformation that COFs are also prone to appear in acidic environment.16,17,34 Imine-based COFs have attracted considerable interest due to their ease of synthesis through the Schiff base reaction and the susceptibility of the imine moiety to protonation.35 However, the influences of protonation and conjugation have not been systematacially explored. Thus, it is crucial to investigate the protonation and conjugation mechanism of photocatalytic H2 evolution in the ideal D–I–D COFs with minimized interference.
In this study, we reasonably selected three D–I–D COFs as relatively pure model materials to thoroughly investigate how protonation and conjugation affect photocatalytic H2 evolution. The absence of heteroatoms and D–A constitutional isomerism in the linkages gives them a distinct advantage. This work of conjugation and protonation engineering provides a significant reference for investigating exciton effects and charge separation, thus enabling the efficient photocatalytic hydrogen evolution in D–I–D COFs.
Results and discussion
By utilizing the described building blocks, we successfully synthesized three imine-linked D–I–D COFs with similar structures but varying degrees of conjugation. Py-COF (Tet-COF, Tpe-COF) underwent post-protonation with 0.1 M ascorbic acid (AA) and the corresponding protonated COF was denoted as Py-hCOF (Tet-hCOF, Tpe-hCOF) (Fig. 1A). | ||
| Fig. 1 (A) The synthetic conditions and chemical structures of COF and hCOF. Structure, and experimental and simulated PXRD patterns of (B) Py-COF, (C) Tet-COF, and (D) Tpe-COF. | ||
The three COFs prepared using this approach exhibit the X-ray diffraction (XRD) pattern of highly ordered mesoporous materials, with prominent diffraction peaks observed at 5.01°, 6.91°, and 6.36° for Py-COF, Tet-COF, and Tpe-COF, respectively. Full profile matching was carried out using Pawley refinement of the PXRD pattern, resulting in excellent match factors and reasonable profile differences (Rwp = 8.21% and Rp = 6.84% for Py-COF, Rwp = 8.04% and Rp = 6.31% for Tet-COF, and Rwp = 7.77% and Rp = 5.91% for Tpe-COF) (Fig. 1B–D). Furthermore, the unit–cell parameters of Py-COF in the P222 space group were determined to be a = 24.5268 Å, b = 26.2976 Å and c = 4.3019 Å (other COFs shown in Tables S1–S3†). After being exposed to a 0.1 M AA (ascorbic acid) aqueous solution, the color of the COFs was noticeably altered, while the XRD patterns of the as-prepared hCOFs remained fully consistent with the original samples (Fig. S1†). In the TEM images, the Py-COF displayed an apparent layered structure. The distorted hexagonal channel pores of Py-COF could be found through Fourier transform. Other COFs have an ideal pore structure (Fig. 2A–C and S2†). Scanning electron microscopy (SEM) was employed to further investigate the morphologies of the samples. The SEM images revealed the presence of crystalline nanosheets in the COFs (Fig. S3†). Fourier transform infrared spectroscopy (FT-IR) was additionally conducted to confirm the successful condensation of imine linkages. The FT-IR spectrum exhibited N–H bands in the range of 3200–3400 cm−1 for Py-NH2 and –CHO stretching vibration bands at 1635–1645 cm−1 for Py-CHO. These bands were significantly attenuated following the condensation reaction, as depicted in Fig. 2D. New peak for –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond stretching (at around 1620 cm−1) appears for Py-COF, indicating the successful synthesis of the imine bond in the COF. After treatment with 0.1 M AA solution, the FT-IR spectrum of the as-prepared Py-hCOF has a new peak at 3300–3600 cm−1, which could be attributed to the –N–H stretching vibration from the protonated COF (Fig. 2D, S4 and S5†). These results indicate that protonation post-treatment does not result in dramatic changes in crystal structure and the initial structure of COFs. The X-ray photoemission spectroscopy (XPS) analysis of the COFs also reveals the presence of C and N elements (Fig. S6†). The N 1s XPS spectrum of Py-COF shows one obvious new N 1s signal centered at 401.73 eV assigned to protonated N (Fig. 2E).36,37 Nitrogen sorption measurements determine the permanent porosities of the samples at 77 K and show a type IV nitrogen sorption isotherm for all of the samples, which indicates a mesoporous character (Fig. 2F and S7†). The Brunauer–Emmett–Teller (BET) surface areas of Py-COF, Tet-COF, and Tpe-COF were determined to be 1681, 1211 and 1224 m2 g−1, respectively, and their corresponding pore sizes were in the range of 1.74–3.92, 1.74–3.62, and 3.83 nm, respectively.
N bond stretching (at around 1620 cm−1) appears for Py-COF, indicating the successful synthesis of the imine bond in the COF. After treatment with 0.1 M AA solution, the FT-IR spectrum of the as-prepared Py-hCOF has a new peak at 3300–3600 cm−1, which could be attributed to the –N–H stretching vibration from the protonated COF (Fig. 2D, S4 and S5†). These results indicate that protonation post-treatment does not result in dramatic changes in crystal structure and the initial structure of COFs. The X-ray photoemission spectroscopy (XPS) analysis of the COFs also reveals the presence of C and N elements (Fig. S6†). The N 1s XPS spectrum of Py-COF shows one obvious new N 1s signal centered at 401.73 eV assigned to protonated N (Fig. 2E).36,37 Nitrogen sorption measurements determine the permanent porosities of the samples at 77 K and show a type IV nitrogen sorption isotherm for all of the samples, which indicates a mesoporous character (Fig. 2F and S7†). The Brunauer–Emmett–Teller (BET) surface areas of Py-COF, Tet-COF, and Tpe-COF were determined to be 1681, 1211 and 1224 m2 g−1, respectively, and their corresponding pore sizes were in the range of 1.74–3.92, 1.74–3.62, and 3.83 nm, respectively.
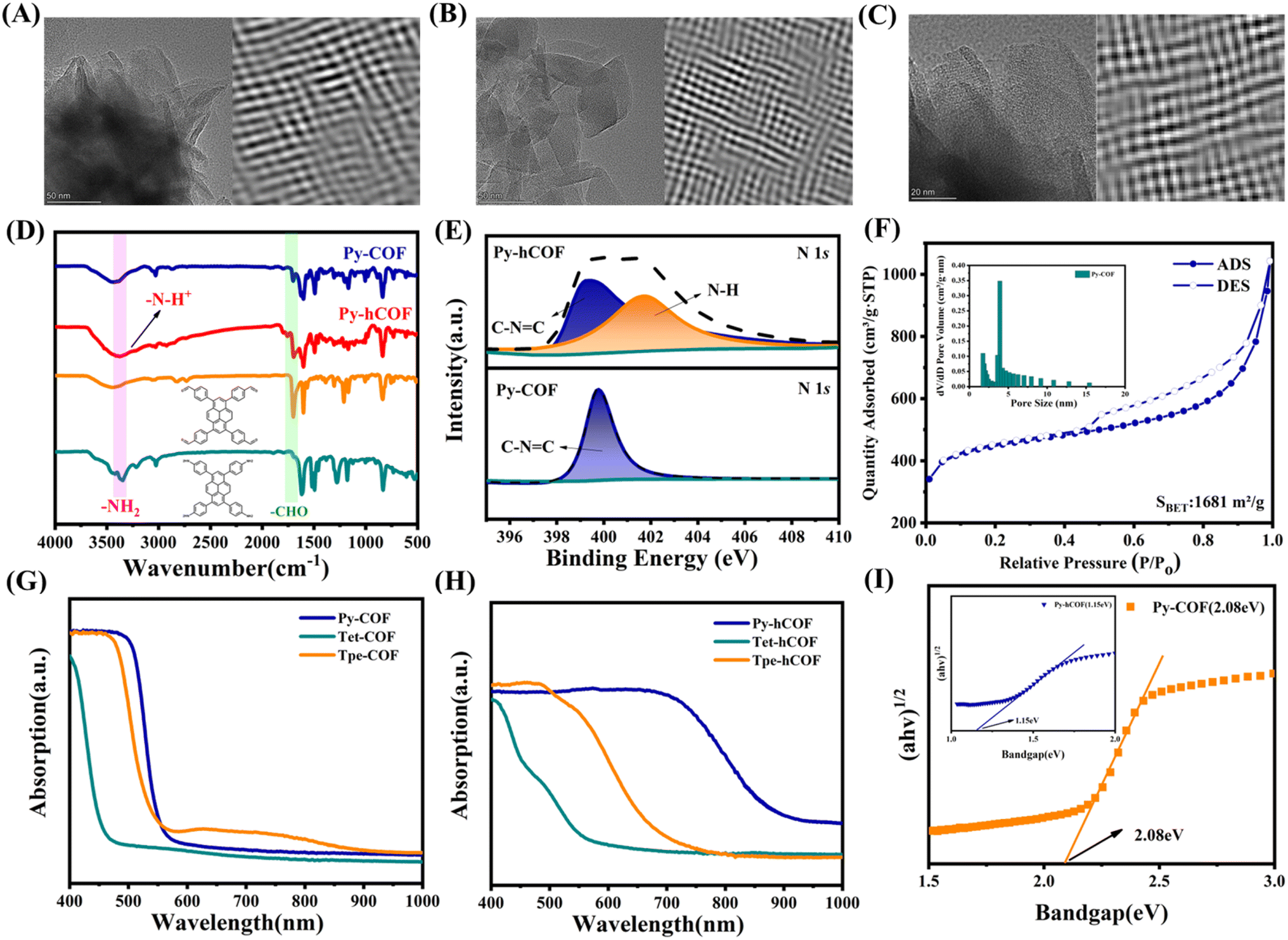 | ||
| Fig. 2 TEM and FFT images of (A) Py-COF, (B) Tet-COF and (C) Tpe-COF. (D) FT-IR spectra of Py-COF and Py-hCOF. (E) The N 1s XPS spectra of Py-hCOF and Py-COF. (F) Nitrogen adsorption and desorption isotherms of Py-COF. Insets: pore size distributions calculated. UV-Vis DRS spectra of COFs (G) before and (H) after AC treatment. (I) Tauc plots of the Py-COF, inset Tauc plots of the Py-hCOF. | ||
Photophysical and electrochemical measurements were conducted to investigate the optical and electronic properties of the photocatalysts. Ultraviolet-visible diffuse reflectance spectroscopy (UV-Vis DRS) was employed to assess the light absorption capacity and bandgap of the photocatalysts. The absorption spectra of all COFs show a distinct slope. This indicates the formation of discrete energy bands.32 The UV-Vis diffuse reflectance spectrum of Py-COF shows strong absorption in the visible region with the edge of 580 nm (Fig. 2G). The determined optical bandgap was 2.08 eV for the Py-COF (the determined optical bandgaps were 2.56 and 2.11 eV for the Tet-COF and Tpe-COF, respectively) (Fig. S8†). Protonation significantly influences the light absorption properties of COFs. Thus, the UV-visible absorption characteristics of the protonated COF were also examined. After protonation, Py-hCOF exhibits enhanced light absorption, accompanied by a red-shifted absorption edge observed at approximately 920 nm. It is noteworthy that this red shift is not attributable to the absorption of the added AA, a colorless compound that does not absorb light in the visible region (Fig. 2H).38 Additionally, a comparison was made between the colors of the COFs before and after protonation (Fig. S8†). The bandgaps of Py-COF and Py-hCOF were estimated to be 2.08 eV and 1.15 eV by Tauc plots (Fig. 2I). The determined optical bandgaps were 1.92 and 1.54 eV for the Tet-hCOF and Tpe-hCOF, respectively (Fig. S9†). These results demonstrate that protonation markedly enhances the light absorption capabilities of COFs. The photoelectric properties of the photocatalysts were compared to reveal the photocatalytic mechanism of the D–I–D COFs. Py-COF displays a greater degree of conjugation than the other two COFs, thereby being more significantly affected by protonation. Subsequent UV absorption calculations of COFs in the S1 excited state further confirm that Py-COF is the most influenced by protonation (Table S7†).
To further explore the effect of conjugation intensity on the photocatalysis, the photocatalytic H2 evolution experiments were conducted by irradiating a suspension of COFs with 3 wt% Pt as cocatalyst and 0.1 M AA solution serving as a sacrificial agent under visible light. Furthermore, the photocatalytic hydrogen evolution activity of the three distinct D–I–D COFs was evaluated (Fig. 3A). Concurrently, the apparent quantum yield of Py-COF was determined. Py-COF exhibited a maximum quantum yield of 9.58% at 420 nm (Fig. S10†). Although the absorption wavelength of Tpe-COF is red-shifted in comparison to that of Tet-COF, the hydrogen evolution activity is still lower than that of Tet-COF. This result can be attributed to the weaker conjugation strength of Tpe-COF compared to Tet-COF. The average H2 evolution rates were 0.5, 18.2, and 44.2 mmol g−1 h−1 for Tpe-COF, Tet-COF, and Py-COF, respectively (Fig. 3B). The photocatalytic activity was subsequently evaluated under various sacrificial conditions. Under alkaline conditions, alterations in conjugation intensity did not significantly impact the photocatalytic efficiency of the COFs. However, under acidic conditions, an increase in conjugation intensity notably affected the photocatalytic performance (Fig. 3C). The long-term photocatalytic experiments of Py-COF, Tet-COF, and Tpe-COF exhibit a stable photocatalytic H2 evolution reaction while refreshing the headspace every 3 h (Fig. 3D). Additionally, photocatalytic hydrogen evolution tests were performed with varying platinum loadings and different COF qualities (Fig. S11†). The XRD analysis after the reaction indicates that it still has a great COF structure (Fig. S12†). The electron mobility of the photogenerated carriers was monitored by electrochemical impedance spectroscopy (EIS), and the EIS spectra for the three COFs are presented in Fig. S13.† Py-COF shows the smallest half-circle, indicating the lowest charge transfer resistance and the highest electron transfer efficiency. The transient photocurrent response (TPR) further elucidates the efficiency of charge separation. The TPR of Py-COF is significantly higher than those of Tet-COF and Tpe-COF. This indicates that the Py-COF allows more efficient charge separation (Fig. 3E). These results were corroborated by surface photovoltage (SPV) measurements (Fig. S14†). Enhanced photoelectric properties were observed with increasing conjugation intensity among the D–I–D COFs, in the following order, Py-COF > Tet-COF > Tpe-COF. The lower conjugation intensity may be responsible for the low photoelectric properties observed in Tpe-COF, despite its increased light harvesting ability compared to Tet-COF. Moreover, the positions of the conduction band (CB) and valence band (VB) of the photocatalysts were estimated using Mott–Schottky analysis (Fig. S15†), VB-XPS measurement (Fig. S16†), and their optical bandgap (Fig. 3F and G). These findings suggest that the conduction band (CB) and valence band (VB) positions are optimal for facilitating hydrogen evolution.39
 | ||
| Fig. 3 (A) Time profiles of photocatalytic hydrogen generation of COFs. (B) Average hydrogen evolution rate. (C) Comparison of the hydrogen evolution activity of different sacrificial agents, inset: amplification of the hydrogen evolution activity comparison of triethanolamine. (D) Cyclic H2 evolution over Py-COF, Tet-COF, and Tpe-COF. (E) Transient photocurrent spectra of the COFs. (F) The band diagram of COFs. (G) The band diagram of the three hCOFs. (H) Low-temperature steady-state fluorescence spectra for Py-hCOF, Tet-hCOF, and Tpe-hCOF. (I) Low-temperature steady-state phosphorescence was recorded at a delayed time of 10 μs for Py-hCOF, Tet-hCOF, and Tpe-hCOF. | ||
The properties of excitons and charge carriers are crucial for understanding the photocatalytic process. To further validate the effect of conjugation intensity on the photocatalytic performance of D–I–D COFs, we measured the steady-state delayed fluorescence and phosphorescence. This measurement enabled us to gain insights into the exciton dissociation processes.40 The intensity of the steady-state delayed fluorescence represents the amount of radiative decay of singlet excitons in the COFs. The steady-state fluorescence properties are attenuated with an increase in the conjugation intensity of the units in the D–I–D COFs (Py-hCOF, Tet-hCOF, and Tpe-hCOF in this order), indicating a significantly reduced quantity of singlet excitons (Fig. 3H).41Fig. 3I also shows that the concentration of triplet excitons decreases as the conjugation strength increases. Additionally, the low exciton concentrations (for both the singlet and triplet) would reduce excitonic interactions, consequently enhancing the quantum yields of the excited species.42 The kinetics of exciton dissociation, measured by the Eb (exciton binding energy) value, is one of the most critical factors affecting the photocatalytic activity of COFs. The lower Eb values represent easier dissociation of excitons, facilitating the photocatalytic reaction. The exciton binding energy after protonation of the highly conjugated Py-hCOF is only 55.1 meV. The results demonstrate that the increase in conjugation and protonation can reduce the exciton binding energy (Fig. 4A–F).
 | ||
| Fig. 4 Integrated PL emission intensity as a function of temperature (inset: temperature-dependent PL spectra from 120 to 300 K): (A) Tpe-COF, (B) Tet-COF, (C) Py-COF, (D) Tpe-hCOF, (E) Tet-hCOF and (F) Py-hCOF. | ||
To investigate the effects of conjugation intensity and protonation on photoelectron separation and transfer during the photocatalytic hydrogen evolution reaction in the D–I–D COFs, femtosecond time-resolved transient absorption spectra (fs-TAS) measurements were performed. The height of the positive absorption band of the COFs increases with the enhancement of conjugation strength at around 700 nm, correlating with the ultrafast separation of carriers and the rapid capture of electrons. The same outcome is similarly observed for COFs with water as the dispersant. When comparing the differences between various dispersants, the protonated COFs (hCOFs) exhibit greater sensitivity to changes in conjugation intensity. Notably, this observation is consistent with the performance experiment data (Fig. 5A–F and S17†). Using Py-COF as an example, the electron transfer mechanism of photocatalytic hydrogen evolution before and after protonation was explored. The fs-TAS kinetics of Py-COF (τ1 = 0.96 ps; τ2 = 15.70 ps) and Py-hCOF (τ1 = 0.43 ps; τ2 = 6.17 ps) can be fitted using biexponential decay processes (Fig. 5G and H). The τ1 of Py-hCOF is slightly smaller than the corresponding value of Py-COF, indicating faster electron trapping in Py-hCOF. The τ2 for Py-hCOF is significantly shorter than that of Py-COF due to the more rapid exciton dissociation-induced charge separation in Py-hCOF.12 The electron paramagnetic resonance (EPR) spin trap technique used DMPO as the spin trap reagent to confirm the identical findings. The COFs exhibited a higher EPR signal when exposed to irradiation compared to when in the dark, suggesting the presence of photogenerated electron–hole pairs in the COF catalysts. The signal intensity of the ˙O2− and ˙OH radicals progressively increases from Tpe-hCOF to Tet-hCOF, with further enhancement in Py-hCOF. This indicates that charge separation and exciton dissociation efficiency substantially increase with enhanced conjugation intensity (Fig. 5I and S18†).
 | ||
| Fig. 5 Fs-TA spectra of (A) Py-COF, (B) Tet-COF, and (C) Tpe-COF in ethylene glycol. Fs-TA spectra of (D) Py-hCOF, (E) Tet-hCOF, and (F) Tpe-hCOF in water. The decay kinetics were monitored for (G) Py-COF in ethylene glycol and (H) Py-hCOF in water. (I) EPR spectra of DMPO–˙O2− for Py-hCOF, Tet-hCOF, and Tpe-hCOF. | ||
To further investigate the conjugation strengths of various COFs, we evaluated the central core of the COFs using the Localized Orbital Locator (LOL).43 Initially, the planar LOL π-electrons were calculated and mapped onto the central core of the COFs (Fig. 6A–C). The results indicate that benzene possesses more LOL π-electrons than ethylene, while pyrene exhibits the highest number of LOL π-electrons. To eliminate the influence of the benzene ring on the central core of the COFs, we conducted a calculation of the non-planar LOL π-electrons for the COF structures. The results are consistent with the planar LOL π-calculations.44 Additionally, LOL evaluations were performed on the minimum fragments of the COFs before and after protonation. We also found the protonated COF to have more LOL-π electrons (Fig. 6D–F). A higher number of LOL π-electrons signifies enhanced π-conjugation and, consequently, increased conjugation intensity.45 The frontier molecular orbitals (HOMO–LUMO) and energy levels of the smallest repeating units of the COFs were calculated using a well-defined density functional theory (DFT) approach to elucidate the effects of varying conjugation intensities. The monolithic HOMO–LUMO has been provided for reference (Fig. S19†). The separation of the HOMO–LUMO energy levels becomes more pronounced with increasing conjugation intensity (Fig. S20†). This HOMO–LUMO distribution originated from the easier charge transfer in D–I–D COFs.46 As illustrated in Fig. 6G–I, the electrostatic potential of the COFs changes significantly after protonation, thereby enhancing electron–hole pair separation. This finding is consistent with the observation of improved photocatalytic performance following protonation.
 | ||
| Fig. 6 LOL-pi distribution of planar graphs over the COF centres of (A) pyrene, (B) benzene, and (C) ethylene. LOL-pi distribution of non-planar graphs over COFs of (D) upper panel: Py-COF and lower panel: Py-hCOF, (E) upper panel: Tet-COF and lower panel: Tet-hCOF, and (F) upper panel: Tpe-COF and lower panel: Tpe-hCOF. The ESP of COFs of (G) upper panel: Py-COF and lower panel: Py-hCOF, (H) upper panel: Tet-COF and lower panel: Tet-hCOF, and (I) upper panel: Tpe-COF and lower panel: Tpe-hCOF. | ||
To further investigate the mechanism underlying the changes in energy states before and after light excitation, the partial density of states (PDOS) of COFs was obtained through DFT calculations. The PDOS of the donor units (Py units, Tet units, and Tpe units) exhibits only minor changes upon excitation, which can be attributed to their inherent electron-rich nature as electron donors. In contrast, the PDOS of the imine bond tends to shift toward the Fermi level, indicating that the imine bond can be effectively activated through continuous electron transfer following photoexcitation (Fig. 7A–C). This trend illustrates the flow of electrons from the donor units to the imine bond upon absorption of solar energy. In summary, COFs with a periodically ordered donor–immobilized–donor (D–I–D) structure demonstrate superior photoelectron transfer capabilities and shorter transfer distances, which theoretically substantiates their potential as excellent photocatalysts.47
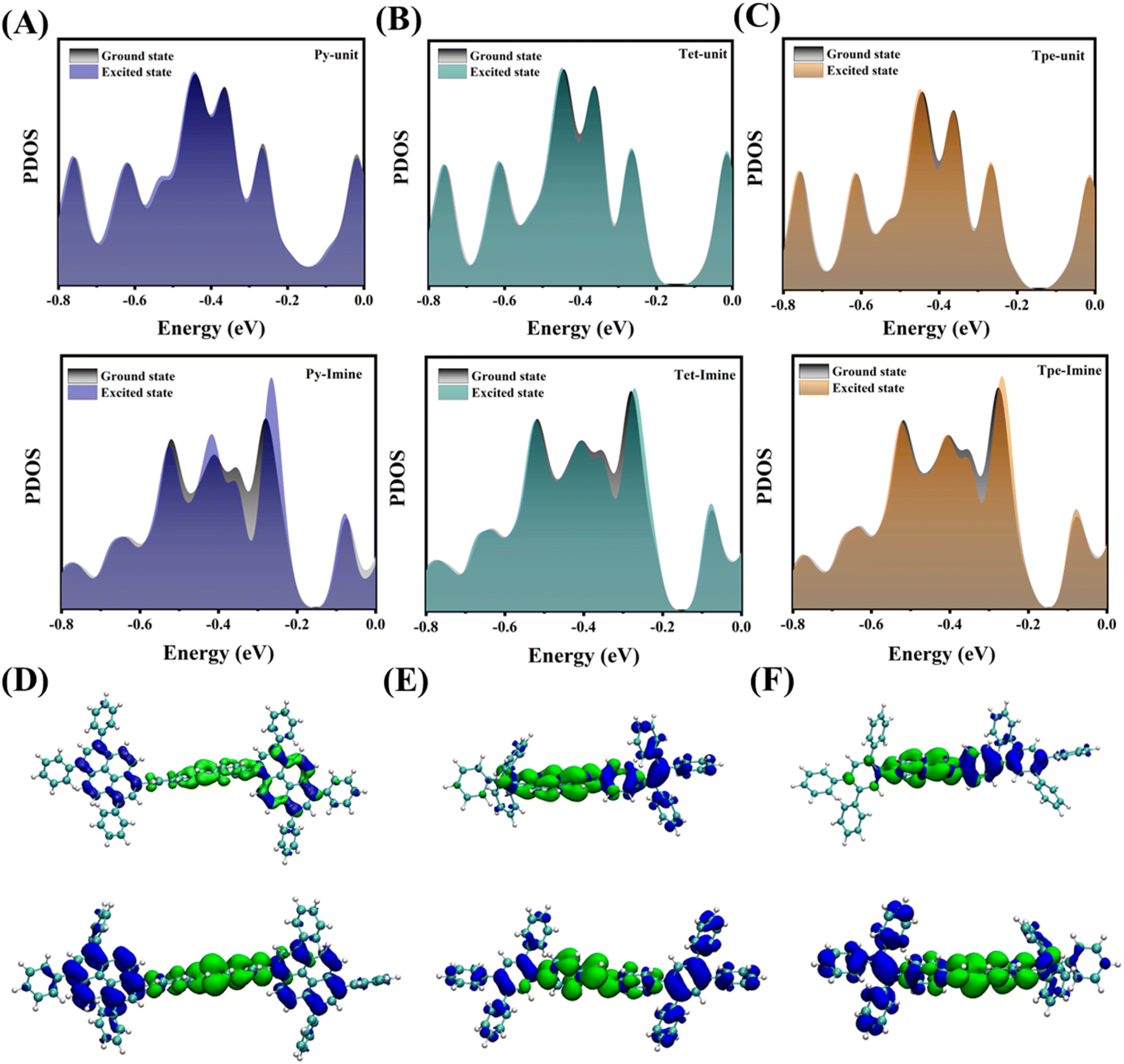 | ||
| Fig. 7 The variation of the PDOS of (A) Py-COF (upper panel: Py units and lower panel: imine units), (B) Tet-COF (upper panel: Tet units and lower panel: Imine units), and (C) Tpe-COF (upper panel: Tpe units and lower panel: imine units) before and after photoexcitation. Real-space hole (blue regions) and electron (green regions) distributions of (D) upper panel: Py-COF and lower panel: Py-hCOF, (E) upper panel: Tet-COF and lower panel: Py-hCOF, and (F) upper panel: Tpe-COF and lower panel: Tpe-hCOF. | ||
The excited state electronic structures of the COFs were further calculated using the time-dependent density functional theory (TD-DFT) method to gain a deeper understanding of the electron transport process in COFs (Fig. 7D–F). With the increase of conjugation strength, the separation of electron holes becomes more obvious. After protonation, this leads to the generation of a greater number of photogenerated carriers by significantly broadening the light absorption range.15 The calculated UV-vis absorption spectra are shown in Fig. S21.† This result also suggests that increased conjugation intensity enhances the visible excitation frequency on S0 → S1 (Tables S4–S6†).48
Conclusions
We have synthesised a series of D–I–D COFs with short electron channels using pure carbon monomers. The enhancement of the conjugation intensity leads to excellent photocatalytic hydrogen evolution performance in D–I–D COFs. The imine bonds in COFs are readily protonated. Therefore, the effects of protonation on photocatalytic hydrogen evolution were explored. The photocatalytic performance of Py-hCOF (44.2 mmol g−1 h−1) with the highest conjugation intensity is 88.4 times that of Tpe-hCOF (0.5 mmol g−1 h−1). Both experimental and theoretical results indicate that higher conjugation intensity facilitates more efficient exciton dissociation and charge transport. The protonation can also enhance the light absorption capacity and electron transport efficiency of D–I–D COFs. This study provides new insights into the design of novel COF systems that utilize structural transformation modulation to improve photocatalytic performance.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Huan He, Rongchen Shen, Yuhao Yan, Dejun Chen, Zhixiong Liu, Lei Hao and Xin Li designed the systems, synthesized the photocatalysts, performed the experimental measurement, conducted DFT calculations, analyzed the data as well as wrote the manuscript. Xin Zhang carried out transient absorption spectroscopy experiments. Peng Zhang helped to revise the language of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (22378148, 524721102, 230082074) and the Natural Science Foundation of Guangdong Province (2024A1515012433) for their support.References
- T. Hisatomi, J. Kubota and K. Domen, Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- X.-J. Li, M.-Y. Qi, J.-Y. Li, C.-L. Tan, Z.-R. Tang and Y.-J. Xu, Visible light-driven dehydrocoupling of thiols to disulfides and H2 evolution over PdS-decorated ZnIn2S4 composites, Chin. J. Catal., 2023, 51, 55–65 CrossRef.
- F. Liu, B. Sun, Z. Liu, Y. Wei, T. Gao and G. Zhou, Vacancy engineering mediated hollow structured ZnO/ZnS S-scheme heterojunction for highly efficient photocatalytic H2 production, Chin. J. Catal., 2024, 64, 152–165 CrossRef CAS.
- H. Yang, J. Guo, Y. Xia, J. Yan and L. Wen, Schottky-assisted S-scheme heterojunction photocatalyst CdS/Pt@NU-1000 for efficient visible-light-driven H2 evolution, J. Mater. Sci. Technol., 2024, 195, 155–164 CrossRef.
- H. Wang, L. Yu, J. Peng, J. Zou and J. Jiang, Strategically designing and fabricating nitrogen and sulfur Co-doped g-C3N4 for accelerating photocatalytic H2 evolution, J. Mater. Sci. Technol., 2025, 208, 111–119 CrossRef.
- G. Liu, R. Chen, B. Xia, Z. Wu, S. Liu, A. Talebian-Kiakalaieh and J. Ran, Synthesis of H2O2 and high-value chemicals by covalent organic framework-based photocatalysts, Chin. J. Catal., 2024, 61, 97–110 CrossRef CAS.
- Y. Zhao, C. Yang, S. Zhang, G. Sun, B. Zhu, L. Wang and J. Zhang, Investigating the charge transfer mechanism of ZnSe QD/COF S-scheme photocatalyst for H2O2 production by using femtosecond transient absorption spectroscopy, Chin. J. Catal., 2024, 63, 258–269 CrossRef CAS.
- Y. Zhang, J. Qiu, B. Zhu, G. Sun, B. Cheng and L. Wang, Hollow spherical covalent organic framework supported gold nanoparticles for photocatalytic H2O2 production, Chin. J. Catal., 2024, 57, 143–153 CrossRef CAS.
- Q. Niu, L. Mi, W. Chen, Q. Li, S. Zhong, Y. Yu and L. Li, Review of covalent organic frameworks for single-site photocatalysis and electrocatalysis, Chin. J. Catal., 2023, 50, 45–82 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, Covalent Organic Frameworks: Chemistry beyond the Structure, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Covalent organic frameworks: an ideal platform for designing ordered materials and advanced applications, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- L. Hao, R. Shen, C. Huang, Z. Liang, N. Li, P. Zhang, X. Li, C. Qin and X. Li, Fluorenone-based covalent organic frameworks with efficient exciton dissociation and well-defined active center for remarkable photocatalytic hydrogen evolution, Appl. Catal. B Environ., 2023, 330, 122581 CrossRef CAS.
- C. Li, D. Li, W. Zhang, H. Li and G. Yu, Towards High-Performance Resistive Switching Behavior through Embedding a D-A System into 2D Imine-Linked Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2021, 60, 27135–27143 CrossRef CAS PubMed.
- H. Yang, M. Hao, Y. Xie, X. Liu, Y. Liu, Z. Chen, X. Wang, G. I. N. Waterhouse and S. Ma, Tuning Local Charge Distribution in Multicomponent Covalent Organic Frameworks for Dramatically Enhanced Photocatalytic Uranium Extraction, Angew. Chem., Int. Ed., 2023, e202303129 CAS.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, Three-Component Donorâ’π–Acceptor Covalent–Organic Frameworks for Boosting Photocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS.
- L. Ascherl, E. W. Evans, J. Gorman, S. Orsborne, D. Bessinger, T. Bein, R. H. Friend and F. Auras, Perylene-Based Covalent Organic Frameworks for Acid Vapor Sensing, J. Am. Chem. Soc., 2019, 141, 15693–15699 CrossRef CAS PubMed.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Solvatochromic covalent organic frameworks, Nat. Commun., 2018, 9, 3802 CrossRef.
- J. Yang, S. Ghosh, J. r. m. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M.-Y. Ye, J. Grüneberg, S. Li, C. Li, R. Schomäcker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Constitutional isomerism of the linkages in donor–acceptor covalent organic frameworks and its impact on photocatalysis, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Isomeric Oligo(Phenylenevinylene)-Based Covalent Organic Frameworks with Different Orientation of Imine Bonds and Distinct Photocatalytic Activities, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS.
- X. Wu, X. Han, Q. Xu, Y. Liu, C. Yuan, S. Yang, Y. Liu, J. Jiang and Y. Cui, Chiral BINOL-Based Covalent Organic Frameworks for Enantioselective Sensing, J. Am. Chem. Soc., 2019, 141, 7081–7089 CrossRef CAS PubMed.
- W.-R. Cui, C.-R. Zhang, W. Jiang, F.-F. Li, R.-P. Liang, J. Liu and J.-D. Qiu, Regenerable and stable sp2 carbon-conjugated covalent organic frameworks for selective detection and extraction of uranium, Nat. Commun., 2020, 11, 436 CrossRef CAS.
- S. Wang, H. Li, H. Huang, X. Cao, X. Chen and D. Cao, Porous organic polymers as a platform for sensing applications, Chem. Soc. Rev., 2022, 51, 2031–2080 RSC.
- N. Keller and T. Bein, Optoelectronic processes in covalent organic frameworks, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- S. Wei, F. Zhang, W. Zhang, P. Qiang, K. Yu, X. Fu, D. Wu, S. Bi and F. Zhang, Semiconducting 2D Triazine-Cored Covalent Organic Frameworks with Unsubstituted Olefin Linkages, J. Am. Chem. Soc., 2019, 141, 14272–14279 CrossRef CAS PubMed.
- J. Xu, Y. He, S. Bi, M. Wang, P. Yang, D. Wu, J. Wang and F. Zhang, An Olefin-Linked Covalent Organic Framework as a Flexible Thin-Film Electrode for a High-Performance Micro-Supercapacitor, Angew. Chem., Int. Ed., 2019, 58, 12065–12069 CrossRef CAS PubMed.
- R. Chen, J.-L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Designed Synthesis of a 2D Porphyrin-Based sp2 Carbon-Conjugated Covalent Organic Framework for Heterogeneous Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Modulating Benzothiadiazole-Based Covalent Organic Frameworks via Halogenation for Enhanced Photocatalytic Water Splitting, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, Metal-Free Thiophene-Sulfur Covalent Organic Frameworks: Precise and Controllable Synthesis of Catalytic Active Sites for Oxygen Reduction, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- X. Ren, C. Li, W. Kang, H. Li, N. Ta, S. Ye, L. Hu, X. Wang, C. Li and Q. Yang, Enormous Promotion of Photocatalytic Activity through the Use of Near-Single Layer Covalent Organic Frameworks, CCS Chem., 2021, 4, 2429–2439 CrossRef.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Two-dimensional sp2 carbon–conjugated covalent organic frameworks, Science, 2017, 357, 673–676 CrossRef CAS.
- S. Wang, X.-X. Li, L. Da, Y. Wang, Z. Xiang, W. Wang, Y.-B. Zhang and D. Cao, A Three-Dimensional sp2 Carbon-Conjugated Covalent Organic Framework, J. Am. Chem. Soc., 2021, 143, 15562–15566 CrossRef CAS PubMed.
- J.-P. Jeon, Y. J. Kim, S. H. Joo, H.-J. Noh, S. K. Kwak and J.-B. Baek, Benzotrithiophene-based Covalent Organic Framework Photocatalysts with Controlled Conjugation of Building Blocks for Charge Stabilization, Angew. Chem., Int. Ed., 2023, 62, e202217416 CrossRef CAS PubMed.
- R. Bao, Z. Xiang, Z. Qiao, Y. Yang, Y. Zhang, D. Cao and S. Wang, Designing Thiophene-Enriched Fully Conjugated 3D Covalent Organic Framework as Metal-Free Oxygen Reduction Catalyst for Hydrogen Fuel Cells, Angew. Chem., Int. Ed., 2023, 62, e202216751 CrossRef CAS.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Dual donor-acceptor covalent organic frameworks for hydrogen peroxide photosynthesis, Nat. Commun., 2023, 14, 5238 CrossRef CAS.
- J. L. Segura, M. a. J. Mancheño and F. l. Zamora, Covalent organic frameworks based on Schiff-base chemistry: synthesis, properties and potential applications, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- B. Luo, Y. Chen, Y. Zhang and J. Huo, Benzotrithiophene and triphenylamine based covalent organic frameworks as heterogeneous photocatalysts for benzimidazole synthesis, J. Catal., 2021, 402, 52–60 CrossRef CAS.
- T. Shi, H. Wang, L. Li, Z. Zhao, C. Wang, X. Zhang and Y. Xie, Enhanced photostability in protonated covalent organic frameworks for singlet oxygen generation, Matter, 2022, 5, 1004–1015 CrossRef CAS.
- J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. r. m. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Protonated Imine-Linked Covalent Organic Frameworks for Photocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS.
- L. Dai, A. Dong, X. Meng, H. Liu, Y. Li, P. Li and B. Wang, Enhancement of Visible-Light-Driven Hydrogen Evolution Activity of 2D π-Conjugated Bipyridine-Based Covalent Organic Frameworks via Post-Protonation, Angew. Chem., Int. Ed., 2023, 62, e202300224 CrossRef CAS PubMed.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, Boosting Hot-Electron Generation: Exciton Dissociation at the Order–Disorder Interfaces in Polymeric Photocatalysts, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, Photocatalytic Molecular Oxygen Activation by Regulating Excitonic Effects in Covalent Organic Frameworks, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS.
- R. Shen, X. Li, C. Qin, P. Zhang and X. Li, Efficient Photocatalytic Hydrogen Evolution by Modulating Excitonic Effects in Ni-Intercalated Covalent Organic Frameworks, Adv. Energy Mater., 2023, 13, 2203695 CrossRef CAS.
- H. L. Schmider and A. D. Becke, Chemical content of the kinetic energy density, J. Mol. Struct., 2000, 527, 51–61 CrossRef CAS.
- T. Lu and Q. Chen, A simple method of identifying π orbitals for non-planar systems and a protocol of studying π electronic structure, Theor. Chem. Acc., 2020, 139, 25 Search PubMed.
- S. N. Steinmann, Y. Mo and C. Corminboeuf, How do electron localization functions describe Ï€-electron delocalization?, Phys. Chem. Chem. Phys., 2011, 13, 20584–20592 RSC.
- X. Yan, B. Wang, J. Ren, X. Long and D. Yang, An Unsaturated Bond Strategy to Regulate Active Centers of Metal-Free Covalent Organic Frameworks for Efficient Oxygen Reduction, Angew. Chem., Int. Ed., 2022, 61, e202209583 CrossRef CAS PubMed.
- Y. He, M. Wang, L. Zhang, Q. Cheng, S. Liu, X. Sun, Y. Jiang, T. Qian and C. Yan, Donor-Site-Acceptor Covalent Organic Frameworks Enable Spontaneous Nitrogen Dissociation for Boosted Photoelectrochemical Ammonia Synthesis, Adv. Funct. Mater., 2024, 2315548 CrossRef CAS.
- Y. Yang, X. Chu, H.-Y. Zhang, R. Zhang, Y.-H. Liu, F.-M. Zhang, M. Lu, Z.-D. Yang and Y.-Q. Lan, Engineering Î2-ketoamine covalent organic frameworks for photocatalytic overall water splitting, Nat. Commun., 2023, 14, 593 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07028c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |