
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–property relationships in renewable composites of poly(lactic acid) reinforced by low amounts of micro- and nano-kraft-lignin†
Sofia P.
Makri
 ab,
Panagiotis A.
Klonos
*ac,
Giacomo
Marra
d,
Alexandros Zoikis
Karathanasis
b,
Ioanna
Deligkiozi
b,
Miguel Ángel
Valera
d,
Ana
Mangas
d,
Nikolaos
Nikolaidis
a,
Zoi
Terzopoulou
a,
Apostolos
Kyritsis
c and
Dimitrios N.
Bikiaris
*a
ab,
Panagiotis A.
Klonos
*ac,
Giacomo
Marra
d,
Alexandros Zoikis
Karathanasis
b,
Ioanna
Deligkiozi
b,
Miguel Ángel
Valera
d,
Ana
Mangas
d,
Nikolaos
Nikolaidis
a,
Zoi
Terzopoulou
a,
Apostolos
Kyritsis
c and
Dimitrios N.
Bikiaris
*a
aLaboratory of Polymer Chemistry and Technology, Department of Chemistry, Aristotle University of Thessaloniki, GR-541 24, Thessaloniki, Greece. E-mail: pklonos@central.ntua.gr; dbic@chem.auth.gr
bCreative Nano PC, 43 Tatoiou, Metamorfosi, 14451 Athens, Greece
cDielectrics Group, Department of Physics, National Technical University of Athens, Zografou Campus, 15780, Athens, Greece
dAIMPLAS, Asociación de Investigación de Materiales Plásticos Y Conexas, Mechanochemistry & Reactive Extrusion, Carrer de Gustave Eiffel, 4, 46980 Valencia, Spain
First published on 7th June 2024
Abstract
We investigate the direct and indirect effects of micro- and nano-kraft lignin, kL and NkL, respectively, at a quite low amount of 0.5 wt%, in poly(lactic acid) (PLA)-based composites. These renewable composites were prepared via two routes, either simple melt compounding or in situ reactive extrusion. The materials are selected and prepared using targeted methods in order to vary two variables, i.e., the size of kL and the synthetic method, while maintaining constant polymer chain lengths, L-/D-lactide isomer ratio and filler amounts. The direct/indirect effects were respectively investigated in the amorphous/semicrystalline state, as crystallinity plays in general a dominant role in polymers. The investigation involves structural, thermal and molecular mobility aspects. Non-extensive polymer–lignin interactions were recorded here, whereas the presence of the fillers led to both enhancements and suppressions of properties, e.g., glass transition, crystallization, melting temperatures, etc. The local and segmental molecular dynamics map of the said systems was constructed and is shown here for the first time, demonstrating both expected and unexpected trends. An interesting discrepancy between the trends in the calorimetric measurement against the dielectric Tg is revealed, providing indications for ‘dynamical heterogeneities’ in the composites as compared to neat PLA. The reactive extrusion as compared to compounding-based systems was found to exhibit stronger effects on crystallizability and mobility, most, probably due to the severe enhancement of the chains’ diffusion. In general, the effects are more pronounced when employing nano-lignin compared to micro-lignin, which is the expected beneficial behaviour of nanocomposites vs. conventional composites. Interestingly, the variety of these effects can be easily manipulated by the proper selection of the preparation method and/or the thermal treatment under relatively mild conditions. The latter capability is actually desirable for processing and targeted applications and is proved here, once again, as an advantage of biobased polyesters such as PLA.
1. Introduction
Polymers and polymer-based materials are in wide use for more than a century. The relatively effective and economic processing along with the superior performance1,2 has made these materials essential in everyday life, industry and academia. Over the last two decades, there has been growing interest in biobased and renewable materials that align with the latest trends of circular and green economy, i.e., sustainability.3,4 The conventional polymers dominating the market over the previous century have been synthesized using fossil-based sources (petroleum-based) and employing significantly non-ecofriendly and quite energy-consuming thermochemical processes.5 Therefore, academia in collaboration with industry has focused on the development of polymers based on renewable resources, such as agricultural products and byproducts, e.g., acids formed during metabolic physical processes (lactic acid, furan-dicarboxylic acid, succinic acid, adipic acid, vanillic acid, etc.).6–11Among the most known and already used sustainable polymers is poly(lactic acid) (PLA), also called polylactide.9,10,12 PLA is an exquisite biobased aliphatic polyester, being mainly derived from renewable resources such as potatoes, corn starch, beets or sugarcane.13,14 So far, the most effective synthesis method for PLA has been the ring opening polymerization (ROP)15 of lactide formed by lactic acid, with the latter being a byproduct of agricultural compounds fermentation.16,17 Next to its ‘green’ character, PLA exhibits high thermal and chemical stability,18 moderate glass transition (Tg ∼ 20–70 °C) and melting (e.g., Tm ∼ 140–190 °C) temperatures.16,19–22 More interestingly, the semicrystalline character of PLA provides the ability to tune a series of properties, such as the mechanical performance23,24 and thermal conductivity, via the easily manipulable crystallinity and semicrystalline morphology (dense/sparse).25–27 Interestingly, depending on the structure, molecular weight and the thermal treatment, PLA can be transformed from completely amorphous to quite highly crystalline form.20,21,25,28 Except for the PLA's performance within the application, there is a general desire to facilitate its physical degradation, for example in soil – compostability, which in the case of bulk PLA is rather slow.8,29 Additionally, PLA exhibits high potential for combination with other polymers (copolymers, blends, interpenetrating polymer networks, and stars)12,22,30–32 as well as with inorganic/organic fillers and nanoparticles.24,33,34 The latter case, i.e., polymer nanocomposites (PNCs), is also at the centre of interest of the present study.
Regarding specific applications, PLA has already found use in a vast range of them, namely, in bio-engineering and bio-medicine (tissue engineering and drug delivery),35–37 in food packaging,9,10,38 and even in 3D-printing devices.39–41 Furthermore, PLA has been proved to be a proper substrate for the development of functional surfaces with enhanced antioxidative and/or antimicrobial properties for several biochemical applications.10,33
A significant number of studies in the literature regarding PLA-based composites deal with PLA reinforced by small and large amounts of inorganic particles of various geometries and surface modifications, e.g., spherical metal oxide particles (silica), nano-clays, graphene and graphene oxide nanosheets and carbon nanotubes. These have mainly aimed to enhance, among others, the mechanical performance, crystallinity, and tune the permeability of small molecules and improve the electrical and thermal conductivities. On the other hand, less attention has been paid to the simultaneous improvements of performance with the enrichment of biocompatibility (non-toxicity) and the ‘green’ character. The latter are not compatible, in general, with the employment of the conventional nano-additives mentioned above. A solution to this need has come quite recently with the employment of particles/compounds originating from bio-resources as well. Such a compound is lignin, being usually addressed as kraft- or soda-lignin.42 Lignin is an aromatic biopolymer of relatively high Tg (>100 °C)43 being isolated from lignocellulosic biomass and is the second most abundant natural polymer after cellulose.44,45 Lignin is characterized by high biocompatibility and low toxicity, and it exhibits a variation in functional groups, while it occupies a large amount of carbon that may be potentially transformed to composites and carbon-rich materials.42 Among others, the addition of lignin in polymers, including PLA, facilitates antioxidation and antibacterial performance.46–49
Despite the vast amount of published works on PLA and related systems, not many of them deal with totally biobased PNCs; moreover, quite a few focus on PLA–lignin PNCs.49–52 Thus, there is still room for such basic research studies. In this basic-research study, we present an investigation of the structure–properties relationship of the addition of kraft Lignin, in particular, in the micro-(as received) and nano-metric form, at the small loading of 0.5 wt% in PLA. Despite the ‘polymeric character’ of lignin, due to its higher Tg compared to that of PLA, along with the low lignin loading, we may consider these systems as composites and nanocomposites of a PLA matrix. Additionally, we comparatively study the PLA/lignin composites and nanocomposites upon their preparation through two individual routes, namely, by melt compounding against in situ polymerization of PLA in the presence of lignin. The direct effects of lignin, the lignin size and the PNC synthesis method are assessed in the amorphous state of the polymer, whereas the indirect effects are investigated in the semicrystalline state, when being available. For the study of these effects, we comparatively seek for any interfacial interactions, alternations in the glass transition, crystallization and melting aspects, the local and segmental molecular mobility, the electrical conductivity and thermal diffusivity. To these aims, we employed the following experimental methods: Fourier transform infra-red spectroscopy (FTIR), differential scanning calorimetry (DSC), polarized light microscopy (PLM), and the sophisticated technique of broadband dielectric spectroscopy (BDS). The data are critically analyzed and evaluated employing widely adopted methods and models. To the best of our knowledge, for the said systems and followed parameters, this is the first time to present results using the abovementioned techniques; furthermore, the molecular-dynamics map is presented for the first time here as well.
2. Experimental section
2.1. Materials
Two series of relatively new PLA–kraft lignin (kL) composites recently prepared by our groups49 are studied here. Each series of polymer composites consists of neat linear PLA and two composites of PLA filled with 0.5 wt% micro-kL and nano-kL. The detailed preparation method along with initial structural characterization can be found in a recent work by Makri et al.,49 while the basic aspects are briefly reported in the following section. Raw kL is microscopic and has been purchased from UPM (Helsinki, Finland, product code name ‘BioPiva300’). The kL was found to consist of particles of a wide range of sizes on few to many tens of μm.49 On the other hand, the nano-lignin ‘NkL’, with an average particle size of ∼650 nm, was kindly provided by Creative Nano PC (Athens, Greece).The first series involves the melt compounding of PLA with lignin (code name: ‘PLA comp.’ + kl/NkL). The used PLA is the Ingeo™ Biopolymer 2003D of NatureWorks (Minneapolis, MN, USA), with the ratio of the L/D-isomers equalling 96/4, a measured molecular weight Mn of ∼72 kg mol−1 and a polydispersity of 2.4,49 that was kindly supplied by Plastika Kritis S.A. (Irakleio, Greece). Obviously, the Mn of PLA is the same in bulk and in the composites and this has been experimentally confirmed.49
For the second series, the PLA polymerization, i.e., ring opening polymerization of L-lactide, took place in the presence of kL and NkL (in situ) in a Brabender® Plasti-Corder® Lab-Station torque rheometer. The L-lactide (of 99% purity and L/D mass ratio 95/5) had been purchased from Corbion N.V. (Gorinchem, The Netherlands). The employed ‘in situ’ method actually imitates the ‘reactive extrusion, Rex’ process;53 thus, this second series is described here by the code name: ‘PLA Rex.’ + kL/NkL. Usually, the in situ polymerization results in a drop of Mn,51,54,55 however, here the Mn differences were minor as the lignin amount was purposely chosen extremely small. Mn of neat PLA Rex. was found to be ∼65 kg mol−1, while the polydispersity was estimated to be ∼2.7.49
So, we study totally six (6) samples listed in Table 1. The samples were initially formed as films and cylindrical disks of ∼1–1.5 mm in thickness, formed within a thermal press employing homemade molds.
| Sample | Scan 1 (cooling at high rates) | Scan 2 (cooling at 10 K min−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CFc (wt%) | T g (°C) | Δcp (J g−1 K−1) | T cc (°C) | CFcc (%) | T c (°C) | CFc (%) | T g (°C) | Δcp (J g−1 K−1) | CFcc (%) | T m (°C) | CFm (%) | |
| PLA comp. | 0 | 59 | 0.43 | 123 | 19 | — | 0 | 59 | 0.42 | 19 | 151 | 19 |
| + 0.5% kL | 0 | 58 | 0.49 | 124 | 16 | — | 0 | 58 | 0.48 | 17 | 150 | 17 |
| + 0.5% NkL | 0 | 58 | 0.49 | 122 | 19 | — | 0 | 58 | 0.48 | 19 | 149 | 20 |
| PLA Rex | 0 | 43 | 0.52 | 86 | 42 | 86 | 18 | 43 | 0.37 | 29 | 163 | 51 |
| + 0.5% kL | 0 | 50 | 0.51 | 90 | 42 | 88 | 12 | 51 | 0.46 | 33 | 166 | 51 |
| + 0.5% NkL | 0 | 48 | 0.50 | 94 | 41 | 87 | 11 | 48 | 0.47 | 37 | 165 | 49 |
2.2. Experimental techniques
Conventional differential scanning calorimetry (DSC) was employed to study the thermal transitions, namely, glass transition, crystallization, and melting, in a nitrogen atmosphere and within the temperature range from −100 to 190 °C. The measurements were performed by means of a TA Q200 calorimeter (TA Instruments, USA). Upon erasing the thermal history by heating at 190 °C, we employed two thermal protocols, i.e., (scan 1) involving fast cooling and heating at 10 K min−1 and (scan 2) standard linear cooling at 10 K min−1 and heating at 10 K min−1.The semicrystalline morphology for all samples was screened via polarized light microscopy (PLM) during melt-crystallization, i.e., during cooling at 10 K min−1 beginning from the melt state. The PLM micro-images were captured using a Nikon Optiphot-1 polarizing microscope equipped with a Linkam THMS 600 heated stage, a Linkam TP91 control unit and a Jenoptic ProgRes C10Plus camera.
FTIR-ATR spectra were recorded on a Tension 27 FTIR spectrometer (Bruker, Billerica, MA, USA) equipped with a diamond ATR accessory at a spectral resolution of 4 cm−1 in the wavenumber range from 600 to 4000 cm−1.
Broadband dielectric spectroscopy (BDS)56 was employed to assess the molecular mobility of PLA in bulk and within the composites, in the initially amorphous state, employing a Novocontrol BDS setup (Novocontrol GmbH, Germany). The sample was in the form of a sandwich-like capacitor, with the sample (polymeric) being placed and melt-quenched, between brash disks (electrodes). Silica spacers of ∼100 μm in thickness were used for that. The measurements were conducted at various temperatures in the range from 10 to 120 °C upon heating and within the broad frequency, f, ranging from 10−1 to 106 Hz. The molecular mobility was investigated and evaluated via the complex dielectric permittivity, ε*(f,T), in particular from its imaginary part, ε′′(f,T) which is related to the dielectric loss.56
3. Results and discussion
Before presenting and discussing the experimental findings, it should be noted that we have made such a choice of polymeric systems, initial materials, amounts and synthetic routes so that some major parameters are kept almost constant, namely, the PLAs’ L-/D-ratio and Mn and the kL loadings. This makes the present study to directly aim the effects imposed by the kL size (micro vs. nano) and the composites synthetic route (‘comp.’ vs. ‘Rex’). Next, the study will discuss the indirect effects via the implementation of crystallinity, in terms of nucleation, crystallinity degree and semicrystalline morphology (dense or sparce).In Fig. 1, we present the ATR-FTIR spectra for all systems, including neat NkL, in the amorphous state. In the composites, as expected, the signal is dominated by the vibrations/stretchings of molecular groups/bonds originating from PLA. The results are in accordance with previous PLA-based systems27,34 in terms of the number and position of the various absorbance peaks (basic origins described within Fig. 1).
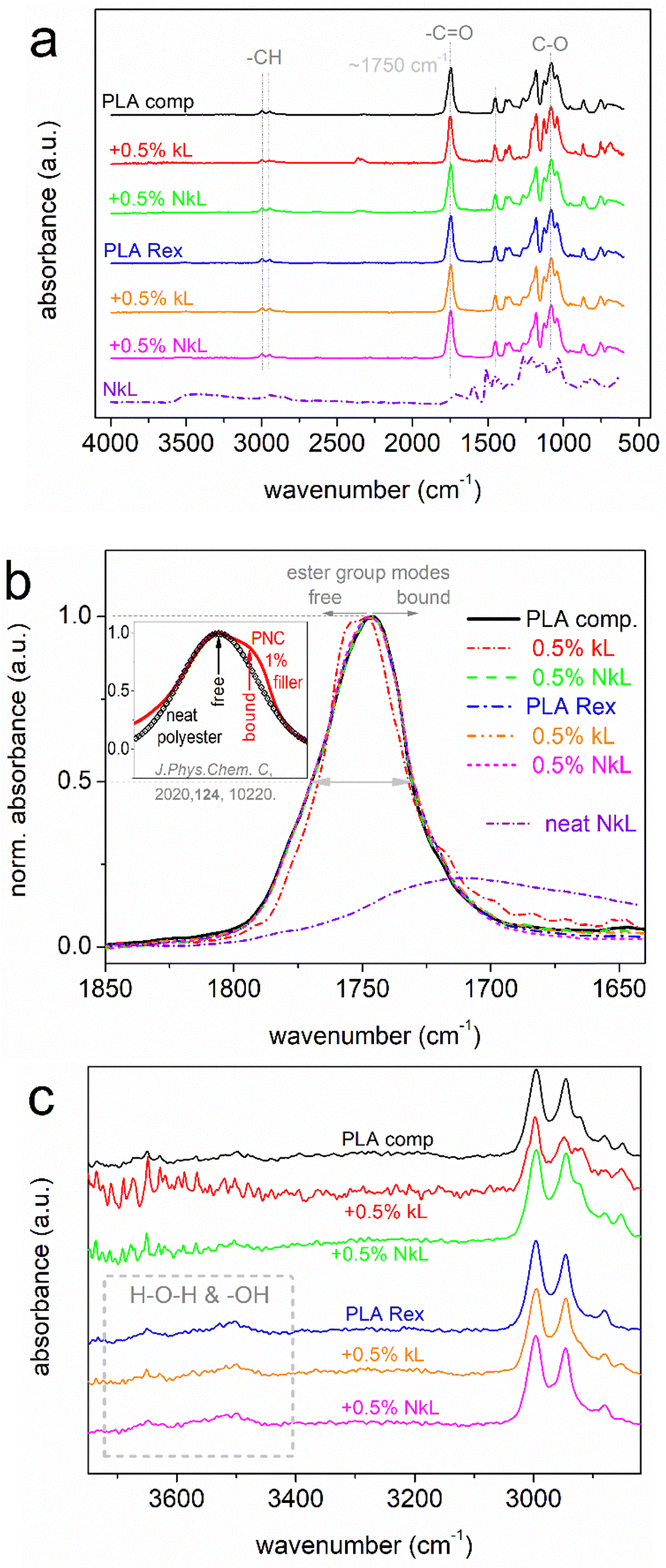 | ||
Fig. 1 (a) ATR-FTIR spectra of PLA and all PLA–lignin composites in the amorphous state, along with neat nano-lignin for comparison. The molecular origins of the main peaks are shown in the plot. (b) Detailed view of the peak corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of PLA, upon shape normalization to the peak maxima. The inset to (b) provides an example of recording of strong interfacial interactions via FTIR in polyester-based PNCs loaded with only 1 wt% nanoparticles, adapted from a previous work.57 (c) Focus on the high-wavenumber range of the FTIR spectra. O vibration of PLA, upon shape normalization to the peak maxima. The inset to (b) provides an example of recording of strong interfacial interactions via FTIR in polyester-based PNCs loaded with only 1 wt% nanoparticles, adapted from a previous work.57 (c) Focus on the high-wavenumber range of the FTIR spectra. | ||
The main reason for employing FTIR here, in particular in the amorphous state of PLA, is the investigation of the existence of kL–PLA interactions. If present, and depending on their amount/strength, these interactions would be expected to involve the ester group of PLA, i.e., of the carbonyl, which is the most polar one,58 for example, affected by hydrogen bonding with the surface hydroxyls of lignin. The effect of such interfacial interaction would be recorded in FTIR via the vibration modes of the ester bond (CO) in the wavenumber range ∼1700–1800 cm−1,41 as a migration or additional contribution of the FTIR peak toward smaller wavenumbers. Such recording has been revealed in the past in PLA- and other polyesters-based composites and is assigned to the formation of more bound ester group modes involved within the interfacial interactions.41,57,59
Herein (Fig. 1(b)), there are no such effects in our composites, at least not easily resolved. Thus, we may conclude that non-worth noting interfacial interactions exist, between PLA and kL or PLA and NkL. This should not be a simple matter of the low kL loadings, as a similar FTIR view was recorded in previous PLA/lignin systems with higher lignin loadings (up to 2.5%).27
On the other hand, in previous PNCs with 1 wt% filler loadings, tremendous effects on ester bond bands have been reported (inset to Fig. 1(b)).34,57
3.1. Glass transition and crystallinity aspects
We proceed with the results by conventional calorimetry. In Fig. 2(a), we present the fast cooling of initially melted samples (i.e., scan 1). This cooling was performed using the command ‘jump to temperature’ of the TA software, and the apparatus performs the fastest possible cooling. The T(t) profile for that (not shown) is not linear, whereas in the temperature range of the expected crystallization of PLA, the cooling rate was roughly estimated as ∼90–100 K min−1. During this fast cooling, none of the polymers exhibited any crystallization exothermal peak. Therefore, the samples have been kept amorphous. According to previous knowledge25,28 despite the absence of spherulites, we expect the existence of crystal nuclei (not recordable by conventional DSC) due to the involvement of strong supercooling.60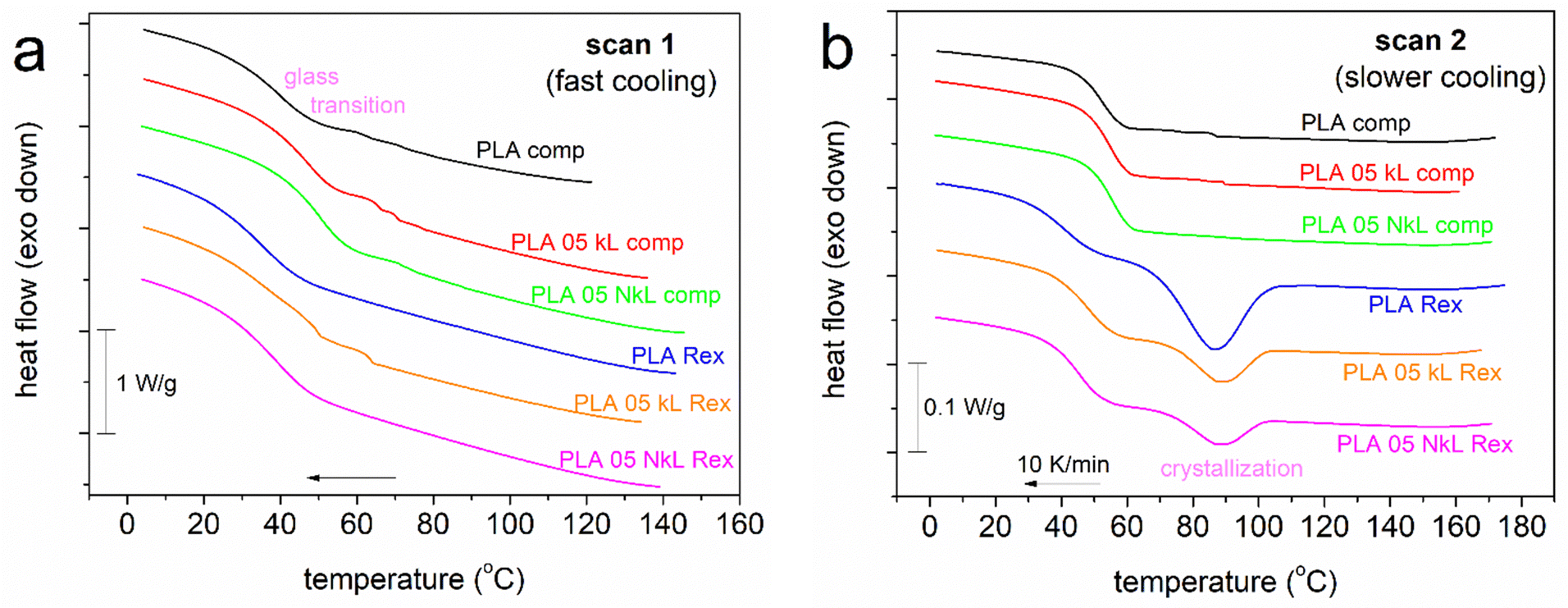 | ||
| Fig. 2 Comparative cooling DSC traces for all samples, shown for (a) scan 1 and (b) scan 2. The heat flow is shown in W g−1 units, i.e., normalized to the mass of the sample. | ||
Interestingly, during cooling at a lower rate, here 10 K min−1 in Fig. 2(b) (scan 2), the PLA-comp. systems still remained amorphous, while the PLA-Rex ones exhibited single melt crystallization peaks between 60 and 110 °C. We will attempt to explain this difference in the following part.
In Fig. 3(a), the subsequent heating traces for both scan 1 and 2 are presented. Therein, all samples exhibit single glass transition steps (30–60 °C) and cold crystallization exotherms. Cold crystallization is indicative of incomplete crystallization during the prior cooling. The temperature ranges of cold crystallization are quite different between the PLA-comp. (100–150 °C) and the PLA-Rex (80–100 °C) systems. Similar is the situation for melting. It is clear that for the two series, interestingly, all thermal transitions are recorded in different temperature regions, with these differences, however, being non systematically lower or higher for a given series. If so, we would suspect the implementation of significant differences in the corresponding Mns, however, this is not the case here as the Mns are comparable. Therefore, other parameters seem to affect the differences, among others, the existence of chain–chain interactions, polymer–lignin associations, alternations in the free volume (away or close to the lignin entities) and/or existence of conformational modifications of the PLA chains. These are expected to be reflected in polymer mobility, as recorded here via the static glass transition (DSC, Fig. 3(b)) and molecular dynamics in BDS (later section).
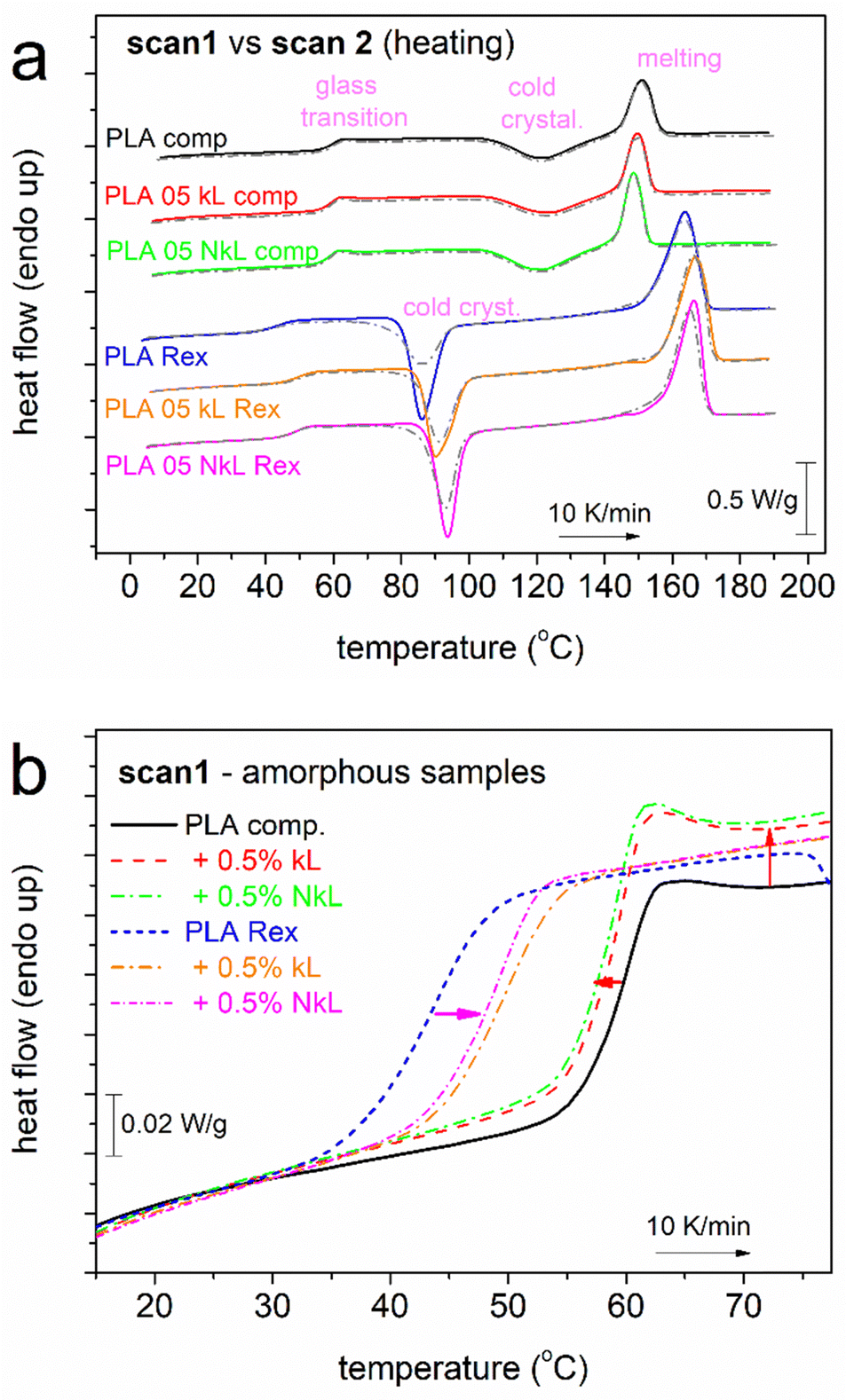 | ||
| Fig. 3 (a) DSC traces during heating at 10 K min−1, for all samples shown comparatively for scan 1 (solid-colored lines, initially amorphous) and scan 2 (dash-dotted grey lines). Heat flow is shown upon normalization to the samples’ mass. (b) Detailed view of the glass transition step of scan 1 (amorphous systems). The added arrows mark the effects on the temperature position and the Δcp of glass transition by the addition of kL and NkL. | ||
The glass transition steps are followed in more detail for the amorphous state in Fig. 3(b), shown comparatively for all compositions. Both qualitative and quantitative differences are recorded.
Given the variety of effects observed, we will discuss them on the basis of the various values estimated for all thermal events, namely, characteristic temperatures (Tg, Tc, Tcc, Tm), heat capacity changes, Δcp, and crystalline fractions, CF. These are described and listed in Table 1. To calculate the CF, we compared the enthalpy change, ΔHi, of the melt- or cold-crystallization, ΔHc or ΔHcc, respectively, with the heat of fusion of fully crystalline PLA (here taken as 93 J g−1).61
In Fig. 4 and 5, the most important values are presented in terms of column diagrams and shown comparatively for the two series of PLA/lignin.
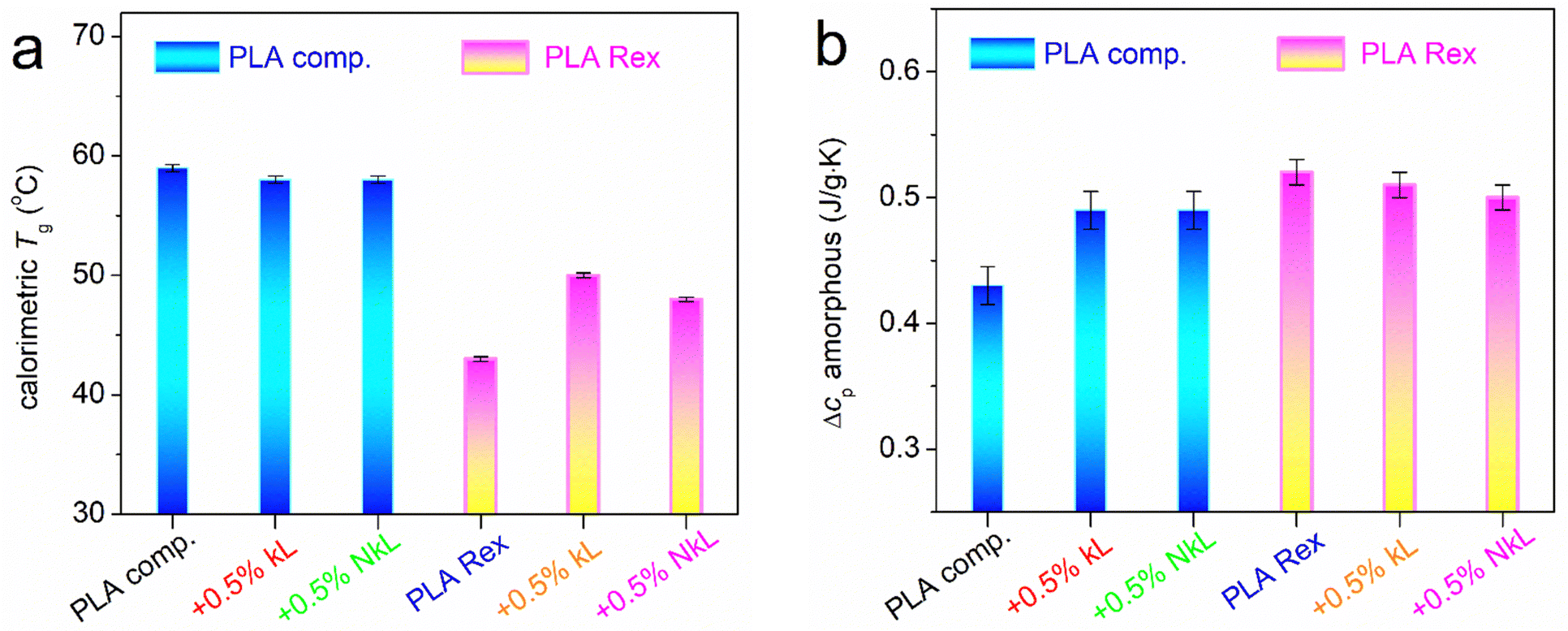 | ||
| Fig. 4 Values related to the glass transition of PLA in the amorphous state (scan 1), in particular, (a) Tg and (b) Δcp, shown in the form of column diagrams for all samples (description on the horizontal scale axis). | ||
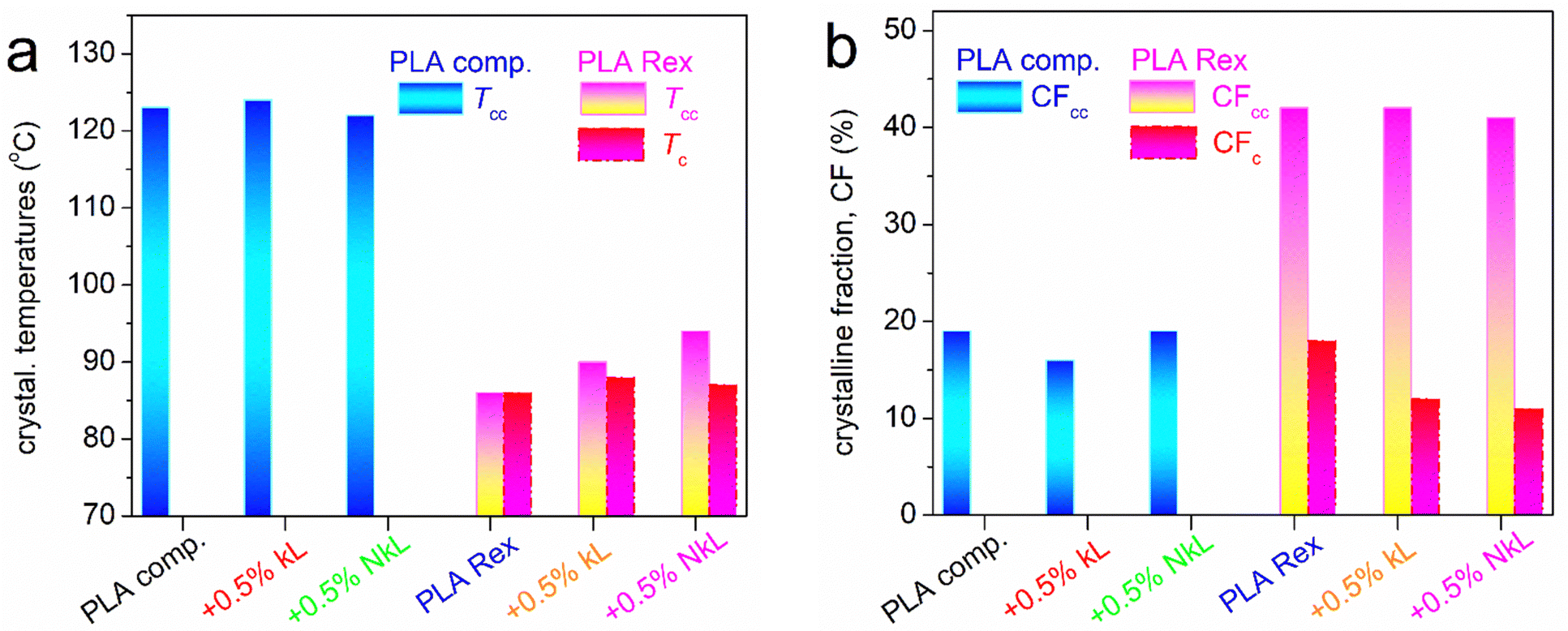 | ||
| Fig. 5 DSC values of interest related to PLA's crystallization, in particular, (a) Tcc and Tc from scan 1 and scan 2, respectively, and (b) CFcc and CFc from scan 1 and scan 2, respectively. | ||
In the amorphous state, neat PLA-comp shows a Tg of 59 °C, while in neat PLA-Rex, Tg equals 43 °C. This suggests the easier mobility of chains (less constrained) in PLA-Rex than in PLA-comp. The same conclusion comes when comparing the corresponding Δcp values (Fig. 4(b) and Table 1), i.e., 0.52 and 0.43 J g−1 K−1, respectively. These differences are not expected to arise from the different Mn values of the two PLAs. For the sake of completeness, based on previous knowledge on the Tg(Mn) dependence for PLA,19,62,63 we report that the expected Mn threshold for chain entanglements is ∼8–25 kg mol−1. Our PLAs are above that threshold, namely, the Tg is stabilized with respect to Mn. Thus, the Tg differences between the neat PLA-Rex and PLA-comp should be due to the fact that PLA-comp is commercial and may potentially contain a plethora of stabilizers, whereas the home-made PLA-Rex does not, which can further suppress chain mobility.
Coming back to the striking effect demonstrated in crystallization (Fig. 2(b)) between the neat PLAs of the two series, we may now conclude that the enhanced crystallizability of PLA-Rex compared to PLA-comp originates from differences in polymer chain kinetics. The lower Tg denotes the existence of more mobile polymer chains and subsequently both easier nucleation and lamellae formations (chain foldings).
Another less likely explanation on that could be sought through structural characterization in combination with the different synthetic routes. We recall that neat PLA-Rex was synthesized under identical conditions (chemistry, time periods, temperatures, etc.) as in the composites, but without the presence of lignin. For these systems, we may look back at the FTIR data, in particular Fig. 1(c). Therein, it seems that the FTIR spectra of vibrations of –OH and H–O–H exhibit stronger or better distinguishable structures in PLA-Rex compared to PLA-comp systems. This could be an indication for the existence of more –OH terminal units (active sites) in PLA-Rex, that are known to favor the chain–chain associations and, subsequently, to potentially facilitate nucleation/crystallization64,65 as happens here. The existence of more terminal –OH could be compatible with the moderately lower Mn of PLA-Rex, as more chain ends are expected for the polymer with the shorter chains. Unfortunately, the quantification of the –OH fractions for these materials is technically quite difficult.
So, it is more likely that the enhanced crystallizability of PLA-Rex is due to its easier chains’ mobility/diffusivity as compared to PLA-comp.
Coming to effects on lignin, in the PLA-comp series, Tg of neat PLA-comp is 59 °C and is barely reduced by 1 K with the addition of lignin (Fig. 4(a)). This denotes a slight acceleration of segmental mobility. The corresponding Δcp of neat PLA-comp equals 0.43 J g−1 K−1 and increases to 0.49 J g−1 K−1 in the two composites (Fig. 4(b)), suggesting the existence of either higher mobile amorphous fraction (MAF)66,67 or, in general, increased chains’ mobility.
Qualitatively opposite effects are recorded in the PLA-Rex series (Fig. 4). The Tg of neat PLA-Rex equals 43 °C and is increased by 7 and 5 K by the addition of kL and NkL, respectively, whereas the Δcp drops from 0.52 to 0.51 and 0.50 J g−1 K−1, respectively. Both effects indicate a worth-noting hindering of polymer chains’ mobility and, most possibly, suppression in the MAF within the composites. Usually, such suppression is observed when a fraction of the polymer chains are immobilized at the interfacial layer with the fillers66–69 due to strong attractive interfacial interactions. The ‘filler-interfacial polymer’ entities may usually act as obstacles to the mobile chains’ diffusion away from the interface, thus, the Tg usually increases, similar to the case of PLA-Rex/kL here. This seems incompatible here based on the findings from FTIR spectra (no strong interactions evidenced). However, we may not exclude the concentration of PLA chains close to or around the kL and NkL, without a high degree of direct interfacial interactions. We will come back to this point, along the light of BDS results on molecular dynamics. In any case, these recordings on glass transition in the amorphous state are another major difference between the composites prepared by in principle different routes.
The attention is now turned on the effects of lignin on crystallinity (Fig. 5). In Fig. 5(a), we present the temperature of crystallization, in particular the melt- and cold-crystallization, Tc and Tcc, respectively. As noted above, the PLA-comp series exhibits uniquely cold crystallization, i.e., only during heating and upon subjection to large supercooling. Tcc barely changes (122–124 °C, Table 1 and Fig. 5(a)), while similar is the situation for the crystalline fraction, CFcc ∼ 16–19 (±1)% (Table 1 and Fig. 5(b)), in PLA-comp, being in general small. Regarding the quality of the crystals, represented in DSC by Tm, this seems to drop since Tm decreases from 151 (PLA-comp) to 150 and 149 °C due to the addition of kL and NkL (Fig. 5(c)). The results suggest minor effects imposed by the presence of lignin.
In the PLA-Rex series, Tc (by scan 2) and Tcc (by scan 1) lie within lower temperature ranges. Both Tc and Tcc of neat PLA-Rex (85 °C) are elevated in the composites. The elevation of Tc usually suggests that lesser supercooling (i.e., the ‘Tm − Tc’ distance) is needed for the initiation of crystals’ development, or else, more crystal nuclei exist at the same high temperature. We recall that during melt crystallization, compared to cold crystallization, polymer chains exhibit high mobility, thus, spherulites are formed quite faster and possibly larger spherulites are made (details supplied later with PLM results). In our case, the role of additional crystal nuclei could be played by the lignins or, at least, some of them. On the other hand, the elevation of Tcc, generally, suggests the opposite effect, as there seems to exist a retardance of crystallization initiation. This is, nevertheless, compatible with the prior recording on retarded chains’ mobility in the composites. In Fig. 5(b), CFcc of PLA-Rex is more than doubled as compared to PLA-comp, while the addition of kL and NkL induces mild suppression. Suppression is also recorded in CFc in the composites. Both effects are compatible with the retarded chains’ mobility and partly with the formation of an interfacial rigid polymer layer around kL and NkL.34,57,67
Finally, opposite effects of lignin are recorded between the two series, regarding the melting temperature (Tm, Table 1). While a mild lowering of the crystals’ quality was recorded in PLA-comp, the Tm of PLA-Rex (163 °C) exhibits an increase of 2–3 K in the presence of lignin, indicating a mild increase of the quality of spherulites. The term quality refers to the size or/and density (lamellar packing) of the spherulites.
To visualize the melt-crystallization of our systems, we performed PLM during cooling of initially melted polymers. The results are shown in Fig. 6. Therein, in accordance to calorimetry, the PLA-comp systems showed no crystallization or, at least, not recordable by PLM. PLA-Rex systems crystallized during cooling and the results were compatible with those obtained by DSC. For example, at the intermediate stages of crystallization, the composites exhibited a larger number of crystallites, with the largest number observed for the PNC as compared to the micro-composite. At the ending stage of crystallization, the overall sample volume is filled with crystals in all cases, whereas the PNC exhibited a denser semicrystalline morphology and higher homogeneity in terms of the size of crystals (many tiny crystals). These results confirm the enhanced nucleation in the PLA-Rex systems, whereas it seems that the corresponding elevation of Tm is not indicative of the crystals size. In addition, the relatively small CFc values (12 and 11% for kL and NkL, respectively, Table 1) are not compatible with the final crystallization views in Fig. 6. Combining together all these data, most probably, Tm here is related to a sparser lamellae chain packing that can be more directly proved using structural techniques such as X-ray diffraction.
 | ||
| Fig. 6 PLM micrographs for (left and middle) PLA-Rex and (right) PLA-comp-based systems. These results correspond to initially melted samples and subsequently subjected to linear cooling at 10 K min−1 (i.e., melt-crystallization for the PLA Rex-based systems). | ||
3.2. Molecular dynamics
The discussion is now turned onto molecular mobility/dynamics, via the analysis of BDS results.56,70 BDS enables the recording of the imaginary part of dielectric permittivity as a function of frequency and temperature, ε′′(f,T). ε′′ is considered to screen the ‘dielectric losses’56,70 and correlate the ε′′(f) peaks arising from specific groups of dipole moments to actual molecular motions and other charge transport/relaxation/trapping. The peak maxima frequencies, fmax, and the corresponding f region are related to the relaxation times (τrel) of the relaxing dipoles at given temperatures. A good example of these can be seen in the present recordings shown in Fig. 7 for all samples.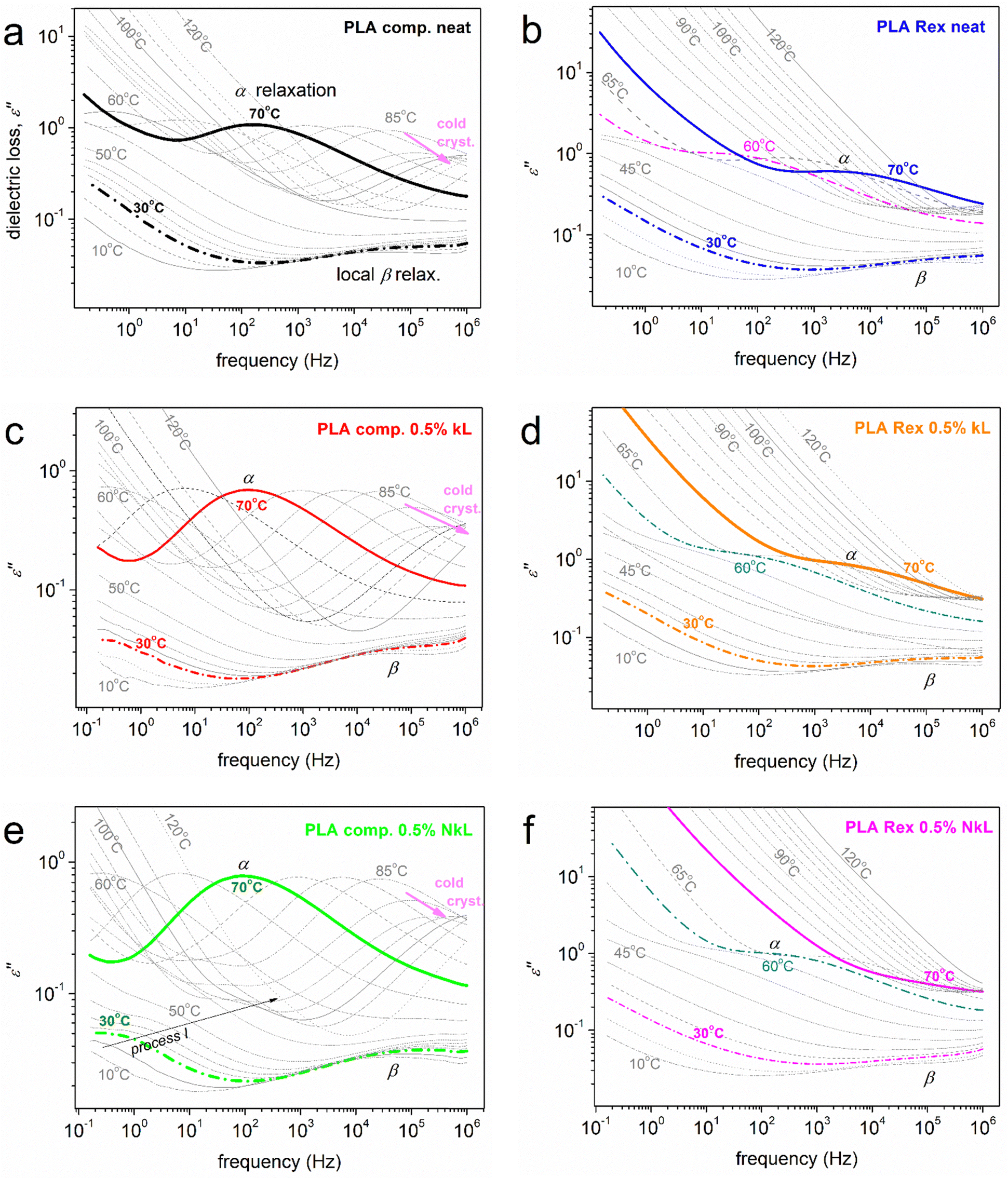 | ||
| Fig. 7 BDS isothermal spectra in the form of frequency dependence of ε′′(f) in the overall range of temperature studied. The various dipolar processes recorded (local β and segmental α) are indicated on selected curves. | ||
With increasing temperature, the relaxations usually migrate towards higher fmax due to increased thermal energy and reduced τrel. This is actually the recording of ‘molecular dynamics’, a precious advantage of BDS among other experimental techniques. Regarding PLA, there is already a rich literature on its molecular dynamics, in simple (bulky)71–74 and complex forms (PNCs, copolymers, under spatial confinement, etc.),21,75–79 which sets the origins of the various recorded relaxation processes mainly known.
At T < Tg, the dielectric response as shown in Fig. 7 is governed by relaxations of local character, herein, the so called local β relaxation. β has been assigned to localized fluctuations71,78 or local twisting motions72 in the polymer chain, in particular, related to the carbonyl group. The process is recorded in all samples, as expected (Fig. 7). At T ≥ Tg, a significant signal uprise is recorded which originates from the relaxation of dipole moments perpendicular to the polymer chain backbone. Obviously, this is related to the segmental dynamics due to the evolution of the glass transition. The corresponding strong ε′′(f) peak recorded is commonly addressed as the main α segmental relaxation.56,70 Here, α is recorded in all cases. At even higher temperatures/lower frequencies, the signal becomes gradually dominated by strong ionic conductivity and interfacial charge effects, recorded by a signal increase of more than one order of magnitude.70
The main focus here is on α relaxation, which is the dielectric analogue of the glass transition. In Fig. 8(a), we show comparatively for all samples the α relaxation at the fixed temperature of 70 °C, at which the overall peak is mainly inside our frequency window. It can be easily seen by bare eye that in the case of PLA-comp, with the addition of lignin, the α relaxation has migrated toward lower frequencies, in other words, it is decelerated. In contrast, in PLA-Rex, the addition of lignin leads to an acceleration of α (transition to higher frequencies). Please note in Fig. 7(a), (c) and (e) that at temperatures around 80 °C, a strong suppression of α relaxation takes place. Keeping in mind that the materials are initially amorphous and combining the DSC data of Fig. 3(a), one can easily conclude that this effect is due to the implementation of cold crystallization, namely, immobilization of a fraction of polymer chains within the newly formed crystals.73
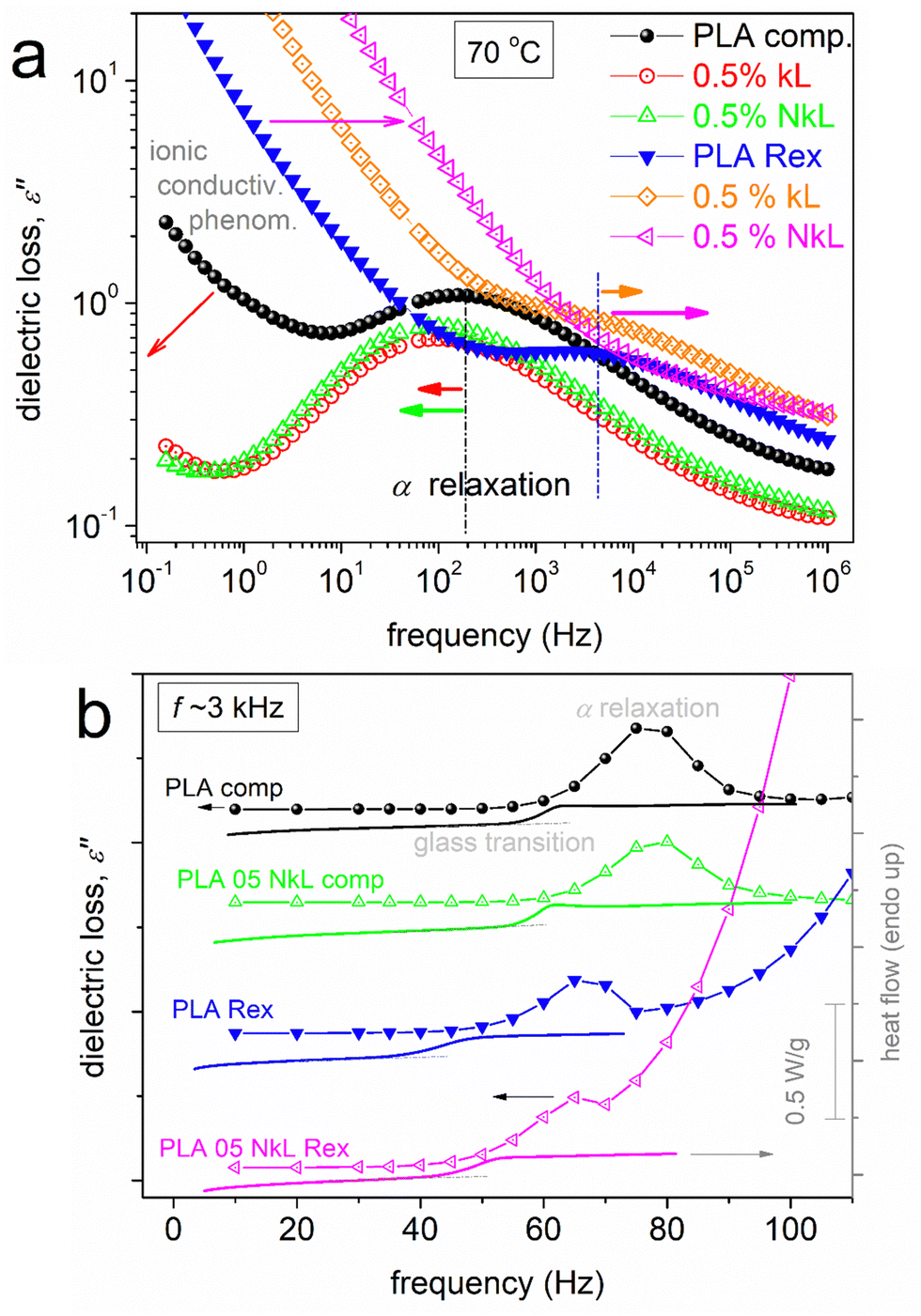 | ||
| Fig. 8 (a) Comparative isothermal curves of ε′′(f) at the selected temperature of 70 °C, showing the segmental α relaxation for all samples, effects being marked by the added horizontal arrows. (b, line-connected points) Comparative isochronal plots of ε′′(T) at the selected frequency of 3.16 kHz for selected compositions, the resolved local and segmental relaxations (peaks) being indicated. Along the isochronal BDS data in (b), we comparatively show the corresponding DSC heating traces of scan 1 (lines). | ||
To facilitate a more direct comparison with the calorimetric findings, for selected samples (neat PLAs and corresponding PNCs), we constructed the so called ‘isochronal ε′′(T)’ plots70 for the selected f of 3.16 kHz and showed the data alongside the DSC heating thermograms (Fig. 8(b)). The temperature regions of glass transition are comparable between the dielectric and calorimetric recordings. Besides that, opposite behaviours of the segmental process are recorded between BDS and DSC as far as the effect of kL addition is concerned. Such discrepancy is more or less usual in the literature, especially when ‘complex’ polymeric systems are involved.68,80 A probable reason for recording such discrepancy has been proposed to be the existence of dynamical heterogeneities, potentially influenced by variations in cooperativity lengths at different temperatures,81,82 as monitored by different physical processes, here, thermally (DSC) vs. dielectrically (BDS).
To provide a more in-depth view of dynamics, the BDS data were critically analyzed by fitting model functions, following widely adopted routes.56,70 Examples of fitting of ε′′(f) peaks employing the known asymmetric Havriliak–Negami (HN) or symmetric Cole–Cole (CC) functions83 are demonstrated in Fig. 9. As usual in polymers, HN and CC were proper and sufficient to fit the α and β relaxations, respectively.70 During the fitting, linear terms were used to mimic the ionic conductivity effects, while in PLA-comp + 0.5% NkL, additional relaxation, named ‘process I’ was resolved. The fitting process enables the evaluation of the relaxations time scale, in terms of the reciprocal temperature dependence of fmax. This is shown in the constructed dielectric relaxation map of Fig. 10, also called as an ‘Arrhenius plot’.
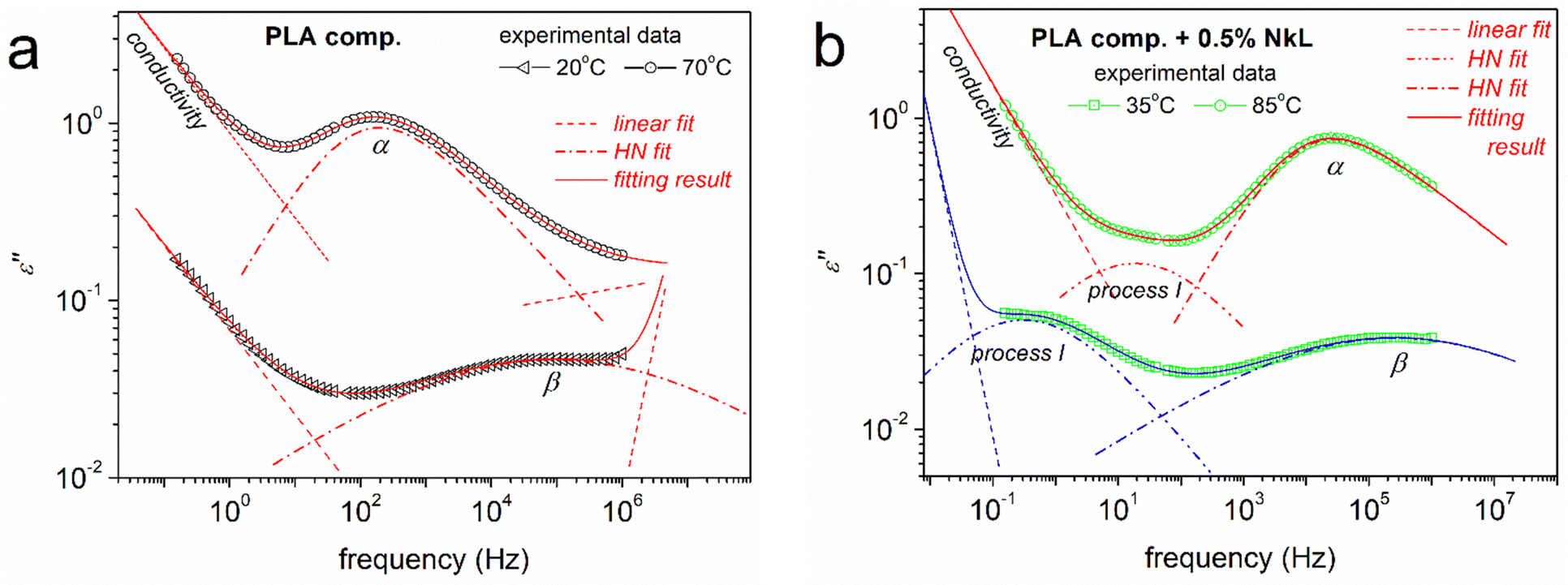 | ||
| Fig. 9 Examples of analysis of the complex ε′′(f) spectra in terms of model functions [here, the Havriliak–Negami (HN)],83 being shown for the indicated samples and temperatures. | ||
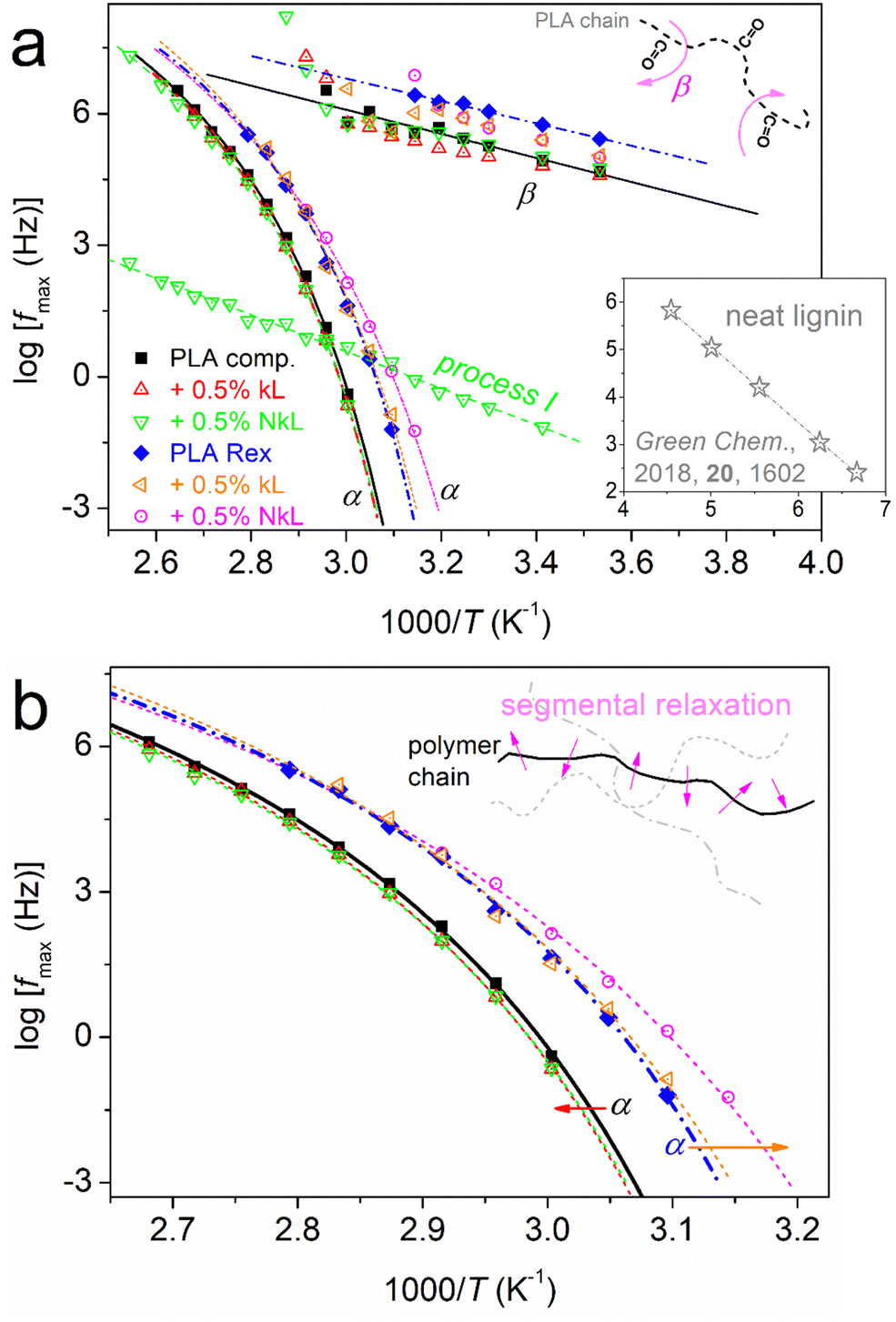 | ||
| Fig. 10 Dielectric relaxation map (Arrhenius plots) for samples/compositions in terms of time scale, shown for (a) the overall mobility and (b) focusing on the segmental relaxation. The added straight and curved lines connecting the experimental points are fittings of the Arrhenius and VFTH equations, respectively. The schematics added to both (a) and (b) are used to explain the molecular origins of the local and segmental relaxations.70,71 The inset to (a) shows the time scale of a relaxation recorded by BDS in a previous study on neat lignin (adapted from ‘D. Vural et al., Green Chem., 2018, 20, 1602’84). | ||
From the overall view provided in Fig. 10(a), two groups seem to exist in both the local and segmental dynamics, the faster and slower ones. This grouping coincides with the ‘comp’ and ‘Rex’ series separation. What is exceptionally interesting here is that the faster local dynamics is accompanied by faster segmental ones as well. This is not the general case for all polymers; however, we have observed such associations for many different polyesters (Chapter 4 in ref. 56). The association has been proposed to be due to the fact that the group responsible for the molecular origin of β is located at the chain backbone rather than a side branch.
As expected, in Fig. 10, β exhibits a linear, or else, an Arrhenius-like time scale, whereas the α relaxation exhibits a ‘curved’ trend described well by the Vogel–Fulcher–Tammann–Hesse (VFTH) equation.70 The second case is characteristic of segmental dynamics, with the curvature actually denoting non constant activation energy and the character of cooperativity.85 In general, for each individual PLA series, β of neat PLA is not seriously affected by the presence of kL or NkL. The situation is different for α relaxation. The data of α relaxation are reproduced and better focused on in Fig. 10(b). Therein, we clearly observe that kL and NkL imply weak deceleration in PLA-comp, whereas, they impose strong acceleration in the case of PLA-Rex. Following previous works56 (and references therein), using the VFTH fitting results (curved lines in Fig. 10), we estimated the ‘dielectric glass transition temperatures, Tg,diel’, and present them in Fig. 11(a). Aside Tg,diel, we also estimated the fragility index, m, of α relaxation employing Böhmer's formalism.85 In our case, m can be used as a measure of cooperativity. The corresponding data are shown in Fig. 11(b).
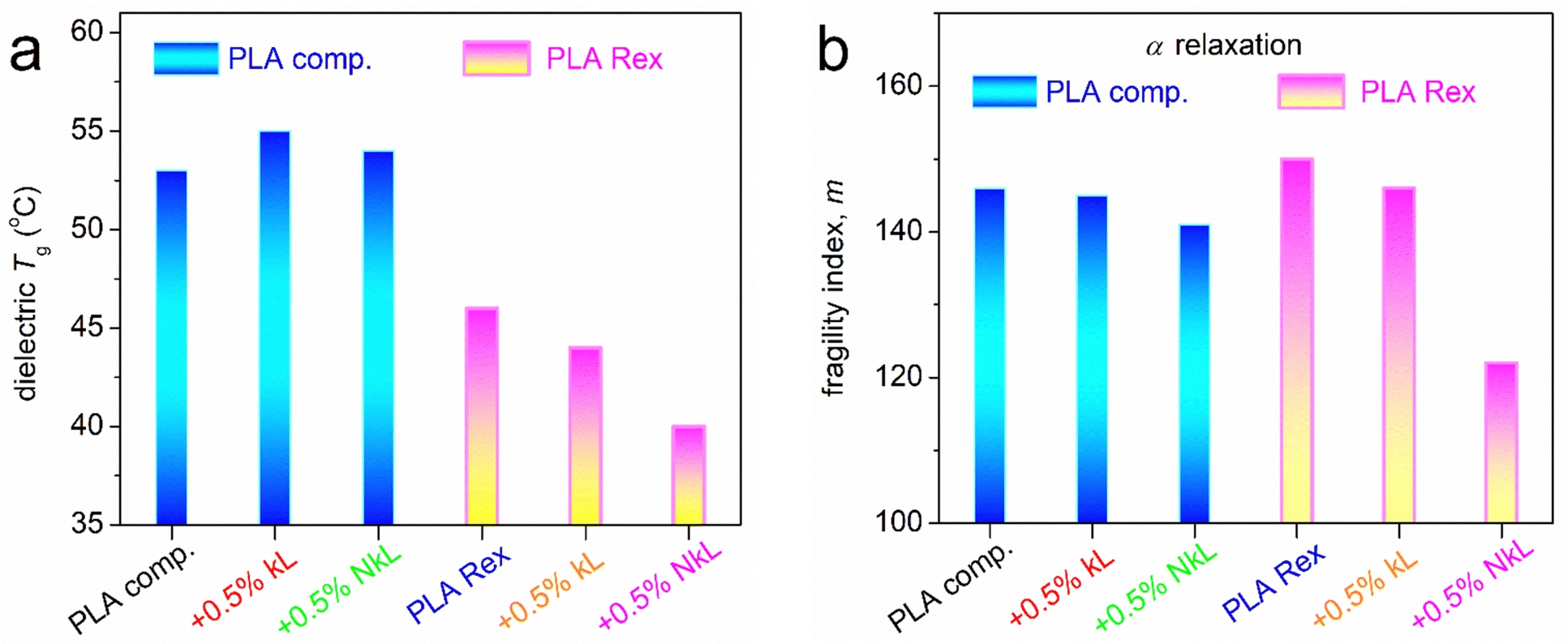 | ||
| Fig. 11 (a) Dielectric glass transition temperature and (b) fragility index of α relaxation for all samples in the form of column diagrams (more details in the main text). | ||
The PLA-comp systems exhibit higher Tg,diel, while kL and NkL impose a mild increase. At the same time, m slightly drops. Partially opposite effects occur in PLA-Rex, as the lower Tg,diel values are further reduced by kL and NkL. Simultaneously, kL and NkL impose a significant drop in m. NkL imposed clearly stronger impacts on both values as compared to kL. The drop in Tg suggests some kind of plasticization of the polymer mobility. This is supported by the drop in fragility which could indicate the increase in cooperativity length, ξ,86 possibly due to the increase in inter-chain distances, or else, increase the free volume of the area away from the lignins. This is compatible with the previously mentioned increase of the polymer concentration around the lignins and subsequently the decrease of polymer density away from them. Such a situation has been observed before in PLA-based systems21,27,30,32 as well as in other polymers.65
A final point regarding dynamics is the recording of the clear presence of process I uniquely in the case of PLA-comp + 0.5% NkL (Fig. 10(a)). Interestingly, this process exhibits a linear time scale of both faster and slower modes compared to those of α, along with a moderately high activation energy (∼0.8 eV). The process does not seem to arise from lignin itself, at least, when compared with the few findings from the literature on neat lignin (inset to Fig. 10(a)).84 A similar process has been recorded in PLA-based PNCs involving strong interfacial attractions27,87 and resembles the dynamics of polymer chains strongly attached to solid surfaces.88 Due to the lack of similar findings from the literature for PLA/lignin, we may not conclude to the definite origins of the said process.
Looking back at the results of Tg and the worth-noting difference between the neat ‘in-house’ PLA-Rex and the ‘commercial’ PLA-comp, we concluded that the differences arise, most probably, from the additives (impurities) interfering within the latter. It would be interesting to follow this point in the near future, for example, by purifying the commercial PLA, perform synthesis of PLA/lignin by the latter and compare the structure/performance results of these systems with those of the present work.
Last but not the least, in the ESI,† we show and discuss results on the electrical conductivity and thermal diffusivity, as recorded by BDS and light flash analysis (LFA, Section S1 in ESI†), respectively. The most significant alternations therein involve the significant roles of the rubbery vs. the glassy state (ionic conductivity) and that of the amorphous vs. the semicrystalline state of the polymer (heat transport).26,89–92 In this connection, the properties of the initial PLA and that of lignin were found to have an indirect effect, namely, on the Tg and the semicrystalline morphology. Please note that the various effects recorded on the ions or heat transfer demonstrate the potential to tune them via processing and the nanocomposite composition. Among others, this may help to mimic in the future, the transfer of small molecules (e.g., oxygen, humidity, other gases, etc.) that occur within certain applications, needing the involvement of renewable polymer-based materials.
4. Conclusions
The two series of sustainable PLA/lignin composites, loaded with micro and nano kraft lignin involving two synthetic routes, were found, in general, homogeneous systems, regarding the thermal transitions and crystallizability. The melt-compound synthesized PLA and the corresponding composites were found to barely crystallize, whereas the reactive extrusion (in situ) synthesized ones exhibited increased nucleation and crystalline fractions. The direct effects of lignin on glass transition and molecular dynamics were investigated in the amorphous state and were found mainly opposite between the two series. A dynamical heterogeneity seems to interfere, as manifested by a discrepancy between the trends in the calorimetric against the dielectric Tgs. The overall results were rationalized in terms of possible effects/alternations in the free volume, or else, in the polymer density around the lignins and away from them. The nano-lignin demonstrated mainly stronger effects as compared to the micro-lignin. We recorded a variety of effects co-affecting the thermal properties (crystallinity, semicrystalline morphology, Tg) and the transport phenomena (both increasing and decreasing). We have successfully employed recently proposed models here to explain the studied physical mechanisms, which is a small additional contribution to basic research. Lastly, we should mark that the variety of effects records sets these other similar ‘green’ materials as suitable means for the manipulation of various properties, always envisaging targeted applications.Author contributions
Sofia P. Makri: resources, investigation, writing – review & editing; Panagiotis A. Klonos: conceptualization, methodology, investigation, formal analysis, writing – original draft; Giacomo Marra: investigation, writing – review & editing; Alexandros Zoikis-Karathanasis: resources, validation, writing – review & editing; Ioanna Deligkiozi: resources, validation, writing – review & editing; Miguel Ángel Valera: resources, validation, writing – review & editing; Ana Mangas: resources, writing – review & editing; Nikolaos Nikolaidis: supervision, investigation, writing – review & editing; Zoi Terzopoulou: supervision, investigation, formal analysis, validation, writing – review & editing; Apostolos Kyritsis: resources, validation, writing – review & editing; Dimitrios N. Bikiaris: supervision, resources, validation, writing – review & editing.Data availability statement
The raw data for the article cannot be made available via an open access source, due to legal or ethical confidentiality requirements. Selected data could be available upon request to the corresponding authors, uniquely in the frame of private communication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union's Horizon 2020 Research and Innovation Programme under grant agreement no. 952941 (BIOMAC Project). The publication of the article in OA mode was financially supported by HEAL-Link.References
- M. Chanda and S. K. Roy, Industrial polymers, specialty polymers, and their applications, CRC Press, Boca Raton, 2008 Search PubMed.
- S. J. Rowan, ACS Macro Lett., 2021, 10, 466–468 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- R. Mori, RSC Sustainability, 2023, 1, 179–212 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- A. Gandini and T. M. Lacerda, Macromol. Eng. DOI:10.1002/9783527815562.mme0019.
- N. M. Ainali, D. Kalaronis, E. Evgenidou, G. Z. Kyzas, D. C. Bobori, M. Kaloyianni, X. Yang, D. N. Bikiaris and D. A. Lambropoulou, Sci. Total Environ., 2022, 832, 155014 CrossRef CAS PubMed.
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. D. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Polymers, 2021, 13, 1822 CrossRef CAS PubMed.
- E. Psochia, L. Papadopoulos, D. J. Gkiliopoulos, A. Francone, M.-E. Grigora, D. Tzetzis, J. Vieira de Castro, N. M. Neves, K. S. Triantafyllidis, C. M. Sotomayor Torres, N. Kehagias and D. N. Bikiaris, Macromol, 2021, 1, 49–63 CAS.
- T. Zhang, B. A. Howell, A. Dumitrascu, S. J. Martin and P. B. Smith, Polymer, 2014, 55, 5065–5072 CrossRef CAS.
- A. Z. Naser, I. Deiab and B. M. Darras, RSC Adv., 2021, 11, 17151–17196 RSC.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- P. Saini, M. Arora and M. N. V. Kumar, Adv. Drug Delivery Rev., 2016, 107, 47–59 CrossRef CAS PubMed.
- O. Coulembier, J. De Winter, T. Josse, L. Mespouille, P. Gerbaux and P. Dubois, Polym. Chem., 2014, 5, 2103–2108 RSC.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- R. Zhang, F. Du, K. Jariyavidyanont, E. Zhuravlev, C. Schick and R. Androsch, Thermochim. Acta, 2022, 718, 179387 CrossRef CAS.
- T. Beslikas, I. Gigis, V. Goulios, J. Christoforides, G. Z. Papageorgiou and D. N. Bikiaris, Int. J. Mol. Sci., 2011, 12, 6597–6618 CrossRef CAS PubMed.
- P. A. Klonos, K. Chronaki, S. Vouyiouka and A. Kyritsis, ACS Appl. Polym. Mater., 2024, 6, 1573–1583 CrossRef CAS.
- T. Zhou, Y. T. Guo, C. Yang, X. B. Meng, F. S. Du and Z. C. Li, Polym. Chem., 2024, 15, 156–165 RSC.
- P. Georgiopoulos, E. Kontou, A. Meristoudi, S. Pispas and M. Chatzinikolaidou, J. Biomater. Appl., 2014, 29, 662–674 CrossRef CAS PubMed.
- M. Rahmanifard, S. M. H. Khademi, R. Asheghi-Oskooee, T. Farizeh and F. Hemmati, RSC Adv., 2024, 14, 794–807 RSC.
- R. Androsch, R. Zhang and C. Schick, Polymer, 2019, 176, 227–235 CrossRef CAS.
- P. A. Klonos, V. Peoglos, D. N. Bikiaris and A. Kyritsis, J. Phys. Chem. C, 2020, 123, 5469–5479 CrossRef.
- K. Pušnik Črešnar, P. A. Klonos, A. Zamboulis, Z. Terzopoulou, E. Xanthopoulou, L. Papadopoulos, A. Kyritsis, K. Kuzmič, L. Fras Zemljič and D. N. Bikiaris, Thermochim. Acta, 2021, 703, 178998 CrossRef.
- A. Toda, R. Androsch and C. Schick, Polymer, 2016, 91, 239–263 CrossRef CAS.
- M. Naddeo, G. Viscusi, G. Gorrasi and D. Pappalardo, Molecules, 2021, 26, 4454 CrossRef CAS PubMed.
- P. A. Klonos, N. D. Bikiaris, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis and Z. Terzopoulou, Soft Matter, 2023, 19, 7846–7858 RSC.
- P. A. Klonos, M. Lazaridou, Ch Samiotaki, A. Kyritsis and D. N. Bikiaris, Polymer, 2022, 259, 125329 CrossRef CAS.
- N. D. Bikiaris, P. A. Klonos, R. O. Ioannidis, P. Saranti, P. Barmpalexis and A. Kyritsis, Polymer, 2024, 292, 126635 CrossRef CAS.
- N. D. Bikiaris, I. Koumentakou, C. Samiotaki, D. Meimaroglou, D. Varytimidou, A. Karatza, Z. Kalantzis, M. Roussou, R. D. Bikiaris and G. Z. Papageorgiou, Polymers, 2023, 15, 1196 CrossRef CAS PubMed.
- Z. Terzopoulou, P. A. Klonos, A. Kyritsis, A. Tziolas, A. Avgeropoulos, G. Z. Papageorgiou and D. N. Bikiaris, Polymer, 2019, 166, 1–12 CrossRef CAS.
- T. Casalini, F. Rossi, A. Castrovinci and G. Perale, Front. Bioeng. Biotechnol., 2019, 7, 259 CrossRef PubMed.
- L. Sha, Z. Chen, Z. Chen, A. Zhang and Z. Yang, Int. J. Polym. Sci., 2016, 2016, 6869154 Search PubMed.
- B. Wang, T. Wen, X. Zhang, A. Tercjak, X. Dong, A. J. Müller, D. Wang and D. Cavallo, Macromolecules, 2019, 52, 6274–6284 CrossRef CAS.
- J. Ahmed and S. K. Varshney, Int. J. Food. Prop., 2011, 14, 37–58 CrossRef CAS.
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Q. Nquyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS.
- A. Constanzo, R. Spotorno, M. V. Candal, M. M. Fernández, A. J. Müller, R. S. Graham, D. Cavallo and C. McIlroy, Addit. Manuf., 2020, 36, 101415 Search PubMed.
- J. Dominguez-Robles, N. K. Martin, M. L. Fong, S. A. Stewart, N. J. Irwin, M. I. Rial-Hermida, R. F. Donnely and E. Larrenta, Pharmaceutics, 2019, 11, 165 CrossRef CAS PubMed.
- C. Ma, T. H. Kim, K. Liu, M. G. Ma, S. E. Choi and C. Si, Front. Bioeng. Biotechnol., 2021, 9, 708976 CrossRef PubMed.
- O. Gordobil, R. Delucis, I. Egüés and J. Labidi, Ind. Crops Prod., 2015, 72, 46–53 CrossRef CAS.
- Y. Zhang and M. Naebe, ACS Sustainable Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- J. Li, W. Liu, X. Qiu, X. Zhao, Z. Chen, M. Yan, Z. Fang, Z. Li, Z. Tu and J. Huang, Green Chem., 2022, 24, 823–836 RSC.
- J. Domínguez-Robles, E. Larrañeta, M. L. Fong, N. K. Martin, N. J. Irwin, P. Mutjé, Q. Tarrés and M. Delgado-Aguilar, Int. J. Biol. Macromol., 2020, 145, 92–99 CrossRef PubMed.
- I. Ullah, Z. Chen, Y. Xie, S. S. Khan, S. Singh, C. Yu and G. Cheng, Int. J. Biol. Macromol., 2022, 208, 819–832 CrossRef CAS PubMed.
- S. Domenek, A. Louaifi, A. Guinault and S. Baumberger, J. Polym. Environ., 2013, 21, 692–701 CrossRef CAS.
- S. P. Makri, E. Xanthopoulou, M. A. Valera, A. Mangas, G. Marra, V. Ruiz, S. Koltsakidis, D. Tzetzis, A. Zoikis Karathanasis, I. Deligkiozi, N. Nikolaidis, D. Bikiaris and Z. Terzopoulou, Polymers, 2023, 15, 2386 CrossRef CAS PubMed.
- K. Shi, G. Liu, H. Sun and Y. Weng, Polymers, 2023, 15, 2807 CrossRef CAS PubMed.
- S. P. Makri, E. Xanthopoulou, P. A. Klonos, A. Grigoropoulos, A. Kyritsis, K. Tsachouridis, A. Anastasiou, I. Deligkiozi, N. Nikolaidis and D. Bikiaris, Polymers, 2022, 14, 5274 CrossRef CAS PubMed.
- O. Gordobil, I. Egüés, R. Llano-Ponte and J. Labidi, Polym. Degrad. Stab., 2014, 108, 330–338 CrossRef CAS.
- K. S. Guiao, A. Gupta, C. Tzoganakis and T. H. Mekonnen, J. Cleaner Prod., 2022, 155, 131840 CrossRef.
- M. Herrero, K. Núñez, R. Gallego, J. C. Merino and J. M. Pastor, Eur. Polym. J., 2016, 75, 125–141 CrossRef CAS.
- D. Kourtidou, P. A. Klonos, L. Papadopoulos, A. Kyritsis, D. N. Bikiaris and K. Chrissafis, Soft Matter, 2021, 17, 5815–5828 RSC.
- A. Schönhals and P. Szymoniak, Dynamics of composite materials, Springer, Cham, Switzerland, 2022 Search PubMed.
- P. A. Klonos, L. Papadopoulos, G. Z. Papageorgiou, A. Kyritsis, P. Pissis and D. N. Bikiaris, J. Phys. Chem. C, 2020, 124, 10220–10234 CrossRef CAS.
- K. Yuniarto, Y. A. Purwanto, S. Purwanto, B. A. Welt, H. K. Purwadaria and T. C. Sunarti, AIP Conf. Proc., 2016, 1725, 020101 CrossRef.
- M. Füllbrandt, P. J. Purohit and A. Schönhals, Macromolecules, 2013, 46, 4626–4632 CrossRef.
- R. Androsch and C. Schick, in Polymer Crystallization I. Advances in Polymer Science, ed. F. Auriemma, G. Alfonso and C. de Rosa, Springer, Cham, 2016, vol. 276, pp. 257–288 Search PubMed.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Kolloid-Z. Z. Polym., 1973, 251, 980–990 CrossRef CAS.
- J. R. Dorgan, J. S. Williams and D. N. Lewis, J. Rheol., 1999, 43, 1141–1155 CrossRef CAS.
- P. A. Klonos, N. D. Bikiaris, P. Barmpalexis and A. Kyritsis, Polymer, 2024, 305, 127177 CrossRef.
- L. Sangroniz, A. Sangroniz, L. Meabe, A. Basterretxea, H. Sardon, D. Cavallo and A. Müller, Macromolecules, 2020, 53(12), 4874–4881 CrossRef CAS.
- P. A. Klonos, N. D. Bikiaris, E. Christodoulou, A. Zamboulis, G. Z. Papageorgiou and A. Kyritsis, Polymer, 2022, 242, 124603 CrossRef CAS.
- A. Sargsyan, A. Tonoyan, S. Davtyan and C. Schick, Eur. Polym. J., 2007, 43, 3113–3127 CrossRef CAS.
- A. Wurm, M. Ismail, B. Kretzschmar, D. Pospiech and C. Schick, Macromolecules, 2010, 43, 1480–1487 CrossRef CAS.
- S. Koutsoumpis, K. N. Raftopoulos, O. Oguz, C. M. Papadakis, Y. Z. Menceloglu and P. Pissis, Soft Matter, 2017, 13, 4580–4590 RSC.
- P. A. Klonos, L. V. Nosach, E. F. Voronin, E. M. Pakhlov, A. Kyritsis and P. Pissis, J. Phys. Chem. C, 2019, 123, 28427–28436 CrossRef CAS.
- F. Kremer and F. Schönhals, Broadband dielectric spectroscopy, Springer-Verlag, Berlin, 2002 Search PubMed.
- J. Ren, O. Urakawa and K. Adachi, Polymer, 2003, 44, 847–855 CrossRef CAS.
- E. Laredo, D. Newman, R. Pezzoli, A. J. Müller and A. Bello, J. Polym. Sci., Part B: Polym. Lett., 2016, 54, 680–691 CrossRef CAS.
- A. R. Brás, M. T. Viciosa, Y. Wang, M. Dionísio and J. F. Mano, Macromolecules, 2006, 39, 6513–6520 CrossRef.
- M. Pluta, J. K. Jeszka and G. Boiteux, Eur. Polym. J., 2007, 43, 2819–2835 CrossRef CAS.
- J. Leng, P. J. Purohit, N. Kang, D. Y. Wang, J. Falkenhagen, F. Emmerling, A. F. Thüneman and A. Schönhals, Eur. Polym. J., 2015, 68, 338–354 CrossRef CAS.
- G. Shi, Y. Guan, G. Liu, A. J. Müller and D. Wang, Macromolecules, 2019, 52, 6904–6912 CrossRef CAS.
- P. A. Klonos, Z. Terzopoulou, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis, P. Pissis and D. N. Bikiaris, Soft Matter, 2022, 18, 3725–3737 RSC.
- J. Leng, N. Kang, D. Y. Wang, J. Falkenhagen, A. F. Thünemann and A. Schönhals, Macrom. Chem. Phys., 2017, 218, 1700232 CrossRef.
- D. E. Martínez-Tong, B. Vanroy, W. Wübbenhorst, A. Nogales and S. Napolitano, Macromolecules, 2014, 47, 2354–2360 CrossRef.
- S. Madkour, H. Yin, M. Füllbrandt and A. Schönhals, Soft Matter, 2015, 11, 7942–7958 RSC.
- X. Monnier and D. Cangialosi, Phys. Rev. Lett., 2018, 121, 137801 CrossRef CAS PubMed.
- C. M. Roland, K. J. McGrath and R. Casalini, Macromolecules, 2006, 39, 3581–3587 CrossRef CAS.
- S. Havriliak and S. Negami, Polymer, 1967, 8, 161–210 CrossRef CAS.
- D. Vural, C. Gainaru, H. O’Neill, Y. Pu, M. D. Smith, J. M. Parks, S. V. Pingali, E. Mamontov, B. H. Davison, A. P. Sokolov, A. J. Ragauskas, J. C. Smith and L. Petridis, Green Chem., 2018, 20, 1602–1611 RSC.
- R. Boehmer, K. Ngai, C. A. Angell and D. J. Plazek, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef CAS.
- N. Delpouve, A. Saiter and E. Dargent, Eur. Polym. J., 2011, 47, 2414–2423 CrossRef CAS.
- F. Wu, S. Zhang, Z. Chen, B. Zhang, W. Yang, Z. Liu and M. Yang, Polymer, 2016, 90, 264–275 CrossRef CAS.
- S. Madkour, P. Szymoniak, M. Heidari, R. Von Klitzing and A. Schönhals, ACS Appl. Mater. Interfaces, 2017, 9, 7535–7546 CrossRef CAS PubMed.
- Z. Han and A. Fina, Prog. Polym. Sci., 2011, 36, 914–944 CrossRef CAS.
- D. Hansen and G. A. Bernier, Polym. Eng. Sci., 1972, 12, 204–208 CrossRef CAS.
- P. A. Klonos, L. Papadopoulos, D. Kourtidou, K. Chrissafis, V. Peoglos, A. Kyritsis and D. N. Bikiaris, App. Nano, 2022, 2, 31–45 CrossRef.
- P. Ding, S. Su, N. Song, S. Tang, Y. Liu and L. Shi, RSC Adv., 2014, 4, 18782–18791 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00622d |
| This journal is © The Royal Society of Chemistry 2024 |