
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modified synthetic peptides: from therapeutics to chemosensors
Conor
Wynne
 ab and
Robert B. P.
Elmes
*ab
ab and
Robert B. P.
Elmes
*ab
aDepartment of Chemistry, Maynooth University, National University of Ireland, Maynooth, Co. Kildare, Ireland. E-mail: robert.elmes@mu.ie
bSynthesis and Solid-State Pharmaceutical Centre (SSPC), Bernal Institute, University of Limerick, Castletroy, Co. Limerick, Ireland
First published on 13th April 2024
Abstract
Modified synthetic peptides have emerged as an exciting avenue for enhancing therapeutic efficacy and expanding the scope of applications in various disease contexts. Indeed, the inherent tunability of synthetic peptides has facilitated the creation of highly selective and responsive sensors capable of detecting specific analytes with precision. More recently, their unique structural diversity and chemical versatility has been elegantly adapted for use in supramolecular sensing platforms. This Perspective article highlights the synergistic interplay between modified synthetic peptides, therapeutic applications, and the sensing technologies that underscore the interdisciplinary nature of contemporary chemistry.
 Conor Wynne | Conor Wynne achieved his BSc (Hons) in Pharmaceutical & Biomedical Chemistry at Maynooth University (MU) in 2019. He is now pursuing his PhD under the supervision of Dr Robert Elmes within the Chemistry Department at MU, where he utilises Solid-Phase Peptide techniques to generate responsive synthetic peptide conjugates, with applications in various disease contexts. His research interests are around the use of modified synthetic peptides as fluorescent/luminescent sensors and novel peptidomimetics. |
 Robert B. P. Elmes | Rob graduated with a B.A. Mod. in Medicinal Chemistry from Trinity College Dublin in 2007 before he was awarded an IRCSET Embark Scholarship to undertake his PhD under the supervision of Prof. Thorri Gunnlaugsson at TCD. After a short postdoctoral tenure at the Trinity Biomedical Sciences Institute in Dublin, Rob moved to The University of Sydney under the guidance of Prof. Kate Jolliffe where he was involved in the development of new platforms for the recognition, sensing and transport of biologically relevant anions. In late 2014, Rob returned to Ireland taking up a lecturing position at Maynooth University where he is currently Associate Professor and Associate Dean of Research for the Faculty of Science and Engineering. He has enjoyed periods as an invited researcher at Universite d'Orleans, in France in the Centre for Molecular Biophysics and at The Scripps Research Institute in La Jolla, California and is a funded investigator at the SSPC, Ireland's Pharmaceutical Research Centre and the Kathleen Lonsdale Institute for Human Health Research at Maynooth University. Rob's research interests lie in the fields of Supramolecular Chemistry and Chemical Biology where the group is trying to use supramolecular chemistry to develop new drug delivery vehicles, diagnostic tools, and novel approaches to treating microbial infections. |
Introduction
Historical notes
Peptides are physiologically active compounds made up of short chains of amino acid (AA) monomers connected by amide bonds (peptide bonds). The beginning of peptide chemistry has long been attributed to the year 1901, when chemists Emil Fischer, and Ernest Fourneau published the first ever “synthetic peptide” – a dipeptide called glycyl-glycine (Gly-Gly).1 One of the most significant scientific successes in peptide drug discovery was the development of insulin, a peptide containing 51 amino acids. It was discovered in 1921 by Frederick Banting and further refined by Frederick and Charles Best.2,3 Peptide chemistry received little attention for the next 30 years, until the medicinal use of peptides really kicked off after World War 2. Following this, oxytocin was synthesized by du Vigneaud in 1953, a major achievement for peptide chemistry and a synthetic milestone.4 However, these methods required a huge amount of time and synthetic effort for even the most trivial of peptides. It wasn't until Bruce Merrifield revolutionised peptide chemistry in 1963 with Solid-Phase Peptide Synthesis (SPPS).5 This technology pioneered the way for quick, easy, and efficient preparation of peptides, and was the basis for his 1984 Nobel Prize in Chemistry. SPPS substantially accelerated progress across the chemical and medicinal sciences. Furthermore, peptide's inherent specificity and excellent efficacy have made them attractive building blocks for the design of novel therapeutics.6Clinical use of peptides
Peptides are frequently involved in human physiology, acting as hormones,7 neurotransmitters,8 growth factors,9 or ion channel ligands.10 Moreover, peptides tend to act as intrinsic signalling molecules for many of these physiological functions, opening up the possibility for peptide-therapeutic mediation that closely resembles the natural process.11 Indeed, utilising peptide-based therapeutics has been commercially proven to help treat many diseases/disorders such as, type 2 diabetes, multiple sclerosis, acromegaly and osteoporosis – with the likes of Eli Lilly (Trulicity™), Novo Nordisk (Victoza™) and Novartis (Sandostatin™) dominating this space (Fig. 1).12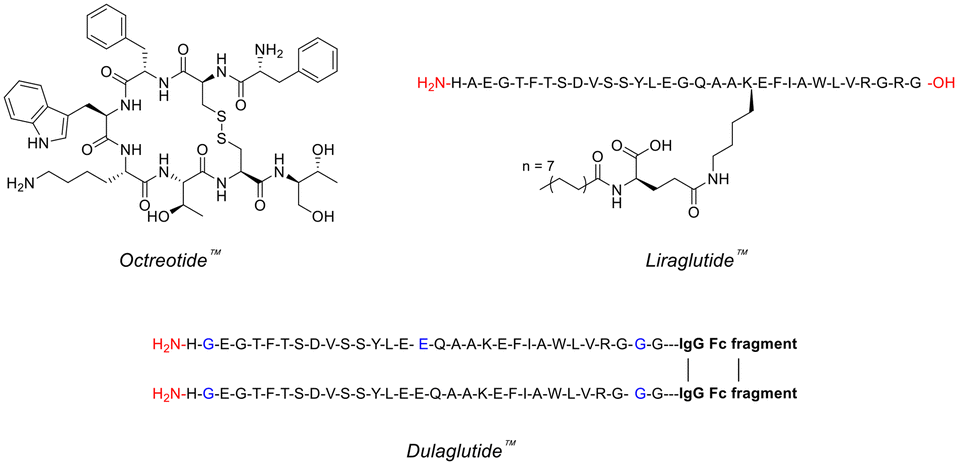 | ||
| Fig. 1 Chemical structures of famous peptide therapeutics (single-letter abbreviations used for amino acids to ensure clarity). | ||
In recent years, over 60 peptide drugs have been approved by the US-FDA, Europe, and Japan: over 150 are in active clinical development, and an additional 260 have been tested in human clinical trials.10,13 This translates to a global peptide therapeutics market currently valued at $37.8bn (US dollars), with projected growth of 9.6% (CAGR) to $91.25bn by 2031 (Fig. 2).14 Over 21 peptide therapeutics are currently being utilised to treat COVID-19, including 15 synthetic peptides in development against SARS-Cov-2 infection-related respiratory disorders.15 Peptide therapeutics are also being examined as a COVID-19 treatment option. As a result, significant industry participants reported a rise in revenue during the COVID-19 epidemic.14 Through the many lockdowns and long working-from-home periods, much of the population experienced more sedentary lifestyles. Coupled with the corresponding bad habits and unhealthy diets, a surge in chronic disease prevalence and incidence may be on the horizon. To this end, the worldwide peptide therapeutics market is likely to be driven by an increase in metabolic disorders forecast for the coming years.16
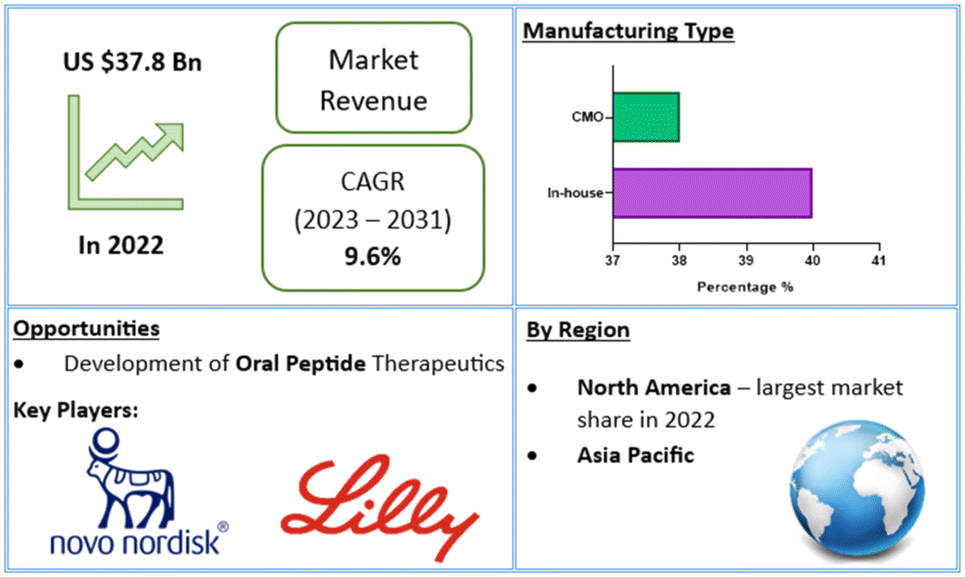 | ||
| Fig. 2 Global peptide therapeutics market value and financial forecasts.14 | ||
Peptides interact with cell-surface receptors and cascade intracellular reactions with high affinity and specificity, similar to large biologics such as proteins and antibodies.17 However, peptide-therapeutics provoke less of an immune response and typically have reduced production costs.18,19 Small-molecule drugs also benefit from low production costs and tend to have good oral bioavailability and membrane permeability.20 On the other hand, the clinical use of small-molecule therapeutics can be restricted due to their low specificity compared to peptide drugs. Peptide-based therapeutics usually have two inherent shortcomings: their ability to penetrate the cell-membrane(s) to reach intracellular targets, and in vivo stability – as their amide bonds can be hydrolysed by domestic enzymes.21 These inherent benefits and drawbacks of peptide therapeutics provide both challenges in peptide drug development, as well as opportunities for peptide design and drug discovery. Peptides remain a distinct family of pharmaceutical compounds that are molecularly positioned somewhere between small molecules and proteins (Fig. 3), while being biochemically and therapeutically distinct from both.22
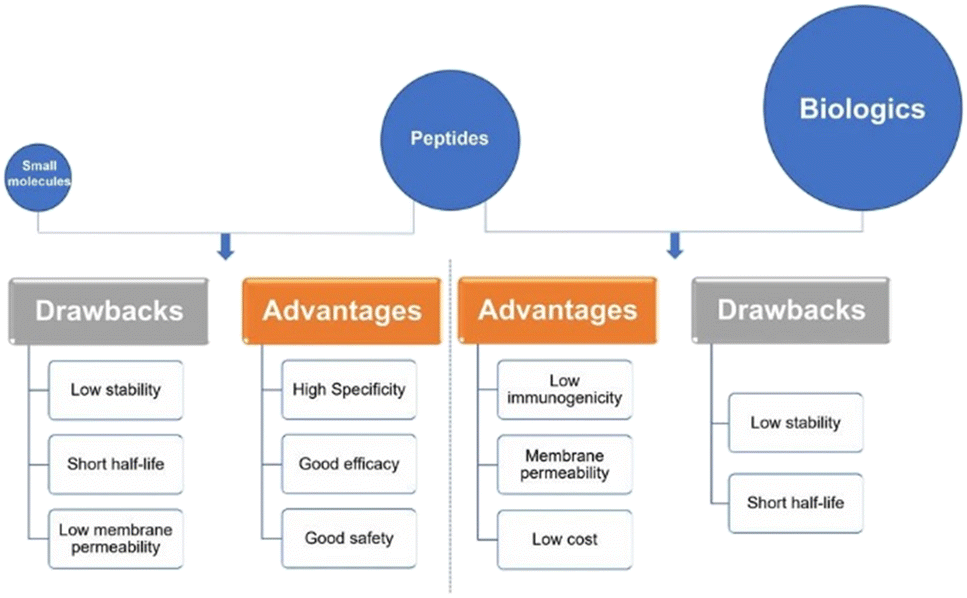 | ||
| Fig. 3 Peptides versus small molecules and biologics. Comparison of advantages and drawbacks between peptides and small molecules or biologics (reprinted from ref. 22 with permission from Springer Nature, copyright 2022).22 | ||
Peptides as building blocks in supramolecular chemistry
Peptides can also play a crucial role as building blocks in supramolecular chemistry – a field that focuses on the study and design of molecular assemblies beyond individual molecules. The importance of peptides in supramolecular chemistry lies within their unique structural and functional properties, which make them versatile and valuable components for creating complex and functional materials.The potential sequence variability provided by the 20 natural amino acids presents a significant opportunity for investigating optimal building blocks conducive to specific self-assembling characteristics, leveraging their diverse attributes in terms of charge, hydrophobicity, and polarity.23 Incorporation of non-natural amino acids into peptide sequences has also been employed to augment the diversity of self-assembling peptides and enhance the complexity of the resulting self-assembled structures.24 In addition to the primary structures determined by the amino acid sequence, peptides also exhibit distinct secondary structures. These inherent secondary structures can function as foundational units for elevated levels of self-assembly.25 Consequently, peptides prove highly adept for hierarchical assembly, representing a potent yet intricate method for producing functional materials. The establishment of secondary (and more advanced peptide structures) is notably influenced by hydrogen bonding, predominantly originating from polar amide-, amino-, and carboxyl-groups present in both the backbone and the side chains of peptides.
Achieving effective peptide self-assembly has been exemplified through the utilisation of amphiphilic peptides, dendritic peptides, polypeptides, cyclic peptides, and aromatic di-peptides (Fig. 4).26,27
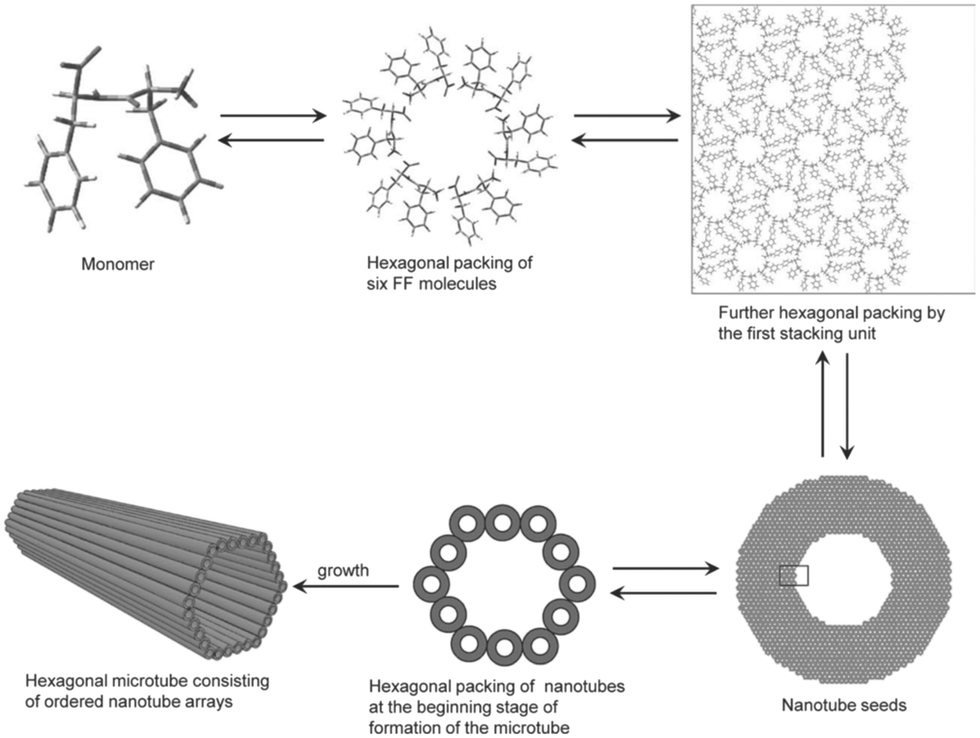 | ||
| Fig. 4 Schematic illustration of the formation of hexagonal peptide microtubes viaL-Phe-L-Phe molecules (reprinted from ref. 28 with permission from John Wiley and Sons, copyright 2011).28 | ||
Frequently, the peptide sequences are crafted by employing biologically-inspired strategies, resulting in self-assembled materials with structures and properties closely resembling those found in nature. Peptide-based supramolecular materials can offer distinct advantages in many biomedical applications, including; tissue-engineering, drug delivery, antibacterial treatments, immunotherapy, and imaging.29
However, before we explore the applications of modified synthetic peptides, we must understand the chemical alterations that are available to amino acids. With that said, the next section will delve into the unique molecular interactions and synthetic versatility that peptides offer, allowing for the precise design and engineering of supramolecular architectures.
Chemical modification of peptides
Since Merrifield introduced us to SPPS back in 1963, research in the peptide space has grown remarkably. Although the classical methods of SPPS provided a vital boost for peptide synthesis, it was limited by the dramatic decrease in purity as the number of coupling steps increased.30 Building on the success of the Boc and Fmoc protecting groups,31 innovative amino acid protecting groups and new methodologies were incorporated to produce high quality peptide products with superb yields. One early-stage method was fragment condensation via prior thiol capture, but this technique suffered from racemisation and other reaction complications.32 An improvement on this coupling reaction was achieved by Kent & co-workers by the chemoselective reaction of unprotected peptides, they coined this – native chemical ligation (NCL).33 Other ligation methods include (but not limited to) expressed protein ligation (EPL),34 the Staudinger ligation,35 and “click” or “switch” peptide ligation.36The biological activity of peptide therapeutics is closely related to their chemical structure. Sometimes the peptide structure may need to be synthetically modified to achieve an optimal secondary structure for improved biological activity, while retaining stability, selectivity and solubility of the peptide product.37 Peptide natural products – often isolated from secondary metabolites produced by plants and microorganisms – have gained the appreciation of many research groups and institutions, due to their fascinating biological activity.38 Most proteins consist of the 20 natural amino acid residues together with some post-translational modifications (phosphorylation or disulfide bridging), however the peptide-containing secondary metabolites frequently incorporate an assortment of unorthodox amino acids.39–41 For this reason, the introduction and/or manipulation of side-chain(s) within a peptide sequence presents a dynamic alternative to traditional peptide chemistry, with the possibility of generating many different analogues from one peptide precursor.42
Introduction of peptidomimetic-elements
The classification of peptidomimetics has been cultivated alongside the progress of synthetic peptides in recent years. The classification that will be used throughout this section is based on the modern taxonomy introduced by Grossmann.43 His categorisation denotes four distinct classes of peptidomimetics – A, B, C and D – depending on their resemblance to the natural substrate.44 Class A peptidomimetics closely resemble the parent peptide, utilising very few modified amino acids to stabilise the bio-active conformation, with modifications being restricted to the peptide backbone or sidechains. Next is class B peptidomimetics, featuring derivatives of class A mimetics with small-molecule insertions, uncanonical amino acids, and considerable backbone modifications. This class is home to peptoids and foldamers, where the backbones are extensively modified, but the side-chain functionalities are retained in the same order as the parent peptide.45 Class C mimetics are more small-molecule in stature, containing an uncanonical framework that almost completely replaces the peptide backbone. The orientation of key residues is retained, and the bio-active conformation remains intact. However, the central scaffold bears little resemblance to that of the native peptide. The final category of peptidomimetics – class D – are a far-cry from the natural peptide. Class D molecules can emulate the mode-of-action of the natural peptide without a remotely similar backbone or side-chain functionalities.Modification of peptide structures has been a growing area of research in recent years, with side-chain manipulations and backbone modifications being the two popular methods.
Some side-chains are more difficult to modify than others, take valine and alanine for example – aliphatic side-chains without an obvious functional group present (Fig. 5) – there are relatively few techniques for derivatisation. However, recent breakthroughs in the direct functionalisation of C–H bonds have generated novel technologies for targeted modifications.49 Of these recent breakthroughs, a notable example by Yu & group demonstrate the Pd-catalysed C–H arylation of N-terminal alanine residues.50 The largest sequence that the group managed to modify using this technique was a tetra-peptide, nonetheless this preliminary study illuminates the (mostly) untapped potential of post-assembly C(sp3)–H derivatisation as an exciting tool for the modification of aliphatic side-chains.
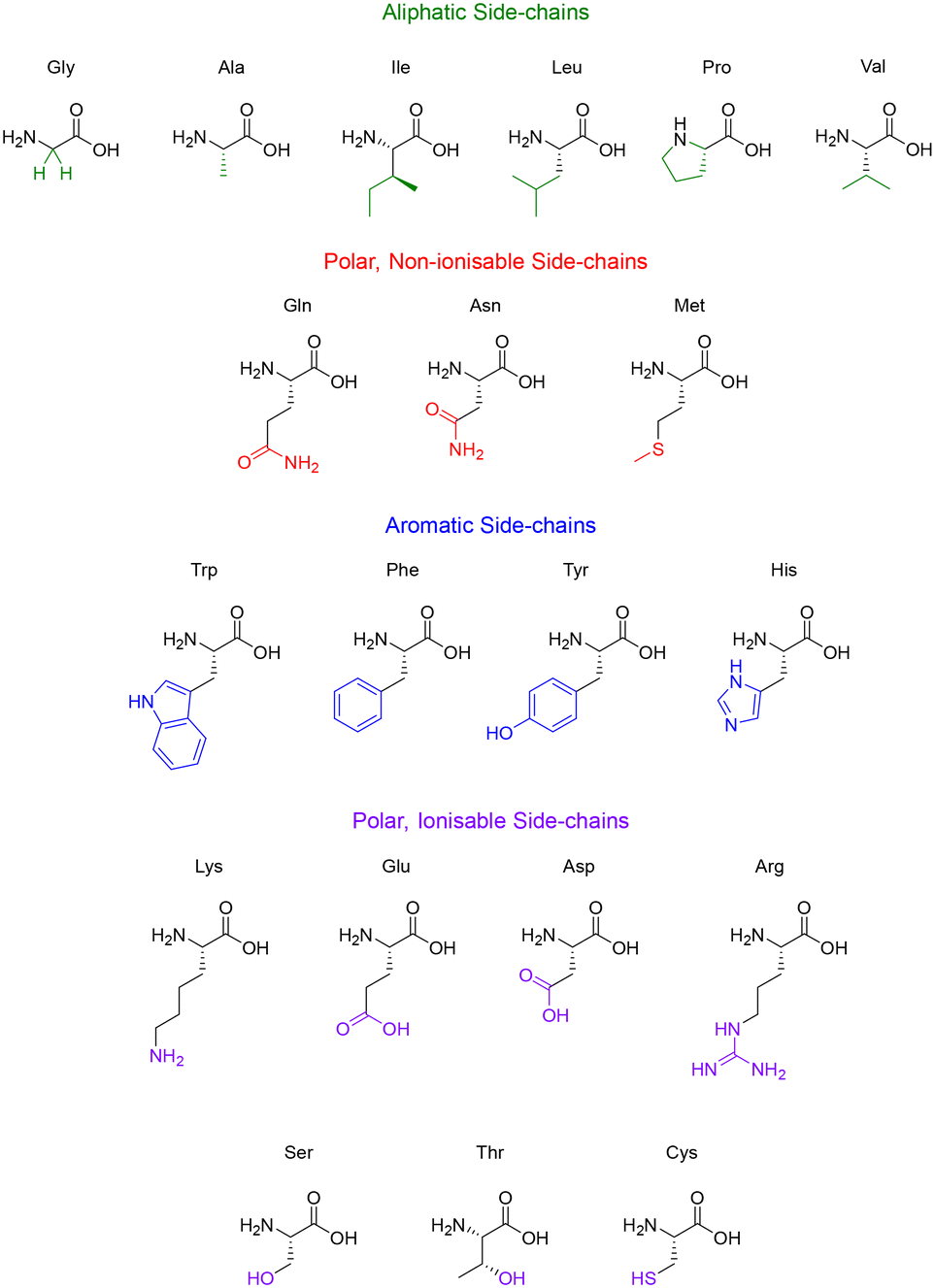 | ||
| Fig. 5 The 20 natural amino acids categorised by their side-chain functionality. | ||
Like the aliphatic residues, the polar non-ionisable side-chains (primary amides) of glutamine and asparagine remain troublesome targets for derivatisation. Popp and Ball however, employed a molecular recognition strategy which facilitated the selective modification of the Gln and Asn side-chains using dirhodium metallo-peptides.51 Reactions pursuing the modification of methionine however, are much more common. Methionine possesses a relatively high oxidation potential, and the reversible oxidation of the thioether is a well-described reaction pathway.52 Interestingly, it is the only natural amino acid residue that can be alkylated under acidic conditions.53
Amino acid residues with aromatic side-chains tend to have a much larger pool of analogues to choose from, such as unnatural heterocycles,54 and derivatives that include β-methyl groups for added conformational rigidity.55 Aromatic side-chains can be further categorised into ionisable (tyrosine & histidine), and non-ionisable (phenylalanine & tryptophan). Barbas introduced us to a powerful new aqueous ene-type reaction that permits click-like Tyr coupling,56 and has paved the way for the functionalisation of diverse handles, including PEG chains and multi-functional linkers.57 Although the side-chain of tryptophan is regarded as non-ionisable, there still exists some potential for synthetic modification, as highlighted by the many specific reaction pathways available to the indole moiety. Indeed, these modifications are currently dominated by C–H functionalisation at the indole C-2 position.58,59 This C–H functionalisation strategy has been used to facilitate alkynylation and arylation of the indole side-chain. However, similar approaches for the C–H functionalisation of the side-chain(s) for His and Phe remain underexplored. This represents an exciting challenge for researchers to develop novel methodologies to produce synthetically modified His and Phe residues.
Unsurprisingly, the amino acid residues with polar ionisable side-chains have been well-studied, with cysteine taking the top spot as the most documented residue within bioconjugation literature.60 There are many properties of cysteine that make it a convenient target for side-chain modification, like its inherently low pKa (∼8.3) and the considerable nucleophilicity of the thiol group. Although the cysteine–maleimide conjugation has been a popular method of thiol modification,61,62 the use of transition metals has gained recent recognition with the likes of Buchwald and Pentelute describing a Pd(II)-mediated arylation of Cys under mild conditions.63 The theory described in this section serves to highlight the potential of side-chain modifications to introduce new functionalities or substituents into a given peptide natural product.
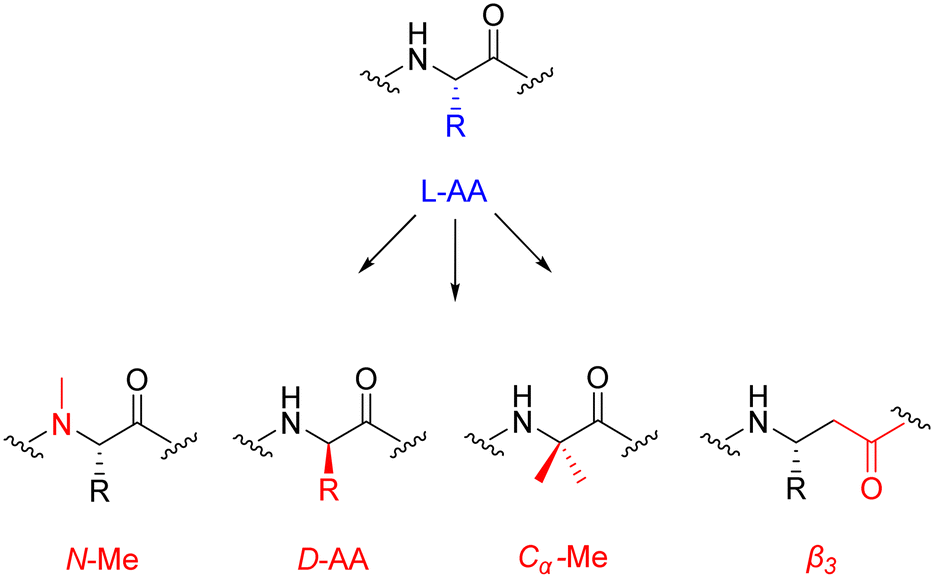 | ||
| Fig. 6 Chemical structures of popular peptide backbone modifications. | ||
The introduction of these structurally diverse amino acids into the peptide sequence – even more so at the site of proteolysis – can be an effective method for increasing the plasma half-life of peptide therapeutics. An interesting example is selepressin – an analogue of vasopressin, containing the backbone modification [Phe(2), Ile(3), Hgn(4), Orn(iPr)(8)] – which was being developed by Ferring Pharmaceuticals for the treatment of vasodilatory hypotension in septic shock.68 Selepressin seen early clinical success, displaying comparable selectivity coupled with an enhanced plasma half-life. However, the phase 2b/3 clinical trial was terminated in February 2018 for futility, as the administration of selepressin, compared with placebo, did not result in any statistically significant improvement.69 Perhaps further research is required to decide if selepressin will play a potential role in other patient-related conditions due to septic shock.
Significant work regarding backbone modifications was pioneered by Seebach et al. who introduced side-chains into small peptides via enolate chemistry.70 One of the groups most impressive applications of this technique is the site-selective alkylation of cyclosporin A,71 where they demonstrate that the deprotonation of amide N–H bonds shields adjacent amino acids from deprotonation and prevents epimerisation. This process allowed the compound to undergo nucleophilic substitution with electrophiles to produce the modified cyclosporins in satisfactory yields (∼90%) with a diastereomeric selectivity ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Re/Si, D/L).
:1 (Re/Si, D/L).
As peptide chemists, the use of transition metals has allowed us access to previously unheard-of bioconjugate transformations, this is particularly true in the ever-expanding area of peptide backbone modification.72,73 A recent example was inspired by known non-ribosomal peptide synthetase (NRPS) pathways, where researchers describe an iron-catalysed oxidative derivatisation.74 This approach was utilised to generate 21 non-natural amino acids from 4 canonical residues while preserving the innate chirality. While this particular use of transition metals is scarce, the opportunities they present for peptide backbone modification, and protein structure/function will surely inspire innovation for novel peptide conjugates. The next section will discuss a few of the main techniques used for the medicinal chemistry optimisation of peptides.
Macrocyclisation
Cyclisation is an essential strategy for the medicinal chemistry optimisation of peptide leads during drug discovery.75,76 Cyclisation is an excellent method for improving the proteolytic stability of a target peptide. This allows medicinal chemists to take advantage of the high selectivity, increased potency, and low toxicity that are intrinsic to peptides, to progress them as potential biotherapeutic agents. An early example on the use of peptide-cyclisation during drug design was a cyclic analogue of somatostatin (Veber–Hirschmann peptide).77 This newly cyclised peptide constrained the sequence into a bioactive conformation while also improving its proteolytic stability, resulting in a peptide-product with increased duration of action and oral bioavailability (Fig. 7). From this discovery, structural studies on other natural peptides were conducted to probe the use of cyclisation to explore novel bioactive conformations.78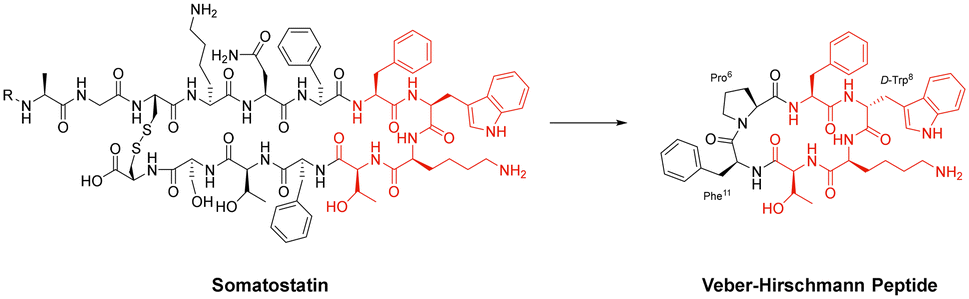 | ||
| Fig. 7 Structural comparison between somatostatin and Veber–Hirschmann analogue. | ||
As can be the case for many novel design scaffolds created by synthetic chemists, Nature did it first. Or at least provided the necessary inspiration to facilitate the discovery. The advantages imparted by cyclisation have been exploited by nature with the many cyclic peptides found in fungi, bacteria, plants, and animals.79 One study by Craik et al. documented the effects of peptide-cyclisation on the structural-activity of sunflower trypsin inhibitor-1 (SFTI-1). This cyclic peptide is comprised of one cross-linking disulfide bond and is the smallest, most potent known inhibitor of trypsin.80 The group observed that cyclisation was essential to its enzymatic stability and inhibitory activity (Fig. 8).81 Many natural peptides like SFTI-1 possess exceptional chemical, thermal, and proteolytic stability, which can be (at least partially) attributed to their cyclic backbones. This section will examine the growing interest in macrocyclic peptides, and the various methods for synthesising cyclic peptides.
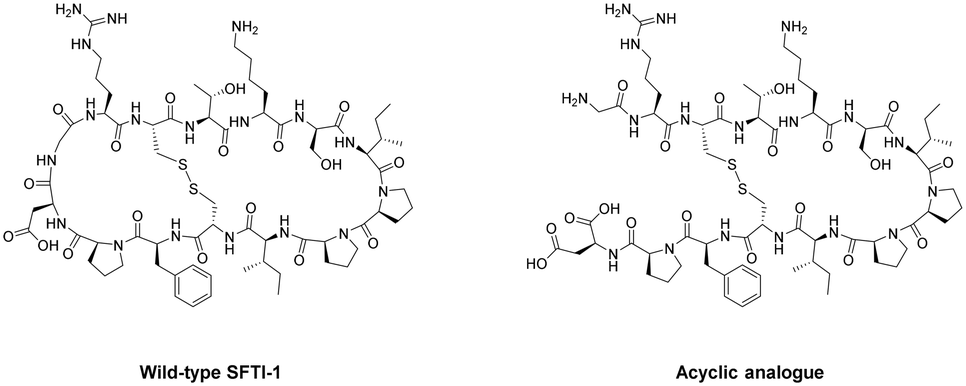 | ||
| Fig. 8 Structural comparison between natural SFTI-1 and its acyclic analogue. | ||
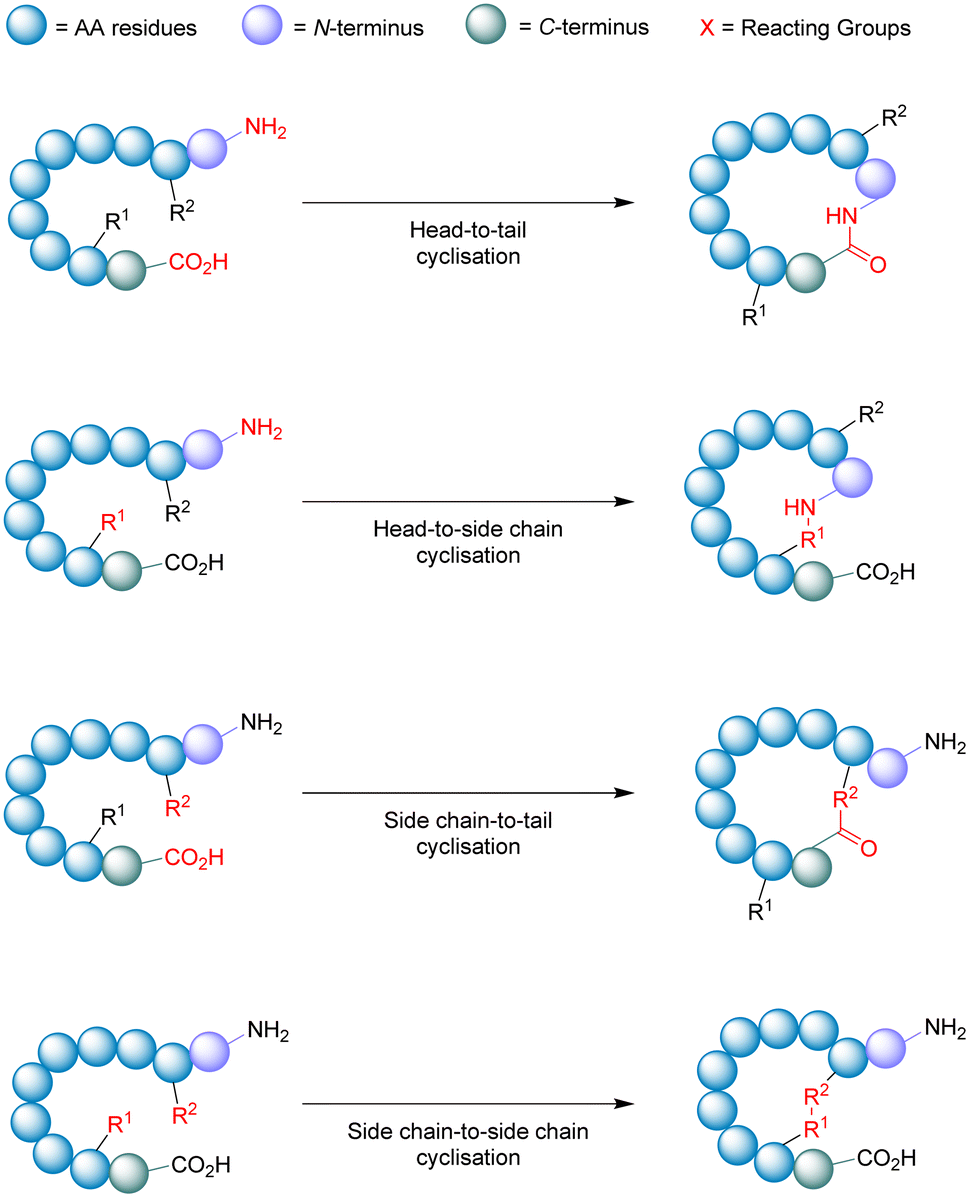 | ||
| Fig. 9 Schematic illustration of cyclisation strategies. | ||
During the synthesis of cyclic peptides, the final ring-closing reaction can often be a lactonisation (cyclic carboxylic ester),82 lactamisation (cyclic amide),83 or produce disulfide-bridge.84 Peptide cyclisations are generally carried out at high-dilution (<mM conc.) to promote intramolecular interactions and minimise troublesome intermolecular processes like polymerisation.
Macrocyclisation using SPPS has been employed as an effective means to generate cyclic peptides.85 While the linear peptide is bound to the solid support (resin), it experiences a sort of pseudo-dilution phenomenon – where the functional groups bound to the resin are less likely to encounter one another in comparison to the free molecules within the solution. This environment promotes the favourable intramolecular interactions necessary for peptide cyclisation. To achieve on-resin cyclisation, the linear peptide is usually bound to the solid support via the side-chain of one of the amino acids in the sequence (e.g. Asp or Glu). At least 3 independent and orthogonal protecting groups (resin included) are required for this strategy. The linear peptide needs to be constructed, N- & C-termini to be deprotected, cyclised from head-to-tail, and then finally cleaved from the resin.86 A noteworthy feature of on-resin cyclisation is that basic washing and filtering is usually sufficient to achieve (relative) purity, circumventing intermediate purification steps and solubility issues.
One of the most important factors that influence the success of peptide cyclisation is ring size. The cyclisation of large peptides is sometimes reported as troublesome, however peptides containing >7 amino acid residues usually cyclise without too much difficulty. This is not the case for smaller peptide structures. During the head-to-tail cyclisation of peptides with <7 amino acid residues, C-terminal epimerisation and cyclodimerisation are common pitfalls.87
The activation free-energy (ΔG‡) of cyclisation is governed by an enthalpy term (ΔH‡) and an entropy term (ΔS‡): ΔG‡ = ΔH‡ − T(ΔS‡). Using the head-to-tail cyclisation approach as an example, the activation entropy (ΔS‡) of the intramolecular interaction is based on the probability that the N- & C-termini approach each other at an angle (∼107°)88 to facilitate conjugation. This probability is reduced as the amino acid chain gets longer. One might assume that this should be of benefit to the smaller linear peptides (<7 AAs), however the loss in entropy is almost completely eclipsed by the enthalpic term. The activation enthalpy (ΔH‡) represents the stress on the molecule during the transition state (‡). This value can be very high for peptides containing less than 7 amino acid residues, as there is significant ring-strain generated by the preferred confirmation of the amide bonds within – as all trans.89
One early study highlighted these challenges as they tried to synthesize numerous naturally occurring cyclic tetra- and penta-peptides using a head-to-tail style ring-closure. However, results were sparse with most cyclisation reactions proving unsuccessful.90 They noted that the ring disconnection (site of cyclisation) had to be chosen very carefully, suggesting that product yields could be improved if the site of bond formation was not sterically hindered by the likes of N-alkylation or β-branched amino acids. They also documented improved macrocyclisation rates between two amino acid residues of opposite stereochemical configuration (D/L), and when linear peptides had a sufficient level of pre-organisation due to certain turn-inducing motifs. This study demonstrates the many difficulties that can arise from an apparently straightforward retrosynthetic analysis of cyclic peptides.
Internal conformational elements. As mentioned above, peptide cyclisation is more successful when the linear peptide can accommodate the angular criteria for both reactive groups being in the transition state with the least amount of strain. Smith and Daidone studied this idea and demonstrated that the cyclisation rate of longer polypeptide chains is influenced by the formation of intra-peptide hydrogen bonds. This generates ephemeral β-sheet like structures that serve to lower the free-energy of macrocyclisation.93 The inverse is also true, slower cyclisation kinetics are observed with the absence of these hydrogen bonds in smaller peptides. Peptides with few amino acid residues lack the structural flexibility to accommodate these intra-peptide formations, thus highlighting their inherent rigidity and reluctance towards cyclisation.
In order to optimise the macrocyclisation of peptides, chemists have yet again sought inspiration from nature to help them overcome their synthetic woes. The secondary structure of proteins – notably, the reverse turn – has inspired the introduction of a cis-amide bond in the middle of the peptide sequence.94 This modification is analogous to a β-turn and provides an elegant way of obtaining sufficient spatial proximity between the reactive groups. Proline has the highest natural occurrence within these reverse turns, with the cis-amide bonds of proline (Fig. 10) being displayed in the crystal structures of many proteins. One classical study took advantage of this fact in the cyclisation of the tri-peptide cyclotri-L-prolyl.95 In a similar fashion, linear precursors containing a di-proline unit with alternating stereochemistry (L-Pro-D-Pro or vice versa) are solid candidates for macrocyclisation due to their impressive β-hairpin inducing features. Robinson & group exploited this to generate cyclic peptides that accurately mimicked canonical conformations of hypervariable loops observed in the crystal structures of antibody fragments.96
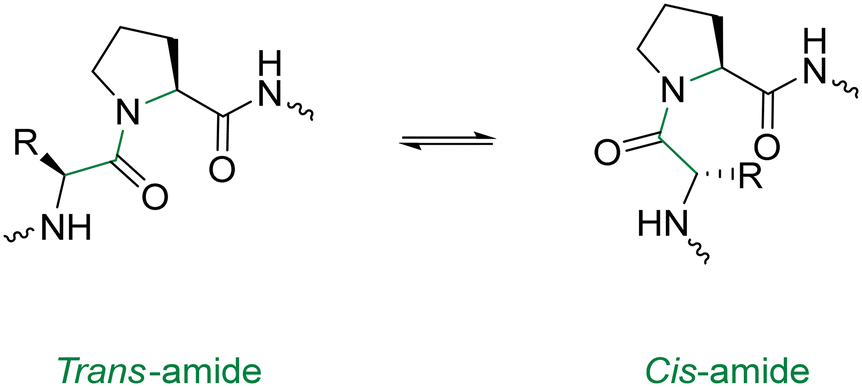 | ||
| Fig. 10 Cis & trans proline isomerisation around the Xaa–Pro amide bond. | ||
N-Methyl AAs have a comparable stereochemical influence on the backbone of peptides to that of the Pro residue. They can also be used to introduce cis-amide bonds into the peptide backbone and are well-equipped to generate β-turns.97 Turn-inducing effects are not exclusive to the Pro residue. Indeed the incorporation of other D-AAs in to L-homopeptides can apply similar contortions, and have been used to improve the yields of several peptide macrocyclisations.98,99
External conformational elements. External elements for promoting peptide cyclisation operate by pseudo-isolating the linear peptide from the bulk solution. This unique micro-environment serves to reduce the chances of polymerisation or cyclo-oligomerisation.100 Metal ions offer a non-covalent ancillary-based strategy for peptide macrocyclisation.101 The inspiration for this strategy comes from the well-documented capability of cyclic peptides to act as ionophores – binding metal cations in solution and in vivo.102,103 Beck and Co. were one of the first groups to demonstrate linear peptide pre-organisation via metal ions for the use of macrocyclisation.104 They constructed a cyclic tetra-peptide through a metal-cation-assisted dimerisation of 2 dipeptide methyl esters under basic conditions (Scheme 1). An interesting caveat for this double head-to-tail lactamisation is that both dipeptides must orientate themselves in a trans fashion around the metal centre before nucleophilic attack can occur. This macrocyclisation strategy can facilitate ring sizes of 12- to 18-membered cycles, can incorporate α- and β-AAs, and does not require protecting groups, coupling reagents, or high-dilution conditions. The cyclic peptide product can then be purified via isolation of the coordinated dianion, and the metal ion can be liberated by acid methanolysis.
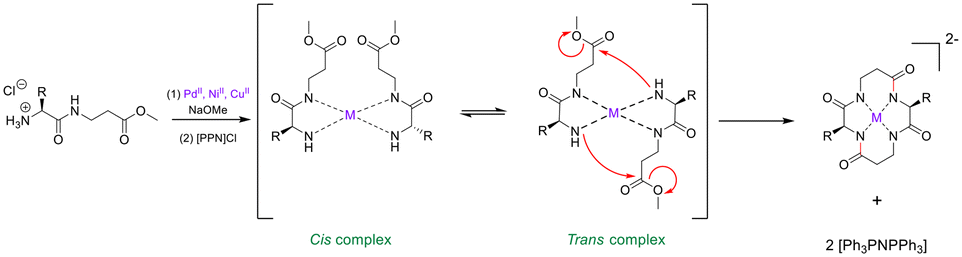 | ||
| Scheme 1 Transition metal-assisted cyclo-dimerisation of dipeptide esters (adapted from ref. 104 with permission from John Wiley and Sons, copyright 1998).104 | ||
An analogous strategy – one that remains underdeveloped and largely unexplored – is the anion templation strategy. Although published reports on this strategy remain scant,105,106 its use suggests that not only cations, but anions can be used for the pre-organisation of linear peptides to promote macrocyclisation. Speranza and Tomišić demonstrate this by using the Cl− anion as a templating agent for the synthesis of cyclic peptides (Fig. 11).107 They prepared 3 novel cyclic homo-lysines and 6 other cyclic peptides using the head-to-tail lactamisation strategy. Experimental yields of cyclic peptide products – that were Cl− anion templated – were found to be significantly higher than those obtained via the cation approach. Indeed, in some instances, only the anion-mediated synthesis yielded the target cyclic peptides. To further support their theory that the Cl− anion played a major role in the macrocyclisation reaction, they studied the corresponding ring-closure reaction kinetics. Macrocyclisation experiments were conducted using increasing concentrations of TEACl (2–100 eq.), with higher concentrations of TEACl inducing faster cyclisation rates and increased yields. This was evidenced by TLC and quantitative 1H NMR analysis.
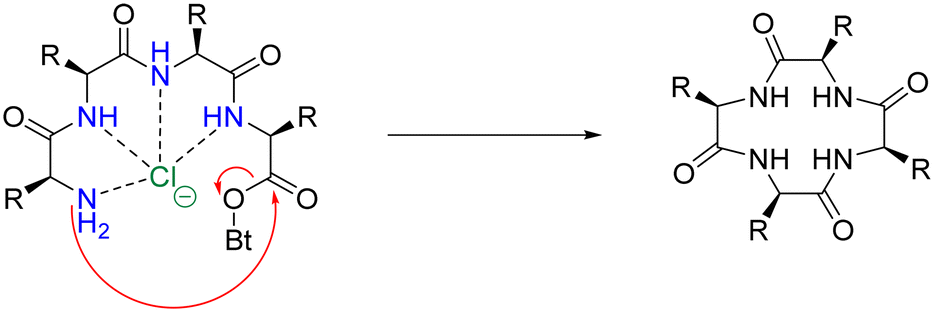 | ||
| Fig. 11 Suggested mechanism for the chloride-mediated macrocyclisation (adapted from ref. 107 with permission from American Chemical Society, copyright 2020).107 | ||
It is understood that anion-recognition by cyclic peptides has been well-documented,108 and many examples exist of their use as templating-agents for the synthesis of organic and inorganic scaffolds,89,109 however – at the time of writing this – no example(s) of chloride-templated synthesis of cyclic peptides have been reported. This novel strategy for pre-organisation of linear peptides may promote further research into this vastly under-appreciated method of macrocyclisation. Peptidomimetic alterations of natural peptides can produce bio-active analogues. Furthermore, macrocyclisation of linear peptides is often used as an effective strategy to provide increased conformational rigidity and more bio-stable products.
Substitution of L-amino acids with D-amino acids
Specific nomenclature has been used to denote the absolute configurations of the 4 substituents around sp3 carbon atoms. The same notation is used for simple sugars and amino acids – the L- and D-system of absolute configuration(s), suggested by Emil Fischer.110 He denoted chiral molecules with a configuration related to that of L-glyceraldehyde – L, while stereoisomers related to D-glyceraldehyde were described as D. It is also worth mentioning that not all L-AAs rotate plane-polarised light to the left, they can also rotate it to the right; and the same holds true for D-AAs. Fischer's notation, L- and D-only refer to the absolute configuration of the substituents about the carbon atom.D-AAs, the enantiomers of the canonical L-AAs, came under investigation in the mid-20th century, prompted by Krebs' discovery of D-AA oxidase.111 However, the use of D-AAs to improve proteolytic stability was not employed until much later. Zisman and Seia showed us that the incorporation of D-AAs into polypeptide antigens could enhance their proteolytic stability.112 Building on this work, Tugyi et al. studied the antigenic properties and proteolytic stability of certain MUC2 peptides partially substituted by D-AAs. Their goal was to generate peptides that were resistant to enzymatic degradation while possessing similar, if not increased, binding kinetics compared to the original L-homopeptides.113 The results generated from this study suggested that the activity of the peptides was sustained even in the presence of 2 D-AA residues at its N-terminal flanking domain, and up to 3 at its C-terminal flanking domain. This novel D-/L-heteropeptide also displayed enhanced proteolytic resistance in lysosomal media and diluted human serum. These observations seem to demonstrate the benefits of appropriate D-AA modification(s) to produce synthetic antigens with comparable recognition properties and resistance to proteolytic degradation.
An interesting study by Werner et al. explored the effect(s) of heterogeneous-backbone modification for enhancing proteolytic protection. They employed the four most common motifs for this kind of modification: D-AA residues, N-Me residues, Cα-Me residues, and β3 residues. From this, the group synthesized a family of compounds – 32 analogues of a control peptide – with residues replaced by the above mentioned motifs.114 These heterogeneous-backbone motifs have already been studied in isolation,64,115 but what makes this report exciting is that it has ventured to compare and contrast their ability to protect the substrate from proteolytic attack. Interestingly, the team discovered that the level of protection was as follows: D-AA > Cα-Me > β3 > N-Me. D-AAs and Cα-Me residues induced proteolytic stability by moving the overall structure away from a conformation that was easily recognised by the enzyme. This imparted a large degree of proteolytic protection, with wide-ranging and synergistic effects when substitutions were combined in a single sequence. N-Me AAs were observed to influence proteolytic stability through the disruption of particular enzyme–substrate points of contact, with limited effect to local folding – resulting in short-range effects of humble magnitude, that were only additive when used in tandem. The protection offered via β3 AAs was rather intricate – imparting only middling levels of stability, yet far-reaching. The authors describe the effects of combined β3 replacement as partially-synergistic, but noted that the quantity of β3 AAs did not directly correlate to enzymatic resistance.
Caution is advised when appropriating these findings in a more general sense, as the results generated by the group come from one single serine protease (chymotrypsin). Nonetheless, the study presents a solid hypotheses that will aid the future development of novel peptides/proteins containing enhanced proteolytic stability with minimal unnatural backbone content. D-AA residues possess folding characteristics that differ greatly from their L-AA brethren, this makes them rather effective turn-inducers. Building on the results highlighted in this study – demonstrating the ample proteolytic stability imparted by D-AAs – they may well be ideal candidates for bio-compatible backbone modifications.
Peptide conjugates
The original design of molecular conjugates can be attributed to the German physician/scientist Paul Ehrlich when he introduced us to the iconic phrase ‘magic bullet’. This magic bullet is described as a cytotoxic payload that would only become armed when selectively delivered to the site of interest by a targeting motif.116 It wasn't until 1958 when the first few examples were reported,117,118 and only in 1983 did the first clinical trial of such a conjugate begin.119 Fast forward another 20 years until the first FDA-approved conjugate (gemtuzumab ozogamicin) – operating under the trade name Mylotarg™ – consisting of an antibody bound to calicheamicin for the treatment of acute myeloid leukaemia.120The conjugation of molecular species to peptides is an elegant strategy to tackle substandard aqueous solubility, premature metabolic degradation, and may also be used to promote cellular uptake. Certain peptide sequences can provide targeted-delivery of small molecules to boost local drug concentration. This in turn, helps to reduce the adverse effects pertaining to systemic exposure and from accumulation in healthy tissues.121 Drug-conjugates that possess peptide carriers can bind to cell-surface receptors with high affinity – similar to that of antibodies – and recognise a whole host of endogenous targets. Clinically, the most famous of these biological receptors are the integrins, tyrosine kinases, and G-protein-coupled receptors. Peptide conjugates have a much smaller molecular size compared to antibodies, and may provide a more efficient delivery to obscure biological targets with decreased immunogenicity.
The pharmacokinetic profile of peptide–drug conjugates is unique, with a shortened circulation time and metabolic half-life in comparison to antibodies. This inherent property gives peptide-based drug conjugates an edge when it comes to the delivery of cytotoxic agents where prolonged exposure is undesirable. However, a significant drawback is encountered when dealing with solid tumours. Treatment of this nature usually requires long-lasting pharmacokinetics, often displayed by antibodies. Nowadays, as peptide chemistry has matured and improved, techniques such as pegylation, lipidation and other modern advancements has made up the difference, making them much more comparable to antibodies in this respect. The peptide–drug conjugate strategy has the potential to simplify commercial synthesis, streamline compliance with regulatory agencies and consequent criteria during manufacturing, and is a powerful means to achieve preferable disease outcomes that can be conveniently administered at a reasonable price.122
Linker and conjugation chemistry
The linker is an essential component of the peptide–drug conjugate strategy, and serves to covalently unite the peptide and the small-molecule moiety. The linker supports both the peptide sequence and the payload (drug) by upholding their structural integrity during administration, until such a time when the conjugate has reached its desired destination. To reduce the likelihood of unwanted side-effects, it is desirable for a peptide–drug conjugate to have a linker that only releases the payload after the target (cancer cells etc.…) has taken up the conjugate intracellularly. However, upon systemic administration – most linkers within peptide conjugates start to break down quickly after being exposed to the blood plasma. The remnants of the intact-conjugate(s) are eventually taken up by the target cells, but this vastly diminished concentration can often prove inadequate.123The linker within peptide–drug conjugates (usually) makes two covalent chemical bonds – one between the peptide/substrate and the linker, and another between the payload and the linker. The bond between the payload and the linker should be cleaved to release the free (unmodified) payload at the target site, and the bond between the peptide and the linker should not interfere with the peptide's affinity for its receptor. Many linker functional groups exist in the literature.124 However, it is generally accepted that they can be arranged into 4 main families: acid-cleavable (carbonate & hydrazone), enzyme-cleavable (carbamate, amide & ester), non-cleavable (oxime, triazole & thioether), and reducible disulfide (Fig. 12). This (rather broad) categorisation was established by monitoring how these functional groups reacted after cellular uptake or in the presence of in vivo stress. Because of this, linker chemistry can often dictate whether or not the overall conjugate will be successful in enhancing efficacy. This section will provide a brief summary of the main linker strategies used in the design and synthesis of peptide–drug conjugates, with a focus on enzyme-cleavable linker chemistries.
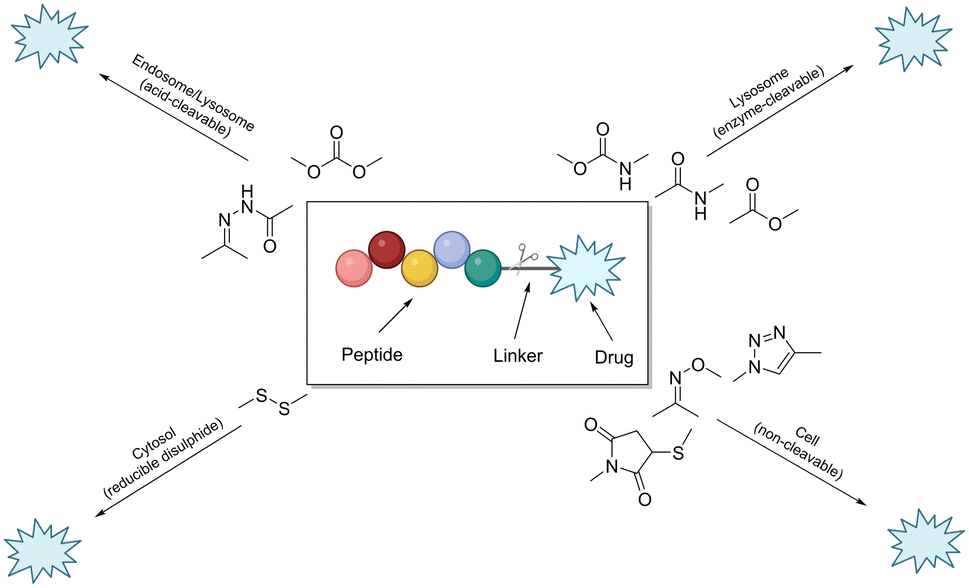 | ||
| Fig. 12 Cartoon schematic of a peptide–drug conjugate, and the associated linker chemistries (adapted from ref. 123 with permission from American Chemical Society, copyright 2021).123 | ||
Linker chemistries that involve enzyme-cleavable amide or ester bonds have become attractive, as they can be manipulated for site-specific cleavage in lysosomes or tumour microenvironments. Cancer cells' intracellular compartments – such as lysosomes and endosomes – have high levels of esterases and amidases, which can be utilised for site-specific release through upregulated expression of these enzymes. More complex amino acid sequences recognised by enzymes like caspase-3 or cathepsin B have been employed as linkers.125,126 It is important to note that these enzyme-cleavable linker chemistries are not foolproof, as many accidental-cleavage opportunities can arise prior to reaching the intended target. Appropriate control of the linker chemistry is essential for ensuring the overall stability of the conjugate until it reaches the target site.
One study employed an ester bond to couple paclitaxel to a peptide called angiopep-2. In this peptide–drug conjugate, the paclitaxel molecules are coupled to the side-chain(s) of two Lys residues and the N-terminal amine of angiopep-2, to form the complete therapeutic (Fig. 13).127 This conjugate (ANG-1005) is currently being used for treating patients that display solid tumour or brain metastasis. It works by overcoming the main drawback of (unmodified) paclitaxel, which struggles with blood–brain barrier permeability due to the presence of multi-drug resistance efflux pumps in brain tumour cells. The ester bond of the peptide–drug conjugate is selectively cleaved by esterases within the lysosomes, delivering paclitaxel to the brain.
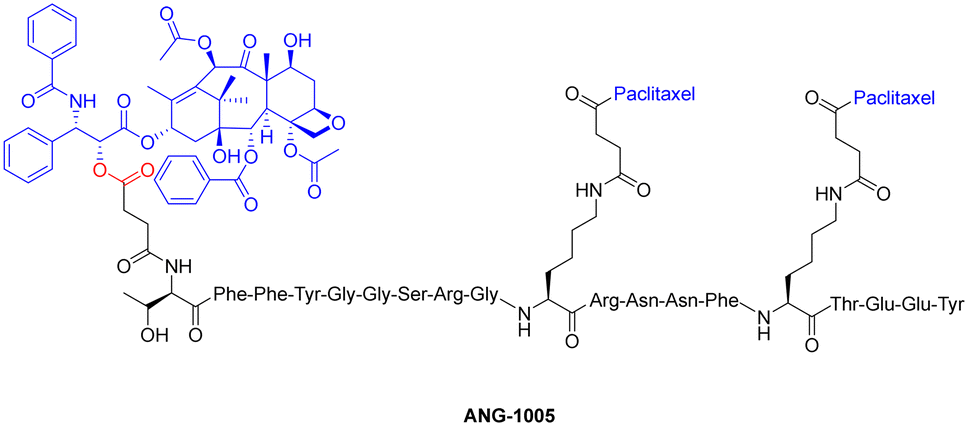 | ||
| Fig. 13 Chemical structure of paclitaxel conjugate ANG-1005 (ester bond highlighted in red). | ||
A radionuclide coupled to the peptide octreotide – called 177Lu DOTA–TATE (Lutathera®) – was approved by the FDA in 2018 for treatment against neuroendocrine tumours (targeting somatostatin receptors).128177Lu DOTA–TATE is administered during peptide-receptor radionuclide therapy (PRRT) – a type of internal radiotherapy, also known as radioligand therapy – and was the first peptide–drug conjugate to be approved by the FDA for treating prostate cancer.129 This conjugate consists of the radionuclide 177Lu chelated to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), and DOTA is coupled to Tyr(3)-octreotate (TATE) by an amide bond (Fig. 14).130
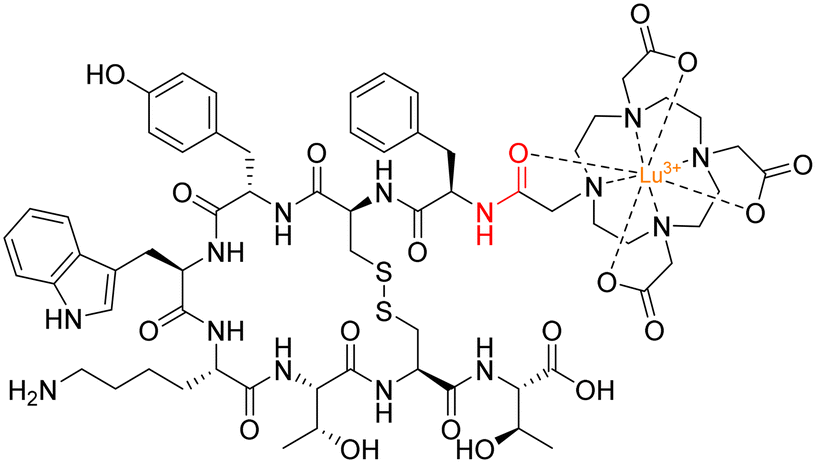 | ||
| Fig. 14 Structure of 177Lu DOTA–TATE with amide bond highlighted in red. | ||
To maximise effectiveness, ester and amide linkers need to be carefully manipulated so that they remain inert until within the tumour tissue/cancer cells. Although research in this area has boomed in recent years, more data is needed to understand what specific manipulations of amide/ester linkers can be employed to enhance chemical stability and efficacy.
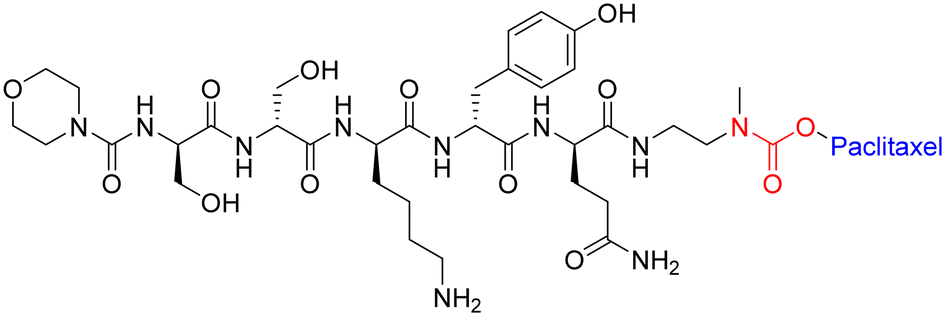 | ||
| Fig. 15 Example of peptide–paclitaxel conjugate with carbamate bond highlighted in red. | ||
This study agrees with other reports that suggest the in vivo linker stabilities can be ranked as follows: amide > carbamate > ester > carbonate.133 However, further studies are required to demonstrate which linker strategy will lead to better efficacy for tumour reduction.
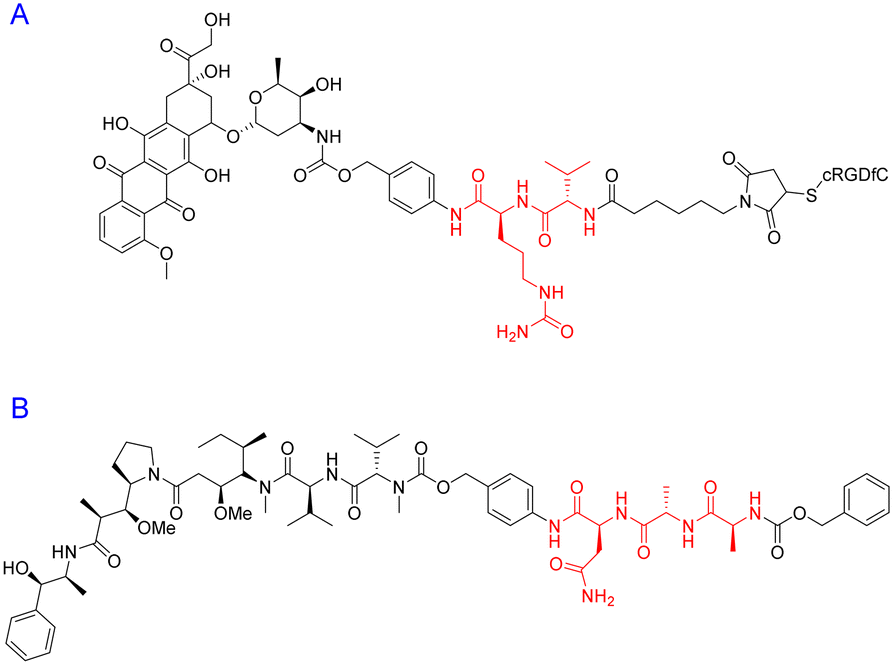 | ||
| Fig. 16 Peptide–drug conjugates with (A) di- and (B) tri-peptide linkers highlighted in red. | ||
The tri-peptide sequence has also been used for the site-specific release of payloads within tumour micro-environments. Ala-Ala-Asn is a cleavable sequence that undergoes degradation via the enzyme legumain.135 The asparaginyl endopeptidase – legumain – is reported to be upregulated in tumour cells and specifically cleaves at the C-terminus of asparagine.136 One group was determined to capitalise on this catalytic property for prodrug activation – with potential application(s) in cancer therapy. To this end, Bajjuri and colleagues synthesized distinct prodrugs containing the cytotoxic payload(s) monomethylauristatin E (MMAE) or didesmethylauristatin E (DDAE) conjugated to the tri-peptide (B) via an amide or carbamate bond (Fig. 16).135 The absence of a targeting peptide within their synthetic design is noted, however these novel prodrugs display considerable synthetic flexibility and could potentially be functionalised with a specific peptide sequence to enhance their site-specificity. Among the Bajjuri-prodrugs that were synthesized, compound B was found to yield the best results, demonstrating 57% inhibition (4T1 breast cancer) in mice when compared to subjects treated with buffer alone. Interestingly, tri-peptide prodrug B managed to overcome one of the main drawbacks of MMAE-based therapeutics, displaying no cytotoxicity, in contrast to the high mortality rate observed in mice that were treated with MMAE alone.
The selection of both the linker and functional group holds significant importance in peptide–drug conjugates, as it profoundly impacts the intended pharmacokinetic and pharmacodynamic characteristics of the conjugate. Notably, factors such as linker length, composition, and flexibility play a pivotal role in governing the conjugate's stability, the pace of drug liberation, and its elimination rate from the system. Moreover, the functional group present on the linker exerts an influence on enzymatic breakdown and cellular absorption of the conjugate, thereby ultimately shaping its effectiveness and safety profile.
Convergence of therapeutics and sensing
Peptide probes with luminescent sensing capabilities are used throughout the chemical and biomedical fields. In cellular imaging, these probes allow real-time visualisation of biological processes without disrupting cellular functions, while also enabling molecular recognition and selective binding to specific biomolecules.137 This aids in disease diagnosis and monitoring, particularly in biosensing applications for early detection of diseases like cancer. Moreover, luminescent peptide probes play a vital role in drug development through high-throughput screening and target validation, expediting the identification of potential therapeutic agents.138Fluorescent peptide probes
The fields of chemical diagnostics and imaging are constantly being developed and updated, with fluorescence imaging becoming an essential technique for observing changes in biomarkers within living systems.139,140 The compounds that generate fluorescent-imaging data are called – fluorogenic probes. These latent fluorophores can modulate their signal in response to environmental fluctuations, analyte interactions, or chemical modifications.141 Fluorogenic probes are synthesized by chemically engineering the parent probe so that its fluorescence profile is sufficiently distinct from the released fluorophore – this activation is usually triggered by a specific event. In the interest of conciseness, this specific introductory section will focus solely on enzyme-activated fluorogenic probes. For more information regarding fluorescent spectroscopic properties and fluorophore chemistry, see the following papers discussed elsewhere.142,143Enzyme-activated fluorogenic probes that take advantage of enzymatic degradation to modulate fluorescence output, can equip researchers with an impressive toolkit for monitoring biological events in cellulo and in vivo. Initially, many enzyme-activated probes were constructed around xanthene dye motifs, for the detection of esterases,144 galactosidases,145 lipases,146 and phosphatases.147 Building on from this work, primitive live-cell imaging was possible via cell-permeable probes,148 paving the way for more advanced fluorometric applications, including enzyme-linked immunosorbent assays (ELISAs),149 diagnostic tests,150 and viability assays.151 Advancements in the design of fluorogenic probes are constantly stimulating the progress of complementary methodologies and their practical applications in the field of chemistry.
The two main strategies employed for fluorogenic probe design and application are; the latent properties of the fluorophore itself, and the method utilised to hide its fluorescence. Some crucial properties of the parent fluorophore may include; excitation and emission wavelength(s), resistance to photobleaching, quantum yield, and effects of pH on fluorescence output. Recently, researchers have begun to focus their attention on generating fluorogenic probes that do not overlap with auto-fluorescence produced by endogenous species, and result in minimal phototoxicity. Probes that fluoresce in the near-infrared and far-red region have an emission wavelength far above (nm) the auto-fluorescence window.152,153 It is also worth noting that optimising the spectroscopic properties and brightness (product of quantum yield and extinction coefficient) is quickly becoming an essential part of the design strategy for the synthesis of fluorogenic probes.
D. Tang and coworkers have elegantly exploited the use of fluorogenic probes as a method for detecting carcinoembryonic antigen (CEA) using a bio-responsive release system (Fig. 17).
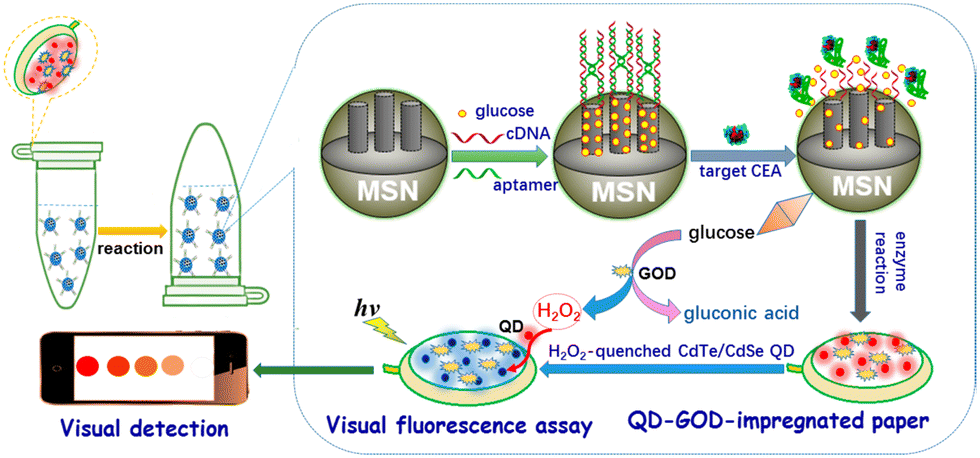 | ||
| Fig. 17 Graphical illustration of the PAD for the visual fluorescence detection of CEA (reprinted from ref. 154 with permission from American Chemical Society, copyright 2017).154 | ||
The group engineered an all-in-one paper-based analytical device (PAD) system that combined DNA-gated mesoporous silica nanocontainers with CdTe/CdSe quantum dots and enzymes on paper.154 Glucose-loaded mesoporous silica nanocontainers with a CEA aptamer and CdTe/CdSe quantum dot-enzyme paper were utilised in a centrifuge tube for this assay, with the fluorescence of the quantum dot-enzyme paper becoming quenched at higher CEA concentrations. This produced a colour change from red to colourless, which facilitated the qualitative evaluation of CEA levels visually, and quantitative determination via a fluorimeter.
A similar study by the same group investigated the NH3-triggered structural change of NH2-MIL-125(Ti) impregnated on paper, for the detection of CEA.155 Gold nanoparticles were functionalised with glutamate dehydrogenase, and secondary antibodies were used to generate wet NH3 in a sandwiched immunoassay format. The PAD coated with NH2-MIL-125(Ti) showed good visible fluorescence intensity through wet NH3-triggered structural change with high accuracy and reproducibility, indicating the device's potential for protein diagnostics and biosecurity.
The inherent properties of a fluorogenic probe are indeed important, however, the strategy used to mask its fluorescence will likely impact its enzymatic target and determine its effectiveness. An efficient fluorescence masking-strategy would see the probe generate a response selectively for the target enzyme, be chemically inert until within sufficient proximity, and terminate absorption and fluorescence output at the initial excitation wavelength (upon activation).
Fluorescence quenching techniques based on Photoinduced Electron Transfer (PET) and Förster Resonance Energy Transfer (FRET), and to a lesser extent – Ratiometric Imaging, have been very popular in recent years (Table 1).159 Indeed, one interesting example involved a novel immunoassay strategy for detecting cancer biomarkers – focusing on alpha-fetoprotein (AFP) as a model analyte.160 This method focused on the enzyme-controlled formation of fluorescent polydopamine (PDA) nanoparticles via the dissolution of manganese dioxide (MnO2) nanoflakes. The in situ synthesis of PDA nanoparticles possessed high specificity and a low detection limit, indicating the potential of this “off → on” fluorescence strategy as a tool for early cancer diagnosis.
| Fluorescence quenching technique | Description |
|---|---|
| Förster Resonance Energy Transfer (FRET) | A non-radiative energy transfer between a donor fluorophore and an acceptor molecule |
| Photoinduced Electron Transfer (PET) | An electron is transferred from the excited state of the fluorophore to a nearby electron acceptor (quencher) |
| Internal-charge Transfer (ICT) | Involves the transfer of an electron or charge within a molecule |
| Through-bond Energy Transfer (TBET) | Involves the transfer of energy between two fluorophores linked by a series of covalent bonds |
| Aggregation-induced Emission (AIE) | Fluorescence enhancement upon aggregation – often associated with restricted intramolecular motions within the luminogenic molecule |
In contrast to this, an “on → off” fluorescence quenching strategy has been employed for the detection of prostate-specific antigen (PSA), without interference from autofluorescence.161 This study detailed the preparation of Mn2+- & Pr3+-doped Zn2GeO4 nanorods with persistent luminescence properties and their application as pH-responsive fluorogenic probes. The fluorescence intensity (533 nm) was observed to decrease with increasing concentrations of PSA, which was verified by control experiments, optimization of experimental conditions, and analytical performance evaluation with PSA standard samples. Overall these studies highlight the potential of fluorescence quenching techniques to produce novel and innovative nanomaterial sensing systems.
To further enhance these quenching techniques, self-immolative linkers have been employed to enhance probe stability and performance.162 Dal Corso and Gennari described self-immolative linkers as “covalent constructs designed to degrade spontaneously in response to a specific stimuli”.163 The two most prevalent self-immolative linker motifs in literature (in regards to enzymatic probes) are elimination (electron cascade) and acyl-transfer (Fig. 18). In this particular context, the linker is being used to modify the fluorescence profile as a means for detecting the presence of a hydrolytic enzyme. It is worth highlighting that self-immolative linkers have demonstrated their utility in both peptide/small molecule and macromolecular drug-delivery and sensor systems. After additional refinement, these systems may have the potential to evolve into commercially feasible solutions.
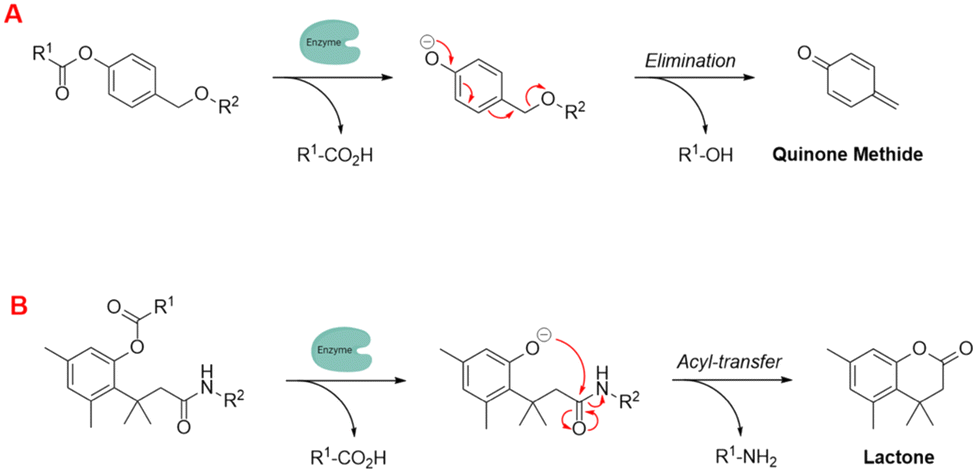 | ||
| Fig. 18 Examples of self-immolative linker strategies – mechanism(s) of activation, (A) elimination and (B) acyl-transfer (adapted from ref. 164 with permission from American Chemical Society, copyright 2018).164 | ||
In the next few sections, we will focus our attention on peptide-based fluorescent probes and build on some of the ideas that were discussed above.
|
|
|||
|---|---|---|---|
| λ ex/λem (nm) | 220/360 | 225/304 | 220/285 |
| Absorptivity (ε) (L mol−1 cm−1) | 5600 | 1400 | 200 |
| Lifetime (τ) (ns) | 3.1 | 3.6 | 6.4 |
| Quantum yield (ΦF) | 0.2 | 0.14 | 0.04 |
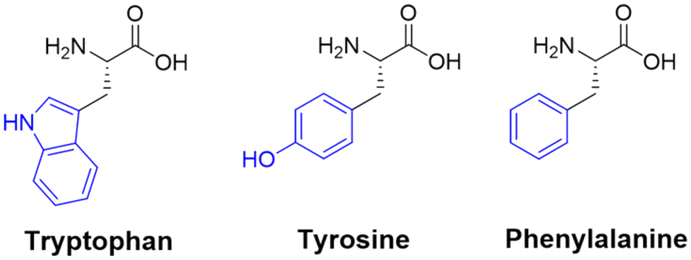
One popular use of these aromatic residues is in the study of self-assembling peptides – employing the endogenous fluorescence as a marker. The fluorescence data may be used to probe the conditions necessary for the covalent synthesis of the peptide itself,167 or to investigate the compound's effect on the micro-environment – by creating self-assembling peptides with fluorescence output that can be altered under different conditions.170 In the context of peptide conformational changes, the accumulation and internal clustering of amino acids within peptides can induce structural modifications, and may potentially influence the fluorescent characteristics of the peptides.
An interesting facet regarding the Trp residue is that the native peptide-bond can act as a weak intermolecular quencher for the fluorescent indole moiety, which is further enhanced (quenching ability) in cyclic peptides.171 In the case of Tyr fluorescent peptides, the tyrosine kinase protein plays a significant role in influencing peptide fluorescence, as it can induce phosphorylation of Tyr residues within peptides, resulting in the suppression of intrinsic fluorescence.172 Fluorescent peptides containing the Phe residue can be used for monitoring the in vitro interactions with circulating-tumour DNA.173 This interaction can cause the fluorescence output of the Phe residue(s) to become quenched, and may provide a novel method for DNA detection.
Despite significant advancements in the development of probes utilising canonical aromatic amino acids to generate fluorescence data, the covalent combination of fluorescent moieties offers an alternative and effective approach for producing peptide structures with tuneable and enhanced optical properties. The synthesis of fluorescent peptides typically involves coupling the fluorophore to a reactive site within the peptide (side-chains, N-/C-termini or incorporated spacer), with initial coupling experiments targeting carboxylic acids, amides and thiols176 – and more recently phenols177 and imidazoles.178 However, the covalent coupling of a fluorophore to a peptide can potentially disrupt and alter the properties of the peptide. Consequently, selecting an appropriate fluorophore is of utmost importance, as different fluorophores exhibit distinct chemical characteristics.
Many organic fluorophores available today possess a wide range of physico-chemical properties, which can impact biologically-active peptides (when coupled together). Antimicrobial peptides (AMPs) are compounds generated by various organisms, including fungi, protozoa, bacteria, plants, and animals as a natural defence mechanism against common human pathogens.179 Functionalising these peptides with fluorophores has become a useful way for researchers to investigate their mechanism of action and design imaging probes for the swift detection of microorganisms at sites of infection.180 AMPs have been modified to contain various synthetic handles (carboxylic acid, sulfonyl chloride, alkyne etc.…) to facilitate facile coupling with fluorophores without hindering their main identification characteristics.181,182 A study by the Vendrell group sought to uncover the optimal fluorophore to label AMPs.183 They found that certain larger fluorophores – like Nile blue and Rhodamine B – and smaller fluorophores – naphthalimide and dansyl – had little effect on their antifungal activity (Fig. 19).
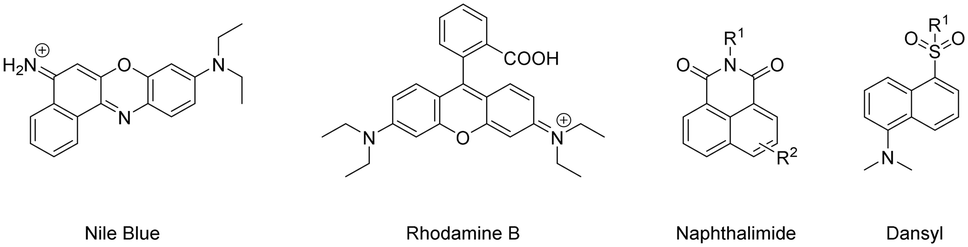 | ||
| Fig. 19 Example fluorophores suitable for coupling to AMPs (counter ions omitted for clarity). | ||
Cell-penetrating peptides (CPPs) serve as efficient delivery carriers, and the investigation of their uptake and transportation mechanisms often involves the use of conjugates labelled with fluorophores.184 Fluorophores tend to be (relatively) planar lipophilic molecules, with a rigid and bulky structure containing a (sometimes extensive) aromatic conjugated double-bond system. Many characteristics of CPPs (cellular uptake, membrane affinity, intracellular transport, and cytotoxicity) can be affected by the inherent properties of the fluorophore.185 However, Birch demonstrated that CPPs coupled to fluorophores possess enhanced membrane association and display a more defined intracellular localisation pattern.186 When choosing a fluorophore for conjugation with biologically active peptides found in organisms, it is essential to consider its potential toxicity and any adverse side-effects, as well as the fluorescence intensity it exhibits during its application.
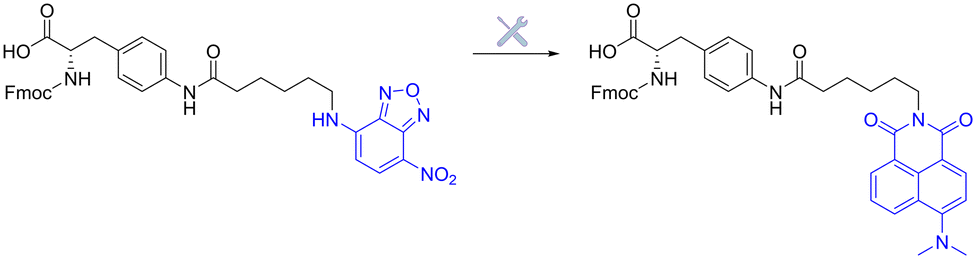 | ||
| Fig. 20 Initial benzoxadiazole- vs. improved naphthalimide-based fluorogenic amino-acid. | ||
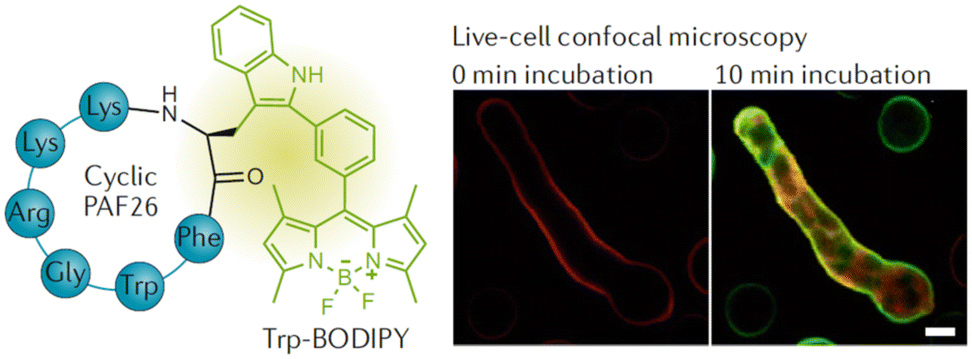 | ||
| Fig. 21 Chemical structure of the Trp-BODIPY-labelled cyclic peptide and live-cell confocal imaging of Aspergillus fumigatus over time (reprinted from ref. 180 with permission from Springer Nature, copyright 2016).180 | ||
Optical imaging studies can also be conducted to obtain substance-related read-outs from cells. For example, fluorescent peptide probes containing coumarin-modified amino acid residues have been designed to report the endogenous phosphatase activity of protein tyrosine phosphatases in live cells.193 The wash-free imaging capabilities of fluorescent peptide probes makes them valuable tools for applications where samples need to be analysed quickly with minimal processing steps, as seen in metabolic engineering and clinical diagnostics.
Lanthanide-based peptide probes
Luminescent lanthanide Ln3+ complexes – particularly those of Eu3+ and Tb3+ – have garnered significant attention in recent years owing to their unique photophysical properties and potential applications in fluoroimmunology, NIR-spectroscopy, and lighting devices.194,195 This class of compounds exhibits narrow emission bands, long luminescence lifetimes, and resistance to photobleaching, making them promising candidates for use in biological imaging, sensing, and opto-electronic devices.196 The prolonged luminescence lifetimes of emissive Ln3+ complexes facilitates the use of time-gated detection techniques (Fig. 22) to remove the inherent autofluorescence present biological fluorophores, thus enhancing the signal-to-noise ratio of the luminescence output.197 Additionally, their ability to tune emission colours, and their high quantum yields make them an attractive alternative to traditional organic fluorophores.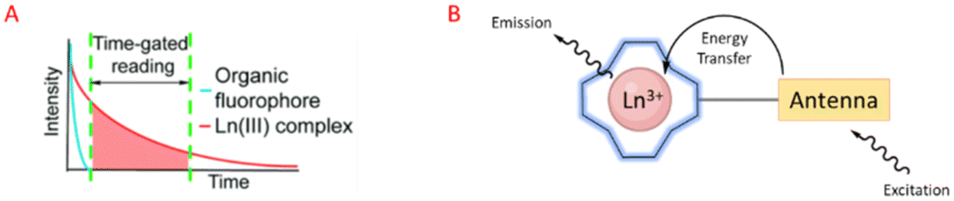 | ||
| Fig. 22 Prolonged luminescence lifetime of the Ln3+ complex, in contrast to traditional organic fluorophores (A) (reprinted from ref. 198 with permission from Royal Society of Chemistry, copyright 2018).198 Cartoon schematic of antenna excitation (B). | ||
Multiple factors must be taken into account when designing a luminescent lanthanide complex. First and foremost, it's important to note that directly exciting Ln3+ ions is highly inefficient, primarily due to the Laporte forbidden character of f–f transitions. Ln3+ ions in aqueous environments usually do not luminesce due to the efficient non-radiative decay process provided by the surrounding H2O molecules. Nevertheless, the luminescent characteristics of lanthanides can be enhanced by chelating the ions with appropriately designed ligands that exhibit strong light-absorbing properties – antennae.199 Ln3+ ions can therefore be shielded from the solvent environment, and the electronic energy in the form of light absorption from the ligands can be transferred to the metal ion. A cartoon schematic describing the photophysical pathway for Ln3+ luminescence is outlined in Fig. 22. This process involves an energy transfer from the excited state of an (appropriately) absorbing antenna to the Ln3+ excited state, which results in metal-centred luminescence.200 The choice of the sensitising group can be especially valuable when creating responsive lanthanide probes, as alterations to the antenna can influence its absorption or energy transfer properties.
The next thing to consider is, Ln3+ ions generally maintain coordination numbers between 8–10 in aqueous environments,201 so the synthesized ligand must be able to facilitate enough hard donors to complete this coordination sphere. There exists many kinetically stable Ln3+ complexes containing heptadentate or octadentate ligands, featuring; the carboxylic acid functionalised cyclen, EDTA, DTPA, and 9N3 macrocycles.202,203 If the Ln3+ coordination sphere is not complete, H2O molecules will coordinate to the remaining site(s). In this case, there may be a luminescence quenching event due to vibrational energy transfer to O–H oscillators.204 This quenching effect of coordinated H2O molecules can be used for the design of responsive Ln3+ complexes, where upon displacement of the coordinated water can lead to an increase in luminescence intensity.194
As we have seen before, amino acids/peptides possess diverse chemical functionalities, and can readily undergo functional-group interchange, with the introduction of groups such as aromatic rings, alkyl chains and heteroatoms. This diversity is valuable for creating peptides with specific chemical, electronic, or steric characteristics. The Gunnlaugsson group elegantly exploited this idea with their recent publication detailing the synthesis of chiral α-amino acid derived ligands for Ln3+ luminescence.205 These novel ligands contained L-phenylalanine and L-tryptophan – as their methyl esters – which underwent supramolecular self-assembly to form 1:3 complexes with Tb3+ under thermodynamic control. Indeed, both complexes were able to evoke Tb3+-centred emission (λex = 255 nm). Fig. 23 highlights the Tb3+-centred transitions of complex [Tb(Phe)3]3+ at λmax = 490, 545, 584, 622, 648 and 645 nm, upon deactivation of the 5D4 state to the 7Fx (x = 6–2) states, respectively.
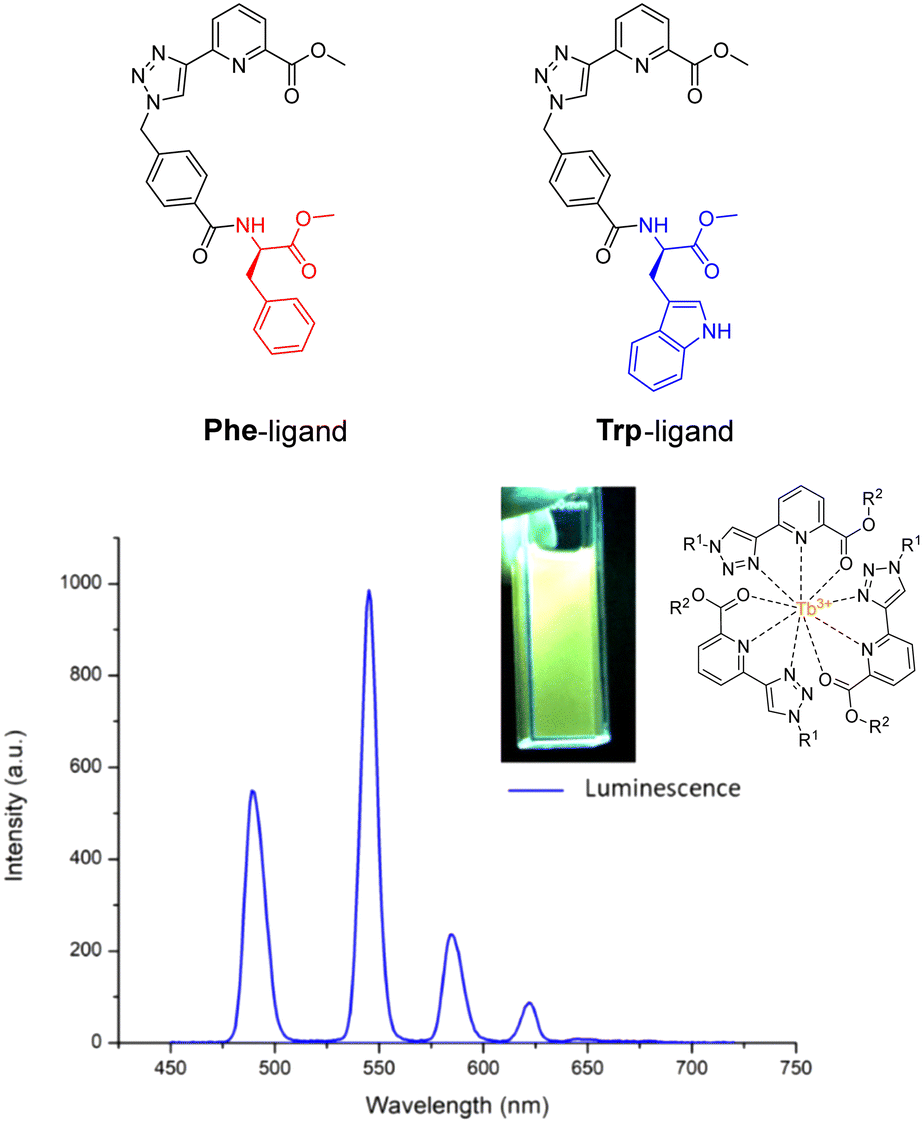 | ||
| Fig. 23 Structures of ligands Phe & Trp, and the delayed luminescence spectra of [Tb(Phe)3]3+ recorded in MeCN. Inset: Picture of complex solution under illumination via UV lamp (adapted from ref. 205 with permission from Royal Society of Chemistry, copyright 2023).205 | ||
Although the Tb3+ coordination sphere was populated by the (1,2,3-triazol-4-yl)-picolinamide motif and did not involve the C-terminal amino acid(s), this functionality may act as a site for further conjugation chemistry. For example, in the production of soft-materials, where the acid-moiety could be used to extend the overall supramolecular assembly.
An early example of a lanthanide-based peptide probe employed a simple DTPA ligand coupled to dipeptides Ser-Trp and phospho-Ser-Trp – utilising the Trp residue as an antenna. Tremblay and coworkers discovered that Tb3+ luminescence was enhanced by the non-phosphorylated (Ser-Trp) ligand, in contrast to the phosphorylated ligand.208 This phenomenon was ascribed to a transition from a monomeric state to a dimeric state, which occurs in the phosphorylated and non-phosphorylated peptides, respectively. This change in luminescence output was employed to track the enzymatic dephosphorylation of the phosphorylated peptide using alkaline phosphatase. Tremblay developed this idea further by using a non-canonical quinolone antenna, and inserting an isoleucine residue between this new antenna and the phosphor-Tyr amino acid. This novel peptide-probe facilitated Eu3+ and Tb3+ luminescence upon phosphorylation (Fig. 24), and was applied to observe the conversion between non-phosphorylated and phosphorylated peptides using tyrosine kinase or tyrosine phosphatase.209
 | ||
| Fig. 24 Lanthanide-based peptide probes to monitor enzyme activity. (A) Observing tyrosine phosphatase and kinase activity using a 5-residue peptide (adapted from ref. 209 with permission from American Chemical Society, copyright 2008).209 (B) Asp-functionalised Eu3+ complex within peptide sequence coordinates to phospho-Ser residue (adapted from ref. 210 with permission from Royal Society of Chemistry, copyright 2012).210 | ||
A lanthanide-based peptide probe containing a DO3A-propylamino ligand was coupled to an aspartic acid residue (resin bound), using typical Fmoc SPPS.210 Interaction from a nearby phospho-Ser residue induced competitive displacement of the β-diketonate antenna from the Eu3+ coordination sphere, causing a subsequent decrease in Eu3+ luminescence output (Fig. 24). This “on–off” change in luminescence was utilised to monitor PKCα-catalysed phosphorylation of the peptide, with the aim of generating Michaelis–Menten kinetic data for the enzyme. On the other hand, the dephosphorylation pathway could be monitored by observing the enhancement in Eu3+ luminescence intensity upon preferential coordination to the β-diketonate.
The Arg-Gly-Asp (RGD) tripeptide motif displays a high affinity for the αvβ3 integrin – which is often associated with tumour metastasis.211 Out of numerous RGD-containing structures, the c(RGDfV) peptide – originally synthesized by Kessler and colleagues in 1992 (ref. 212) – stands out as a remarkably potent and selective αvβ3 antagonist.213 This cyclic-pentapeptide sequence has since become a well-established vehicle for targeted drug-delivery and cancer imaging.214,215 Indeed, Gillaizeau and coworkers utilised it as a benchmark to showcase the efficacy and versatility of their design strategy.216
The group employed this cRGD-peptide – as a targeting agent – in a methodological attempt to generate NIR-imaging probes, by combining a DO3A-based ligand and an azobenzene-antenna to promote lanthanide luminescence (Fig. 25).
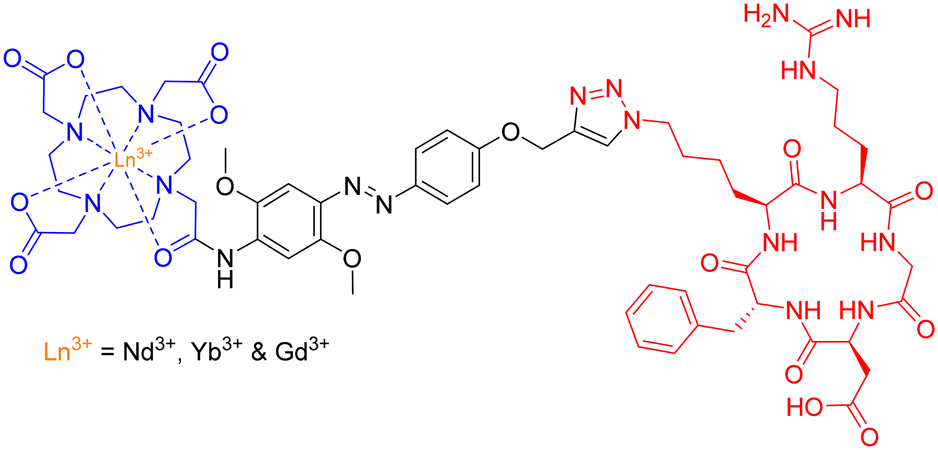 | ||
| Fig. 25 Structure of Ln3+-based peptide probe for potential NIR cancer-imaging. | ||
Photophysical evaluation of this design strategy demonstrated the sensitisation of both Nd3+ and Yb3+ ions in aqueous environments with moderate NIR-emitting efficiency. The presence of a coordinated H2O molecule (q = 0.9 and 1.0, respectively) – providing a non-radiative decay pathway – is partially to blame for the observed modest NIR quantum yields. Nevertheless, this synthetic scaffold may possess potential application in the conjugation of Ln3+-based luminescent probes to various biomolecules.
Labelling amino acids and peptides enables the real-time monitoring of endogenous biological reactions and allows us to use low reagent concentrations. The practice of labelling peptides with conventional organic fluorophores is widespread in many biological assays, and is readily being expanded to harness the numerous benefits offered by luminescent lanthanide complexes. It is essential to exercise caution when choosing the coupling site and size of the lanthanide complex – to minimise any disruption to the enzymatic reaction. However, considering the extensive use of fluorescently labelled peptides, this is unlikely to pose a significant obstacle to the adoption of this technology. Anticipated progress in the development of lanthanide-based enzyme assays is on the horizon, offering potential attractive alternatives to current commercial assay formats, which may aid in the drug discovery process.
Peptide-probes with therapeutic and sensing capabilities
Peptide probes with dual therapeutic and sensing capabilities have the potential to address a critical need at the intersection of medicine and diagnostics. In the area of therapeutics, these probes offer a targeted approach for treating diseases. Simultaneously, the sensing capabilities of these peptide probes are invaluable for diagnostics and monitoring. By incorporating luminescent or other sensing properties, these probes can detect specific biomarkers indicative of diseases or physiological changes. This dual-functionality allows for real-time monitoring of treatment efficacy, enabling adjustments to therapeutic strategies based on the observed molecular responses.In the context of personalised medicine, the combination of therapeutic and sensing capabilities in peptide probes provides a tailored and adaptable approach, allowing clinicians to optimise treatment-plans based on individual patient responses.217 Overall, the integration of therapeutic and sensing capabilities in peptide probes addresses the growing demand for precision medicine, offering more effective and personalized treatments while advancing our ability to monitor and understand complex biological processes.
The group of J. S. Kim and colleagues have reported two fluorescent naphthalimide-based peptide theragnostic probes Naph-1 and Naph-2, for the treatment of cancer (Fig. 26).218,219
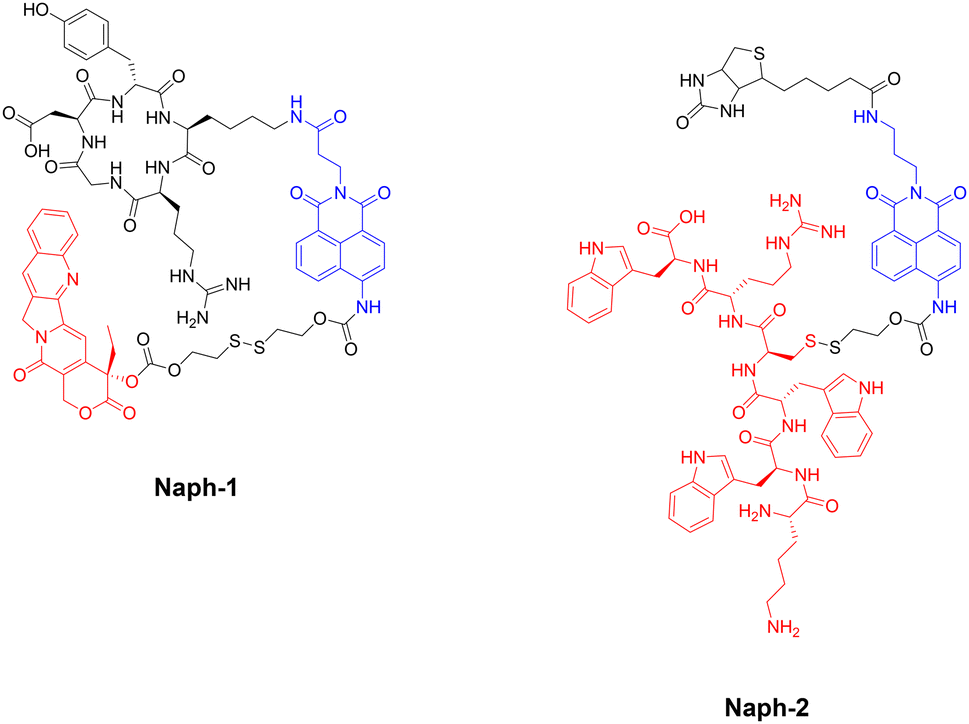 | ||
| Fig. 26 Structures of fluorescent naphthalimide-based peptide theragnostic probes. Note: Sensing moiety highlighted in blue, and therapeutic agent in red. | ||
Naph-1 utilised an RGD peptide sequence as its tumour-targeting motif, and employed camptothecin as its therapeutic agent – an anti-tumour inhibitor of topoisomerase I. This was also composed of a disulfide-linker coupled to a naphthalimide fluorophore, that produced a bathochromic fluorescence shift upon cleavage via glutathione or thioredoxin (species overexpressed in cancer cells). Indeed, the results indicated that Naph-1 was selectively up-taken by U87 tumour cells via a αvβ3 integrin-mediated mechanism, evidenced by competitive okadaic acid treatment. An “off–on” fluorescence change was observed (λem = 535 nm) upon reaction with glutathione, which released the camptothecin warhead into the nucleus of the cell. This could serve as an excellent theragnostic delivery platform, offering precise tumour targeting while allowing the monitoring of free drug concentration through changes in fluorescence signalling.
The group then employed a similar design strategy for the generation of Naph-2, which consisted of biotin (as the cancer targeting motif), a fluorescent naphthalimide reporter, and a Holliday Junction inhibitor peptide (KWWCRW) as the therapeutic. Like before, fragmentation occurred via disulfide-bond cleavage leading to the release of the inhibitor peptide, with the fluorescent signal being monitored in real-time. Although Kim et al. expertly showcase the application of the naphthalimide fluorophore in generating fluorogenic theragnostic prodrugs (Naph-1 & Naph-2), further research is needed to evaluate the translational potential of these compounds for clinical use, including pharmacokinetics, toxicity profiles, and efficacy in animal models.
In regard to lanthanide luminescence, Chau et al. synthesized DO3A-based Eu3+ and Yb3+ complexes, with appendaged P19 peptides (Pra-KAhx-K-LDLALK-FWLY-K-IVMSDKW-K-RrRK) designed to selectively bind to the latent-membrane protein I (LMP1) for visible and near-IR imaging and cancer monitoring (Epstein–Barr virus).220
Due to the pivotal role of the FWLY sequence in regulating the signalling pathway, peptide P19 was synthesized to emulate the essential amino acid residues found in the transmembrane region of LMP1. Propargyl-glycine (Pra) was introduced into the peptide-backbone to facilitate conjugation between P19 and the Ln3+ complex via click chemistry.
The subcellular localisation of LMP1 expression was then investigated by immunoluminescence imaging (Fig. 27). Overlapping of the LMP1 (green) signal with the EuP19 (red) signal was observed in LCL3 and AFB1 cells after 12 hours, with EuP19 colocalising with LMP1 in the LCL3 cell-membrane – highlighted by the overlayed yellow signals. Similar images were recorded for EuP19 in C666-1 cells, but with dramatically decreased luminescence intensity. No luminescent signal was observed for both EuP19 and LMP1 in LMP1-negative HeLa cells, suggesting that EuP19 is selective for cells that express LMP1. Although the emission quantum yield of EuP19 was 8.3 (±0.8%), YbP19 was relatively low at 0.05 (±0.005%), indicating room for improvement in terms of enhancing the efficiency of the NIR emission. Be that as it may, this study discloses a novel method for live-visualisation of LMP1 in Epstein–Barr virus tumour cells, with subsequent selective cytotoxicity for LMP1-positive cells by suppressing the NF-KB pathway.
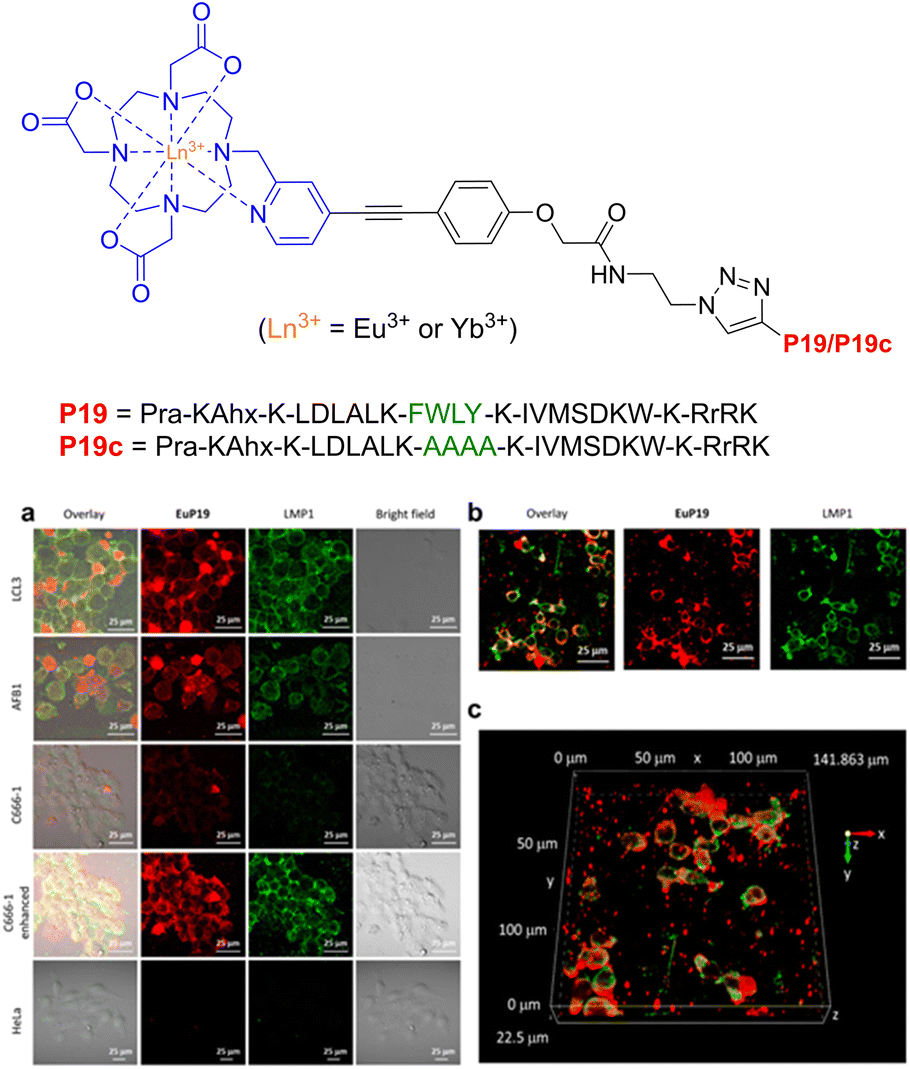 | ||
| Fig. 27 Structure of lanthanide-based peptide probe, containing Eu3+ and Yb3+. (a) Immunoluminescence imaging of the LMP1 protein and EuP19 in LCL3, AFB1, C666-1, and HeLa cells after 12 h of incubation. (b) Immunoluminescence images of LMP1 and EuP19 in LCL3 in the xy plane. (c) Z-Stacks of 2D images of LMP1 and EuP19 in LCL3 cells (adapted from ref. 220 with permission from American Chemical Society, copyright 2021).220 | ||
This theragnostic approach may have clinical importance as tumours expressing LMP1 are recognised for their heightened aggressiveness compared to LMP1-negative tumours, making them more susceptible to lymph node metastasis.221 Additionally, LMP1-positive tumours are linked to poorer overall survival, with LMP1 serving as a robust risk factor for unfavourable prognosis in patients with nasopharyngeal cancer.222
Other fluorescent/luminescent anti-cancer peptide probes have been reported for targeting discrete proteins.223,224 However, these larger supramolecular structures will be discussed in more detail in the next section. Thus, we limit the current selection to small-molecule theragnostic peptide probes.
Peptide-modulated self-assembly of luminescent structures
The L-Phe-L-Phe (FF) dipeptide motif represents the most basic sequence displaying self-assembling traits. The self-assembly of FF draws inspiration from the development of amyloid plaques formed by polypeptides – containing FF – during the progression of Alzheimer's disease.225 The FF dipeptide has proven to be a versatile building-block for the supramolecular construction of well-organized nanostructures.226,227 FF self-assemblies can also evoke different morphologies,228 induce controlled patterning,229 and facilitate dimensional control over assembled nano-structures.230 In contrast to other self-assembling peptides, aromatic dipeptides offer the advantages of synthetic versatility, small molecular structure(s), and reduced experimental costs. These attributes are highly valuable for both elucidating inherent self-assembly mechanisms and tailoring properties for specific applications. Indeed, Q. Zou231 and K. Tao232 (amongst others) have produced fantastic review articles documenting many of these applications, and have provided in-depth explanations for this ubiquitous self-assembly phenomenon. We highly recommend reading their work for a more detailed account of peptide-based supramolecular chemistry. With that said, this section will solely focus on select examples of peptide-modulated self-assembly of luminescent supramolecular structures.Self-assembled peptide nanostructures have been used in the construction of biological and biomedical applications, leveraging their unique attributes such as biocompatibility and tuneable self-assembly.233 In drug delivery, these nanostructures serve as targeted carriers, enabling controlled release with minimal side-effects. They play a vital role in imaging and diagnosis, acting as contrast agents and biosensors for various modalities.234
The Zhang group demonstrated the self-assembly of L-Trp-L-Phe nanoparticles that shifted the native peptide's fluorescent output from the UV- to the visible-region.235 The essence of their design strategy was inspired by two natural phenomena; the molecular mechanism responsible for the red-shift in the yellow fluorescent protein (YFP) and the enhanced fluorescence intensity observed in the green fluorescent mutant protein BFPms1. The bathochromic shift in YFP is a direct result of π–π stacking,236 with the fluorescent enhancement of BFPms1 arising from the structural rigidification by Zn(II).237 Following the reaction of the (L-Trp-L-Phe) dipeptides with ZnCl2 (dissolved in a mixture of methanol and aq. sodium hydroxide), atomic force microscopy (AFM) and transmission electron microscopy (TEM) revealed the formation of uniform spherical nanoparticles (Fig. 28).
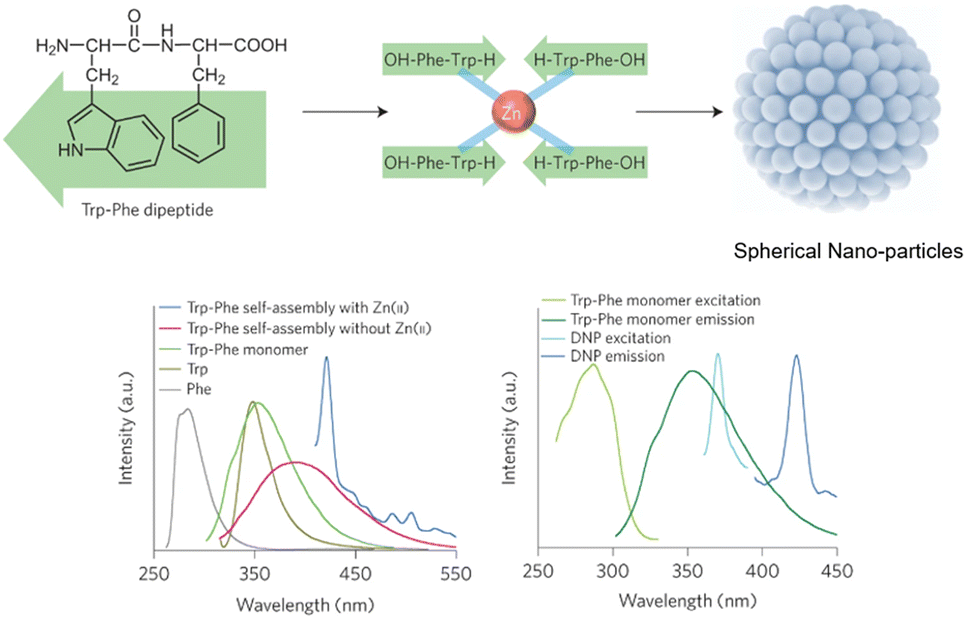 | ||
| Fig. 28 Design strategy for fluorescent nanoparticles viaL-Trp-L-Phe self-assembly. (Bottom left) Various fluorescent emission spectra. (Bottom right) Fluorescent excitation and emission spectra of dipeptide monomers vs. self-assembled nanoparticles.235 | ||
The photophysical properties of the nanoparticles were then investigated. The Zn2+-chelated (L-Trp-L-Phe) nanoparticles possessed a fluorescence emission maxima at 423 nm, which was red-shifted 33 nm compared to the dipeptide assemblies without Zn2+ coordination (390 nm). Fluorescence microscopy images of the Zn-bound nanoparticles were recorded, with bright-blue fluorescence being observed. The group then went on to compare the photostability and biocompatibility of the novel peptide nanoparticles against a fluorescent rhodamine dye (Rh6G) and CdSe quantum dots in NIH-3T3 cells, respectively. After continuous irradiation, the nanoparticles remained stable, indicating enhanced photostability compared to the Rh6G dye. The MTT assay suggested that the nanoparticles were far more biocompatible than the CdSe quantum dots.
A theragnostic approach utilising these L-Trp-L-Phe nanoparticles was then explored for the potential visualisation of drug-release in real-time. Doxorubicin (DOX) has been shown to adsorb onto nanoparticles with appropriate functionalities,238 with π–π stacking and N–H–π electrostatic forces dominating this attractive interaction.239 The interaction between DOX and the nanoparticles was characterised by their fluorescent and absorbance spectra, with the authors claiming that the reduced absorbance at 480 nm and fluorescent quenching at 595 nm pertained to the electrostatic interactions between the nanoparticles and DOX. This was further investigated with fluorescent imaging in A549 cells. Minimal red fluorescence was observed from the cells incubated with DOX/nanoparticle-conjugates, compared to cells treated with only DOX. This indicated that fluorescent-quenching was likely due to contact with the L-Trp-L-Phe nanoparticles (Fig. 29).
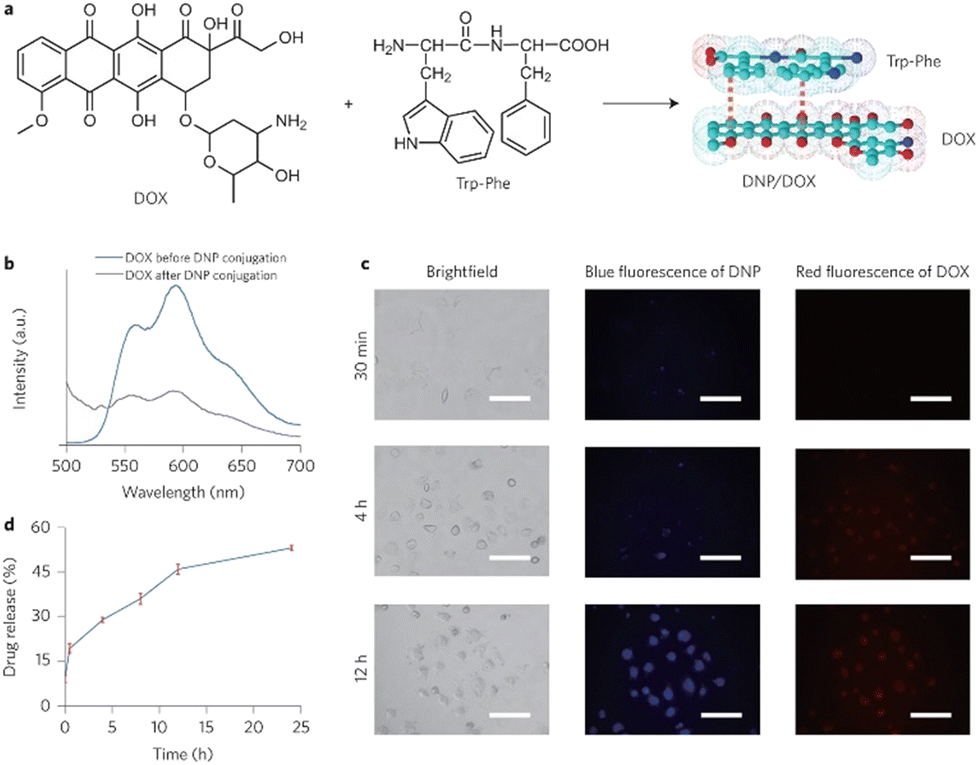 | ||
| Fig. 29 (a) DOX coordinated to the L-Trp-L-Phe nanoparticles via π–π stacking. (b) Fluorescence emission spectra of DOX before and after nanoparticle-coordination. (c) Fluorescence imaging of the DOX/nanoparticle-conjugates in A549 cells over time. (d) Time-dependent drug-release of DOX from the DOX/nanoparticle-conjugates.235 | ||
After an extended period of incubation (12 hours), more DOX was gradually released, leading to an increase in fluorescence signal for both free DOX and nanoparticle. These results indicate that this DOX/Trp-Phe nanoparticle-conjugate design strategy may have promising potential to act as a peptide–drug nanostructure for the visualisation of DOX release in real-time. In comparison to organic fluorophores – which are usually perturbed by photo-bleaching and broad emission bands – and quantum dots (solubility and biocompatibility concerns),240 Zhang and coworkers have demonstrated that their (L-Trp-L-Phe) nanoparticles are biocompatible, photostable and possess visible fluorescent characteristics.
Another interesting example of self-assembled peptide supramolecular structures comes from the group of Xiao and colleagues, in which they report the synthesis of luminescent lanthanide–collagen peptide-hybrid nanofibers, for pH-controlled drug delivery.241 This strategy takes inspiration from the biomimetic-scaffold – which has been often employed in tissue-engineering and regenerative medicine.242,243
The synthesized collagen peptides all contained a Gly-Pro-Hyp (GPO) repeating unit, which is understood to be one of the most-stabilising sequences for triple-helix structures.244 Aspartic acid residues were then functionalised at each peptide-terminus to form the coordination sphere around the Ln3+ ions and between the collagen-mimetic peptides. The initial synthesized peptide (DDColDD) contained an Asp-Asp dipeptide at each terminus for Ln3+ coordination. The design strategy behind the next synthetic peptide (DWColWD) was two-fold; replacement of D-residues with W-residues to probe how the composition of amino acids at each terminus affects the self-assembly, and W-residues can be used an antenna for Ln3+ luminescence.245 A third and final synthetic peptide was constructed (HWColWD) by replacing the N-terminal D-residue with a H-residue as an additional coordination-site for the Ln3+ ions (Fig. 30).
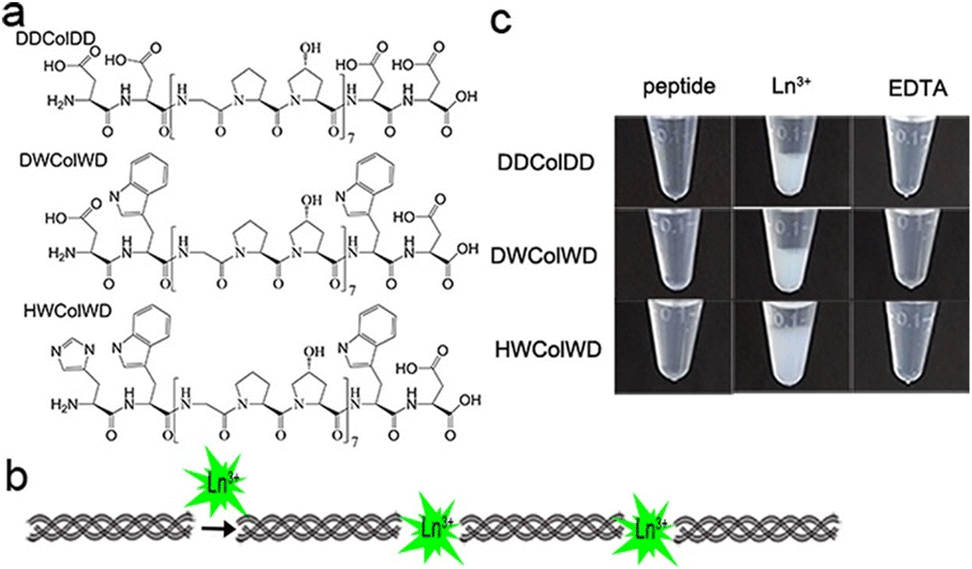 | ||
| Fig. 30 Ln3+-triggered assembly of collagen-mimetic peptides. (a) Amino-acid sequences of three collagen-mimetic peptides. (b) Schematic illustration of the self-assembly reaction. (c) Changes of the peptide-solution with, and without Ln3+ ions/EDTA (reprinted from ref. 241 with permission from American Chemical Society, copyright 2019).241 | ||
Various Ln3+ ions (Tb3+, Ce3+, Eu3+, La3+, Tm3+, Er3+ and Yb3+) were mixed with the DDColDD, DWColWD and HWColWD peptides to investigate their self-assembly. Fig. 30 demonstrated that all the Ln3+ ions caused the peptide solutions to become turbid, suggesting lanthanide-induced self-assembly. The reversibility of this assembly was also highlighted by the clear solution upon addition of EDTA, which effectively sequestered the Ln3+ ions.
The pH-responsive characteristics of the collagen-mimetic peptide–lanthanide structures were explored for their potential application as drug-carriers. Camptothecin, a commonly employed substance for evaluating the delivery efficiency of anticancer drugs,246 served as a model drug in this investigation (Fig. 31).
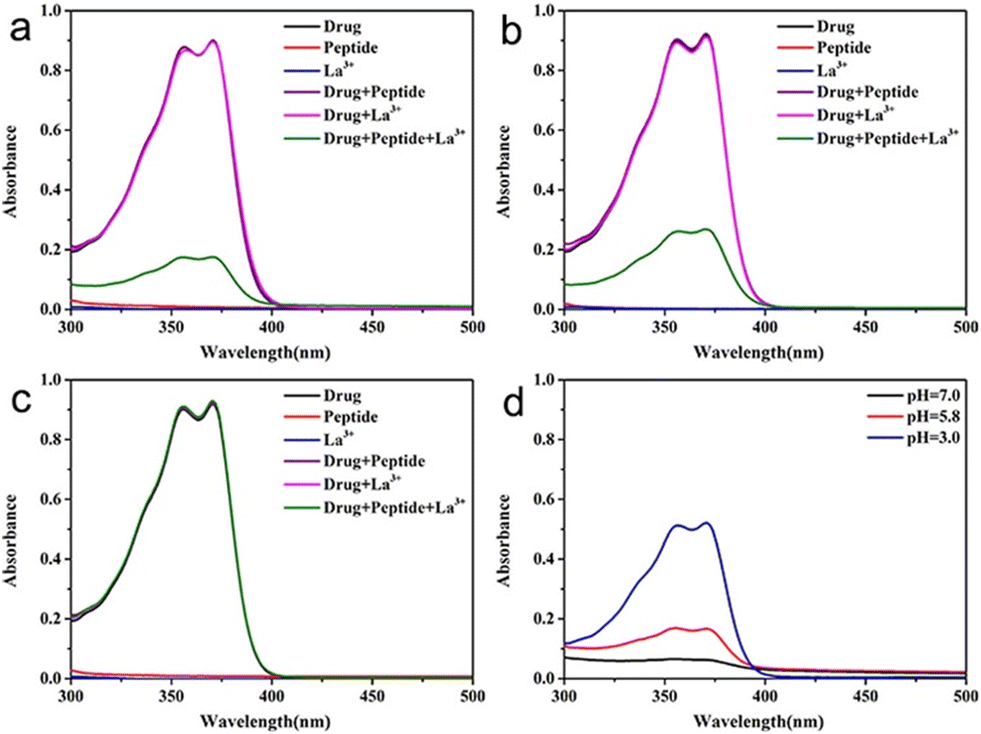 | ||
| Fig. 31 Drug-loading and release of camptothecin via DDColDD–La3+. Absorbance spectra at (a) pH = 7.0, (b) pH = 5.8, (c) pH = 3.0. Absorbance spectra of camptothecin release within the supernatant after 24 h (d) (reprinted from ref. 241 with permission from American Chemical Society, copyright 2019).241 | ||
The peptide chosen for this study was DDColDD. Mixtures of peptide DDColDD and camptothecin were generated at pH 7.0, 5.8, and 3.0. Upon the addition of La3+ ions to the mixtures at pH = 7.0 and pH = 5.8, aggregates were promptly formed, enveloping the camptothecin. The supernatant – obtained by removing the aggregates – was then collected, and the camptothecin content was determined using UV-analysis, with the highest camptothecin-loading value being observed at pH 7.0. Complementary camptothecin-release studies were then conducted for the same peptide under the same pH conditions (7.0, 5.8 & 3.0). The drug-release results were as follows: pH 3.0 > 5.8 > 7.0, with the highest amount being released at pH = 3.0 (34.3 mg g−1). Cefoperazone sodium – a popular antibiotic247,248 – was also investigated as an example drug-model. The absorbance spectra of the solution containing cefoperazone alongside the synthetic peptide or La3+ ions (or both) closely resembled the absorbance profile of the free-drug. This similarity suggested that these species would not interfere in the absorbance measurements of cefoperazone. When subject to the same pH-mediated drug-loading and release experiments, maximum loading was achieved a pH = 7.0, with the largest drug-release being observed at pH = 3.0 (111.3 mg g−1). These findings suggest that a diverse range of drugs can be effectively loaded using the collagen peptide–lanthanide scaffolds.
The self-assembly of all three collagen-peptides (DDColDD, DWColWD, and HWColWD) facilitated by Ln3+ ions has been successfully demonstrated, and found to be reversibly controlled by pH. The pH-dependent variations of these assemblies can be modulated by the specific functionality of the terminal amino acids. Employing camptothecin and cefoperazone as example drugs, the loading and releasing efficiency of the collagen peptide–lanthanide scaffolds were also examined. While the in vitro results are promising, future studies could focus on in vivo experiments to evaluate the efficacy and safety of the luminescent lanthanide–collagen peptide scaffolds for drug delivery in living systems, considering factors like tissue distribution, metabolism, and long-term effects.
There are many other interesting and noteworthy studies that have been documented regarding peptide-probes as luminescent supramolecular sensors.249–252 However, this brief perspective was not intended to be an exhaustive report, more a succinct portrayal of the extensive applications available to synthetically modified peptides.
Conclusions
The evolution of chemical methods for peptide synthesis has not only expanded our capabilities for generating complex peptide structures but has also paved the way for innovative applications in various scientific domains. The strategic chemical modification of peptides has improved their overall performance and biocompatibility, enabling their integration into diverse research and therapeutic endeavours.Moreover, the emergence of fluorescent and luminescent peptide conjugates has brought a new dimension to the field, with these conjugates serving as valuable tools for creating highly sensitive and specific sensing technologies. As peptides continue to play an indispensable role in uniting the realms of chemistry and biology, their potential for future discoveries and applications remains boundless. This brief perspective, from the therapeutic history of peptides to their luminescent application in supramolecular scaffolds, underscores their enduring significance in shaping the interdisciplinary landscape of modern chemistry and sets the stage for exciting progress in the years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding from Science Foundation Ireland (SFI), grant number 12/RC/2275/P2, which is co-funded under the European Regional Development Fund. This article is based upon discussions emanting from COST Action CA22131 Supramolecular LUminescent Chemosensors for Environmental Security (LUCES), supported by COST (European Cooperation in Science and Technology).References
- R. H. A. Plimmer, The Chemical Constitution of the Proteins Part II, Read Books, 2008 Search PubMed.
- D. A. Scott and C. H. Best, Ind. Eng. Chem., 1925, 17, 238–240 CrossRef CAS.
- M. Bliss, F. G. Banting, C. H. Best and J. B. Collip, Bull. Hist. Med., 1982, 56, 554–568 CAS.
- V. du Vigneaud, C. Ressler, J. M. Swan, C. W. Roberts and P. G. Katsoyannis, J. Am. Chem. Soc., 1954, 76, 3115–3121 CrossRef CAS.
- B. Merrifield, Science, 1986, 232, 341–347 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- D. F. Steiner, in Encyclopedia of Endocrine Diseases, ed. L. Martini, Elsevier, New York, 2004, pp. 545–555 Search PubMed.
- M. A. Gonzalez-Lozano and K. W. Li, in eLS, 2020, pp. 1–9 Search PubMed.
- A. U. Dignass and A. Sturm, Eur. J. Gastroenterol. Hepatol., 2001, 13, 763–770 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- A. Muheem, F. Shakeel, M. A. Jahangir, M. Anwar, N. Mallick, G. K. Jain, M. H. Warsi and F. J. Ahmad, Saudi Pharm. J., 2016, 24, 413–428 CrossRef PubMed.
- A. T. Zizzari, D. Pliatsika, F. M. Gall, T. Fischer and R. Riedl, Drug Discovery Today, 2021, 26, 1097–1105 CrossRef CAS PubMed.
- S. S. Usmani, G. Bedi, J. S. Samuel, S. Singh, S. Kalra, P. Kumar, A. A. Ahuja, M. Sharma, A. Gautam and G. P. S. Raghava, PLoS One, 2017, 12, e0181748 CrossRef PubMed.
- T. M. Research, Peptide Therapeutics Market Outlook 2031, https://www.transparencymarketresearch.com/peptide-therapeutics-market.html, (accessed October, 2022).
- J. N. Shah, G.-Q. Guo, A. Krishnan, M. Ramesh, N. K. Katari, M. Shahbaaz, M. H. Abdellattif, S. K. Singh and K. Dua, Therapies, 2022, 77, 319–328 CrossRef PubMed.
- J. H. Park, J. H. Moon, H. J. Kim, M. H. Kong and Y. H. Oh, Korean J. Fam. Med., 2020, 41, 365–373 CrossRef PubMed.
- K. Imai and A. Takaoka, Nat. Rev. Cancer, 2006, 6, 714–727 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- J. Davda, P. Declerck, S. Hu-Lieskovan, T. P. Hickling, I. A. Jacobs, J. Chou, S. Salek-Ardakani and E. Kraynov, J. Immunother. Cancer, 2019, 7, 105 CrossRef PubMed.
- A. J. Smith, SLAS Discovery, 2015, 20, 437–453 CrossRef CAS PubMed.
- L. Diao and B. Meibohm, Clin. Pharmacokinet., 2013, 52, 855–868 CrossRef CAS PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- J. Zhou, X. Du, N. Yamagata and B. Xu, J. Am. Chem. Soc., 2016, 138, 3813–3823 CrossRef CAS PubMed.
- T. A. Martinek, A. Hetényi, L. Fülöp, I. M. Mándity, G. K. Tóth, I. Dékány and F. Fülöp, Angew. Chem., Int. Ed., 2006, 45, 2396–2400 CrossRef CAS PubMed.
- H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1–18 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- X. Yan, J. Li and H. Möhwald, Adv. Mater., 2011, 23, 2796–2801 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- S. Chandrudu, P. Simerska and I. Toth, Molecules, 2013, 18, 4373–4388 CrossRef CAS PubMed.
- G. W. Anderson and A. C. McGregor, J. Am. Chem. Soc., 1957, 79, 6180–6183 CrossRef CAS.
- D. S. Kemp, S.-L. Leung and D. J. Kerkman, Tetrahedron Lett., 1981, 22, 181–184 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705–6710 CrossRef CAS PubMed.
- B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS PubMed.
- M. Skwarczynski and Y. Kiso, Curr. Med. Chem., 2007, 14, 2813–2823 CrossRef CAS PubMed.
- L. Di, AAPS J., 2015, 17, 134–143 CrossRef CAS PubMed.
- A. Schaller, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 2001, vol. 25, pp. 367–411 Search PubMed.
- B. S. Davidson, Chem. Rev., 1993, 93, 1771–1791 CrossRef CAS.
- N. Fusetani and S. Matsunaga, Chem. Rev., 1993, 93, 1793–1806 CrossRef CAS.
- Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441–4482 CrossRef CAS PubMed.
- U. Kazmaier and J. Deska, Curr. Org. Chem., 2008, 12, 355–385 CrossRef.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- M. Pasco, C. Dolain and G. Guichard, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, pp. 89–125 Search PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- E. Masri, Ahsanullah, M. Accorsi and J. Rademann, Org. Lett., 2020, 22, 2976–2980 CrossRef CAS PubMed.
- K. Wiśniewski, R. Galyean, H. Tariga, S. Alagarsamy, G. Croston, J. Heitzmann, A. Kohan, H. Wiśniewska, R. Laporte, P. J. M. Rivière and C. D. Schteingart, J. Med. Chem., 2011, 54, 4388–4398 CrossRef PubMed.
- A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, Chem. Sci., 2011, 2, 690–695 RSC.
- N. Brot and H. Weissbach, Trends Biochem. Sci., 1982, 7, 137–139 CrossRef CAS.
- J. B. Jones and D. W. Hysert, Can. J. Chem., 1971, 49, 3012–3019 CrossRef CAS.
- V. Frey, J. Viaud, G. Subra, N. Cauquil, J.-F. Guichou, P. Casara, G. Grassy and A. Chavanieu, Eur. J. Med. Chem., 2008, 43, 966–972 CrossRef CAS PubMed.
- C. Haskell-Luevano, K. Toth, L. Boteju, C. Job, A. M. D. L. Castrucci, M. E. Hadley and V. J. Hruby, J. Med. Chem., 1997, 40, 2740–2749 CrossRef CAS PubMed.
- H. Ban, J. Gavrilyuk and C. F. Barbas, III, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas, III, Bioconjugate Chem., 2013, 24, 520–532 CrossRef CAS PubMed.
- Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef CAS PubMed.
- M. B. Hansen, F. Hubálek, T. Skrydstrup and T. Hoeg-Jensen, Chem. – Eur. J., 2016, 22, 1572–1576 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- A. Abbas, B. Xing and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
- R. W. Cheloha, T. Watanabe, T. Dean, S. H. Gellman and T. J. Gardella, ACS Chem. Biol., 2016, 11, 2752–2762 CrossRef CAS PubMed.
- J. A. Schneider, T. W. Craven, A. C. Kasper, C. Yun, M. Haugbro, E. M. Briggs, V. Svetlov, E. Nudler, H. Knaut, R. Bonneau, M. J. Garabedian, K. Kirshenbaum and S. K. Logan, Nat. Commun., 2018, 9, 4396 CrossRef PubMed.
- X. Wei, C. Zhan, X. Chen, J. Hou, C. Xie and W. Lu, Mol. Pharmaceutics, 2014, 11, 3261–3268 CrossRef CAS PubMed.
- R. Laporte, A. Kohan, J. Heitzmann, H. Wiśniewska, J. Toy, E. La, H. Tariga, S. Alagarsamy, B. Ly, J. Dykert, S. Qi, K. Wiśniewski, R. Galyean, G. Croston, C. D. Schteingart and P. J. M. Rivière, J. Pharmacol. Exp. Ther., 2011, 337, 786 CrossRef CAS PubMed.
- P.-F. Laterre, S. M. Berry, A. Blemings, J. E. Carlsen, B. François, T. Graves, K. Jacobsen, R. J. Lewis, S. M. Opal, A. Perner, P. Pickkers, J. A. Russell, N. A. Windeløv, D. M. Yealy, P. Asfar, M. H. Bestle, G. Muller, C. Bruel, N. Brulé, J. Decruyenaere, A.-M. Dive, T. Dugernier, K. Krell, J.-Y. Lefrant, B. Megarbane, E. Mercier, J.-P. Mira, J.-P. Quenot, B. S. Rasmussen, H.-C. Thorsen-Meyer, M. Vander Laenen, M. L. Vang, P. Vignon, I. Vinatier, S. Wichmann, X. Wittebole, A. L. Kjølbye, D. C. Angus and for the SEPSIS-ACT Investigators, JAMA, J. Am. Med. Assoc., 2019, 322, 1476–1485 CrossRef CAS PubMed.
- H. G. Bossler and D. Seebach, Helv. Chim. Acta, 1994, 77, 1124–1165 CrossRef CAS.
- D. Seebach, A. K. Beck, H. G. Bossler, C. Gerber, S. Y. Ko, C. W. Murtiashaw, R. Naef, S.-I. Shoda, A. Thaler, M. Krieger and R. Wenger, Helv. Chim. Acta, 1993, 76, 1564–1590 CrossRef CAS.
- S. Datta, A. Bayer and U. Kazmaier, Org. Biomol. Chem., 2012, 10, 8268–8275 RSC.
- L. Zhao, O. Baslé and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106–4111 CrossRef CAS PubMed.
- T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Nature, 2016, 537, 214–219 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- C. Heinis, Nat. Chem. Biol., 2014, 10, 696–698 CrossRef CAS PubMed.
- D. F. Veber, R. M. Freidinger, D. S. Perlow, W. J. Paleveda, F. W. Holly, R. G. Strachan, R. F. Nutt, B. H. Arison, C. Homnick, W. C. Randall, M. S. Glitzer, R. Saperstein and R. Hirschmann, Nature, 1981, 292, 55–58 CrossRef CAS PubMed.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1982, 21, 512–523 CrossRef.
- C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS PubMed.
- M. L. J. Korsinczky, H. J. Schirra, K. J. Rosengren, J. West, B. A. Condie, L. Otvos, M. A. Anderson and D. J. Craik, J. Mol. Biol., 2001, 311, 579–591 CrossRef CAS PubMed.
- M. L. Colgrave, M. J. L. Korsinczky, R. J. Clark, F. Foley and D. J. Craik, Pept. Sci., 2010, 94, 665–672 CrossRef CAS PubMed.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911–939 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- F. Albericio, R. P. Hammer, C. GarcÍA-EcheverrÍA, M. A. Molins, J. L. Chang, M. C. Munson, M. Pons, E. Giralt and G. Barany, Int. J. Pept. Protein Res., 1991, 37, 402–413 CrossRef CAS PubMed.
- C. F. McCusker, P. J. Kocienski, F. T. Boyle and A. G. Schätzlein, Bioorg. Med. Chem. Lett., 2002, 12, 547–549 CrossRef CAS PubMed.
- S. A. Kates, N. A. Solé, C. R. Johnson, D. Hudson, G. Barany and F. Albericio, Tetrahedron Lett., 1993, 34, 1549–1552 CrossRef CAS.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- H. B. B:urgi, J. D. Dunitz, J. M. Lehn and G. Wipff, Tetrahedron, 1974, 30, 1563–1572 CrossRef.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- U. Schmidt and J. Langner, J. Pept. Res., 1997, 49, 67–73 CrossRef CAS PubMed.
- P. M. Hardy, G. W. Kenner and R. C. Sheppard, Tetrahedron, 1963, 19, 95–105 CrossRef CAS.
- R. W. Hoffmann, Angew. Chem., Int. Ed. Engl., 1992, 31, 1124–1134 CrossRef.
- I. Daidone, H. Neuweiler, S. Doose, M. Sauer and J. C. Smith, PLoS Comput. Biol., 2010, 6, e1000645 CrossRef PubMed.
- J. A. Smith, L. G. Pease and K. D. Kopple, Crit. Rev. Biochem., 1980, 8, 315–399 CrossRef CAS PubMed.
- M. Rothe, K. D. Steffen and I. Rothe, Angew. Chem., Int. Ed. Engl., 1965, 4, 356–356 CrossRef.
- M. Favre, K. Moehle, L. Jiang, B. Pfeiffer and J. A. Robinson, J. Am. Chem. Soc., 1999, 121, 2679–2685 CrossRef CAS.
- Y. Takeuchi and G. R. Marshall, J. Am. Chem. Soc., 1998, 120, 5363–5372 CrossRef CAS.
- H. Kessler and B. Haase, Int. J. Pept. Protein Res., 1992, 39, 36–40 CrossRef CAS PubMed.
- M. Tamaki, S. Akabori and I. Muramatsu, J. Am. Chem. Soc., 1993, 115, 10492–10496 CrossRef CAS.
- M. Liu, Y. C. Tang, K. Q. Fan, X. Jiang, L. H. Lai and Y. H. Ye, J. Pept. Res., 2005, 65, 55–64 CrossRef CAS PubMed.
- J. Wu, J. Tang, H. Chen, Y. He, H. Wang and H. Yao, Tetrahedron Lett., 2018, 59, 325–333 CrossRef CAS.
- M. R. Eshelman, A. R. Aldous, K. P. Neupane and J. A. Kritzer, Tetrahedron, 2014, 70, 7651–7654 CrossRef CAS PubMed.
- A. Kotynia, J. S. Pap and J. Brasun, Inorg. Chim. Acta, 2018, 472, 3–11 CrossRef CAS.
- K. Haas, W. Ponikwar, H. Nöth and W. Beck, Angew. Chem., Int. Ed., 1998, 37, 1086–1089 CrossRef CAS PubMed.
- I. Alfonso, M. Bolte, M. Bru, M. I. Burguete, S. V. Luis and J. Rubio, J. Am. Chem. Soc., 2008, 130, 6137–6144 CrossRef CAS PubMed.
- V. Martí-Centelles, M. I. Burguete and S. V. Luis, J. Org. Chem., 2016, 81, 2143–2147 CrossRef PubMed.
- N. Vidović, G. Horvat, D. Riva, T. Rinkovec, N. Cindro, V. Tomišić and G. Speranza, Org. Lett., 2020, 22, 2129–2134 CrossRef PubMed.
- R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2015, 51, 4951–4968 RSC.
- P. G. Young, J. K. Clegg, M. Bhadbhade and K. A. Jolliffe, Chem. Commun., 2011, 47, 463–465 RSC.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1891, 24, 2683–2687 CrossRef.
- H. A. Krebs, Biochem. J., 1935, 29, 1620–1644 CrossRef CAS PubMed.
- M. Seia and E. Zisman, FASEB J., 1997, 11, 449–456 CrossRef PubMed.
- R. Tugyi, K. Uray, D. Iván, E. Fellinger, A. Perkins and F. Hudecz, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 413–418 CrossRef CAS PubMed.
- H. M. Werner, C. C. Cabalteja and W. S. Horne, ChemBioChem, 2016, 17, 712–718 CrossRef CAS PubMed.
- S.-i. Wada, H. Tsuda, T. Okada and H. Urata, Bioorg. Med. Chem. Lett., 2011, 21, 5688–5691 CrossRef CAS PubMed.
- P. Ehrlich, in The Collected Papers of Paul Ehrlich, ed. F. Himmelweit, Pergamon, 2013, pp. 596–618 Search PubMed.
- G. Mathé, T. B. Loc and J. Bernard, C. R. Hebd. Seances Acad. Sci., 1958, 246, 1626–1628 Search PubMed.
- T. Ghose, M. Cerini, M. Carter and R. C. Nairn, Br. Med. J., 1967, 1, 90 CrossRef CAS PubMed.
- C. H. Ford, C. E. Newman, J. R. Johnson, C. S. Woodhouse, T. A. Reeder, G. F. Rowland and R. G. Simmonds, Br. J. Cancer, 1983, 47, 35–42 CrossRef CAS PubMed.
- P. F. Bross, G. Beitz, X. H. Chen, E. Chen, L. Duffy, S. Kieffer, R. Roy, A. Sridhara, G. Rahman, R. Williams and R. Pazdur, Clin. Cancer Res., 2001, 7, 1490–1496 CAS.
- A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS PubMed.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed.
- M. Alas, A. Saghaeidehkordi and K. Kaur, J. Med. Chem., 2021, 64, 216–232 CrossRef CAS PubMed.
- T. L. Huang, A. Székács, T. Uematsu, E. Kuwano, A. Parkinson and B. D. Hammock, Pharm. Res., 1993, 10, 639–648 CrossRef CAS PubMed.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- A. Régina, M. Demeule, C. Ché, I. Lavallée, J. Poirier, R. Gabathuler, R. Béliveau and J. P. Castaigne, Br. J. Pharmacol., 2008, 155, 185–197 CrossRef PubMed.
- D. L. Bushnell and K. L. Bodeker, Hematol./Oncol. Clin. North Am., 2020, 29, 317–326 Search PubMed.
- O. Rogoza, K. Megnis, M. Kudrjavceva, A. Gerina-Berzina and V. Rovite, Int. J. Mol. Sci., 2022, 23, 1447 CrossRef CAS PubMed.
- S. Banerjee, M. R. A. Pillai and F. F. Knapp, Chem. Rev., 2015, 115, 2934–2974 CrossRef CAS PubMed.
- F. Vacondio, C. Silva, M. Mor and B. Testa, Drug Metab. Rev., 2010, 42, 551–589 CrossRef CAS PubMed.
- S. K. Kumar, S. A. Williams, J. T. Isaacs, S. R. Denmeade and S. R. Khan, Bioorg. Med. Chem., 2007, 15, 4973–4984 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- Y. Liang, S. Li, X. Wang, Y. Zhang, Y. Sun, Y. Wang, X. Wang, B. He, W. Dai, H. Zhang, X. Wang and Q. Zhang, J. Controlled Release, 2018, 275, 129–141 CrossRef CAS PubMed.
- K. M. Bajjuri, Y. Liu, C. Liu and S. C. Sinha, ChemMedChem, 2011, 6, 54–59 CrossRef CAS PubMed.
- C.-W. Mai, F.-L. F. Chung and C.-O. Leong, Curr. Drug Targets, 2017, 18, 1259–1268 CAS.
- D. Maity, Beilstein J. Org. Chem., 2020, 16, 2971–2982 CrossRef CAS PubMed.
- W. D. Lubell, Biomedicines, 2022, 10, 2037 CrossRef PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- J. B. Grimm, L. M. Heckman and L. D. Lavis, in Progress in Molecular Biology and Translational Science, ed. M. C. Morris, Academic Press, 2013, vol. 113, pp. 1–34 Search PubMed.
- Q. Zheng and L. D. Lavis, Curr. Opin. Chem. Biol., 2017, 39, 32–38 CrossRef CAS PubMed.
- G. G. Guilbault and D. N. Kramer, Anal. Chem., 1965, 37, 120–123 CrossRef CAS PubMed.
- B. Rotman, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS PubMed.
- D. N. Kramer and G. G. Guilbault, Anal. Chem., 1963, 35, 588–589 CrossRef CAS.
- B. Rotman, J. A. Zderic and M. Edelstein, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 1–6 CrossRef CAS PubMed.
- B. Rotman and B. W. Papermaster, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 134–141 CrossRef CAS PubMed.
- F. Coutlee, R. P. Viscidi and R. H. Yolken, J. Clin. Microbiol., 1989, 27, 1002–1007 CrossRef CAS PubMed.
- S. Xu, Q. Wang, Q. Zhang, L. Zhang, L. Zuo, J.-D. Jiang and H.-Y. Hu, Chem. Commun., 2017, 53, 11177–11180 RSC.
- K. H. Jones and J. A. Senft, J. Histochem. Cytochem., 1985, 33, 77–79 CrossRef CAS PubMed.
- Y. Tan, L. Zhang, K. H. Man, R. Peltier, G. Chen, H. Zhang, L. Zhou, F. Wang, D. Ho, S. Q. Yao, Y. Hu and H. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 6796–6803 CrossRef CAS PubMed.
- S. Biswas, B. S. McCullough, E. S. Ma, D. LaJoie, C. W. Russell, D. Garrett Brown, J. L. Round, K. S. Ullman, M. A. Mulvey and A. M. Barrios, Chem. Commun., 2017, 53, 2233–2236 RSC.
- Z. Qiu, J. Shu and D. Tang, Anal. Chem., 2017, 89, 5152–5160 CrossRef CAS PubMed.
- S. Lv, Y. Tang, K. Zhang and D. Tang, Anal. Chem., 2018, 90, 14121–14125 CrossRef CAS PubMed.
- F. Zu, F. Yan, Z. Bai, J. Xu, Y. Wang, Y. Huang and X. Zhou, Microchim. Acta, 2017, 184, 1899–1914 CrossRef CAS.
- S. Doose, H. Neuweiler and M. Sauer, ChemPhysChem, 2009, 10, 1389–1398 CrossRef CAS PubMed.
- M. Sikorski, E. Krystkowiak and R. P. Steer, J. Photochem. Photobiol., A, 1998, 117, 1–16 CrossRef CAS.
- C. Geraghty, C. Wynne and R. B. P. Elmes, Coord. Chem. Rev., 2021, 437, 213713 CrossRef CAS.
- Z. Lin, M. Li, S. Lv, K. Zhang, M. Lu and D. Tang, J. Mater. Chem. B, 2017, 5, 8506–8513 RSC.
- Z. Yin, L. Zhu, Z. Lv, M. Li and D. Tang, Talanta, 2021, 233, 122563 CrossRef CAS PubMed.
- A. G. Gavriel, M. R. Sambrook, A. T. Russell and W. Hayes, Polym. Chem., 2022, 13, 3188–3269 RSC.
- A. Dal Corso, S. Arosio, N. Arrighetti, P. Perego, L. Belvisi, L. Pignataro and C. Gennari, Chem. Commun., 2021, 57, 7778–7781 RSC.
- W. Chyan and R. T. Raines, ACS Chem. Biol., 2018, 13, 1810–1823 CrossRef CAS PubMed.
- F. W. J. Teale and G. Weber, Biochem. J., 1957, 65, 476–482 CrossRef CAS PubMed.
- G. Pandit, K. Roy, U. Agarwal and S. Chatterjee, ACS Omega, 2018, 3, 3143–3155 CrossRef CAS PubMed.
- K.-I. Min, G. Yun, Y. Jang, K.-R. Kim, Y. H. Ko, H.-S. Jang, Y.-S. Lee, K. Kim and D.-P. Kim, Angew. Chem., Int. Ed., 2016, 55, 6925–6928 CrossRef CAS PubMed.
- K. R. Grigoryan and H. A. Shilajyan, Proceedings of the YSU B: Chemical and Biological Sciences, 2017, 51, 3–7 Search PubMed.
- A. B. T. Ghisaidoobe and S. J. Chung, Int. J. Mol. Sci., 2014, 15, 22518–22538 CrossRef CAS PubMed.
- A. Handelman, N. Kuritz, A. Natan and G. Rosenman, Langmuir, 2016, 32, 2847–2862 CrossRef CAS PubMed.
- P. D. Adams, Y. Chen, K. Ma, M. G. Zagorski, F. D. Sönnichsen, M. L. McLaughlin and M. D. Barkley, J. Am. Chem. Soc., 2002, 124, 9278–9286 CrossRef CAS PubMed.
- E. Faggi, Y. Pérez, S. V. Luis and I. Alfonso, Chem. Commun., 2016, 52, 8142–8145 RSC.
- S. Biswas, S. Samui, A. Chakraborty, S. Biswas, D. De, U. Ghosh, A. K. Das and J. Naskar, Biochem. Biophys. Rep., 2017, 11, 112–118 Search PubMed.
- L. Merkel, M. G. Hoesl, M. Albrecht, A. Schmidt and N. Budisa, ChemBioChem, 2010, 11, 305–314 CrossRef CAS PubMed.
- P. Talukder, S. Chen, B. Roy, P. Yakovchuk, M. M. Spiering, M. P. Alam, M. M. Madathil, C. Bhattacharya, S. J. Benkovic and S. M. Hecht, Biochemistry, 2015, 54, 7457–7469 CrossRef CAS PubMed.
- A. Fernandez, E. J. Thompson, J. W. Pollard, T. Kitamura and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 16894–16898 CrossRef CAS PubMed.
- M. H. Y. Cheng, H. Savoie, F. Bryden and R. W. Boyle, Photochem. Photobiol. Sci., 2017, 16, 1260–1267 CrossRef CAS PubMed.
- P. N. Joshi and V. Rai, Chem. Commun., 2019, 55, 1100–1103 RSC.
- C. López-Abarrategui, A. Alba, O. N. Silva, O. Reyes-Acosta, I. M. Vasconcelos, J. T. A. Oliveira, L. Migliolo, M. P. Costa, C. R. Costa, M. R. R. Silva, H. E. Garay, S. C. Dias, O. L. Franco and A. J. Otero-González, Biochimie, 2012, 94, 968–974 CrossRef PubMed.
- L. Mendive-Tapia, C. Zhao, A. R. Akram, S. Preciado, F. Albericio, M. Lee, A. Serrels, N. Kielland, N. D. Read, R. Lavilla and M. Vendrell, Nat. Commun., 2016, 7, 10940 CrossRef CAS PubMed.
- Y. Zhu, S. Mohapatra and J. C. Weisshaar, Biophys. J., 2019, 116, 138a CrossRef.
- B.-H. Gan, T. N. Siriwardena, S. Javor, T. Darbre and J.-L. Reymond, ACS Infect. Dis., 2019, 5, 2164–2173 CrossRef CAS PubMed.
- C. Zhao, A. Fernandez, N. Avlonitis, G. Vande Velde, M. Bradley, N. D. Read and M. Vendrell, ACS Comb. Sci., 2016, 18, 689–696 CrossRef CAS PubMed.
- R. Fischer, T. Waizenegger, K. Köhler and R. Brock, Biochim. Biophys. Acta, Biomembr., 2002, 1564, 365–374 CrossRef CAS PubMed.
- F. Illien, N. Rodriguez, M. Amoura, A. Joliot, M. Pallerla, S. Cribier, F. Burlina and S. Sagan, Sci. Rep., 2016, 6, 36938 CrossRef CAS PubMed.
- D. Birch, M. V. Christensen, D. Staerk, H. Franzyk and H. M. Nielsen, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 2483–2494 CrossRef CAS PubMed.
- M. Sainlos, W. S. Iskenderian and B. Imperiali, J. Am. Chem. Soc., 2009, 131, 6680–6682 CrossRef CAS PubMed.
- W. Wang, T. Uzawa, N. Tochio, J. Hamatsu, Y. Hirano, S. Tada, H. Saneyoshi, T. Kigawa, N. Hayashi, Y. Ito, M. Taiji, T. Aigaki and Y. Ito, Chem. Commun., 2014, 50, 2962–2964 RSC.
- W. Wang, L. Zhu, Y. Hirano, M. Kariminavargani, S. Tada, G. Zhang, T. Uzawa, D. Zhang, T. Hirose, M. Taiji and Y. Ito, Anal. Chem., 2016, 88, 7991–7997 CrossRef CAS PubMed.
- V. Y. Postupalenko, O. M. Zamotaiev, V. V. Shvadchak, A. V. Strizhak, V. G. Pivovarenko, A. S. Klymchenko and Y. Mely, Bioconjugate Chem., 2013, 24, 1998–2007 CrossRef CAS PubMed.
- H. Chen, J. Scherkenbeck, T. Zdobinsky and H. Antonicek, Protein Pept. Lett., 2010, 17, 431–436 CrossRef CAS PubMed.
- A. R. Akram, N. Avlonitis, E. Scholefield, M. Vendrell, N. McDonald, T. Aslam, T. H. Craven, C. Gray, D. S. Collie, A. J. Fisher, P. A. Corris, T. Walsh, C. Haslett, M. Bradley and K. Dhaliwal, Sci. Rep., 2019, 9, 8422 CrossRef PubMed.
- J. Ge, L. Li and S. Q. Yao, Chem. Commun., 2011, 47, 10939–10941 RSC.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- J.-C. G. Bünzli, J. Lumin., 2016, 170, 866–878 CrossRef.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- Y. Jia, C. M. Quinn, A. I. Gagnon and R. Talanian, Anal. Biochem., 2006, 356, 273–281 CrossRef CAS PubMed.
- S. H. Hewitt and S. J. Butler, Chem. Commun., 2018, 54, 6635–6647 RSC.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- J. C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef PubMed.
- A. K. R. Junker, M. Tropiano, S. Faulkner and T. J. Sørensen, Inorg. Chem., 2016, 55, 12299–12308 CrossRef CAS PubMed.
- S. Petoud, S. M. Cohen, J.-C. G. Bünzli and K. N. Raymond, J. Am. Chem. Soc., 2003, 125, 13324–13325 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- I. N. Hegarty, C. S. Hawes and T. Gunnlaugsson, Org. Chem. Front., 2023, 10, 1915–1926 RSC.
- J. Peuralahti, H. Hakala, V.-M. Mukkala, K. Loman, P. Hurskainen, O. Mulari and J. Hovinen, Bioconjugate Chem., 2002, 13, 870–875 CrossRef CAS PubMed.
- E. Pazos and M. E. Vázquez, Biotechnol. J., 2014, 9, 241–252 CrossRef CAS PubMed.
- M. S. Tremblay, Q. Zhu, A. A. Martí, J. Dyer, M. Halim, S. Jockusch, N. J. Turro and D. Sames, Org. Lett., 2006, 8, 2723–2726 CrossRef CAS PubMed.
- M. S. Tremblay, M. Lee and D. Sames, Org. Lett., 2008, 10, 5–8 CrossRef CAS PubMed.
- E. Pazos, M. Goličnik, J. L. Mascareñas and M. Eugenio Vázquez, Chem. Commun., 2012, 48, 9534–9536 RSC.
- H. Jin and J. Varner, Br. J. Cancer, 2004, 90, 561–565 CrossRef CAS PubMed.
- M. Gurrath, G. Müller, H. Kessler, M. Aumailley and R. Timpl, Eur. J. Biochem., 1992, 210, 911–921 CrossRef CAS PubMed.
- D. Arosio and C. Casagrande, Adv. Drug Delivery Rev., 2016, 97, 111–143 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Préat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- K. Temming, R. M. Schiffelers, G. Molema and R. J. Kok, Drug Resistance Updates, 2005, 8, 381–402 CrossRef CAS PubMed.
- M. Cieslikiewicz-Bouet, S. V. Eliseeva, V. Aucagne, A. F. Delmas, I. Gillaizeau and S. Petoud, RSC Adv., 2019, 9, 1747–1751 RSC.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668–12674 CrossRef CAS PubMed.
- T. Kim, H. M. Jeon, H. T. Le, T. W. Kim, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 7690–7693 RSC.
- H.-F. Chau, Y. Wu, W.-Y. Fok, W. Thor, W. C.-S. Cho, P. A. Ma, J. Lin, N.-K. Mak, J.-C. G. Bünzli, L. Jiang, N. J. Long, H. L. Lung and K.-L. Wong, JACS Au, 2021, 1, 1034–1043 CrossRef CAS PubMed.
- H. Y. Shair Kathy, M. Bendt Katharine, H. Edwards Rachel, N. Nielsen Judith, T. Moore Dominic and N. Raab-Traub, J. Virol., 2012, 86, 5352–5365 CrossRef CAS PubMed.
- J. J. Juarez-Vignon Whaley, M. Afkhami, S. Sampath, A. Amini, D. Bell and V. M. Villaflor, Curr. Treat. Options Oncol., 2023, 24, 845–866 CrossRef PubMed.
- C.-F. Chan, R. Lan, M.-K. Tsang, D. Zhou, S. Lear, W.-L. Chan, S. L. Cobb, W.-K. Wong, J. Hao, W.-T. Wong and K.-L. Wong, J. Mater. Chem. B, 2015, 3, 2624–2634 RSC.
- W. He, J. Yan, L. Wang, B. Lei, P. Hou, W. Lu and P. X. Ma, Biomaterials, 2019, 206, 13–24 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chem. – Eur. J., 2010, 16, 3176–3183 CrossRef CAS PubMed.
- X. Liu, J. Fei, A. Wang, W. Cui, P. Zhu and J. Li, Angew. Chem., Int. Ed., 2017, 56, 2660–2663 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195–200 CrossRef CAS PubMed.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- Z. A. Arnon, A. Vitalis, A. Levin, T. C. T. Michaels, A. Caflisch, T. P. J. Knowles, L. Adler-Abramovich and E. Gazit, Nat. Commun., 2016, 7, 13190 CrossRef CAS PubMed.
- Q. Zou, K. Liu, M. Abbas and X. Yan, in Supramolecular Chemistry of Biomimetic Systems, ed. J. Li, Springer Singapore, Singapore, 2017, pp. 135–163 Search PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- C. A. E. Hauser and S. Zhang, Nature, 2010, 468, 516–517 CrossRef CAS PubMed.
- Z. Fan, L. Sun, Y. Huang, Y. Wang and M. Zhang, Nat. Nanotechnol., 2016, 11, 388–394 CrossRef CAS PubMed.
- R. M. Wachter, M.-A. Elsliger, K. Kallio, G. T. Hanson and S. J. Remington, Structure, 1998, 6, 1267–1277 CrossRef CAS PubMed.
- D. P. Barondeau, C. J. Kassmann, J. A. Tainer and E. D. Getzoff, J. Am. Chem. Soc., 2002, 124, 3522–3524 CrossRef CAS PubMed.
- M. L. Contreras, C. Torres, I. Villarroel and R. Rozas, Struct. Chem., 2019, 30, 369–384 CrossRef CAS.
- S. Yi, Y. Wang, Y. Huang, L. Xia, L. Sun, S. C. Lenaghan and M. Zhang, J. Biomed. Nanotechnol., 2014, 10, 1016–1029 CrossRef CAS PubMed.
- B. Gidwani, V. Sahu, S. S. Shukla, R. Pandey, V. Joshi, V. K. Jain and A. Vyas, J. Drug Delivery Sci. Technol., 2021, 61, 102308 CrossRef CAS.
- L. Yao, Y. Hu, Z. Liu, X. Ding, J. Tian and J. Xiao, Mol. Pharmaceutics, 2019, 16, 846–855 CrossRef CAS PubMed.
- R. C. Dutta, Ther. Delivery, 2011, 2, 231–234 CrossRef CAS PubMed.
- A. Vats, N. S. Tolley, J. M. Polak and J. E. Gough, Clin. Otolaryngol. Allied Sci., 2003, 28, 165–172 CrossRef CAS PubMed.
- A. V. Persikov, J. A. M. Ramshaw and B. Brodsky, J. Biol. Chem., 2005, 280, 19343–19349 CrossRef CAS PubMed.
- F. Cisnetti, C. Gateau, C. Lebrun and P. Delangle, Chem. – Eur. J., 2009, 15, 7456–7469 CrossRef CAS PubMed.
- J. Liang, X. Susan Sun, Z. Yang and S. Cao, Sci. Rep., 2017, 7, 37626 CrossRef CAS PubMed.
- A. J. Gordon and M. Phyfferoen, Rev. Infect. Dis., 1983, 5, S188–S199 CrossRef PubMed.
- İ. Gürsel, F. Korkusuz, F. Türesin, N. Gürdal Alaeddinoǧlu and V. Hasırcı, Biomaterials, 2000, 22, 73–80 CrossRef PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- X. Wang, W. Zhou, R. Xu, Y. Xu, H. Song, H. Li and J. Wang, J. Colloid Interface Sci., 2023, 645, 466–471 CrossRef CAS PubMed.
- R. Pugliese, M. Montuori and F. Gelain, Nanoscale Adv., 2022, 4, 447–456 RSC.
- K. Liu, R. Zhang, Y. Li, T. Jiao, D. Ding and X. Yan, Adv. Mater. Interfaces, 2017, 4, 1600183 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |