
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2D capsid formation within an oscillatory energy landscape: orderly self-assembly depends on the interplay between a dynamic potential and intrinsic relaxation times†
Jessica K.
Niblo
 a,
Jacob R.
Swartley
a,
Zhongmin
Zhang
b and
Kateri H.
DuBay
*a
a,
Jacob R.
Swartley
a,
Zhongmin
Zhang
b and
Kateri H.
DuBay
*a
aDepartment of Chemistry, University of Virginia, McCormick Road, PO Box 400319, Charlottesville, VA 22903-4319, USA. E-mail: dubay@virginia.edu
bDepartment of Chemistry, University of North Carolina at Chapel Hill, Campus Box 3290, Chapel Hill, NC 27599-3290, USA
First published on 18th July 2024
Abstract
Multiple dissipative self-assembly protocols designed to create novel structures or to reduce kinetic traps have recently emerged. Specifically, temporal oscillations of particle interactions have been shown effective at both aims, but investigations thus far have focused on systems of simple colloids or their binary mixtures. In this work, we expand our understanding of the effect of temporally oscillating interactions to a two-dimensional coarse-grained viral capsid-like model that undergoes a self-limited assembly. This model includes multiple intrinsic relaxation times due to the internal structure of the capsid subunits and, under certain interaction regimes, proceeds via a two-step nucleation mechanism. We find that oscillations much faster than the local intrinsic relaxation times can be described via a time averaged inter-particle potential across a wide range of interaction strengths, while oscillations much slower than these relaxation times result in structures that adapt to the attraction strength of the current half-cycle. Interestingly, oscillation periods similar to these relaxation times shift the interaction window over which orderly assembly occurs by enabling error correction during the half-cycles with weaker attractions. Our results provide fundamental insights to non-equilibrium self-assembly on temporally variant energy landscapes.
1 Introduction
Self-assembly is the process by which a disordered system forms ordered patterns or nanostructures without external intervention due to the interactions that are encoded within the assembling components and their environment. The driving forces behind self-assembly can organize lipids into bilayers,1 gather capsomers into viral capsids,2–6 and arrange block copolymers into a wide range of microphase topologies.7Several studies have worked to uncover governing principles that would enable the design of interparticle interactions that lead to well-ordered, self-assembled equilibrium states. As a result, we now know that the strength, placement, and specificity of the interactions between the assembling components,8–13 their shapes,8,9,12,14 and their concentrations5,15 can all be tuned to stabilize a specific equilibrium target structure. However, the ability to reach these equilibrium assemblies is highly dependent upon the assembly kinetics.5,16 Strengthening the interactions that lower the free energy of the target structure often increases the kinetic barriers to its formation, making it a challenge to design components that reliably self-assemble on a reasonable timescale.10,16–18 Insightful work has been done to characterize the different dynamic pathways a system can take during self-assembly and the various types of kinetic traps that may emerge.18,19 One recent article has even been able to optimize interactions that not only select a target equilibrium structure, but also simultaneously control specific kinetic features along its assembly pathway.20
Alternatively, dissipative self-assembly processes can result in well-ordered structures by driving a system out of equilibrium and either creating new assembly routes towards equilibrium structures or forming non-equilibrium steady-states (NESSs).21,22 These non-equilibrium self-assembly pathways couple an assembling system to an energy source, such as when particles are self-propelled with a constant directional force13,23,24 or when assembly occurs within a shear flow.25,26 Recently, large deviation theory has been successfully employed to optimize both interactions and external shear forces in order to target specific steady states.25
The energy source in dissipative self-assembly may be temporally modulated through the change of an internal or external parameter, such as environmental changes that modify the interactions between assembling particles27–29 or that change their interactions with an external field.30–35 The self-assembly of many biological structures occurs within complex and ever-changing environments, and even certain naturally-forming non-biological materials appear to require time-variant environments to assemble – recent investigations have revealed that cycling between undersaturation and supersaturation may be necessary for the formation of naturally-occurring dolomite.36,37
One way to optimize a time-dependent dissipative self-assembly pathway is to employ feedback control, in which the assembly process is monitored in real time and the driving forces are adjusted on the fly, based on that feedback, to guide the process towards a desired outcome. One such approach adjusted the inter-particle interactions during assembly simulations based on the ratio of correlation and response functions and the degree to which they indicated an optimal balance of local microscopic reversibility (to avoid kinetic traps) and overall global irreversibility (towards assembly).38 More generally, it has been shown that high dimensional non-equilibrium time-dependent forces are able to guide an assembly towards a desired structure, however there is an unavoidable energetic cost to doing so.39 Bevan and coworkers constructed an experimental system to demonstrate the feasibility of feedback control: the 2D assembly of charged colloids were monitored in real time via optical microscopy while being subjected to a tunable electric potential. Within this set-up, the electric potential was adjusted based on feedback from the structural order parameter in order to construct perfectly crystalline configurations.40–42 Even so, it is clear that the need to monitor and adjust assembly conditions in real time would present significant difficulties to widely implementing feedback control approaches.
As an alternative to monitoring and adjusting each assembly process in real-time, information can be gathered from multiple simulations or experimental realizations of a dissipative assembly process and used to construct time-dependent protocols that are optimized for the ensemble of likely assembly pathways. One such approach employed evolutionary reinforcement learning to determine time-dependent temperature and chemical potential protocols for the efficient assembly of patchy disks into desired polymorphs.43 Similarly, Markov state models have been constructed from simulations of colloidal oligomers and capsids in order to design optimal time-dependent interaction profiles for the finite-time folding and assembly of those systems.44 However, significant data is required in order to optimize each time-dependent protocol, which will depend on the specifics of the system, and the implementation of these time-dependent protocols in real systems may prove challenging.
Oscillatory or cyclic changes to the environment in which assembly proceeds may provide a more experimentally accessible avenue to design new dissipative assembly pathways. A number of studies have shown that the cyclical exposure of oil droplets to an external magnetic field can facilitate local relaxation processes in the resulting aggregates, thereby enabling them to overcome kinetic barriers to equilibration.30–35,45 In simulations of photosensitive nanoparticles, light-induced aggregation proceeded more rapidly after short pauses in the light irradiation.46 Simulations by Risbud and Swan found that toggling inter-particle depletion interactions on and off at a timescale that allowed for sufficient particle diffusion relaxed kinetic traps and resulted in the more rapid formation of low-defect colloidal crystals.47 Finally, joint experiment and simulation work showed that the cyclic toggling of an external electric field on a timescale close to the characteristic melting time could anneal defects in colloidal crystals.48
Temporal environmental oscillations may also provide experimentally accessible ways to create and maintain long-lived non-equilibrium steady states (NESSs). Tagliazucchi and coworkers used simulations and theory to show that novel non-equilibrium steady state phases of binary pH-sensitive colloids can form when pH oscillations are faster than the colloid's characteristic diffusional timescale,27,28 and a similar effect was observed with three dimensional close packed colloids.29 Additional simulations have shown that oscillations in inter-particle interactions and external fields can result in ellipsoids adopting a non-equilibrium chiral smectic phase49 and in the formation of non-equilibrium lamellar structures in homopolymer mixtures.50
Prior work has focused on the assembly of extended NESS structures or the annealing of defects in extended colloidal crystals. In this work, however, we investigate the assembly of a self-limited capsid-like model with multiple inherent length scales and relaxation timescales that arise from the internal structure of the capsid building blocks. Specifically, we probe the impact of oscillatory time-dependent interactions on the assembly mechanisms over a range of oscillation timescales and amplitudes. In Section 2, we outline the coarse grained viral capsid-like model. In Section 3.1, we discuss assembly with static interactions. Then, in Section 3.2, we perform simulations with oscillatory interactions that are faster than, slower than, or similar to inherent relaxation timescales within the model to probe their influence on assembly. Finally, in Section 3.3, we vary oscillation amplitude to investigate how these temporal oscillations can act as an error correction technique. As summarized in Section 4, we find that the inherent timescales of the model system are critical in determining the effect of fast, intermediate, and slow oscillatory interactions on the resulting assembly process.
2 Modeling
Viral capsid models have proven essential in the theoretical and computational study of self-assembly, due in part to their organization into self-limiting ordered structures.5,15,19,51–58 Inspired by earlier studies on capsid assembly, in this work we investigate the assembly of two-dimensional, rigid triangular particles that assemble into capsid-like hexamers, as shown in Fig. 1a. The model was based upon a similar one in Mallory and Cacciuto's work on the role of self-propulsion in capsid-like colloidal assembly.13 The placement of attractive particles (green) on only two edges of the triangles causes these monomer units to assemble either into distinct hexamers or into extended snake-like structures (see Fig. 1a).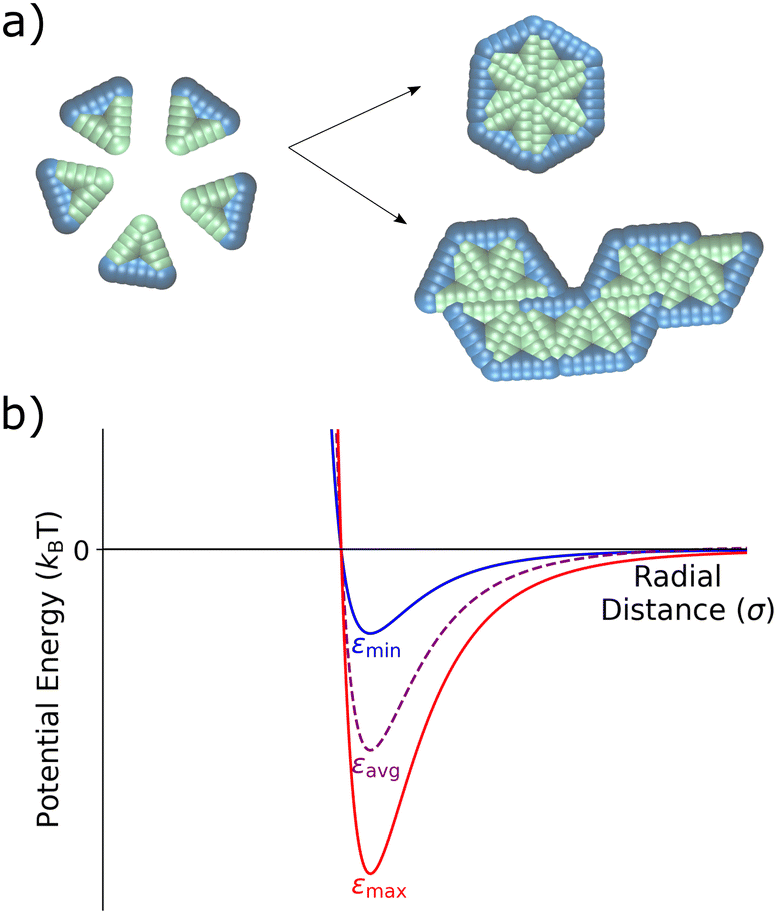 | ||
| Fig. 1 Schematic of the coarse-grained capsid-like model and inter-particle potential. (a) Triangular particles with two type A edges (green) and one type B edge (blue) start in a random distribution within the simulation box. Type A – type A interactions are defined via a Lennard-Jones potential, while the type A – type B and type B – type B particles interact via a Weeks–Chandler–Andersen potential.59 As the simulation proceeds, triangles can assemble into hexameric capsid-like structures or into larger snake-like aggregates. (b) The Lennard-Jones potential for the type A – type A interaction, showing the two different values of ε – εmin and εmax – with the time-averaged εavg shown in purple. | ||
The triangular monomers are composed of fifteen partially overlapping circular subparticles that are rigidly held together, as shown in Fig. 1a. Each subparticle is assigned one of two different types, A or B, which determines its interparticle potential. The subparticles on two edges of the equilateral triangle are assigned type A (green) and interact via a Lennard-Jones (LJ) potential, the strength of which can be tuned by changing ε, the LJ well-depth. The subparticles on the third edge are assigned type B (blue) and interact via the Weeks–Chandler–Andersen (WCA) potential.59 Thus, type B subparticles are purely repulsive, and interactions between type A and type B subparticles are also defined by the WCA potential. The placement of these differently-interacting subparticles introduces an overall anisotropic interaction between the individual triangular particles. Fig. 1b illustrates the attractive interactions between the type A subparticles of different triangular particles for various ε values. A more detailed descriptions of the particles and interactions can be found in the ESI.†
Oscillatory interactions are implemented by switching the ε value of the type A-type A interactions between εmin and εmax in a square wave pattern. We investigate the effect on capsid formation of variations in both the oscillation period and its amplitude (defined as the magnitude of the shift in each direction from the central, εavg-value). Variations in ε are considered here as a way to investigate the effects of a temporally-dependent interaction strength on assembly.
Simulations begin with a randomly distributed system of 150 triangular particles in a periodic box with no particle overlap. Langevin dynamics is utilized to evolve the system in time, which also provides a thermostat to maintain a constant temperature over the simulation and mimics the drag and random fluctuations associated with the dynamics of solvated particles. The system is first allowed to equilibrate with the type A interactions turned off and all subparticles interacting via the WCA potential, as this ensures that there is no particle overlap while establishing a random initial distribution of the triangles, both spatially and orientationally. After the equilibration period, the type A attractive interactions are turned on, and the system is progressed for 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000τ, where τ is time in reduced units. See ESI† for additional details.
000τ, where τ is time in reduced units. See ESI† for additional details.
3 Results and discussion
3.1 Capsid assembly is non-monotonic with attraction strength
We first model the formation of the complete capsid-like hexamers within a series of static environments, each with fixed effective interactions, ranging from ε = 0.75kBT to ε = 2.25kBT. Fig. 2a shows the resulting capsid yields and sample configurations as a function of ε for three different simulation times, and Fig. 2b shows how the presence of differently-sized aggregates changes with ε and simulation time.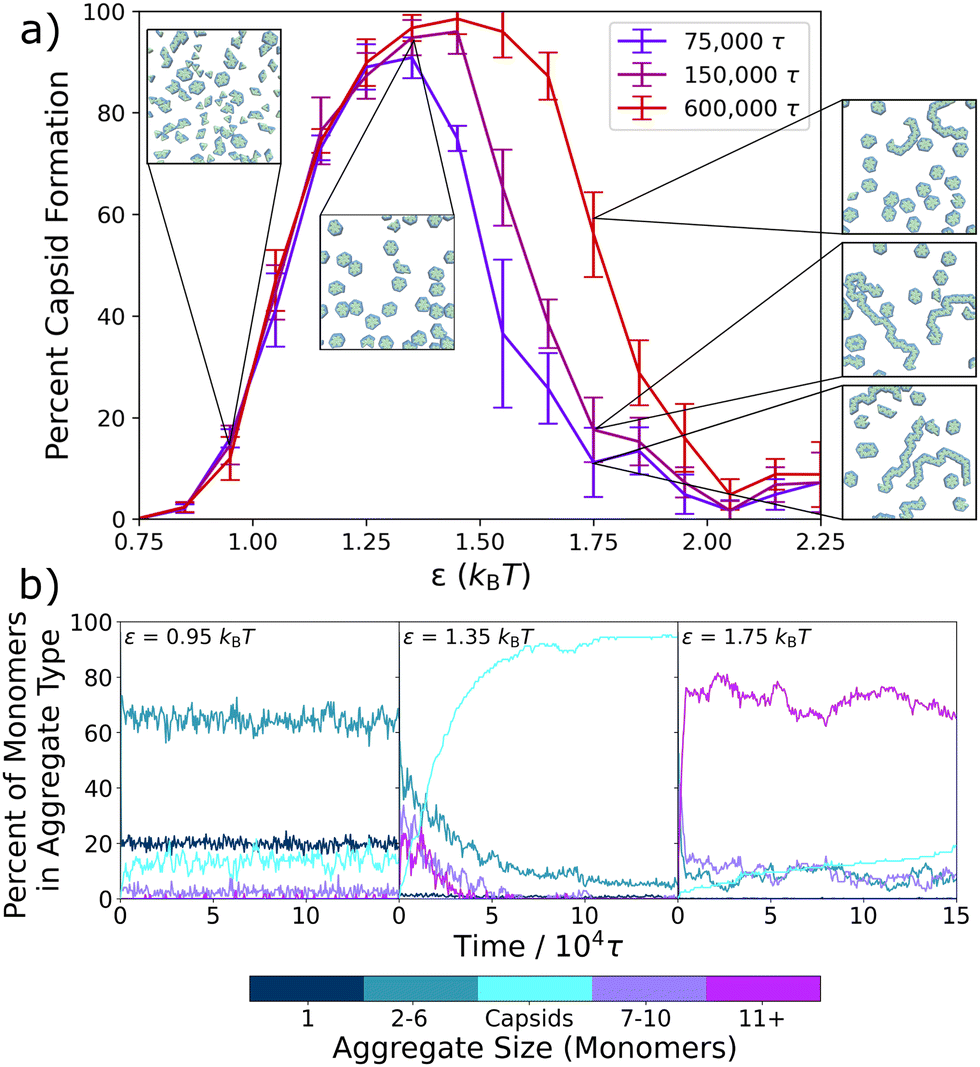 | ||
| Fig. 2 Capsid yield and aggregate formation within the static system. (a) Capsid yield curves are plotted after three different simulation times vs. the attraction strength, ε. As simulation time increases, the capsid yield does not change on the left hand side of the curve. However, on the right hand side, capsid yield increases with longer simulation time. Inset are final snapshots of the system at ε = 0.95kBT, ε = 1.35kBT, and ε = 1.75kBT. (b) The percentage of triangular monomers within each group of different sized aggregates is plotted over 150 000τ for three different ε values (ε = 0.95kBT, ε = 1.35kBT, and ε = 1.75kBT). Aggregates of varying size are shown with the color bar, with the hexameric structure shown in cyan. Capsid yields and kinetic traces are averaged over five independent trajectories, and the error bars in (a) display the standard deviation. | ||
In keeping with prior work on similar models of viral capsids,5,13,19,52,54,56 we observe in Fig. 2a that capsid assembly is non-monotonic with interaction strength. As ε increases from 0.75kBT to 1.35kBT, capsid yield also increases. However, above ε = 1.35kBT, capsid yield decreases. This non-monotonic trend is due to a transition from thermodynamically equilibrated systems on the left-hand side of the curve to kinetically constrained systems on the right-hand side as interactions become too strong for full equilibration to occur within the simulation time. Sampling for various lengths of time verifies this transition from thermodynamic to kinetic products, as the left-hand side of the curve remains unchanged for sampling times ranging from 75000τ to 600000τ, while the right-hand side shifts to higher values as sampling times lengthen and the kinetically constrained structures have additional time to relax.
The inset snapshots in Fig. 2a provide further evidence of this transition. At low ε interaction strengths, such as shown for ε = 0.95kBT, most triangles exist as free monomers or in small aggregates, and only a few complete capsids form. By ε = 1.35kBT, the snapshot shows almost complete capsid formation. However, at ε = 1.75kBT, well-formed capsids appear alongside larger, snake-like and kinetically trapped aggregates, with the number of snake-like aggregates decreasing and the number of capsids increasing as simulation times lengthen.
To better probe capsid assembly kinetics, in Fig. 2b we track the percent of the triangular monomers that are assembled into differently sized aggregates as a function of simulation time. At a weaker interaction strength of ε = 0.95kBT, the system almost instantaneously assembles into small aggregates of 2–6 monomers (turquoise) in length, which slightly decreases over the first 1 × 104τ of the simulation as a small number of capsids (cyan) assemble. At an intermediate attraction strength of ε = 1.35kBT, a significant number of longer aggregates composed of 7–10 (purple) and 11+ (magenta) monomers rapidly form initially, but then decrease on the same timescale as capsid structures emerge. At a stronger attraction of ε = 1.75kBT, where the capsid yield curve has crossed into the kinetic regime, most monomers rapidly assemble into the largest aggregates (magenta), which only slightly and gradually convert into capsids over the remainder of the simulation time. By 150000τ, most monomers remain in these very large aggregates due to the difficulties in overcoming the high energetic barriers associated with strong inter-particle interactions.
Interestingly, at the intermediate and stronger attraction values, the assembly process in Fig. 2b follows a non-classical, two-step assembly pathway.60,61 During non-classical assembly pathways, which have been seen in protein11,62,63 and colloidal particle crystallization,64–66 the system assembles into a condensed, disordered phase that has a lower free energy barrier to nucleation than the disassembled system, so that the final assembled structure grows from pre-formed aggregates.63,65,66 This process is clearly in evidence at both ε = 1.35kBT and ε = 1.75kBT in Fig. 2b, where capsid counts increase as the initially assembled larger aggregates decrease. To probe exactly the ε values at which this changeover in assembly mechanism occurs, we expand Fig. 2b in Fig. S2 (ESI†) and plot the results for values ranging from ε = 0.75kBT to ε = 2.15kBT at 0.10kBT intervals. Evidence for this two-step mechanism can be seen as early as ε = 1.25kBT, and a clear cross-over is seen around ε = 1.45kBT, where non-capsid monomers are present in smaller and larger aggregates in about equal numbers, with both types of aggregates decreasing as capsids form. At higher ε values, the two-step pathway dominates, with most monomers assembling rapidly into the larger, snake-like aggregates.
Having established the assembly behavior of the 2D model capsid system within a series of static environments, we now temporally vary the inter-monomer attractions by switching the strength of the Lennard-Jones potential between two values during assembly. We define the stronger attraction strength as εmax and the weaker interaction strength as εmin. The oscillation amplitude defines the distance of εmax and εmin from a central εavg value, where εavg = (εmax + εmin)/2 see Fig. 1. We also define the period of oscillation, τosc, as the time it takes to complete a full cycle, with half the cycle at εmax and the other half at εmin. In the rest of the paper, we investigate how these oscillatory interactions influence capsid formation across a variety of oscillation periods (Section 3.2) and oscillation amplitudes (Section 3.3).
3.2 Assembly depends on the oscillation frequency
First, we consider how different oscillation periods, τosc, affect the assembly process. During the εmax half-cycle, triangular monomers are more strongly attractive and assemble together, while in the εmin half-cycle, such structures may rearrange or be broken apart. The degree to which assembly and disassembly occurs depends upon the time spent in each half-cycle, and how that time compares to the time required for the system to equilibrate.Previous studies investigating oscillations in inter-colloidal potentials compare τosc to a characteristic diffusional time-scale, td = σ2/D, where σ specifies the size of the assembling colloids and D is the diffusion coefficient.27,47 Oscillation periods that are significantly shorter than the characteristic diffusional time-scale (τosc ≪ td) are considered to be at the fast oscillation limit, while at the slow oscillation limit (τosc » td), periods are long enough to allow the system to relax to different equilibrium structures during each oscillation half-cycle.27,28,47
As can be seen in the structure shown in Fig. 1, more than one length-scale is needed to fully describe the assembling particles; the diameter of the circular subparticles is σLJ = 0.25σ, while the edge length of the triangular particles is 1σ. In addition, at the higher attraction strengths we probe, capsid formation proceeds via the initial formation of a condensed disordered phase (see Fig. 2a) and its subsequent relaxation. The multiple length-scales and assembly pathways within this model are expected to influence how capsid formation varies with oscillation frequency.
To better understand the interplay of the oscillation period and these inherent length-scales, we list in Table 1 a set of key distances that characterize important energetic and structural changes, ranging from the distance required to reduce the attractive interaction to a tenth of its maximum strength to the full edge length of a single triangular particle. To estimate how long it would take for the triangular particles to traverse these distances, we plot the mean squared displacement vs. time for a single triangular particle in Fig. S3a (ESI†). Since the relevant length scales span the ballistic and diffusive time regimes, we estimate the time it takes for the particle to move over these critical distances directly from the results in Fig. S3a (ESI†). Additionally, we plot in Fig. S3b (ESI†) the rotational autocorrelation function vs. time for a single triangular particle and calculate the mean rotational lifetime using an exponential decay fit. Below, we first consider oscillations at the fast limit, where the period is shorter than the timescales associated with these key distances (τosc ≪ {td}), and show that the assembly behavior in this regime can be described by the ε value averaged over a single oscillation period, εavg. Next, we investigate slow oscillations, where the period is longer than the timescales that correspond to the distances in Table 1 (τosc ≫ {td}), such that the system has time to at least partially equilibrate to the current ε value during each half period of the oscillation. Lastly, we simulate oscillations in the intermediate regime (τosc ≈ {td}) and show how the window of capsid assembly shifts with the oscillation period.
| Characteristic distance | Distance (σ) | t d (τ) |
|---|---|---|
| a For the rotational relaxation, td describes the mean lifetime for the exponential decay from the rotational correlation function in Fig. S3b (ESI). | ||
| 0.72σLJ (−ε → −0.1ε) | 0.18 | 0.59 |
| Diameter of subparticle | 0.25 | 0.91 |
| Edge of triangle | 1.00 | 10.33 |
| Rotational relaxationa | — | 7.10 |
To test the behavior of our system at the fast oscillation limit, in Fig. 3 we compare the degree of capsid formation at static ε values to that at the corresponding εavg values for various oscillation amplitudes, monomer concentrations, and simulation times. An oscillation period of 0.02τ was chosen (see Fig. 3a), which is expected to yield fast limit behavior since it is much shorter than the calculated time-scales of interest in Table 1. In Fig. 3b, we confirm that this period reproduces the static non-monotonic yield curve across three different oscillation amplitudes. In Fig. 3c, we test the correspondence between the static and effective potentials across changes in particle density. The static yield curves (solid lines) at volume fractions of ϕ = 0.1 and ϕ = 0.005 differ, which is expected since a decreased volume fraction requires a stronger ε for capsid formation. However, at both densities, the oscillatory potential with a period of 0.02τ and an amplitude of 0.2kBT (dashed lines) returns the corresponding static yield curve for εavg. Finally, in Fig. 3d, we investigate the timescales of relaxation at the fast oscillation limit as compared to those in the static system by comparing the oscillatory and static capsid yield curves after simulation times of 75000τ, 150000τ, and 600000τ. We find that capsid formation at the fast oscillation limit increases with simulation time in precisely the same way as capsid formation does within the corresponding static potential. This result suggests that kinetic traps affect the system dynamics at the fast oscillation limit in the same way as in the static system, supporting the prior claim that even a system's non-equilibrium dynamics can be described by an appropriate time-averaged potential at the fast oscillation limit28 and explaining the observation that in both Fig. 3b and c, the correspondence between the oscillatory and static curves is as good for the kinetically constrained right-hand side of the curve as it is for the thermodynamically equilibrated left-hand side. In summary, when the Lennard-Jones potential is oscillated at a very short period of 0.02τ, the system organizes in the same manner as with the non-oscillatory potential across a number of tested variations.
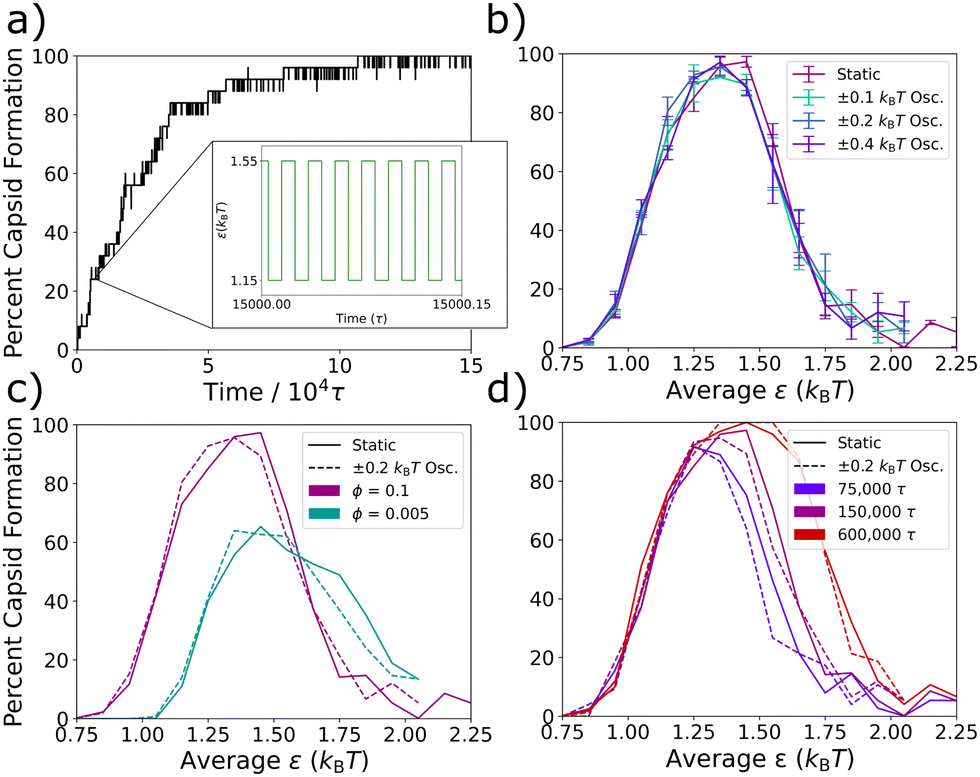 | ||
| Fig. 3 Assembly at the fast oscillation limit. (a) A kinetic trace is shown from a single 150000τ trajectory of the assembling capsids at an attraction strength of εavg = 1.35kBT, an oscillation amplitude of 0.2kBT, and an oscillation period of 0.02τ. Inset displays a zoomed in schematic of the oscillation waveform to compare the oscillation frequency to the timescale of capsid assembly. (b) Capsid yield curves at 150000τ are plotted for three different amplitudes as well as the non-oscillatory system at a period of 0.02τ. (c) Capsid yield curves at 150000τ are plotted for the oscillatory system at a period of 0.02τ and the static potential are shown at two different densities. (d) Capsid yield curves at three different simulation times are plotted for the static system (solid lines) and compared to the results for the system with interactions oscillated for a period of 0.02τ with an amplitude of 0.2kBT (dashed lines). Percent capsid formation is averaged over three independent trajectories, and the average standard deviations for the capsid yield measurements were ±3.2% for the static systems and ±3.8% for the oscillatory systems. | ||
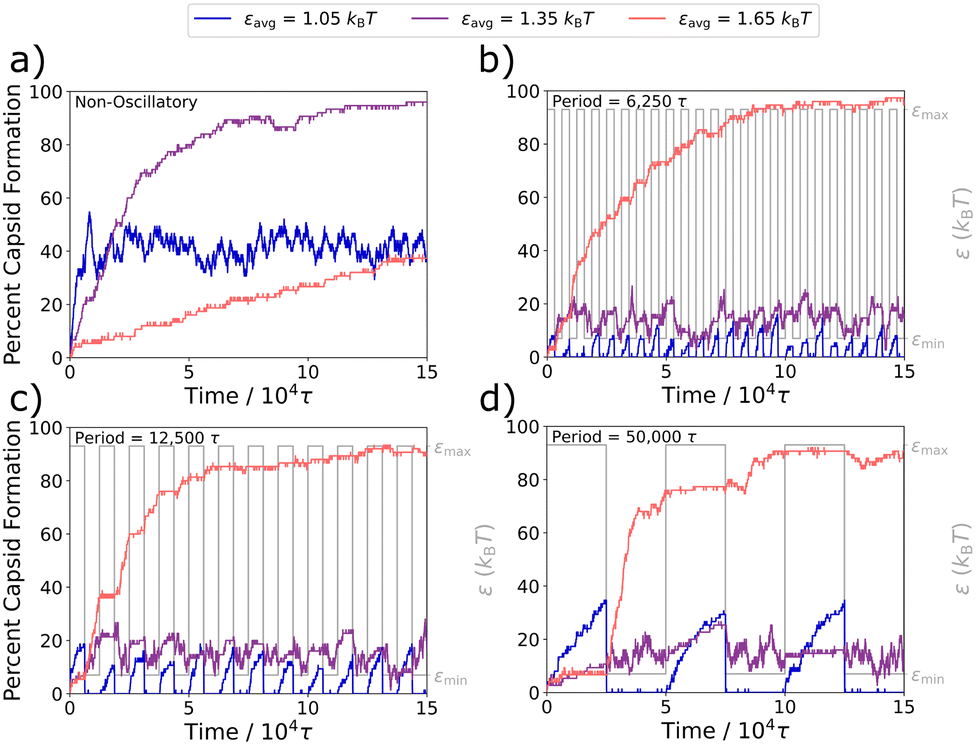 | ||
| Fig. 4 Capsid formation changes with each half-cycle. Capsid formations at different εavg values are shown at an oscillation amplitude of 0.4kBT for three longer periods, as compared to the non-oscillatory capsid formation curves in (a). We show (b) τosc = 6250τ, (c) τosc = 12500τ, and (d) τosc = 50000τ. Each period has the waveform overlaid in gray to show the oscillation period. The kinetic traces are averaged over three independent trajectories. | ||
In the Fig. 4a static case, a weaker attraction strength of εavg = 1.05kBT results in the capsid yield quickly plateauing to about 40%, a moderate attraction strength of εavg = 1.35kBT results in nearly complete capsid assembly over a slightly longer time-scale, and a stronger interaction strength of εavg = 1.65kBT results in kinetic trapping and slow capsid assembly.
In Fig. 4b–d, we plot capsid formation along with the oscillation profiles for periods of 6250τ, 12500τ, and 50000τ, where τosc ≫ {td}. For the weakest interaction strength of εavg = 1.05kBT (in blue), at the given amplitude of 0.4kBT, the simulation oscillates between a well-depth of εmin = 0.65kBT and εmax = 1.45kBT. For all three τosc periods, capsids rapidly assemble in the εmax half-cycle and rapidly disassemble in the εmin half-cycle, where interactions are too weak for the structures to remain intact. As with the weakest εavg value, simulations at the strongest εavg value investigated here of 1.65kBT (in orange) also display significantly different assembly behaviors in each half-cycle. At these stronger attraction strengths, capsids no longer rapidly fall apart during the εmin half-cycles, however the kinetically trapped snake-like structures can still relax to form additional capsids. As a result, significant capsid formation occurs during the εmin half-cycles of εavg = 1.65kBT. These capsids remain intact during the εmax half-cycles, however the stronger attractive forces keeping the subparticles together also largely arrest the relaxation of kinetic traps and thus the formation of additional capsids. Behavior with the intermediate εavg of 1.35kBT (in purple) is more complex. The system oscillates between εmin = 0.95kBT and εmax = 1.75kBT, both of which result in only modest capsid yields in the static system (see Fig. 2a), and we observe that same modest yield in all three longer oscillation periods in Fig. 4b–d, despite the fact that a high yield is obtained for ε = 1.35kBT in the static case in Fig. 4a.
Overall, when considering oscillations that are slow compared to the characteristic displacement times in Table 1, we see that the system substantially adapts to each half-cycle, which can result in capsid yields that grow and shrink over each oscillation or in capsid yields that remain static during most εmax half-cycles and then further increase during most εmin half-cycle. It is important to note that even the very long oscillation periods investigated here are still shorter than the time needed to fully relax the kinetic traps of the system, which can take longer than 75000τ even at ε = 1.45kBT, just slightly above the capsid yield curve's peak (see Fig. 2a).
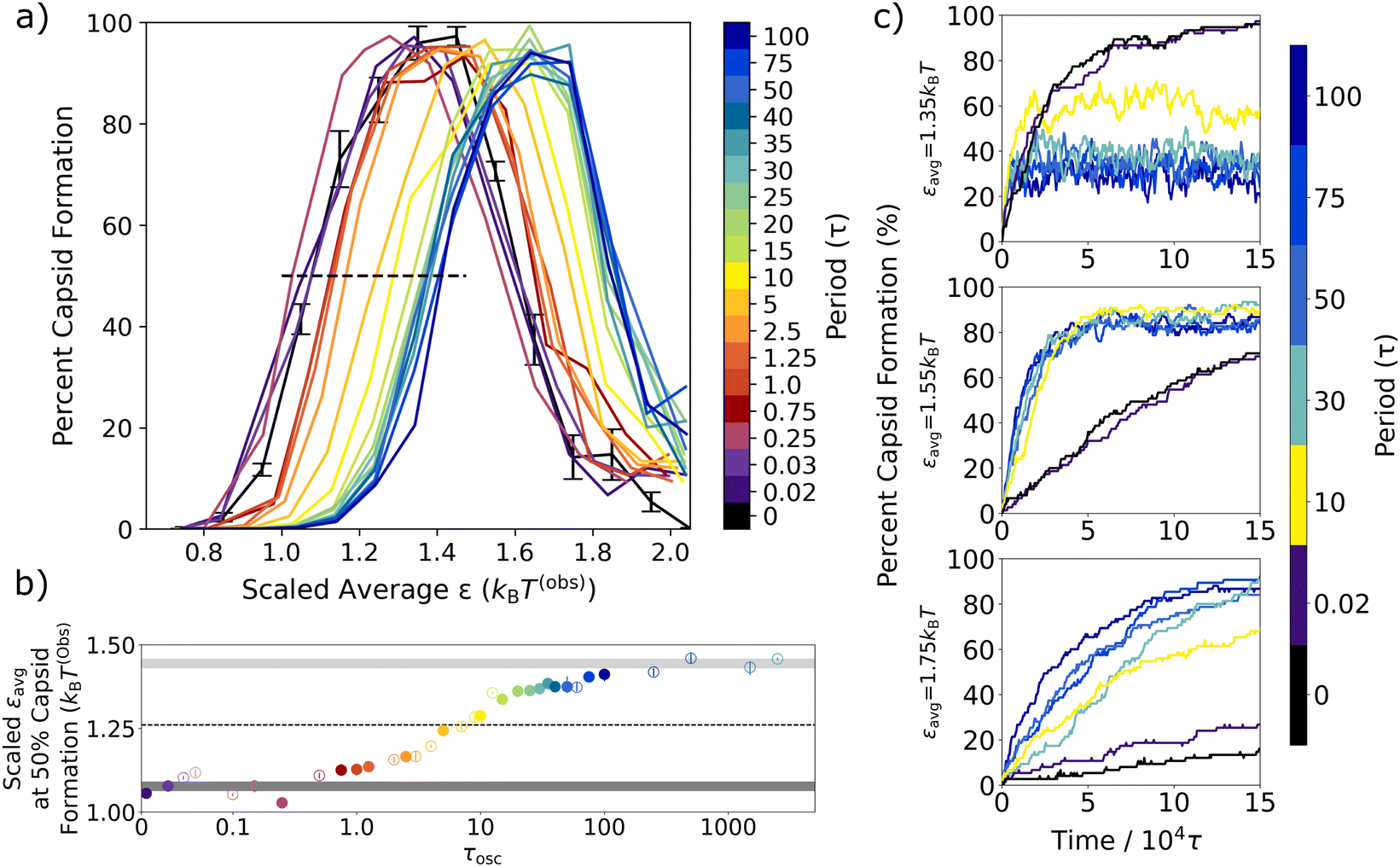 | ||
| Fig. 5 Capsid yield curves for different periods of oscillation. (a) Capsid yield curves after 150000τ are compared to the scaled εavg value (in units of kBT(obs)) for different oscillation periods at an amplitude of 0.4kBT. The black, dashed line indicates 50% capsid formation on the left hand side of the yield curves. (b) The scaled εavg values that result in 50% capsid formation on the left hand side of the capsid yield curve are plotted vs. τosc. The dark gray bar indicates the non-oscillatory regime, the light gray bar plots the upper asymptotic limit of a sigmoidal fit, and the dotted line indicates the midpoint between the two. Points in (b) that are shown in (a) are represented by a closed circle, while additional periods not shown in (a) are denoted by an open circle. (c) The percentage of assembled capsids is plotted over 150 000τ for three different εavg values (εavg = 1.35kBT, εavg = 1.55kBT, εavg = 1.75kBT) for six different oscillation periods and the static case are shown with the color bar. Capsid yield and kinetic growth curves are averaged over three independent trajectories, and the error bars on the static yield curve show its standard deviation. | ||
For all periods, capsid assembly remains non-monotonic with interaction strength, and the window of orderly assembly is approximately the same width for systems undergoing oscillatory interactions as for those with static interactions. However, as the period of oscillation increases in Fig. 5a, the capsid yield curves shift to higher εavg values, which is similar to results from simulations on colloidal crystal grown under toggled interactions.67 Our results show that the window of orderly assembly can be expanded into interaction strengths that are typically infeasible due to their long relaxation times.
One technical complication arises as the oscillation period increases slightly from the fast oscillation limit period of 0.02τ. From about 0.03τ to about 5τ, where τosc is similar to the timescale of the thermostat relaxation, we find that the Langevin thermostat is not able to maintain the target temperature due to the rate of energy transfer as the interparticle potentials oscillate. To enable comparisons that include these oscillation frequencies, the εavg values in Fig. 5 were simply scaled by reporting ε values in units of kBT(obs), where T(obs) is the actual observed temperature at each oscillation period. A plot of the observed temperatures vs. oscillation period is shown in Fig. S1 in the ESI.†
To quantify the capsid yield curve shift, in Fig. 5b we plot the appropriately scaled εavg value that results in 50% capsid formation on the left hand side of the yield curves in Fig. 5a (i.e., where it intersects the black dashed line) vs. the oscillation period, τosc. The ε value that results in 50% capsid formation in the static system is indicated by the dark gray bar, while the corresponding εavg value for the long oscillation periods is indicated by the light grey bar.
The result for the fast oscillation limit (τosc = 0.02τ) overlaps with the non-oscillatory dark gray bar, as previously observed in Fig. 3. For oscillation periods between 0.02τ and about 0.5τ, the scaled εavg values are clustered around the dark gray bar and there is no sustained shift away from the static ε value. After 0.5τ, however, the scaled εavg value at 50% capsid formation steadily increases with oscillation period, crosses the dotted line that indicates the halfway point between the fast and slow oscillation limits at a period of about τosc = 7τ, and then plateaus at the slow oscillation value (light grey bar).
To make sense of the shift with oscillation period in Fig. 5, we return to the inherent length-scales of our model and their corresponding local relaxation time-scales, as described in Table 1. After aggregates nucleate, there are three important local relaxation processes that facilitate the second stage of the hexagonal capsid formation from either smaller or larger aggregates: (1) the diffusion of subparticles away from one another; (2) the sliding of triangular particle edges along one another; and (3) the rotation of a triangular particle away from or towards another. All three of these movements are more likely to occur during the εmin half-cycle when attractions are weaker. Table 1 provides estimates for the timescales of these local relaxation processes, based on simulations of a single triangular particle (see Fig. S3, ESI†). First, the diffusion of one subparticle out of the LJ attractive well of a neighboring subparticle is characterized by the length-scale of the LJ attraction. The distance a subparticle must move for the attractive interaction to be reduced to a tenth of its full strength is 0.18σ, which is estimated to take approximately 0.6τ. Second, the edge length of a single triangular particle is 1.0σ, and the corresponding diffusion time for that distance is 10.3τ. Third, the mean lifetime for the rotational degree of freedom is estimated to be 7.1τ, based on the rotational correlation function calculated in Fig. S3 (ESI†). These intrinsic local relaxation timescales aid our interpretation of the shift in the capsid yield curves with oscillation period in Fig. 5.
At a period of τosc = 0.5τ, where the steady shift towards higher εavg values starts, there is just enough time during each εmin half-period for a subparticle to move away from a neighbor so that their attractive LJ interaction is reduced by two thirds. At a period of about 1.2τ, the εmin half-cycle is just slightly longer than the time it takes for a subparticle's LJ interaction to be reduced to a tenth of its maximum strength, on average. That is, particles that were originally interacting are likely to diffuse far enough from their original configurations so that they no longer experience a significant attraction to their original neighbors.
The black dashed line indicates the half-way mark between the fast and slow oscillation behaviors, which happens at an oscillation period of about τosc = 7τ. From Table 1, we see that several local relaxation processes that are helpful in the formation of capsids are achievable within an εmin half-period of 3.5τ. Within this half-cycle, particles can diffuse far enough from their original configurations so that they no longer experience a significant attraction to their original neighbors, even shifting the full diameter of a subparticle. If a particle located in an aggregate can shift a subparticle away from their original configuration, this could lower the energetic barrier for the triangle to diffuse from an aggregate structure.
After a period of 20τ, we observe the start of a plateau in the scaled εavg values that corresponds to 50% capsid formation. Interestingly, a period of 20τ corresponds to a half-cycle of 10τ, which is about the time needed for a triangular particle to diffuse 1σ, or the distance of one full edge of the triangle. This edge length is an important distance in disrupting longer aggregates, since a particle in the middle of a snake-like aggregate would need to diffuse about 1σ to leave the aggregate and thereby break up a longer aggregate. In addition, this length is central to the motions of a trimer of particles that enable full capsid formation from a half-capsid structure on the end of a snake-like aggregate, as can be seen in Fig. 1. Given the mean rotational lifetime of approximately 7τ, it is clear that by an oscillation period of 20τ, triangular particles can fully diffuse from aggregate structures and rotate to better align with other particles to facilitate capsid formation during the εmin half-cycles.
To probe the effect of oscillations on capsid assembly kinetics, in Fig. 5c we track the percent capsid formation as a function of simulation time for several oscillation periods and εavg values. As expected from prior theoretical work,28 the kinetics under the static potential and at the fast oscillation limit are indistinguishable for all εavg values. In contrast, intermediate oscillation periods result in faster capsid assembly rates relative to the static potential case for all εavg values. This increase in assembly rates has been previously observed in other oscillatory protocols.48,65 We find it is most dramatic at the higher εavg values (Fig. 5c, bottom two panels) since, at intermediate periods, the oscillations shift the capsid yield curve into the interaction regime where static assembly slows due to kinetic trapping. However, this increased rate holds even in the case of εavg = 1.35kBT, where the oscillatory cases with intermediate periods (10τ and longer) plateau more rapidly than the static case – even though they plateau at lower capsid yields.
A closer look at the εavg = 1.35kBT case (Fig. 5c, top panel) provides helpful insight as to why the capsid yield curve shifts to the right as the oscillation period increases but does not broaden. The oscillation amplitude in Fig. 5 is 0.4kBT, so the εavg = 1.35kBT case oscillates between an εmin value of 0.95kBT and an εmax value of 1.75kBT. At the fast oscillation limit, the capsid yield after 150 000τ should be equal to that of the static yield at ε = 1.35kBT (96%), which we observe. At the slow oscillation limit, we expect the yield to switch back and forth between the capsid yields expected for the εmin and εmax values, which are 12% and 14%, respectively. In the intermediate oscillation regime, we see that the system forms a non-equilibrium steady state where the steady-state capsid yield depends on the oscillation period. As the period increases, the capsid yield will eventually transition away from the fast oscillation limit value of 96% to the slow oscillation limit values of 12% (an equilibrium value) and 14% (a kinetically-determined value, which will depend on the initial configuration and the relaxation time). At that slow oscillation limit, it will no longer be in a steady-state, and the number of capsids will shift depending on the oscillation half-cycle. Overall, the observed yield curve on the left-hand side shifts to the right with increased oscillation period. On the right-hand side of the shifted curves, when εmin becomes too strong, the weaker half-cycle can no longer facilitate error correction, which results in the oscillatory capsid yield curves reproducing the static yield curve's non-monotonic behavior.
3.3 Shift in capsid yield with amplitude provides evidence for the critical role of error correction
Previously, Risbud and Swan47 and others30–35,45 observed that oscillating attractions could result in local relaxation, since the system was able to relax kinetic barriers via diffusion when the attractions were turned off. In our model, the formation of capsid-like structures is also limited by kinetic traps at stronger attractions, as demonstrated in Fig. 2a by the increase in capsid yield with longer simulation times. In addition, at these higher ε values, the initial formation of snake-like structures that can convert into capsid-like hexamers makes it clear that error correction – the ability of sub-optimally assembled particles to rearrange themselves into a more favorable structure – plays an important role in the total capsid yield, both in the static and oscillatory interaction cases. Indeed, in Fig. 4b–d, at the slow oscillation limit with an amplitude of 0.4kBT, we directly observed the effect of error correction on the capsid formation process during the lower εmin half-cycles for the case where εavg = 1.65kBT. Capsid yields almost always remained static in the εmax half-cycles, but ratcheted up to higher levels in the εmin half-cycles. In this section, we further probe how the εmin half-cycles affect error correction within this model system by varying the oscillation amplitude. As the amplitude increases, εavg stays the same, but attractions oscillate between a weaker εmin and stronger εmax. Results are shown in Fig. 6 for an intermediate oscillation period of 100τ, which is long enough for most local relaxation processes to occur – see Table 1 and the almost-completed shift towards the long-time oscillation value at 100τ in Fig. 5b.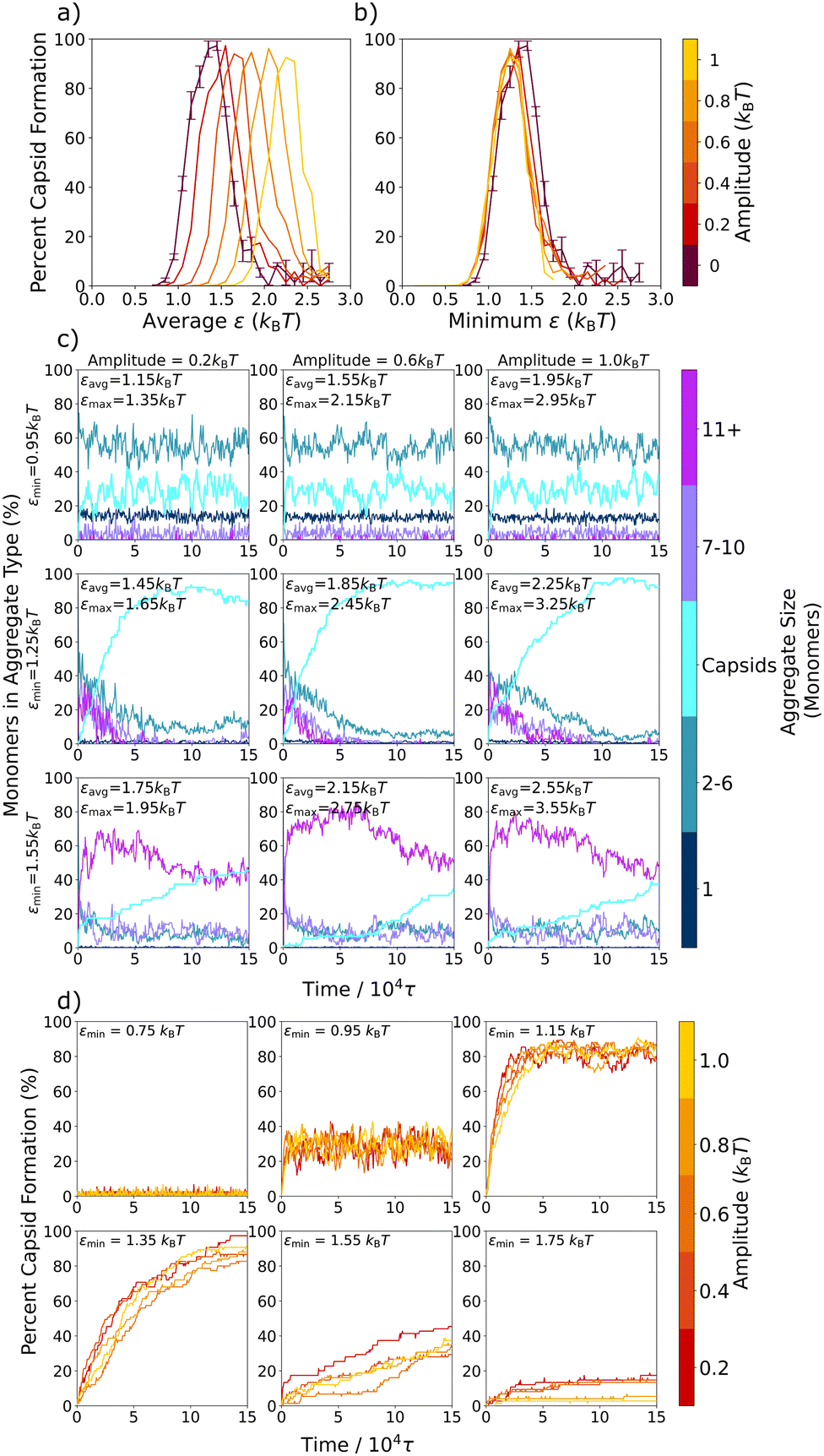 | ||
| Fig. 6 Capsid yield and aggregate formation for different oscillation amplitudes. Capsid yield curves are plotted in (a) vs. εavg and in (b) vs. εmin for five different oscillation amplitudes with an oscillation period of 100τ and the static potential case. The formation of different sized aggregates is plotted vs. time in (c) for three different amplitudes at a period of 100τ. Although they have different εavg values, εmin = 0.95kBT for all amplitudes in row 1, εmin = 1.25kBT for all amplitudes in row 2, and εmin = 1.55kBT for all amplitudes in row 3. (d) The capsid growth curves are plotted over 150 000τ for six different εmin values at five different oscillation amplitudes (shown with the color bar). Capsid yield curves and monomer counts in the different aggregate types are averaged over three independent trajectories. In the static case in (a) and (b), the error bars represent the standard deviation. | ||
In Fig. 6a, we show a series of capsid yield curves at different oscillation amplitudes plotted vs. εavg. As amplitude increases, yield curves shift to the right to higher εavg values – values where orderly assembly is not observed in the static system. However, when we plot these same yield curves vs. εmin instead of εavg in Fig. 6b, the curves for all amplitudes collapse into one curve, which is very close to the capsid yield curve for the static potential. This collapse of the yield curves indicates that, at oscillation periods sufficiently longer than the local intrinsic relaxation processes in the system, the main determinant of capsid yield in the oscillatory system is simply the value of εmin. Indeed, this full shift from the yields observed at εavg to those observed at εmin as the oscillation periods lengthen can be clearly seen in Fig. 5a.
To further probe this shift to the εmin value fully determining the yield at oscillation times longer than the local relaxation processes, in Fig. 6c we show the percent of the triangular monomers in each aggregate size as a function of time for a series of simulations with varying amplitudes and εmin values. From the top row to the bottom row, εmin increases, while within each row, εmin is held fixed while the amplitude increases from left to right. Notably, we find that the kinetic assembly traces for a given εmin value are essentially the same across the three amplitudes. In Fig. 6d we overlay the kinetic traces for full capsid assembly over more amplitudes and εmin values. Regardless of oscillation amplitude, the capsid yield curves collapse onto one another for each εmin value, demonstrating that εmin largely determines capsid assembly even as εavg changes. Similar kinetic curve collapses have been observed in experiments of polarizable colloids exposed to toggled magnetic fields32 and simulations of nanoparticles with toggled potentials.67 This universal εmin dependence shows that, as long as oscillation periods are longer than the local intrinsic relaxation processes and εmax is above the threshold for kinetic trap formation, the strength of εmin is what determines the degree to which kinetically trapped aggregates are able to relax into fully formed capsids.
Building on this insight, in Fig. 7, we plot capsid yield curves for static and oscillatory protocols where εmin is fixed at different attraction strengths, εmax is varied, and the results are plotted vs. εavg. On the left-hand size, there is a slight increase in the yield as the εavg value increases from the corresponding static case (indicated by the dot). In contrast, on the right-hand side, there is a drop off in the yield as εavg increases from the static case. In both cases, the yield appears to plateau at some point as εavg increases further. Essentially, further increases in attraction strength past some value of εmax do not change the resulting yield, which then depends only on the error correction enabled by the εmin value, as shown in Fig. 6. As a result, fixing εmin to a value just below the peak of the static capsid yield curve removes the non-monotonicity of the static yield curve.
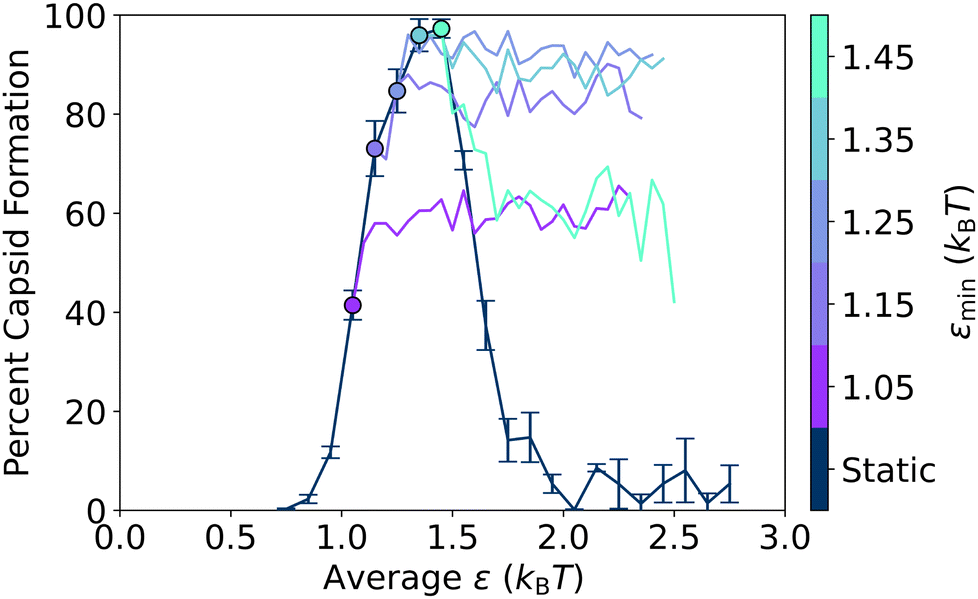 | ||
| Fig. 7 Capsid yield for oscillation protocols where εmin is held constant. Capsid yields are plotted at an oscillation period of 100τ for five fixed εmin conditions while εmax is varied, and the results are plotted vs. εavg. The static yield curve is shown for comparison, and the static result corresponding to each εmin series is indicated with the solid colored dot. All capsid yields plotted have been averaged over three independent trajectories. The error bars show the standard deviations on the static curve. | ||
4 Conclusion
In this work, we investigate how temporal oscillations influence the dissipative self-assembly of anisotropic 2D triangular particles that assemble into self-limited capsid-like structures. At stronger attractions, the formation of these hexamers occurs through a non-classical, two-step nucleation pathway and is prone to kinetic trapping, resulting in a non-monotonic dependence of capsid yield on attraction strength in both the static and oscillatory cases.Temporal oscillations in inter-particle attractions offer experimentally accessible dissipative self-assembly protocols for a wide range of materials. Such oscillations may be implemented by changes in temperature, external applied fields, light irradiation, pH changes, or other mechanisms, and in our study we implement them by changing the strength of the attractions in time by switching between two LJ well-depths. As in previous studies, we find that tuning the oscillations enables orderly assembly across a broader range of time-averaged interactions, as can be seen in the envelope of capsid yield curves in Fig. 5 and the full region of εavg that they span, which provides routes to substantive capsid yields at any of these time-averaged interaction strengths. In addition, we generally observe faster capsid formation under oscillatory conditions, providing a route to more rapidly produce self-limited assembled structures.33,48,67
In keeping with prior work,47,48 our results highlight the correspondence between the different oscillation timescales and the particle motions that govern local relaxation processes – in the case of our more complex anisotropic particles, these include both translations and rotations. At the limit of oscillations that are very fast compared to these intrinsic time-scales, assembly proceeds as if the system were subject to the static attraction that is the time-averaged strength over a single oscillation period, defined by the averaged LJ well-depth, εavg. At the slow oscillation limit, the system evolves according to the attractive forces at play within the current half-cycle. In between these extremes, the assembly yield curve shifts from one determined by εavg to one determined by εmin, the attraction strength during the weaker attraction half-cycles, since it is the error correction made possible at those times that determines the degree to which kinetic traps can be overcome.
These findings provide insights that could aid in the design of time-dependent assembly protocols for novel materials. Advanced materials may be composed of more complex, hierarchically ordered components with multiple intrinsic lengths, and thus multiple relaxation time-scales. Even without closed feedback loops or detailed simulations of the specific system of interest, we show that designing protocols with interactions that oscillate over times similar to the intrinsic relaxation times of that material may provide efficient assembly routes. Estimating the translational and rotational timescales that corresponds to important energetic and structural length scales within the assembling system will serve as a starting point for creating oscillation protocols that can relax kinetically trapped structures. In addition, the choice of an oscillation amplitude may be guided by evaluating the interaction regimes where moderate assembly occurs under static conditions, as we find that error correction within a system can be enhanced by periodically switching to attraction strengths that promote a moderate yield and are therefore weak enough to allow critical relaxation processes to proceed.
Author contributions
Jessica K. Niblo: conceptualization, investigation, methodology, data curation, software, formal analysis, visualization, writing – original draft. Jacob R. Swartley: methodology, writing – reviewing. Zhongmin Zhang: methodology, writing – reviewing. Kateri H. DuBay: conceptualization, methodology, supervision, formal analysis, writing – original draft.Data availability
The data presented in this study, along with submission and analysis scripts, are openly available on GitHub at https://github.com/dubayresearchgroup/2024_VCO_1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by a startup grant from the University of Virginia. The authors also acknowledge Research Computing at the University of Virginia (https://rc.virginia.edu) for providing computational resources and technical support that have contributed to the results reported within this publication.Notes and references
- N.-J. Cho, L. Y. Hwang, J. J. R. Solandt and C. W. Frank, Materials, 2013, 6, 3294–3308 CrossRef CAS PubMed.
- A. Zlotnick, J. Mol. Biol., 1994, 241, 59–67 CrossRef CAS PubMed.
- A. Zlotnick, R. Aldrich, J. M. Johnson, P. Ceres and M. J. Young, Virology, 2000, 277, 450–456 CrossRef CAS PubMed.
- S. D. Hicks and C. L. Henley, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74, 031912 CrossRef PubMed.
- M. F. Hagan and D. Chandler, Biophys. J., 2006, 91, 42–54 CrossRef CAS PubMed.
- C. Sigl, E. M. Willner, W. Engelen, J. A. Kretzmann, K. Sachenbacher, A. Liedl, F. Kolbe, F. Wilsch, S. A. Aghvami, U. Protzer, M. F. Hagan, S. Fraden and H. Dietz, Nat. Mater., 2021, 20, 1281–1289 CrossRef CAS PubMed.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- Z. Zhang and S. C. Glotzer, Nano Lett., 2004, 4, 1407–1413 CrossRef CAS PubMed.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- S. Hormoz and M. P. Brenner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5193–5198 CrossRef CAS PubMed.
- T. K. Haxton and S. Whitelam, Soft Matter, 2012, 8, 3558 RSC.
- M. Grünwald and P. L. Geissler, ACS Nano, 2014, 8, 5891–5897 CrossRef PubMed.
- S. A. Mallory and A. Cacciuto, Phys. Rev. E, 2016, 94, 022607 CrossRef CAS PubMed.
- P. F. Damasceno, M. Engel and S. C. Glotzer, Science, 2012, 337, 453–457 CrossRef CAS PubMed.
- M. F. Hagan and O. M. Elrad, Biophys. J., 2010, 98, 1065–1074 CrossRef CAS PubMed.
- J. Grant, R. L. Jack and S. Whitelam, J. Chem. Phys., 2011, 135, 214505 CrossRef PubMed.
- A. B. Pawar and I. Kretzschmar, Macromol. Rapid Commun., 2010, 31, 150–168 CrossRef CAS PubMed.
- S. Whitelam and R. L. Jack, Annu. Rev. Phys. Chem., 2015, 66, 143–163 CrossRef CAS PubMed.
- M. F. Hagan, O. M. Elrad and R. L. Jack, J. Chem. Phys., 2011, 135, 104115 CrossRef PubMed.
- C. P. Goodrich, E. M. King, S. S. Schoenholz, E. D. Cubuk and M. P. Brenner, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024083118 CrossRef CAS PubMed.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- M. Fialkowski, K. J. M. Bishop, R. Klajn, S. K. Smoukov, C. J. Campbell and B. A. Grzybowski, J. Phys. Chem. B, 2006, 110, 2482–2496 CrossRef CAS PubMed.
- S. E. Ilse, C. Holm and J. de Graaf, J. Chem. Phys., 2016, 145, 134904 CrossRef PubMed.
- H. Löwen, Europhys. Lett., 2018, 121, 58001 CrossRef.
- A. Das and D. T. Limmer, J. Chem. Phys., 2021, 154, 014107 CrossRef CAS PubMed.
- A. Das and D. T. Limmer, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217242120 CrossRef CAS PubMed.
- M. Tagliazucchi, E. A. Weiss and I. Szleifer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9751–9756 CrossRef CAS PubMed.
- M. Tagliazucchi and I. Szleifer, Faraday Discuss., 2016, 186, 399–418 RSC.
- C. Long, Q.-l Lei, C.-l Ren and Y.-q Ma, J. Phys. Chem. B, 2018, 122, 3196–3201 CrossRef CAS PubMed.
- J. H. E. Promislow and A. P. Gast, Langmuir, 1996, 12, 4095–4102 CrossRef CAS.
- J. H. E. Promislow and A. P. Gast, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 642–651 CrossRef CAS.
- J. W. Swan, P. A. Vasquez, P. A. Whitson, E. M. Fincke, K. Wakata, S. H. Magnus, F. De Winne, M. R. Barratt, J. H. Agui, R. D. Green, N. R. Hall, D. Y. Bohman, C. T. Bunnell, A. P. Gast and E. M. Furst, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16023–16028 CrossRef CAS PubMed.
- J. W. Swan, J. L. Bauer, Y. Liu and E. M. Furst, Soft Matter, 2014, 10, 1102–1109 RSC.
- J. L. Bauer, Y. Liu, M. J. Kurian, J. W. Swan and E. M. Furst, J. Chem. Phys., 2015, 143, 074901 CrossRef PubMed.
- H. Kim, M. Sau and E. M. Furst, Langmuir, 2020, 36, 9926–9934 CrossRef CAS PubMed.
- J. M. García-Ruiz, Science, 2023, 382, 883–884 CrossRef PubMed.
- J. Kim, Y. Kimura, B. Puchala, T. Yamazaki, U. Becker and W. Sun, Science, 2023, 382, 915–920 CrossRef CAS PubMed.
- D. Klotsa and R. L. Jack, J. Chem. Phys., 2013, 138, 094502 CrossRef PubMed.
- S. Chennakesavalu and G. M. Rotskoff, J. Chem. Phys., 2021, 155, 194114 CrossRef CAS PubMed.
- J. J. Juárez and M. A. Bevan, Adv. Funct. Mater., 2012, 22, 3833–3839 CrossRef.
- Y. Xue, D. J. Beltran-Villegas, X. Tang, M. A. Bevan and M. A. Grover, IEEE Trans. Control Syst. Technol., 2014, 22, 1956–1963 Search PubMed.
- X. Tang, B. Rupp, Y. Yang, T. D. Edwards, M. A. Grover and M. A. Bevan, ACS Nano, 2016, 10, 6791–6798 CrossRef CAS PubMed.
- S. Whitelam and I. Tamblyn, Phys. Rev. E, 2020, 101, 052604 CrossRef CAS PubMed.
- A. Trubiano and M. F. Hagan, J. Chem. Phys., 2022, 157, 244901 CrossRef CAS PubMed.
- J. Martín-Roca, M. Horcajo-Fernández, C. Valeriani, F. Gámez and F. Martínez-Pedrero, Nanomaterials, 2023, 13, 397 CrossRef PubMed.
- P. K. Jha, V. Kuzovkov, B. A. Grzybowski and M. Olvera de la Cruz, Soft Matter, 2012, 8, 227–234 RSC.
- S. R. Risbud and J. W. Swan, Soft Matter, 2015, 11, 3232–3240 RSC.
- P.-K. Kao, B. J. VanSaders, S. C. Glotzer and M. J. Solomon, Sci. Rep., 2021, 11, 11042 CrossRef CAS PubMed.
- Z.-Q. Chen, Y.-W. Sun, Y.-L. Zhu, Z.-W. Li and Z.-Y. Sun, Soft Matter, 2022, 18, 2569–2576 RSC.
- L. Pigard and M. Müller, Phys. Rev. Lett., 2019, 122, 237801 CrossRef CAS PubMed.
- R. L. Jack, M. F. Hagan and D. Chandler, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 021119 CrossRef PubMed.
- H. D. Nguyen, V. S. Reddy and C. L. Brooks, Nano Lett., 2007, 7, 338–344 CrossRef CAS PubMed.
- D. C. Rapaport, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 051905 CrossRef CAS PubMed.
- D. C. Rapaport, Phys. Rev. Lett., 2008, 101, 186101 CrossRef CAS PubMed.
- V. Krishna, G. S. Ayton and G. A. Voth, Biophys. J., 2010, 98, 18–26 CrossRef CAS PubMed.
- J. D. Perlmutter and M. F. Hagan, Annu. Rev. Phys. Chem., 2015, 66, 217–239 CrossRef CAS PubMed.
- A. J. Pak, J. M. A. Grime, A. Yu and G. A. Voth, J. Am. Chem. Soc., 2019, 141, 10214–10224 CrossRef CAS PubMed.
- M. F. Hagan and G. M. Grason, Rev. Mod. Phys., 2021, 93, 025008 CrossRef CAS PubMed.
- J. D. Weeks, D. Chandler and H. C. Andersen, J. Chem. Phys., 1971, 54, 5237–5247 CrossRef CAS.
- D. Kashchiev, P. G. Vekilov and A. B. Kolomeisky, J. Chem. Phys., 2005, 122, 244706 CrossRef PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- J. Dogan, S. Gianni and P. Jemth, Phys. Chem. Chem. Phys., 2014, 16, 6323–6331 RSC.
- C. J. Wilson, A. S. Bommarius, J. A. Champion, Y. O. Chernoff, D. G. Lynn, A. K. Paravastu, C. Liang, M.-C. Hsieh and J. M. Heemstra, Chem. Rev., 2018, 118, 11519–11574 CrossRef CAS PubMed.
- Z. M. Sherman and J. W. Swan, ACS Nano, 2016, 10, 5260–5271 CrossRef CAS PubMed.
- Z. M. Sherman and J. W. Swan, ACS Nano, 2019, 13, 764–771 CrossRef CAS PubMed.
- J. R. Savage and A. D. Dinsmore, Phys. Rev. Lett., 2009, 102, 198302 CrossRef CAS PubMed.
- Z. M. Sherman, H. Rosenthal and J. W. Swan, Langmuir, 2018, 34, 1029–1041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00455h |
| This journal is © The Royal Society of Chemistry 2024 |