
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visually distinguishing between tumor tissue and healthy tissue within ten minutes using proteolytic probes†
Debora
Reinhardt‡
a,
Björn
ter Mors‡
a,
Marc D.
Driessen‡
ab,
Marcus
Gutmann
a,
Julian
Faber
c,
Lukas
Haug
d,
Anna-Maria
Faber
de,
Anna
Herrmann
a,
Prisca
Hamm
 a,
Tessa
Lühmann
a,
Christian
Linz
*bf and
Lorenz
Meinel
*ag
a,
Tessa
Lühmann
a,
Christian
Linz
*bf and
Lorenz
Meinel
*ag
aInstitute for Pharmacy and Food Chemistry, University of Würzburg, Am Hubland, DE-97074 Würzburg, Germany. E-mail: lorenz.meinel@uni-wuerzburg.de
bInstitute for Molecular Medicine I, Heinrich-Heine-University Duesseldorf, DE-40225 Duesseldorf, Germany
cDepartment of Oral and Maxillofacial Plastic Surgery, University Hospital Würzburg, D-97070 Würzburg, Germany
dDepartment of Pathology, University of Würzburg, DE-97080 Würzburg, Germany
eDepartment of Dermatology and Venerology, University of Cologne, DE-50937, Cologne, Germany
fDepartment for Oral and Craniomaxillofacial and Plastic Surgery, University Hospital Cologne and Faculty of Medicine, University of Cologne, DE-50937, Cologne, Germany. E-mail: christian.linz@uk-koeln.de
gHelmholtz Center for Infection Research (HZI), Helmholtz Institute for RNA-Based Infection Research (HIRI), DE-97080 Würzburg, Germany
First published on 17th July 2024
Abstract
Accurately identifying tumor tissue is crucial during surgery, especially when removing head and neck squamous cell carcinomas (HNSCC). Our tumor-responsive probes are tailored for ex vivo diagnostics, streamlining today's complex surgical workflows and potentially enabling pathologists and surgeons to rapidly and objectively distinguish between healthy and tumor tissue. Designed based on insights from biological furin substrates and cleavage site screening, the probes detect HNSCC-associated protease activity. Within ten minutes of incubation, tumor tissue is differentiated from healthy tissue by visible fluorescence in biopsy supernatant.
Introduction
Head and neck squamous cell carcinomas (HNSCC) result in approximately 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year.1–3 These malignancies commonly originate from epithelial cells in the oral cavity, pharynx, and larynx, leading to limitations in swallowing, speaking, and breathing. The primary risk factors for HNSCC are tobacco use, alcohol use, and human papillomavirus infections.2,4 HNSCC tumors are surgically removed, followed by stage-related radiotherapy or chemotherapy.5 A tumor resection with tumor-free margins is a prerequisite for curative treatment.6 In the head and neck area, extensive tumor resections often result in severe swallowing, speaking, and breathing limitations. These limitations reduce the quality of life for patients and are suspected to contribute to one of the highest suicide rates among cancer survivors.7 Hence, surgical removal of HNSCC tumors is a finely balanced choice: incomplete removal in the surgical margin is a significant cause of relapse and death, while overly generous resection dramatically curtails the patient's quality of life.8 To help balance this situation, the standard practice includes intra-operative pathological assessment of tissue sections, which is time-consuming and logistically demanding.9 Diagnostic probes targeting protease activity in tumor tissue are gaining attention as alternative methods due to their simple and rapid application.10–13 The design of these probes, based on a protease-sensitive peptide core, necessitates the identification of tumor-associated proteases and determination of their cleavage specificities. Hamm et al. recently described an introductory guide to the design of protease-sensitive linkers, starting with target selection and protease cleavage site profiling.14 However, fluorescence-based strategies are often hindered by sample autofluorescence or the limited spectral range of fluorophores, especially in multiplexed analyses, which are less error-prone and more reliable than “singleplex” analyses. To address these challenges our method is designed for ex vivo analysis of small tissue samples, with fluorescence detection in the supernatant, separate from the tissue. This approach eliminates interference from tissue autofluorescence and allows for a much broader method spectrum and multiple uses of small tissue slices. By reusing small tissue samples for multiple probes through sequential exposure, results can be reaffirmed, which is especially important due to the detrimental effect of removing too much healthy tissue. At the same time, our approach at its simplest, can be read out with virtually no equipment but test tubes and a UV lamp. As tissue re-use and multiplexing are a core idea, fluorescence overlap is eliminated, and the detection limit is raised (determined supernatant volume).
000 deaths each year.1–3 These malignancies commonly originate from epithelial cells in the oral cavity, pharynx, and larynx, leading to limitations in swallowing, speaking, and breathing. The primary risk factors for HNSCC are tobacco use, alcohol use, and human papillomavirus infections.2,4 HNSCC tumors are surgically removed, followed by stage-related radiotherapy or chemotherapy.5 A tumor resection with tumor-free margins is a prerequisite for curative treatment.6 In the head and neck area, extensive tumor resections often result in severe swallowing, speaking, and breathing limitations. These limitations reduce the quality of life for patients and are suspected to contribute to one of the highest suicide rates among cancer survivors.7 Hence, surgical removal of HNSCC tumors is a finely balanced choice: incomplete removal in the surgical margin is a significant cause of relapse and death, while overly generous resection dramatically curtails the patient's quality of life.8 To help balance this situation, the standard practice includes intra-operative pathological assessment of tissue sections, which is time-consuming and logistically demanding.9 Diagnostic probes targeting protease activity in tumor tissue are gaining attention as alternative methods due to their simple and rapid application.10–13 The design of these probes, based on a protease-sensitive peptide core, necessitates the identification of tumor-associated proteases and determination of their cleavage specificities. Hamm et al. recently described an introductory guide to the design of protease-sensitive linkers, starting with target selection and protease cleavage site profiling.14 However, fluorescence-based strategies are often hindered by sample autofluorescence or the limited spectral range of fluorophores, especially in multiplexed analyses, which are less error-prone and more reliable than “singleplex” analyses. To address these challenges our method is designed for ex vivo analysis of small tissue samples, with fluorescence detection in the supernatant, separate from the tissue. This approach eliminates interference from tissue autofluorescence and allows for a much broader method spectrum and multiple uses of small tissue slices. By reusing small tissue samples for multiple probes through sequential exposure, results can be reaffirmed, which is especially important due to the detrimental effect of removing too much healthy tissue. At the same time, our approach at its simplest, can be read out with virtually no equipment but test tubes and a UV lamp. As tissue re-use and multiplexing are a core idea, fluorescence overlap is eliminated, and the detection limit is raised (determined supernatant volume).
Where fluorescence guided surgery would clearly mark tumor margins, it also requires injection or oral administration of the probes before surgery as well as to allow some time for the probes to accumulate in the tumor tissue.15,16 The multiplexable ex vivo approach shown here, requires no preliminary preparation of the patient and can in theory be expanded to include potential risk factors present in the surrounding tissue (inflammation, pre-cancerous tissue alteration, etc.) that cannot be detected or targeted through common guided surgery approaches.
In the present study, we introduce diagnostic probes targeting the altered protease activity in HNSCC tissue based on furin as a surrogate marker. Furin is upregulated in several tumor types, and increased levels of furin have been linked to greater malignancy and poorer prognosis in head and neck cancer.17–20 When exposed to tissue samples, the probes are susceptible to multiple proteases, including proteases that share common substrate motifs or are highly specific for distinct amino acids.21,22 The broad spectrum of specificities in biological samples makes it unlikely to design a probe that is sensitive and specific to a single present protease. In consequence, probe design must strike a balance between sensitivity for the target protease and specificity in context of competitive proteases in the patients' sample. Therefore, we present two probes: the first probe (Probe a) exhibits rapid cleavage in tumor tissue but is also susceptible to some cleavage in healthy tissue, while the second probe (Probe 1) is less accessible to healthy tissue, but also less responsive to furin (Fig. 1). For probe design, we leveraged insights from the well-described furin substrates, the common cleavage motif RXXR↓ (with a preference for positively charged or small amino acids at X sites (the downward arrow↓ indicates the cleavage sites), as well as from furin cleavage site profiling.23 The derived furin-sensitive peptides were evaluated for their cleavage efficiency, and the two most effective peptides were transformed into fluorescence resonance energy transfer (FRET) probes. Both probes were verified ex vivo on tissue samples, successfully differentiating HNSCC tissue from healthy tissue in the tissue supernatant.
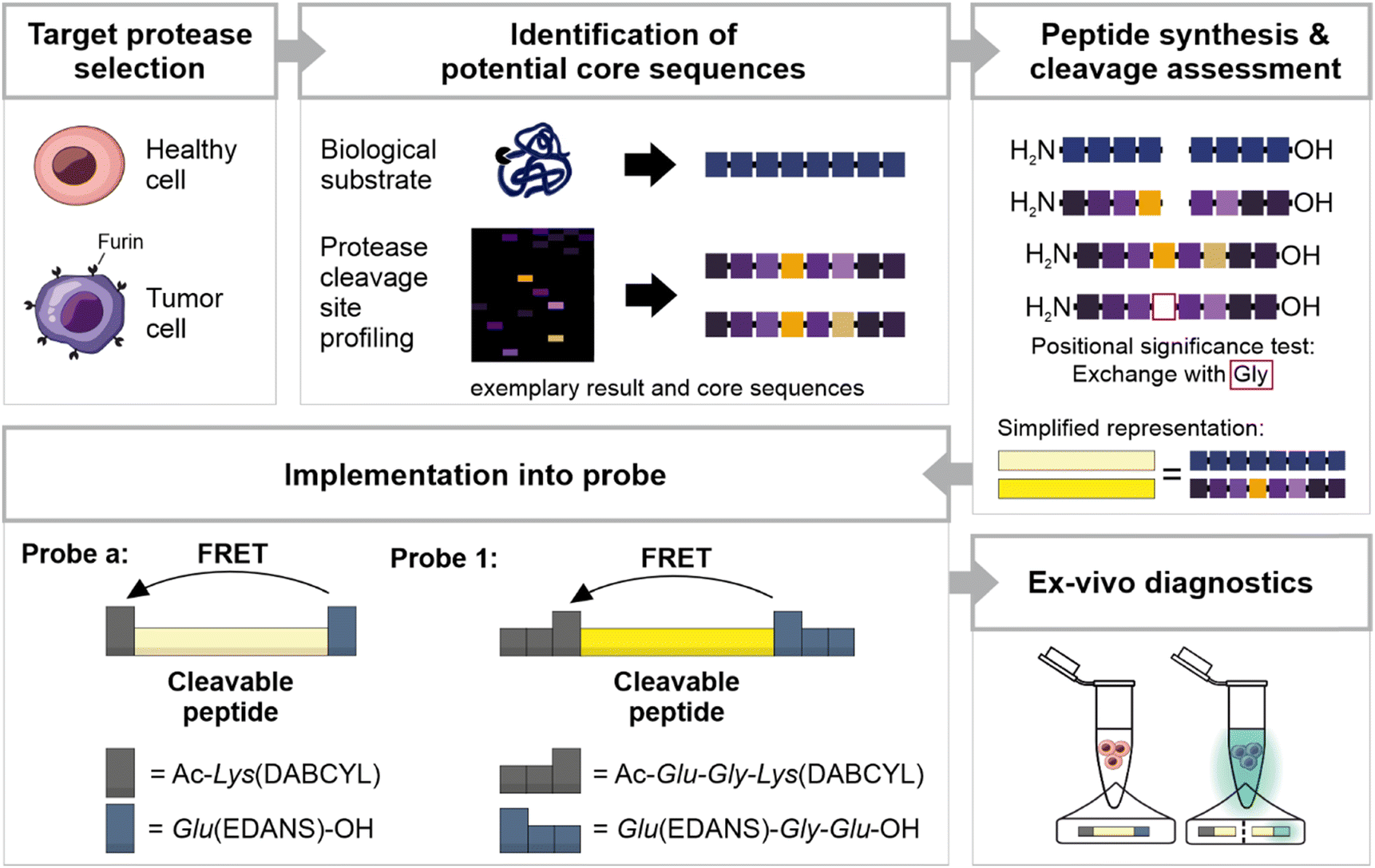 | ||
| Fig. 1 Schematic overview of probe development and design. We selected furin as diagnostic marker and searched for potential core sequences. Our sequences were adapted from biological substrates or derived from protease cleavage site profiling via Proteomic Identification of Cleavage Sites (PICS). To facilitate comprehension, we present a simplified PICS result and exemplary core sequences. These sequences were synthesized and compared for their susceptibility to furin cleavage. The significance of positional preferences was tested by additional sequences in which single positions were replaced by Gly. Two final core sequences were implemented as probes, modified with N-terminal acetylation (Ac) and flanked by a FRET donor (DABCYL) and a FRET quencher (EDANS). Probe 1 was further extended with additional amino acids to improve solubility. In first ex vivo assays, the probes were able to differentiate healthy from tumor tissue by fluorescence in the supernatant. | ||
Material and methods
Proteomic Identification of Cleavage Sites (PICS)
Protease cleavage sites were identified following PICS as described by Schilling et al.24,25 PICS libraries were derived from E. coli proteome (Thermo Fisher Scientific, Waltham, USA, 14 N and 15 N). Three different PICS libraries were generated, and either trypsin (14 N and 15 N E. coli) or GluC (14 N E. coli) was used for specific digestion. For furin cleavage assays, 300 μg of the peptide library was incubated with 5 U of furin in 100 mM 4-(2-hydroxyethyl) piperazine-1-ethane sulfonic acid (HEPES) with a pH of 7.5 and 5 mM CaCl2 at 37 °C for 16 h. Cleavage was stopped by heating to 70 °C for 20 min. Prime-side cleavage fragments were tagged with cleavable biotin and were isolated using a streptavidin-modified resin. After library digestion and purification, the samples were analyzed via LC-MS/MS. LC-MS/MS results were processed via PMI-Byos software by Protein Metrics, Inc. (United States). Cleavage sites were reconstructed via the WebPICS online tool, and cleavage specificities were presented as relative occurrences.22 All relative occurrences of amino acids below 11% were considered inconclusive. A detailed description of the library generation and the processing after library digestion by furin is provided in the ESI.†Solid-phase peptide and probe synthesis
The PICS-derived peptides and probes were synthesized using Fmoc-protected amino acids in solid-phase peptide synthesis. The FRET donor-quencher pair was incorporated into the probes through standard solid-phase peptide synthesis with modified amino acids, Fmoc-Lys(DABCYL)-OH and Fmoc-Glu(EDANS)-OH (Fig. S1†). Probe 1 was further extended by Glu and Gly to improve solubility. Purification and analysis were performed with FPLC, HPLC, and LC-MS. Both methods have been described in detail before.26Peptide cleavage
Lyophilized peptides were solubilized in a furin buffer (i.e., 100 mM HEPES with a pH of 7.5 and 5 mM CaCl2) to 1 mM stock solutions. Cleavage was performed at 37 °C under rigorous shaking in a 30 μL furin buffer that contained 0.1 mM of the respective peptide and 1 U of furin per reaction. The reaction was stopped by adding EDTA (5 mM of final concentration). Experiments were performed in three independent replicates. Cleavage was analyzed via HPLC chromatography. The percentage of the cleavage resulted from comparing the area under the curve of the major peak of each peptide incubated with furin with a negative control analyzed without furin.Cleavage of FRET probes by recombinant furin
10 μM of Probe a or Probe 1 was incubated with 1.67 U of furin in 50 μL of 100 mM HEPES with a pH of 7.5 and 5 mM CaCl2 for 12 h at 37 °C. Negative controls were performed without furin. Experiments were performed in three independent replicates. The samples were analyzed using an Infinite M Plex (Tecan Trading AG, Männedorf, Switzerland) with an excitation wavelength of λ = 340 nm and an emission wavelength of λ = 490 nm. To test whether the illumination of Probe a was visually perceptible, Probe a was incubated with furin for 60 min and illuminated via a universal UV lamp (CAMAG, Germany) with λ = 350 nm. Cleavage rates were calculated using fluorescence intensity and a linear equation resulting from a concentration range of cleaved Probe a (Fig. S5†).Tissue collection and assessment
The study protocol was prepared in accordance with the Declaration of Helsinki and the criteria of good clinical practice. It was approved by the Ethics Committee of the University of Würzburg (file number 280/19). Informed consent was obtained from all human subjects. All included patients had been diagnosed with primary HNSCC. Patients suffering from other tumor entities – as well as inoperable cases – were excluded from the study.A routine histopathological assessment confirmed the diagnosis in every case (dissection margins, tumor stage etc.). Subsequently the specimens were fixated in formalin overnight for histopathological assessment the next day. The following day, the specimens were paraffin-embedded, sectioned and stained with hematoxylin and eosin. HE sections were scanned using a Pannoramic Scan II slide scanner from 3DHistech® and were displayed with CaseViewer Software from 3DHistech®. In addition to tumor resection, a small amount of corresponding healthy oral mucosa of about 5 mm (clinically non-suspicious and at a sufficient distance from the primary tumor resection to ensure safety) was excised separately. The resected tumor tissue and healthy oral mucosa were placed in RPMI-1640 (Dutch modification, 10% FCS, 1% penicillin/streptomycin, nystatin). For further experiments, the tissue samples were cut into smaller specimens and incubated in RPMI-1640 overnight at 37 °C.
Tissue incubation with FRET probes
Before incubation with FRET probes, tissue specimens were washed with reaction buffer (100 mM HEPES with a pH of 7.5 and 1 mM CaCl2). The progress of the probe cleavage over time was monitored with a fluorescence plate reader (Infinite M Plex, Tecan Trading AG, Männedorf, Switzerland) with an excitation wavelength of λ = 340 nm and an emission wavelength of λ = 490 nm. The tissue specimens (6–9 mg) were incubated in 200 μL of reaction buffer containing 10 μM of Probe a or Probe 1. 100 μM of Furin Inhibitor I was added as a negative control. The tissues were incubated at 37 °C. 100 μl of the supernatant, containing the respective probe, was briefly removed and separately analyzed at t = 0, 5, 10, 15, 20, 25, 30, 40, 50, and 60 min. After each measurement the supernatant was returned the tissue. At least n = 3 experiments were performed.Visual detection was proved by probe cleavage and illumination. 22 ± 3.1 mg of tissue specimens were incubated in a 500 μl reaction buffer containing 10 μM of Probe a or Probe 1. The reaction was performed at 37 °C for 1 h. Illumination was induced via an LED unit (16 × 60 mm) with three 3× Luminus SST-10 UV 365–370 nm on a SYSTEM Slider (https://led-tech.com) controlled by an SLT6 – 350 IFG (self-electronics) control unit using 365 nm and 350 mA as control settings. Pictures were taken with a Sony Alpha 58 camera and an 18-55 SAM II objective.
In-silico specificity check
Ferrall-Fairbanks et al. developed an algorithm that identifies putative cleavage sites of a protease in a protein or peptide.Specificity is shown as amino acid preference at each position ranging from P4 to P4′, referred to as the MEROPS specificity matrix.27 We used an in-house programmed extension (Prot-Agonist) that deciphers the proteases (output) that cleave any desired set of primary amino acid sequences (input variable).14
MEROPS provides a specificity matrix ranging from P4 to P4′ when the exact cleavage site of at least ten substrates is known. The database displays how often each amino acid occurs at each position in the documented substrates of the enzyme. Every peptide bond of the linker is defined once as the scissile bond. The four residues before and after this scissile bond are used to estimate the cleavage likelihood. The algorithm initially sets the first N-terminal amino acid at P4′. The resulting first segment for Sequence a (RRARSVAS, identical to the positioning code P4–P3–P2–P1↓P1′–P2′–P3′–P4′, ↓ indicating the cleavage site) is consequently P4–P3–P2–P1↓P1′–P2′–P3′–R. The number that indicates how often this amino acid occurs at P4′ in the substrates documented in MEROPS for each MEROPS listed protease is then checked in the matrix, with a value of 0 being assigned to empty slots. The resulting counts for this iterative process are plotted as a cumulative score for all eight positions (P4–P3–P2–P1↓P1′–P2′–P3′–P4′). As these absolute counts depend on the number of substrates listed in MEROPS, each retrieved matrix entry is normalized by dividing the counts by the sum of its column. Columns are defined as follows and using the example P4–P3–P2–P1↓P1′–P2′–P3′–R again: To normalize the R in P4′, the app counts the sum of substrates for which the amino acid is known in P4′. In the next step, when going for P3′, the app repeats the process. Typically, the number of substrates with known amino acids in P3′ is higher than in P4′; hence, the value in the denominator for normalization is higher for P3′ than P4′. The sum results in the normalized score (norm score). Next, the large 8-residue window is shifted one position further (i.e., P4–P3–P2–P1↓P1′–P2′–R–R for Sequence a). The first amino acid is in P3′, the second is in P4′, and the rest of the cleavage site is empty. The scores are calculated, and the procedure is repeated until the final amino acid is in position P4 (i.e., S–P3–P2–P1↓P1′–P2′–P3′–P4′ for Sequence a). This procedure is repeated for all proteases in MEROPS with an available specificity matrix.
Finally, a plausibility check is (manually) conducted based on literature research, evaluating the relevance of proteases identified by the app. Potentially relevant proteases were defined as those with differential expression in HNSCC tissue and localization at the plasma membrane or secretion.
Statistical analysis
Statistical significance was evaluated using a one-way ANOVA followed by a post-hoc Tukey test or an unpaired t-test (GraphPad Software, San Diego, Californian, USA). Data are presented as mean and standard deviation. Results were considered statistically significant at p ≤ 0.05(*).Results and discussion
A peptide-based furin-activity probe was designed that consists of a peptide core sequence (furin-cleavable linker) flanked by a N-terminal quencher and a C-terminal fluorophore (Fig. 1 and S1†). Peptide core sequences were designed from libraries exposed to recombinant furin (PICS) by using a previously published tool, WEB-PICS, in the final analysis.22,24 The E. coli proteome-derived peptide libraries were generated via trypsin or GluC digestion. The libraries differed in their peptides because trypsin cleaves C-terminal to Arg and Lys, whereas GluC preferentially cleaves C-terminal to Gln.PICS, to some extent, causes chemical modification of the amino acids lysine and cysteine (methylation, alkylation).14,24 We mitigated a potential impact on our study, by incorporating several controls. On the one hand, we controlled our peptide libraries for off-target alkylation or missed cleavages (data not shown). Furthermore, the outcome was congruent with previously published furin specificities, suggesting that chemical modification of lysines and methionines during PICS did not jeopardize the ability to successfully identify cleavage sites. Furin is reported to cleave C-terminal to Arg (RXKR or RXRR), with a preference for Arg at P1′ (Fig. 2A and Table S1†).28–31 Arg was found as the preferred amino acid in positions P4 to P1 and P1′ showed a strong preference for Arg with an high occurrence of 49%. At P2, Ala had the same occurrence as Arg, and in P1 Lys was also found but with lower occurrence. At P2′ and P3′ Ala was preferred. These findings resulted in a GluC-derived furin specificity motif of XRR/AR↓RAAX. Next, we tested furin on the trypsin pre-processed PICS library, distinct from the GluC library in its peptide composition. Trypsin cleaves next to Arg and Lys. Thus, furin motifs with Arg at the cleavage sites – preferentially seen with the GluC pre-processed library – cannot be found with the trypsin-generated library. Due to the variance, there is potential for the discovery of additional furin-specific sequences within this data set. Using the trypsin-generated libraries with furin resulted in more than 560 identified peptides (Fig. 2B and Table S2†). In the absence of internal Lys and Arg, we identified Met as preferred P1 amino acid at the furin cleavage site (Fig. 2B). In position P1′, Thr had the highest relative occurrence, followed by Ser, Ala and Gly, respectively. Position P2′ had Ala, Val, Leu, and Pro, in descending order of occurrence. Therefore, we observed a novel furin specificity motif of XXXM↓S/TAXX.
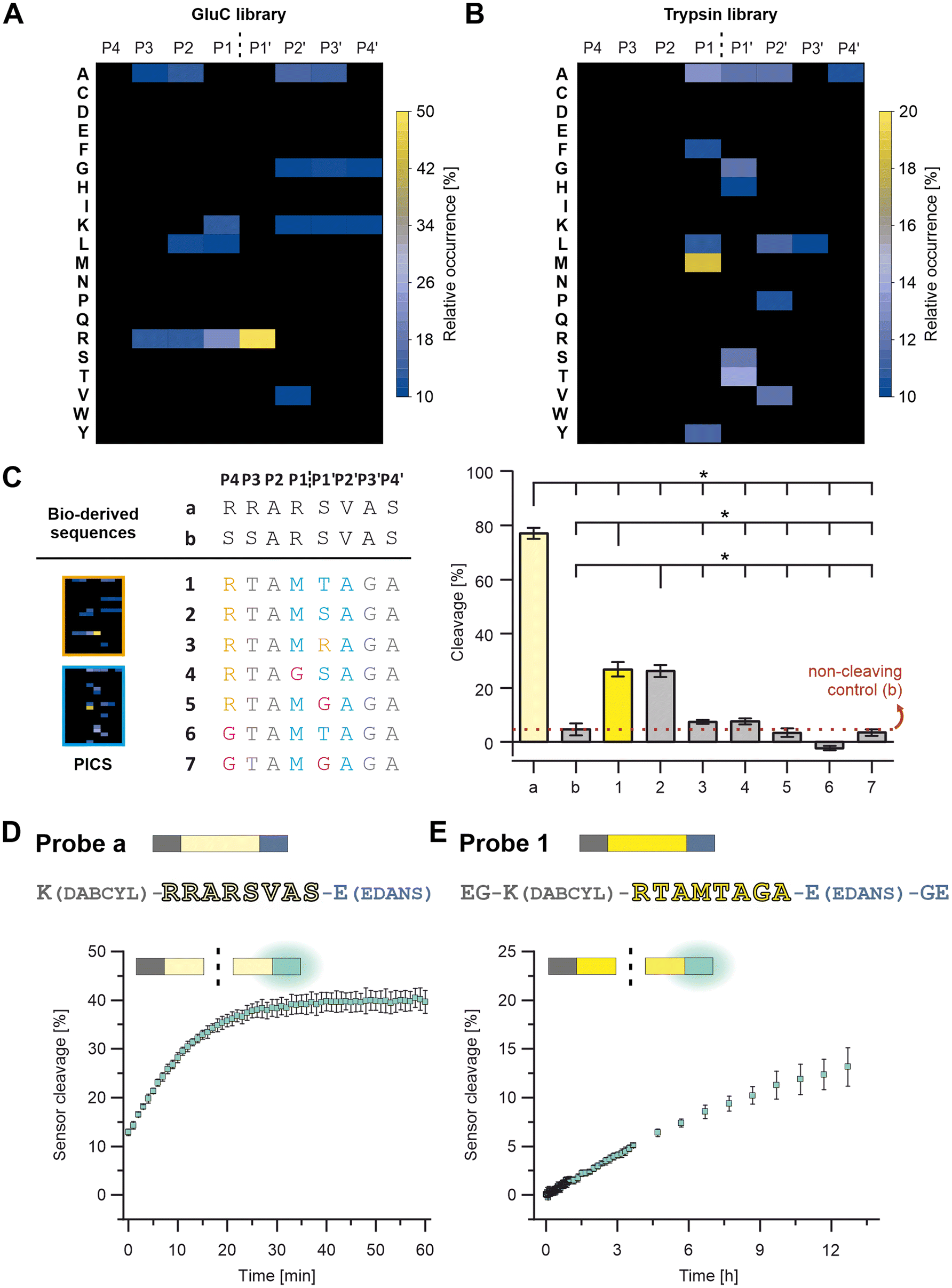 | ||
| Fig. 2 Identifying furin-cleavable core sequences. PICS outcome for amino acids at P4 to P4′ from the (A) GluC and (B) trypsin-digested library. (C) Selected core sequences (from N-to-C terminus), with a from the SARS spike protein, b as non-cleavable mutation of a, and 1 to 7 from the PICS results. Blue amino acids were from the trypsin PICS-derived motif XXXM↓S/TAXX, and orange amino acids were from the GluC PICS. The grey amino acids were selected based on less specific PICS results and information on known furin specificities. Pink amino acids were inserted to probe positions for their relevance in specificity. Furin cleavage efficiency is shown for (C) the core sequences with Sequence b as non-cleaving control, (D) Probe a, and (E) Probe 1. Data were analyzed via one-way ANOVA and post hoc Tukey tests; means were ± standard deviation, n = 3; p ≤ 0.05 was considered statistically significant and is highlighted by asterisks. | ||
Based on the insights of the PICS screens and known cleavage sites, we selected potential peptide core sequences for further characterization, namely the SARS-COV-2 spike protein cleavage site RRAR↓SVAS (referred to as “a”) and a recently described mutated site, that was reported to show no processing by furin as a negative control (SSARSVAS, referred to as “b”; Fig. 2C and Table S3†).32 Furthermore, three alternative peptide core sequences were derived from the peptide library screens (referred to as “1” and “3”; Fig. 2C and Table S3), which in turn were modified in specific core positions, to determine the validity of the PICS findings (“4” to “7”; Fig. 2C; Table S3). The Sequences 1 and 2 were designed as follows: The trypsin PICS library suggested a furin mitigated preference for M↓T/SA in P1 to P2′ (Fig. 2B and C, blue). In the remaining positions, the trypsin PICS results were less specific (Fig. 2B). Therefore, we also considered natural furin substrates (grey) and knowledge obtained from our GluC library outcomes (orange; Fig. 2A): P4 was filled with Arg because it had a high occurrence in the GluC library and was known from most furin cleavage sites (Fig. 2C, orange). In the trypsin library, the highest occurrences at P3 were Ala, Thr, and Val, but we selected Thr because the MEROPS database indicated that polar amino acids are favored at P3 (Fig. 2C, grey).29 P2 was filled with Ala, as suggested by the trypsin PICS library and as known from other substrates (Fig. 2C, grey). For P3′ to P4′, the trypsin and the GluC library had little information aside from a general trend toward non-polar, small amino acids, such as Ala, Val, Leu, and Gly. We chose Gly in P3′ and Ala in P4′ to avoid immediate amino acid repeats (e.g., GG or AA; Fig. 2C, grey). Because of the high occurrence of P1′: Arg in the GluC PICS, we also considered Arg instead of Ser and Thr, resulting in Sequence 3 (Fig. 2C and Table S1†).
These potential core sequences (“a, b” and “1” to “3”, Fig. 2C) were synthesized, characterized by HPLC and LC-MS, and tested for cleavage by recombinant furin (Fig. 2C, S2 and S3 and Table S3†). Cleavage is shown as a percentage decrease in the signal for the respective intact peptide on HPLC (Fig. 2C, right). The same furin activity, peptide concentrations and reaction conditions were used for all peptides and allow direct comparison of cleavage efficiency within the selected peptides. Within 15 minutes, Sequence a was the most efficiently cleaved sequence with a cleavage of 77 ± 2%, followed by 1 and 2 with 26 ± 2% and 27 ± 3%, respectively. (Fig. 2C).
Sequence 3 was a poor furin substrate compared to the other sequences and was not considered further. As expected, Sequence b showed virtually no cleavage and is referred to as the non-cleaving control.
To test the positional significance of the PICS-derived selection at P4, P1 and P1′, further sequences were synthesized and analyzed as described before (“4” to “7”; Fig. 2C and S3 and Table S3†). Here, a replacement with Gly (Fig. 2C, pink) caused a clear reduction in cleavage efficiency and confirms a positional preference for P4: Arg (see 1vs.6 & 7), P1: Met (see 2vs.4) and P1′: Ser or Thr (see 1 & 2vs.5 & 7). We selected peptide core Sequences a and 1 for further probe development (Fig. 1 and S1†).
The peptide core sequences were transformed into FRET turn-on/off probes by attaching DABCYL and EDANS (Fig. 2D and E and Table S3†). One challenge was the solubility of Probe 1, which was met by extending the probe terminally with Gly and Glu (Fig. 1 and S1†). The resulting probes – Probe a and Probe 1 – were characterized by HPLC and LC-MS (Fig. S4†).
Again, both probes were exposed to the same activity of recombinant furin to compare cleavage efficiency. As expected, Probe a was more rapidly cleaved by furin, resulting in 13 ± 1% digestion at the first measurement time point (which was defined as t = 0; Fig. 2D and S5†). Digestion increased to about 40 ± 2% within 40 min. When irradiated after 60 min of incubation, Probe a fluorescence was visually detectable under UV light (Fig. 3A). Probe 1 was more stable, reaching the cleavage rate of 13 ± 2% after 12 h (Fig. 2E and S5†).
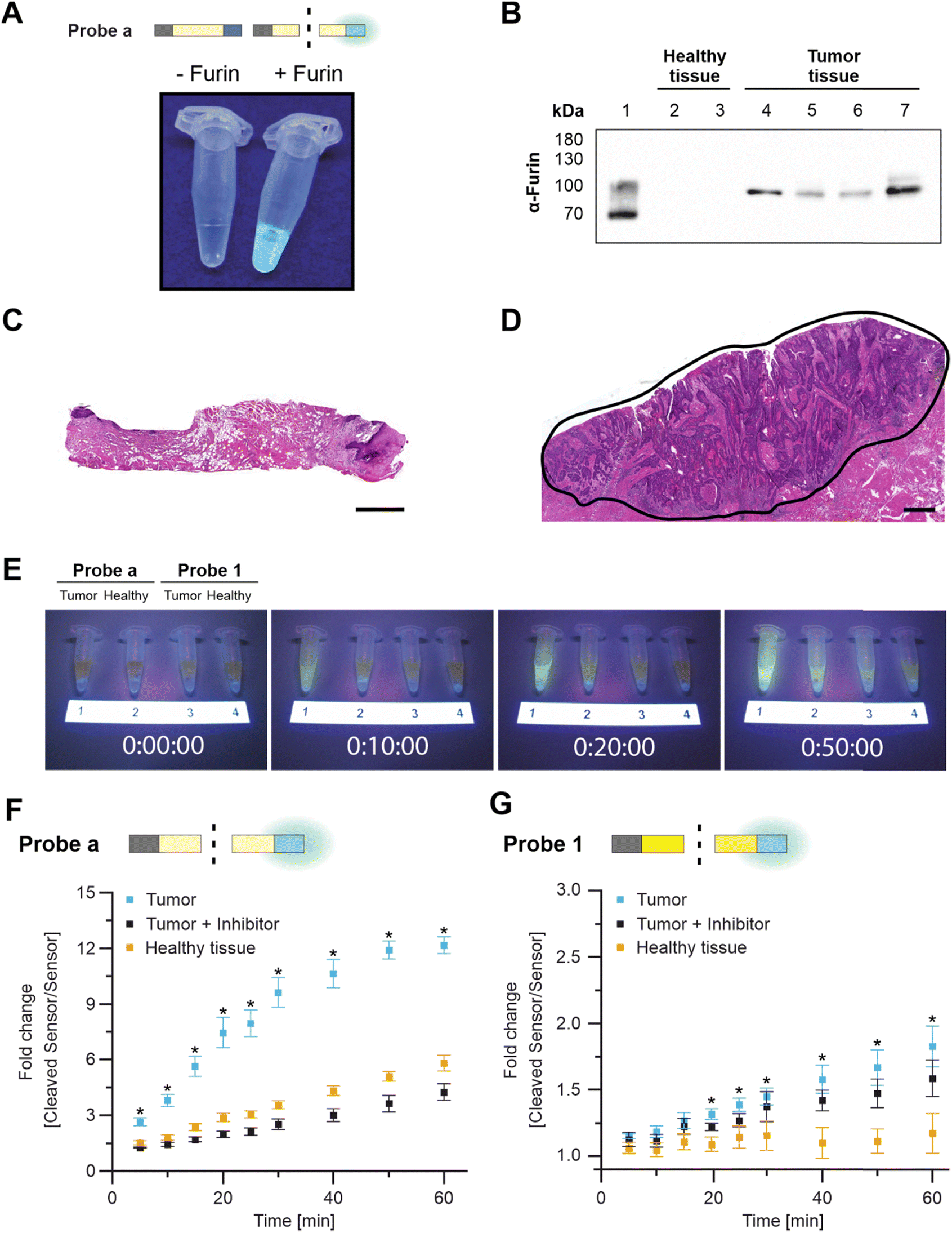 | ||
| Fig. 3 Visually detectable probe cleavage. (A) Probe a without furin (left) and with furin incubation under UV light (right). (B) Furin western blots on HNSCC tissue lysates (Lane 1: recombinant furin; Lanes 2 + 3: healthy tissue lysates; Lanes 4–7: HNSCC tissue lysates). (C) Healthy tissue was located next to (D) HNSCC tissue (encircled area in black, scale bar = 1 mm). (E) Probe cleavage over time (incubation with the tumor in Lanes 1 and 3 and healthy tissue in Lanes 2 and 4). (F) Time-dependent cleavage of Probe a and (G) Probe 1 with tumor tissue, tumor tissue with furin inhibitor, and in healthy tissue. Pairwise comparisons of tumor and healthy tissue were made via Student t-test; means ± standard derivation, n = 3; p ≤ 0.05 was considered statistically significant and highlighted by asterisks. | ||
We next evaluated the probe performances with patient biopsies (Fig. S6 and Table S4†).
Tumor tissue from HNSCC patients – but not specimens from healthy tissues – were furin-positive (Fig. 3B and S7†).18 Healthy tissue and the diagnosis of HNSCC were confirmed via pathological evaluation, with the tumor tissue being characterized by irregular squamous epithelial proliferations with variable and irregular keratinization (Fig. 3C and D and S6†). Within 10 min, Probe a fluorescence was sufficient to distinguish tumor tissue (FRET-on) from healthy tissue (FRET-off) under UV light by eye (Fig. 3E and S8†). The same differentiation by eye was possible for Probe 1, but after 40 min.
Tumor-driven cleavage was confirmed by a plate reader in three additional patients for Probe a (all within 5 min, Fig. 3F and S9†) and Probe 1 (within 10, 20, 40 min, Fig. 3G and S10†). Here, tissue specimens were incubated with the respective probes in reaction buffer. At various time points, a fixed volume of supernatant was briefly removed and analyzed separately from the tissue for fluorescence. This procedure was chosen to make our analysis independent of tissue autofluorescence.
We further corroborated these findings via cross-over incubation to remove possible idiosyncratic effects. To that end, tissue specimens from one patient were first incubated with Probe a, followed by a second incubation with Probe 1 and a third incubation, again with Probe a (Fig. S11†). Each incubation lasted 30 min and was followed by extensive washing. This “bracketing” approach was successful, leading to comparable results for the first and last incubation with Probe a. In addition, we repeatedly incubated two specimens with Probe a for 30 min (four times), and no statistical differences in cleavage rates were observed (Fig. S12†). This paired approach confirmed the faster cleavage of Probe a compared with Probe 1.
We then addressed the furin-specificity of the probes. Although cleavage of Probe a was primarily driven by furin, specific inhibition of furin only reduced cleavage to healthy control results (off-target cleavage). Still, some cleavage occurred over time in healthy tissue, which we will refer to as “off-target cleavage” (Fig. 3F). Therefore, we tested Probe a with a broad-range protease inhibitor, which reduced overall cleavage with tumor tissue or with healthy tissue (Fig. S13†). These results suggested that the off-target cleavage in healthy tissue had been caused by proteolytic cleavage, most likely by proteases other than furin. Cleavage by competitive proteases is referred to as “non-specific” cleavage in this manuscript. The response of Probe 1 to furin inhibition during incubation with tumor tissue showed high variability across replicates with almost no inhibition of Probe 1 cleavage in Fig. 3G, partial inhibition in Fig. S10† (left), and complete inhibition in Fig. S10† (right). The observed variances stem from inherent differences among tissue samples obtained from different patients and suggest that Probe 1 is more susceptible to non-specific cleavage than Probe a. However, the off-target cleavage by healthy tissue was negligible (Fig. 3G and S10†) and we assume that the non-specific cleavage by HSCC tissue is caused by other tumor-related proteases. These findings suggested that Probe a is more susceptible to off-target cleavage by healthy tissue than Probe 1.
Further research needs to be conducted to reveal the identity and impact of competitive proteases, which is beyond the scope of this study. As first approach, we aimed to identify the proteases involved in non-specific probe cleavage to understand whether they were tumor-related. To do so, we programmed a search algorithm for the MEROPS database, which we named Prot-Agonist.14 MEROPS holds experimental information on more than 3000 proteases.29,33 Prot-Agonist was developed on the basis of another app that Ferrall-Fairbanks had coded.27 The Ferrall-Fairbanks app probes the cleavage of one peptide sequence by one protease. Prot-Agonist was coded for automated searches to simultaneously run one peptide or protein of any length against all proteases listed in the MEROPS database, while the app by Ferrall-Fairbanks does not run automated searches across the whole set of MEROPS proteases. Our app can perform a screening of long proteins as substrates (not only peptides) against all MEROPS proteases simultaneously.14 The resulting app suggested a set of other proteases that could potentially recognize core Sequences a and 1. These raw data were checked for relevance (e.g., some “hits” for intracellular proteases were irrelevant for our purposes) for the 18 highest-ranking proteases (Table S5 and S6†). Prot-Agonist suggested that Sequence a may be cleaved by other HNSCC-related proteases in addition to furin, including proprotein convertase Subtilisin/Kexin Type 6 (PCSK6), matriptase (ST14)), and proteinases that are present in the healthy oral mucosa (e.g., hepsin (HPN), transmembrane protease serine 11E (TMPRSS11E), and kallikrein-13 (KLK13); Table S5†). This finding explains why Probe a is also processed by healthy tissue (Fig. 3F, S9, S11 and S13†). Derived from Prot-Agonist, the likelihood of Sequence 1 cleavage by proteases other than furin was much lower (based on the comparison of norm scores) and included the HNSCC-related proteases PCSK6, ubiquitin-specific peptidase 4 (USP4), and methionine aminopeptidase 2 (METAP2) (Table S6†). The identified HNSCC-related proteases support the observation that Probe 1 is still processed despite furin inhibition, and the low norm scores generally fit well with our experimental result, which showed almost no cleavage by healthy tissue (Fig. 3G and S10†).
Therefore, using furin as a surrogate marker for tumor tissue, we successfully developed probes that could differentiate between tumor tissue and healthy tissue. Future studies might combine multiple probes. For example, Probe a (faster cleavage, limited by off-target cleavage) could be multiplexed with Probe 1 (lower off-target cleavage, limited by non-furin-specific cleavage and slower cleavage), or different proteases could be addressed simultaneously by a set of probes that are developed for use with other proteases related to HNSCC (e.g., matrix metalloproteinases or dipeptidyl peptidase IV).34,35 In general, our approach of combining information gained from biological substrates (which usually stem from the interaction of proteases with full-length proteins) and from peptide-library-based analyses of cleavage sites (i.e., less constrained, shorter sequences) proved useful. Even for a comparably long and well-studied protease such as furin, we found a novel cleavage specificity (M↓S/TA). While Probe 1 is not cleaved as fast as the natural sequence-based Probe a, we are able to address a new specificity of the same protease with less off-target cleavage by healthy tissue, as discussed above.
Conclusions
Here, we developed protease responsive peptide-probes by combining biological and analytical information. We started our search using furin, a protease associated with HNSCC. Although the resulting probes were not strictly Furin selective, they successfully differentiated tumor and healthy tissues in the supernatant of human biopsies. The approach may be further fine-tuned to HNSCC or other tumors by multiplexing with additional probes. A direct measurement on tissues has the downside of autofluorescence interfering with the read-out of certain dyes. However, we avoided this challenge by designing our probes for measurements in the supernatant. These probes, used alone or in combination with other probes, have the potential to substantially reduce surgery times for tumor removal. In addition, they offer a complementary approach to fluorescence-guided surgery by being non-invasive and enabling the testing of peripheral tissue surrounding the tumor. In summary, such approaches can enhance surgical outcomes, thereby improving patient well-being.Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Original data sets are available from the corresponding authors, Lorenz Meinel and Christian Linz, upon reasonable request.Author contributions
D. R., B. t. M., and M. D. contributed equally to: investigation, conceptualization, writing original draft, methodology. M. G.: Formal Analysis, Investigation. J. F.: Resources. L. H.: Resources. A.-M. F.: Resources. P. H.: Software. T. L.: Writing – Review & Editing. C. L.: Funding acquisition, Supervision, Resources, Writing – Review & Editing. L. M.: Funding acquisition, Supervision, Writing – Review & Editing.Conflicts of interest
Lorenz Meinel, Tessa Lühmann, Debora Reinhardt, Marc Driessen, Björn ter Mores, and Marcus Gutmann are inventors of patent applications related to the present research.Acknowledgements
We acknowledge the financial support of the collaborative research center TRR225, project B06, Andreas Schlosser and Stephanie Lamer for acquiring MS, and Ryan DeLaney for proof-reading. Original data sets are available from the corresponding authors, LM and CL, upon reasonable request.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68(6), 394–424, DOI:10.3322/caac.21492.
- D. E. Johnson, B. Burtness, C. R. Leemans, V. W. Y. Lui, J. E. Bauman and J. R. Grandis, Head and neck squamous cell carcinoma, Nat. Rev. Dis. Primers, 2020, 6(1), 92, DOI:10.1038/s41572-020-00224-3.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Cancer statistics, 2022, Ca-Cancer J. Clin., 2022, 72(1), 7–33, DOI:10.3322/caac.21708.
- L. Q. M. Chow, Head and Neck Cancer, N. Engl. J. Med., 2020, 382(1), 60–72, DOI:10.1056/NEJMra1715715.
- J. S. Cooper, T. F. Pajak, A. A. Forastiere, J. Jacobs, B. H. Campbell, S. B. Saxman, J. A. Kish, H. E. Kim, A. J. Cmelak and M. Rotman, et al., Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck, N. Engl. J. Med., 2004, 350(19), 1937–1944, DOI:10.1056/NEJMoa032646.
- S. G. Hakim, R. von Bialy, M. Falougy, D. Steller, L. Tharun, D. Rades, P. Sieg and U. Alsharif, Impact of stratified resection margin classification on local tumor control and survival in patients with oral squamous cell carcinoma, J. Surg. Oncol., 2021, 124(8), 1284–1295, DOI:10.1002/jso.26655.
- N. Osazuwa-Peters, M. C. Simpson, L. Zhao, E. A. Boakye, S. I. Olomukoro, T. Deshields, T. M. Loux, M. A. Varvares and M. Schootman, Suicide risk among cancer survivors: Head and neck versus other cancers, Cancer, 2018, 124(20), 4072–4079, DOI:10.1002/cncr.31675.
- M. L. Hinni, A. Ferlito, M. S. Brandwein-Gensler, R. P. Takes, C. E. Silver, W. H. Westra, R. R. Seethala, J. P. Rodrigo, J. Corry and C. R. Bradford, et al. Surgical margins in head and neck cancer: a contemporary review, Head Neck, 2013, 35(9), 1362–1370, DOI:10.1002/hed.23110.
- S. E. Singletary, Surgical margins in patients with early-stage breast cancer treated with breast conservation therapy, Am. J. Surg., 2002, 184(5), 383–393, DOI:10.1016/s0002-9610(02)01012-7.
- M. J. Whitley, D. M. Cardona, A. L. Lazarides, I. Spasojevic, J. M. Ferrer, J. Cahill, C. L. Lee, M. Snuderl, D. G. Blazer, , 3rd and E. S. Hwang, et al. A mouse-human phase 1 co-clinical trial of a protease-activated fluorescent probe for imaging cancer, Sci. Transl. Med., 2016, 8(320), 320–324, DOI:10.1126/scitranslmed.aad0293.
- B. L. Smith, M. A. Gadd, C. R. Lanahan, U. Rai, R. Tang, T. Rice-Stitt, A. L. Merrill, D. B. Strasfeld, J. M. Ferrer and E. F. Brachtel, et al. Real-time, intraoperative detection of residual breast cancer in lumpectomy cavity walls using a novel cathepsin-activated fluorescent imaging system, Breast Cancer Res. Treat., 2018, 171(2), 413–420, DOI:10.1007/s10549-018-4845-4.
- B. L. Smith, C. R. Lanahan, M. C. Specht, B. N. Kelly, C. Brown, D. B. Strasfeld, J. M. Ferrer, U. Rai, R. Tang and T. Rice-Stitt, et al. Feasibility Study of a Novel Protease-Activated Fluorescent Imaging System for Real-Time, Intraoperative Detection of Residual Breast Cancer in Breast Conserving Surgery, Ann. Surg. Oncol., 2020, 27(6), 1854–1861, DOI:10.1245/s10434-019-08158-1.
- H. Li, Q. Yao, W. Sun, K. Shao, Y. Lu, J. Chung, D. Kim, J. Fan, S. Long and J. Du, et al. Aminopeptidase N Activatable Fluorescent Probe for Tracking Metastatic Cancer and Image-Guided Surgery via in Situ Spraying, J. Am. Chem. Soc., 2020, 142(13), 6381–6389, DOI:10.1021/jacs.0c01365 , From NLM Medline.
- P. Hamm, L. Meinel and M. D. Driessen, An Introductory Guide to Protease Sensitive Linker Design Using Matrix Metalloproteinase 13 as an Example, ACS Biomater. Sci. Eng., 2024, 10(6), 3693–3706, DOI:10.1021/acsbiomaterials.4c00407.
- M. Schwake, S. Schipmann, M. Müther, M. Köchling, A. Brentrup and W. Stummer, 5-ALA fluorescence–guided surgery in pediatric brain tumors—a systematic review, Acta Neurochir., 2019, 161(6), 1099–1108, DOI:10.1007/s00701-019-03898-1.
- J. S. D. Mieog, F. B. Achterberg, A. Zlitni, M. Hutteman, J. Burggraaf, R. J. Swijnenburg, S. Gioux and A. L. Vahrmeijer, Fundamentals and developments in fluorescence-guided cancer surgery, Nat. Rev. Clin. Oncol., 2022, 19(1), 9–22, DOI:10.1038/s41571-021-00548-3.
- D. E. Bassi, H. Mahloogi, R. Lopez De Cicco and A. Klein-Szanto, Increased furin activity enhances the malignant phenotype of human head and neck cancer cells, Am. J. Pathol., 2003, 162(2), 439–447, DOI:10.1016/S0002-9440(10)63838-2.
- D. E. Bassi, H. Mahloogi, L. Al-Saleem, R. Lopez De Cicco, J. A. Ridge and A. J. Klein-Szanto, Elevated furin expression in aggressive human head and neck tumors and tumor cell lines, Mol. Carcinog., 2001, 31(4), 224–232, DOI:10.1002/mc.1057.
- M. Mbikay, F. Sirois, J. Yao, N. G. Seidah and M. Chretien, Comparative analysis of expression of the proprotein convertases furin, PACE4, PC1 and PC2 in human lung tumours, Br. J. Cancer, 1997, 75(10), 1509–1514, DOI:10.1038/bjc.1997.258.
- M. Cheng, P. H. Watson, J. A. Paterson, N. Seidah, M. Chretien and R. P. Shiu, Pro-protein convertase gene expression in human breast cancer, Int. J. Cancer, 1997, 71(6), 966–971, DOI:10.1002/(sici)1097-0215(19970611)71:6<966::aid-ijc10>3.0.co;2-4.
- U. Eckhard, P. F. Huesgen, O. Schilling, C. L. Bellac, G. S. Butler, J. H. Cox, A. Dufour, V. Goebeler, R. Kappelhoff, U. A. D. Keller, T. Klein, P. F. Lange, G. Marino, C. Morrison, A. Prudova, D. Rodriguez, A. E. Starr, Y. Wang and C. M. Overall, Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses, Matrix Biol., 2016, 49, 37–60, DOI:10.1016/j.matbio.2015.09.003.
- O. Schilling, U. auf dem Keller and C. M. Overall, Factor Xa subsite mapping by proteome-derived peptide libraries improved using WebPICS, a resource for proteomic identification of cleavage sites, Biol. Chem., 2011, 392(11), 1031–1037, DOI:10.1515/BC.2011.158.
- S. Tian, A 20 Residues Motif Delineates the Furin Cleavage Site and its Physical Properties May Influence Viral Fusion, Biochem. Insights, 2009, 2 DOI:10.4137/bci.S2049.
- O. Schilling and C. M. Overall, Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites, Nat. Biotechnol., 2008, 26(6), 685–694, DOI:10.1038/nbt1408.
- O. Schilling, P. F. Huesgen, O. Barre, U. Auf dem Keller and C. M. Overall, Characterization of the prime and non-prime active site specificities of proteases by proteome-derived peptide libraries and tandem mass spectrometry, Nat. Protoc., 2011, 6(1), 111–120, DOI:10.1038/nprot.2010.178.
- B. Ter Mors, M. D. Driessen, A. Seher, I. R. Haubitz, M. Raschig, M. Nowak, Y. Jockel-Schneider, C. Linz and L. Meinel, The development of matrix-metalloproteinase responsive sensors for the machine-independent detection of oral inflammation, Royal Society of Chemistry (RSC), 2023, vol. 4, 10.1039/d3sd00031a.
- M. C. Ferrall-Fairbanks, Z. T. Barry, M. Affer, M. A. Shuler, E. W. Moomaw and M. O. Platt, PACMANS: A bioinformatically informed algorithm to predict, design, and disrupt protease-on-protease hydrolysis, Protein Sci., 2017, 26(4), 880–890, DOI:10.1002/pro.3113.
- Y. Zhang, L. Zhang, J. Wu, Y. Yu, S. Liu, T. Li, Q. Li, R. Ding, H. Wang and J. Nie, et al. A second functional furin site in the SARS-CoV-2 spike protein, Emerging Microbes Infect., 2022, 11(1), 182–194, DOI:10.1080/22221751.2021.2014284.
- N. D. Rawlings, A. J. Barrett, P. D. Thomas, X. Huang, A. Bateman and R. D. Finn, The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database, Nucleic Acids Res., 2018, 46(D1), D624–D632, DOI:10.1093/nar/gkx1134.
- S. Tian, Q. Huang, Y. Fang and J. Wu, FurinDB: A Database of 20-Residue Furin Cleavage Site Motifs, Substrates and Their Associated Drugs, Int. J. Mol. Sci., 2011, 12(2), 1060–1065, DOI:10.3390/ijms12021060.
- A. Basak, M. Zhong, J. S. Munzer, M. Chretien and N. G. Seidah, Implication of the proprotein convertases furin, PC5 and PC7 in the cleavage of surface glycoproteins of Hong Kong, Ebola and respiratory syncytial viruses: a comparative analysis with fluorogenic peptides, Biochem. J., 2001, 353(3), 537–545, DOI:10.1042/0264-6021:3530537.
- S. Xia, Q. Lan, S. Su, X. Wang, W. Xu, Z. Liu, Y. Zhu, Q. Wang, L. Lu and S. Jiang, The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin, Signal Transduction Targeted Ther., 2020, 5(1), 92, DOI:10.1038/s41392-020-0184-0.
- N. D. Rawlings, MEROPS the Peptidase Database, https://merops.sanger.ac.uk/ (accessed 18 February 2023) Search PubMed.
- T. Mizushima, S. Ohnishi, Y. Shimizu, Y. Hatanaka, K. C. Hatanaka, Y. Kuriki, M. Kamiya, A. Homma, K. Yamamoto and S. Ono, et al. Rapid detection of superficial head and neck squamous cell carcinoma by topically spraying fluorescent probe targeting dipeptidyl peptidase-IV, Head Neck, 2018, 40(7), 1466–1475, DOI:10.1002/hed.25126.
- Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(9), 4317–4322, DOI:10.1073/pnas.0910261107.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, PICS results from P6 to P6′ as a table, peptide and probe characterization via HPLC and LC-MS, concentration series of cleaved Probe a, Ponceau S staining of furin western blot, microscopy images of collected tissues, probe-cleavage experiments, stability of Probe a cleavage, cleavage experiment in the presence of a broad-range inhibitor, and potential competitive proteases identified by Prot-Agonist (PDF). See DOI: https://doi.org/10.1039/d4sd00047a |
| ‡ D. R., B. t. M., and M. D. contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |