
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLowest gelation concentration in a complex-coacervate-driven self-assembly system, achieved by redox-RAFT synthesis of high molecular weight block polyelectrolytes†
Aleksander
Guzik
 ab,
Fabrice
de Maere d'Aertrycke
a,
Marc C. A.
Stuart
c and
Patrizio
Raffa
*a
ab,
Fabrice
de Maere d'Aertrycke
a,
Marc C. A.
Stuart
c and
Patrizio
Raffa
*a
aSmart and Sustainable Polymeric Products, Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4 9747 AG, The Netherlands. E-mail: p.raffa@rug.nl
bDPI, P.O. Box 902, 5600 AX Eindhoven, The Netherlands
cElectron Microscopy, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
First published on 24th September 2024
Abstract
The objective of this work was to synthesize high molecular weight polyelectrolyte complex (PEC) micelles that are effective in controlling the rheology of aqueous solutions at low concentrations, paving the way for industrial applications of thickeners based on the principle of electrostatic self-assembly. Redox-initiated RAFT (reversible addition–fragmentation chain-transfer) polymerization was used to obtain anionic block polyelectrolytes based on poly(sodium 2-acrylamido-2-methylpropane sulfonate) and poly(acrylamide)–poly(AMPS)-block-poly(AM) (di-block) and poly(AMPS)-block-poly(AM)-block-poly(AMPS) (tri-block), with molecular weights of 237 kDa and 289 kDa and polydispersities of 1.29 and 1.34, respectively. A random poly(AMPS)-co-poly(AM) copolymer was also synthesized for comparison. PEC micelles were obtained upon mixing with cationic poly(N-[3-(dimethylamino)propyl]methacrylamide hydrochloride) – poly(DMAPMA), forming viscoelastic gels at unprecedented low concentrations of <3 wt% for the di-block and <1 wt% for the tri-block, which to date is the lowest demonstrated gelation concentration for a synthetic PEC micelle system. Differences between tri-block and di-block architectures are discussed, with the former being more affected by the addition of salt, which is attributed to percolated network breakdown. The random co-polymer was shown not to be an effective thickener but displayed a surprising lack of phase separation upon coacervation. The assemblies were characterized using dynamic light scattering (DLS) and cryo transmission electron microscopy (cryoTEM), revealing spherical micelles with a diameter of approximately 200 nm for the diblock and a mixture of spherical micelles and network particles for the tri-block PEC micelles. The micelles were not affected by dilution down to a polymer concentration of 7.8 × 10−4% (approx. 0.03 μM). Responsiveness to salinity, pH, and temperature was studied using DLS, revealing a critical NaCl concentration of 1.1 M for the block copolymer micelles.
Introduction
Complex coacervation is a mechanism of polymer aggregation, where two oppositely charged polyelectrolyte chains coalesce together in solution, driven by the gain of entropy from the release of counter-ions as well as by coulombic interactions.1 When the polyelectrolyte homopolymers are mixed, typically a macroscopic phase separation occurs. However, by using controlled polymerization methods such as RAFT,2 charged blocks can be incorporated into controlled co-polymer architectures like block copolymers to confine the phase separation into the colloidal domain, leading to what is known in the literature as PEC (polyelectrolyte complex) micelles.3,4 In such a system, typically, a hydrophilic electrically neutral polymer chain is responsible for the stabilization of the micelles, similar to amphiphilic block copolymer micelles.The unique feature of PEC micelles is that the core, despite being phase-separated from the rest of the water solution, remains hydrophilic and can therefore be used to embed hydrophilic compounds like small molecule drugs, DNA, RNA, and proteins5 for targeted therapeutics, which seems to be the primary application of these systems.6–8 Since the forces keeping the core together are dependent on the presence of charges, such assemblies are naturally highly responsive and tuneable, for example due to changes in the ionic strength or pH. This fact, along with the multitude of shapes, sizes and available chemistries, has led to the adoption of PEC micelles in various other branches of industry. PEC micelles can be used as nanoreactors for synthesis of hydrophilic oxide nanoparticles,9 and the strong adsorption of coacervates to solid surfaces makes them suitable for applications in adhesives and coatings.10 The various applications of PEC outside of biomedical applications have been summarized in a recent review.11
In general, PEC systems are much more intricate than the hydrophobically driven micelle assemblies, with factors like water content, solid vs. liquid nature of the coacervates, influence of salt, pH, and charge stoichiometry, and mechanisms of micellization,12 all being crucial for the given applications.
By analogy to amphiphilic block copolymers, ionic self-assembly can be used to influence the rheology of solutions. For instance, a tri-block copolymer with polyelectrolyte-end blocks can form a percolated network in solution upon mixing with oppositely charged polyions, leading to viscosity increase and gelation. First examples of utilizing this approach to compose hydrogels for biomedical applications have already been published,13–16 where the tunability, responsiveness, and the ability to embed compounds are attractive features. Since the dynamics and relaxation of a coacervate core tend to be fast,17 the viscosifying power of ionically driven self-assembled copolymers is rather low (compared to hydrophobically modified copolymers), with gelation concentrations close to 10 wt%.
The viscosity control afforded by ionic self-assembly could potentially be useful for industrial applications as well, such as in paints, the oil industry, drilling fluids, and other related fields. It could constitute an interesting alternative to typically applied hydrophobically modified polymers, potentially offering easier formulation preparation (dissolution of fully hydrophilic copolymers), and more dynamic behaviour with no kinetically frozen aggregates. The inherent aggregate responsiveness, and ability to stabilize colloids,18,19 could be additional advantages. However, in order to consider these applications, the viscosifying power of PEC micelle-forming copolymers would need to be increased, so that lower and more industrially relevant concentrations could be utilized.
Many aspects of the electrostatically driven hydrogels are a subject of ongoing research with the aim of improving applicability: micelle morphology, tunable viscoelasticity, biocompatibility/biodegradability, increasing the shear strength, and broadening the range of available stimuli.20 Interestingly, however, there has not yet been a clear effort to tackle the issue of high gelation concentration/low viscosifying power.
In this work, we attempt to address this challenge by utilizing recent advances in RAFT controlled polymerization, which allow achieving high molecular weights while preserving the control over the structure of the copolymers.21–25 Molecular weights up to MDa can be obtained through the application of various radical generation strategies, such as redox initiation or photoinitiation, which have recently been applied to obtain ultra-high molecular weight coacervate-forming block copolymers.26 Since the viscosifying capacity is known to increase with the molecular weight of the unimers forming the micelles,27 here we aimed to demonstrate a high molecular weight block polyelectrolyte system synthesized using redox-RAFT (Scheme 1) based on poly(acrylamide) PAM, and poly(sodium 2-acrylamido-2-methylpropane sulfonate) poly(AMPS) complexed with poly(N-[3-(dimethylamino)propyl]methacrylamide hydrochloride) (poly(DMAPMA)), capable of much lower gelation concentrations than previously reported. These monomers have been chosen for their relevance to industrially applicable copolymers; additionally, the strong polyelectrolyte nature of poly(AMPS) makes our system significantly different from previously reported acrylic acid-based complex coacervates. The responsive features afforded by the coacervate nature were preliminarily studied.
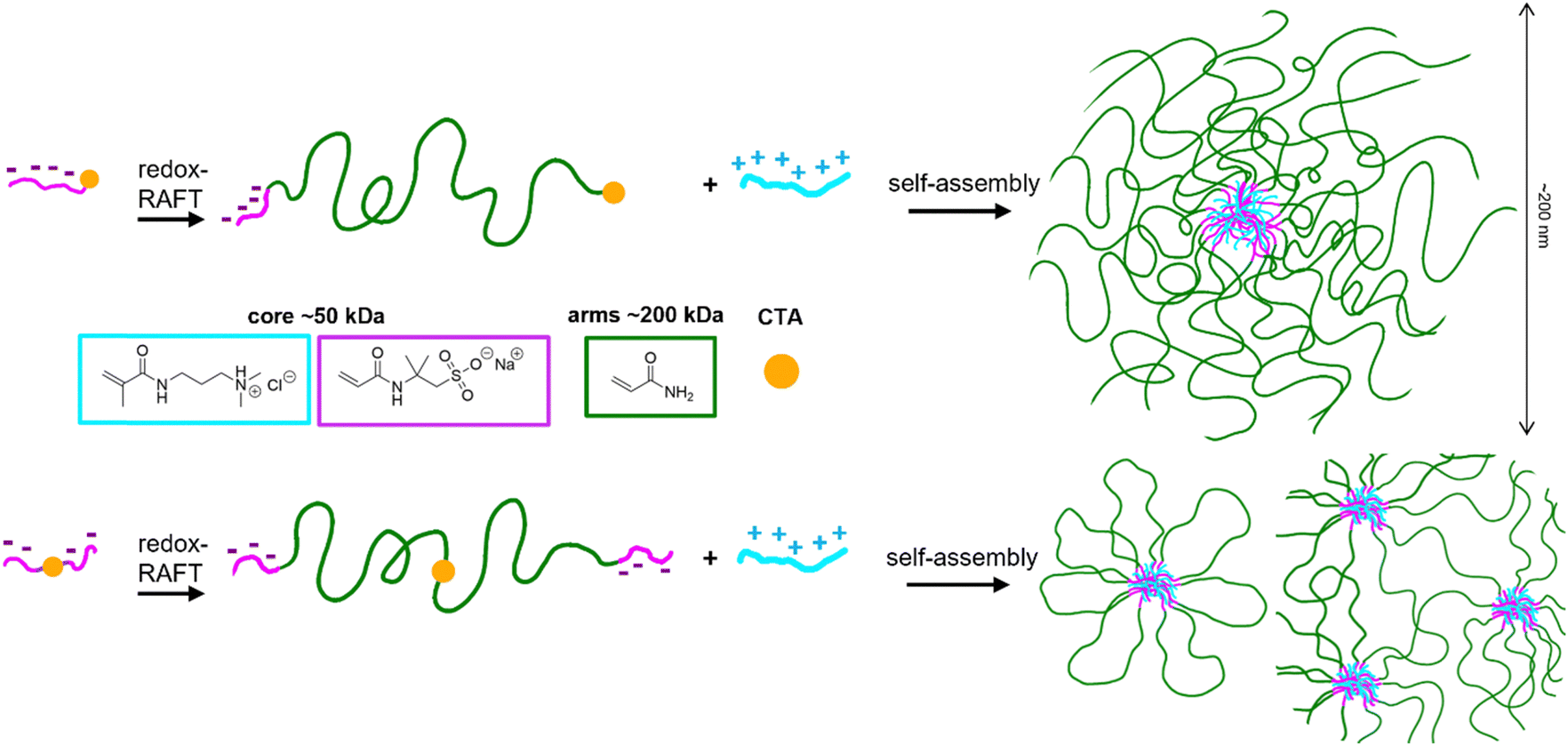 | ||
| Scheme 1 Illustration of the synthesized polymers and self-assembly into PEC micelles. | ||
Experimental
Materials
2-Acrylamido-2-methylpropane sulfonic acid (AMPS, Sigma-Aldrich, ≥99%), acrylamide (AM, Sigma-Aldrich, ≥99%), sodium hydroxide (NaOH, Sigma-Aldrich, ≥98%), hydrochloric acid (HCl, Sigma-Aldrich, 37%), potassium persulfate (K2S2O8, KPS, Sigma-Aldrich, ≥99%), ammonium persulfate((NH4)2S2O8, APS, Sigma-Aldrich, ≥99%), and ascorbic acid (Sigma-Aldrich, ≥99%) were used as received. N-[3 (dimethylamino)propyl]methacrylamide (DMAPMA, Sigma-Aldrich, ≥99%) was filtered through a basic-Al2O3 column prior to use in polymerization reactions. Removal of the inhibitor was confirmed by 1H NMR. 4,4-Azobis(4-cyanovaleric acid) (ACVA, Sigma-Aldrich, ≥98%) was recrystallized from methanol. The RAFT chain transfer agents (CTAs) used, BDMAT28 and DMAcTTC,29 were synthesized according to known literature protocols. Water used for all synthetic and analytical steps was obtained from a Merck Milli-Q filtration system.Polymer synthesis
1.01 g (2.19 × 10−5 mol) of the BDMAT–(NaAMPS)200 macroCTA was added to a round-bottom flask along with 4.67 g (6.57 × 10−2 mol) of acrylamide, and sufficient amount of water to ensure the desired [M]0. The flask was sealed, and the solution was then flushed by bubbling argon gas for 30 minutes. Polymerization was then initiated by the addition of air-free, freshly prepared solutions of 1 mg mL−1 ascorbic acid (0.77 mL, 0.77 mg, 4.38 × 10−6 mol) and potassium persulfate (2.37 mL, 2.37 mg, 8.76 × 10−6 mol), maintaining a [CTA]/[INI] ratio of 5 and an [ox]/[red] ratio of 2. The reaction mixture was then heated to 45 °C and left to react for 24 hours. The reaction progress was monitored by 1H NMR. Upon full conversion, the reaction mixture was diluted in twice the volume of Milli-Q water, before being dialyzed against Milli-Q water for two days using an 8k MWCO RC membrane. The solid was recovered by evaporating the water overnight at 80 °C.
Preparation of coacervate solutions
Coacervate solutions were prepared at a specified anionic/cationic molar ratio (typically 1.0, and 0.9–1.1 for experiments shown in Fig. 3) by assuming the theoretical polymer compositions derived from monomer conversion measurements. The solutions were added by weight, measured on a 4-decimal analytical scale. The solution of the cationic and anionic component was dissolved in MQ water at equal wt% concentrations. The wt% concentrations of PEC micelles reported throughout the text refer to total polymer concentrations. For example,. to reach a 3 wt% PEC micelle concentration, a 3 wt% solution of the anionic co-polymer would be mixed with a 3 wt% solution of the cationic homopolymer in a weight ratio resulting from the charge stoichiometry calculation. NaCl produced during the coacervation process was not removed from the solutions.Analytical methods
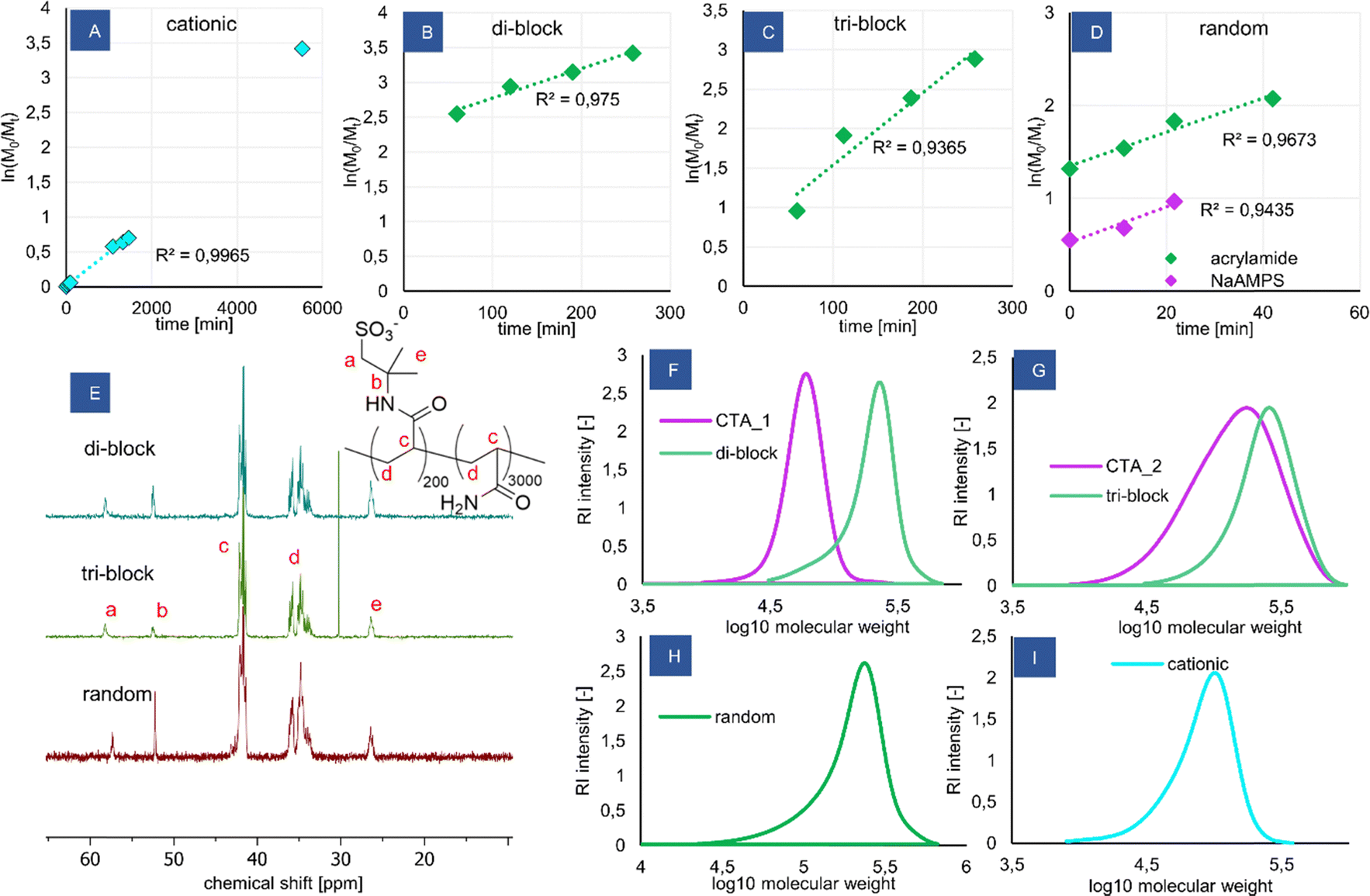 | ||
| Fig. 1 Synthesis of the copolymers and analysis of the products. (A)–(D) Kinetic plots for the polymerization reactions determined using 1H NMR, (E) quantitative 13C NMR spectra of the products recorded in D2O with an inverse-gated decoupling pulse sequence, (F)–(I) molecular weight distributions determined using aqueous SEC. | ||
Results & discussion
Synthesis and molecular characterization of the copolymers
As outlined in Table 1, three different anionic copolymer architectures were synthesized for the study (di-block, tri-block and random) to compare the coacervation behaviour and the influence on the solution rheology.| Sample | RAFT agent | [M]0 [mol L−1] | [M]0/[CTA] | Monomer conversiona [%] | Theoretical degree of polymerizationb | Theor. mol. wt.b [kg mol−1] | SEC Mn [kg mol−1] | SEC D | AMPSc [mol%] | AMPSd [mol%] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Based on 1H NMR. b Based on monomer conversion. c Based on elemental analysis. d Based on 13C NMR. e Theoretical value. | ||||||||||
| CTA_1 | BDMAT | 1.4 | 200 | 100 | AMPS200 | 46.10 | 52.68 | 1.18 | — | — |
| Di-block | CTA_1 | 1.85 | 3000 | 89.7 | AMPS200-b-AM2691 | 237.37 | 161.7 | 1.29 | 6.38 (6.92e) | 7.72 (6.92e) |
| CTA_2 | DMAcTTC | 1 | 400 | 84.5 | AMPS338 | 77.73 | 98.48 | 1.78 | — | — |
| Tri-block | CTA_2 | 2.17 | 3000 | 99.1 | AMPS169-b-AM2973-b-AMP169 | 289.08 | 198.2 | 1.34 | 9.91 (10.21e) | 13.19 (10.21e) |
| Cationic | BDMAT | 1.4 | 200 | 96.7 | DMAPMA193 | 40.15 | 67.03 | 1.37 | — | — |
| Random | BDMAT | AM = 2.32; AMPS = 0.16 | AM = 3000, AMPS = 200 | Total = 88.4%, AM = 88.4% AMPS = 90.2% | AM2653-co-AMPS180 | 229.95 | 164.5 | 1.30 | — | 7.18 (6.37e) |
The copolymers were synthesized using redox-initiated RAFT polymerization according to Schemes 2 and 3 (Experimental section), using RAFT agents BDMAT for the diblock and symmetrical RAFT agent CVPTTC for the triblock (Fig. S1, ESI†). The targeted amount of AMPS ionic units was 200 per block to maintain a similar magnitude of the driving force for self-assembly in the di-block and tri-block copolymers. In the random copolymer, the same amount of 200 AMPS units was randomly distributed along the chain. In all cases, the targeted amount of neutral co-monomer acrylamide was kept constant, at 3000 units per chain, placing the total molecular weight of the copolymers in the range of 200–250 kDa. Separately, a P(DMAPMA) homopolymer was synthesized using RAFT polymerization to serve as the cationic component in coacervate preparation, targeting a chain length equal to that of the ionic blocks of the block copolymers to provide the strongest ionic association.
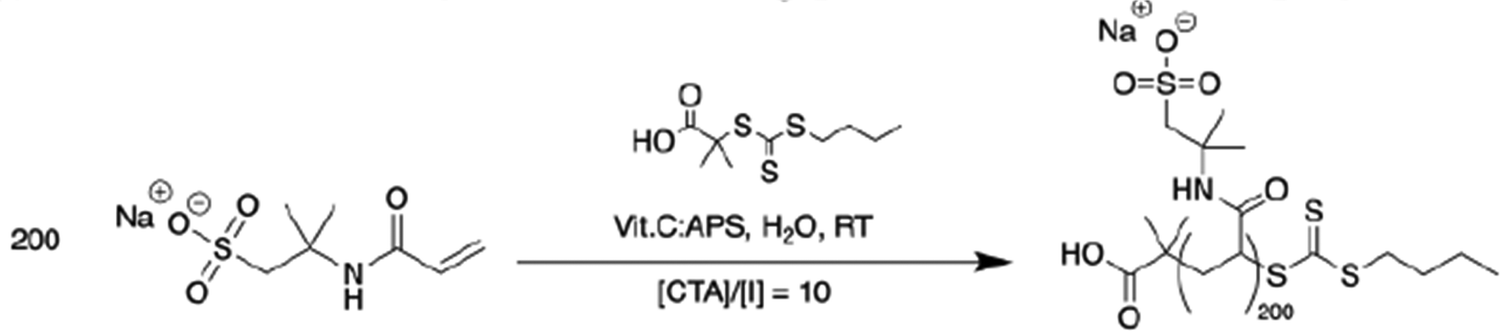 | ||
| Scheme 2 Redox-RAFT polymerization leading to anionic macroCTA. | ||
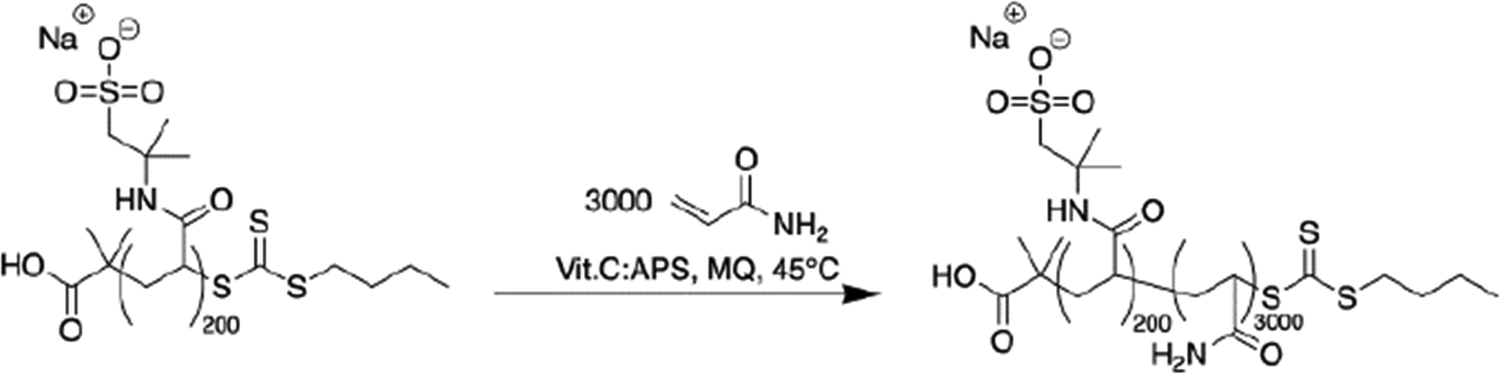 | ||
| Scheme 3 Redox-RAFT polymerization yielding a high molecular weight anionic block copolymer. | ||
The reactions proceeded quickly, adhering to the 1st order kinetics (Fig. 1A–D) and leading to high monomer conversions (Table 1). Additional kinetic plots for the polymerization of P(NaAMPS) macroCTA and its chain extension with acrylamide are shown in Fig. S2 and S3 (ESI†), respectively, along with a full kinetic plot for the random copolymer (Fig. S4, ESI†). The dispersities reached were reasonably low (Table 1), even though the molecular weight of the copolymers is well beyond the practical limit for thermally initiated RAFT polymerization, showing that the redox-RAFT at low-to-moderate temperature was an effective strategy to obtain controlled block copolymers with high molecular weights. The SEC traces are monomodal and shifted to higher molecular weights during chain extension. The measured dispersity of CTA_2 (1.78) is high, which is likely caused by the less suitable RAFT agent CVPTTC possessing no Z-group. However, the chain extension of the significantly polydisperse CTA_2 resulted in a decrease of the dispersity again to an acceptable value (triblock, 1.34), indicating that this particular RAFT agent may work better with the acrylamide monomer. This should be considered in interpreting the observed micelles’ properties, as it could mean that the obtained triblock probably has a high polydispersity of the ionic blocks, containing fractions significantly exceeding the targeted 200 unit chain length.
The molecular weights measured by SEC are consistently lower than the theoretical prediction for the co-polymers, which may be attributed to the relative calibration of the column with PEG standards of different hydrodynamic coil radii than the studied acrylate-based copolymers. Furthermore, unlike the macroCTA poly(AMPS) samples, the block copolymer samples have not been acidified prior to injection, to minimize the risk of poly(acrylamide)hydrolysis. Thus, it is possible that some column adsorption occurred, shifting the measured molecular weights to lower values. Due to this reason, the theoretical molecular weight values for the co-polymers are considered more accurate than the ones obtained from SEC. This is further supported by the AMPS content measurements from 13C NMR and elemental analysis, which show much better agreement with the theoretical molecular weights.
The copolymers were further studied using NMR and elemental analysis. The content of AMPS in the copolymers was derived independently using 13C NMR (Fig. 1E) and elemental analysis, finding the values obtained are in agreement with each other as well as with the theoretical prediction (Table 1). 1H NMR spectra were not useful for analysis due to lack of relevant peaks in poly(acrylamide). The spectra for all products are shown in Fig. S5 (ESI†). 1H DOSY measurements were attempted (see Fig. S6, ESI† for the di-block spectrum) but were found to be limited by the capabilities of the applied diffusion probe to measure polymers with high molecular weight and high viscosity of solutions. Even though the diffusion coefficients of the backbone peaks and AMPS peaks were similar, which could be taken as evidence for a co-polymer nature of the products, the observed peak intensity decrease was too small to classify the 1H DOSY results as reliable.
Polyelectrolyte complex micelles’ morphology
The complex coacervate micelles were prepared from anionic poly(acrylamide)–poly(NaAMPS) block copolymers by directly mixing their solutions with a solution of oppositely charged homopolymer P(PMADMA) at the same concentration. The wt% values reported throughout the text are total solids’ concentrations. The self-assembly was accompanied by the appearance of cloudiness in the solutions (Fig. S7, ESI†), a rise in scattering intensity measured by DLS (Fig. S8, ESI†) and generally a significant increase in viscosity (see the rheology section later). The hydrodynamic sizes measured by DLS also grew significantly compared to the distributions obtained for the solutions of the polymers before mixing (Fig. 2A and B). The DLS measurement for these “unimers” provides an estimate of the hydrodynamic size of the polymer coil, which is relatively close to the value of 35.4 nm calculated by us from an excluded volume polymer coil model with DP = 3200. DLS size distribution for the cationic polymer is shown in Fig. S9 (ESI†). The hydrodynamic diameter values obtained for the PEC micelles are almost an order of magnitude higher than for the unimers, suggesting significant stretching of the PAM chains in the micelle coronas. Macroscopic phase separation was prevented, in line with the previous observation that a corona block three times longer than the core block, yields stable PEC micelles.30 From the CryoTEM observations (Fig. 2F and G), it is evident that the block copolymers self-assembled into spherical micelles with large coacervate cores of size approx. 27.3 ± 8.1 nm for the diblock and 39.9 ± 10.3 nm for the triblock, suggesting that the number of end blocks involved in the di-block and tri-block micelles is similar.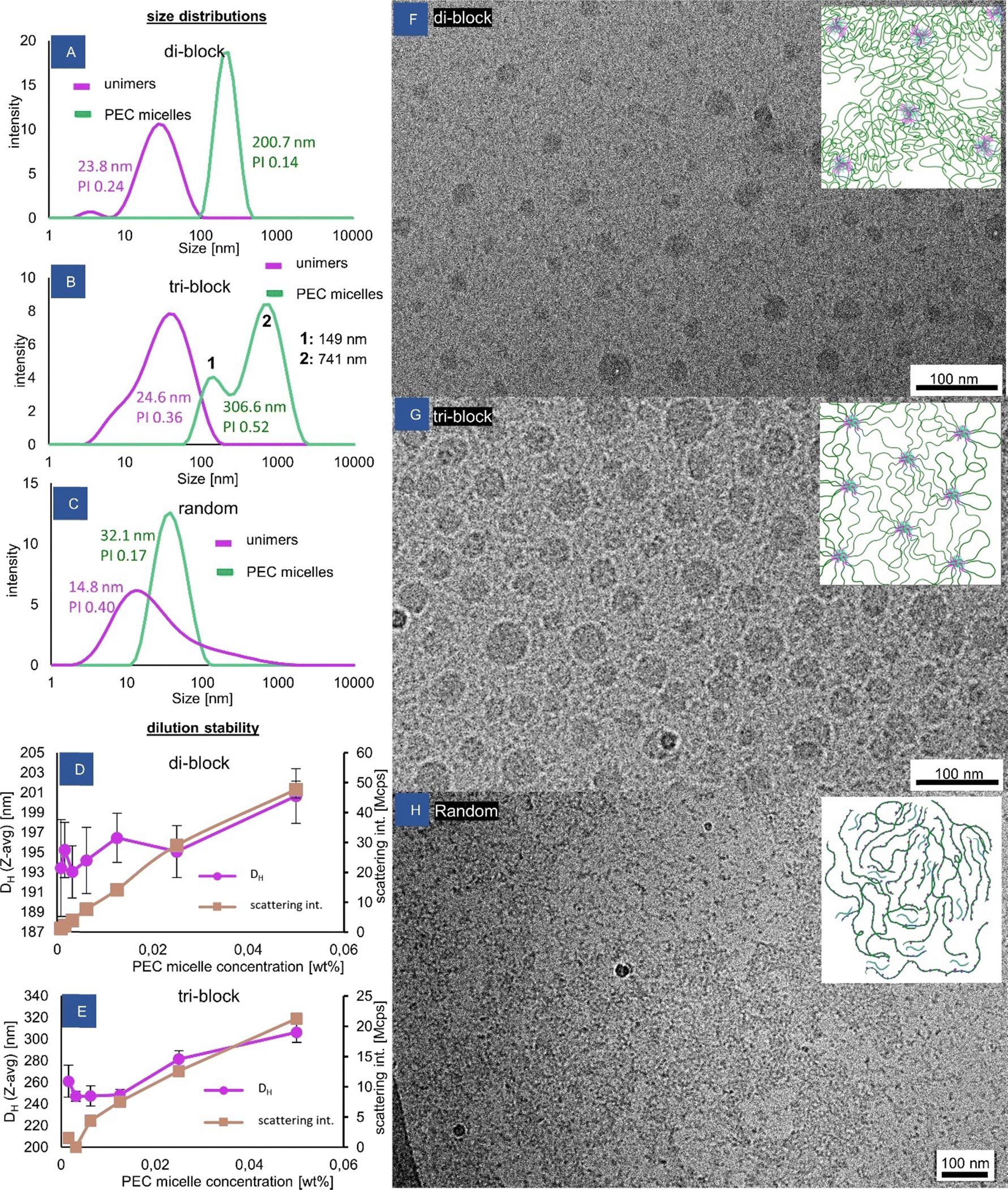 | ||
| Fig. 2 Self-assembly of the complex-coacervate core micelles. (A)–(C) Size distributions obtained from DLS comparing the copolymers before and after complexation with the cationic component. The Z-average size and PI (polydispersity index) values given are derived from the analysis of cumulants. For tri-block (B), mean size values for each population in the size distribution are also given (derived using the NNLS algorithm), which are a better representation of the actual size of nanoobjects. Concentrations are: 0.1 wt% for unimers, 0.05 wt% for PEC micelles. (D) and (E) Dependence of micelle size and scattering intensity on PEC micelle concentration, (F)–(H) cryoTEM pictures of the self-assembled structures (all solutions at 1 wt% in DI water). | ||
The aggregates were stable across a large range of concentration, which is shown in Fig. 2D and E. Even at the lowest tested concentration of 7.8 × 10−4% (equivalent to approx. 0.03 μM), the large micelles were still present, signifying that all tested concentrations were above the critical micellar concentration (CMC). In general, the notion of a CMC is not as straightforward to define for polyelectrolyte complexation-driven micelles as it is for surfactant micelles, and it is known to heavily depend on the polymer structure and ionic strength.31 Some simulations have shown that the presence of PEC aggregates is expected even at experimentally inaccessible low concentrations.32 However, the present system seems to be significantly more stable than some other PEC micelle systems reported to have CMCs of 2 × 10−3%33 and 20 μM.17
Presumably, the relatively high DP (200) of the ionic blocks is responsible for the increased stability.34 In this way, the application of redox-RAFT to reach high molecular weights of the corona is indirectly a new way of increasing the micelle stability, as the long coronal chains enable stabilization against the phase separation of micelles with much longer polyelectrolyte blocks than typically used, resulting in their increased stability. The measured micelle size remains unchanged until the concentration of 2.5 × 10−2%, where it begins to increase slightly (Fig. 2D and E), likely indicating the onset of viscosity increase of the solution (affecting the hydrodynamic size reading), while the scattering intensity increases in proportion to concentration. The application of z-average as a measure of size in a multi-population mixture is not recommended, but is acceptable to demonstrate the existence of micelles and general changes to the system.
The block copolymer architecture had a strong impact on the morphology of the PEC micelles formed. In the di-block, a uniform dispersion of isolated micelles is created, as reflected in a monomodal and narrow DLS size distribution (Fig. 2A), which is also clearly visible in the cryoTEM picture (Fig. 2F). On the other hand, the tri-block copolymer displays a multimodal size distribution. The smaller size population in the DLS size distribution of the triblock (Fig. 2B peak “1”) corresponds to isolated “flowerlike” micelles (Fig. 2G), with two poly(AMPS) blocks residing inside the same micelle, and the neutral poly(acrylamide) chain forming a loop, causing this population to display a lower hydrodynamic diameter than the di-block micelles with star-like corona chains. The larger size population (Fig. 2B peak “2”) is attributed to the spontaneously created nanogel particles, likely consisting of a few PEC cores bridged by poly(acrylamide) chains, forming a network. The presence of a network (at a concentration of 1 wt%) is also suggested by the more tightly packed PEC cores in the CryoTEM picture (Fig. 2G). Such a mixture of isolated micelles and network particles is consistent with previous observations made for ionically associating triblock copolymers, where network formation has been observed to be in equilibrium with isolated micelles even under extreme dilution.33,35 This behaviour (which is completely different from hydrophobically associating triblock copolymers where network formation is observed only above a certain critical concentration) is linked to the lower driving force of a hydrophilic end block entering into a hydrophilic PEC core unable to overcome the entropic penalty associated with the neutral middle block forming a loop, exacerbated by a possible repulsion of polyelectrolyte end blocks from the like-charged PEC cores, leading to a favored inter- rather than intra-micellar connections.
As is shown in the later sections, the tri-block's micelle size distribution can transform to a monomodal one upon various stimuli, e.g. addition of salt, pH cycling, and altering charge stoichiometry. It is speculated that these factors affect the association strength inside the coacervate cores, leading to changes in the preferred self-assembled nanoobject composition. Finally, it cannot be excluded that the nanogel particles observed here could be remaining frozen aggregates after the process of aggregate formation, which progresses through a stage of non-equilibrium nanoobjects followed by equilibration,36 especially given the polydisperse nature of the triblock end blocks.
The coacervation behaviour of the random copolymer is completely distinct from the behaviour of the block copolymers. The expected behaviour of the anionic random copolymer would be to form an insoluble complex upon the addition of the cationic component at the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 charge ratio, due to the lack of long neutral stabilizing chains.37 Surprisingly, no such effect is observed. In fact, the appearance of the coacervate solution changed very little compared to the solution of the unimeric copolymer. Even at concentrations up to 10 wt%, the sample remains transparent, homogenous and does not phase-separate. Upon investigation with DLS, only a slight increase in size can be seen upon complexation, along with a reduction in polydispersity (Fig. 2C). The scattering intensity increases by approximately 3 times for the coacervate sample (Fig. S10, ESI†). As is evident by TEM, at a concentration of 1 wt%, the solution consists of large micron-sized pieces of a continuous network with water pockets, mixed with sub-100 nm irregular objects, which are understood to be soluble macrosalt ion complexes (Fig. 2H). The fact of weak light scattering by these objects suggests that they are extremely loosely aggregated and contain high water content.
:1 charge ratio, due to the lack of long neutral stabilizing chains.37 Surprisingly, no such effect is observed. In fact, the appearance of the coacervate solution changed very little compared to the solution of the unimeric copolymer. Even at concentrations up to 10 wt%, the sample remains transparent, homogenous and does not phase-separate. Upon investigation with DLS, only a slight increase in size can be seen upon complexation, along with a reduction in polydispersity (Fig. 2C). The scattering intensity increases by approximately 3 times for the coacervate sample (Fig. S10, ESI†). As is evident by TEM, at a concentration of 1 wt%, the solution consists of large micron-sized pieces of a continuous network with water pockets, mixed with sub-100 nm irregular objects, which are understood to be soluble macrosalt ion complexes (Fig. 2H). The fact of weak light scattering by these objects suggests that they are extremely loosely aggregated and contain high water content.
Preliminary study on the influence of charge ratio on the structure of the polyelectrolyte complex micelles
The lack of phase separation in the random copolymer coacervate mentioned above may be caused by the extreme asymmetry between molecular weights and linear charge densities between anionic and cationic components. The cationic short and high-charge-density DMAPMA193 chains contain approximately the same amount of chargeable units per chain as the long and low-charge-density AMPS180-co-AM2653, despite the expected radius of gyration of the latter being approximately 5 times larger. This could introduce extreme steric frustration into the aggregates, resulting in the inability to complex all negative charges even though the global charge ratio is 1:1. The uncomplexed charges would then contribute to the colloidal stability.
To investigate this behaviour of the random copolymer further, experiments at varying cationic/anionic ([+]/[−]) charge ratios were conducted. Bulk precipitation of the white solid coacervate phase accompanied by the extreme growth of the size populations upon DLS was induced by raising the [+]/[−] to 1.3 and above (Fig. 3, random), as the excess positive charge could now reach the anionic domains. When the ratio is reversed with [+]/[−] < 1, the system experiences very little change, i.e. precipitation is not observed and the DLS size distribution remains relatively unchanged, which is in line with the above explanation of a sterically frustrated system. It is likely that the excess negative charge can be localized at the periphery of the aggregates, similar to what has been observed for blocky structures under deviation from charge stoichiometry,38 further contributing to aggregate stabilization at [+]/[−] < 1.
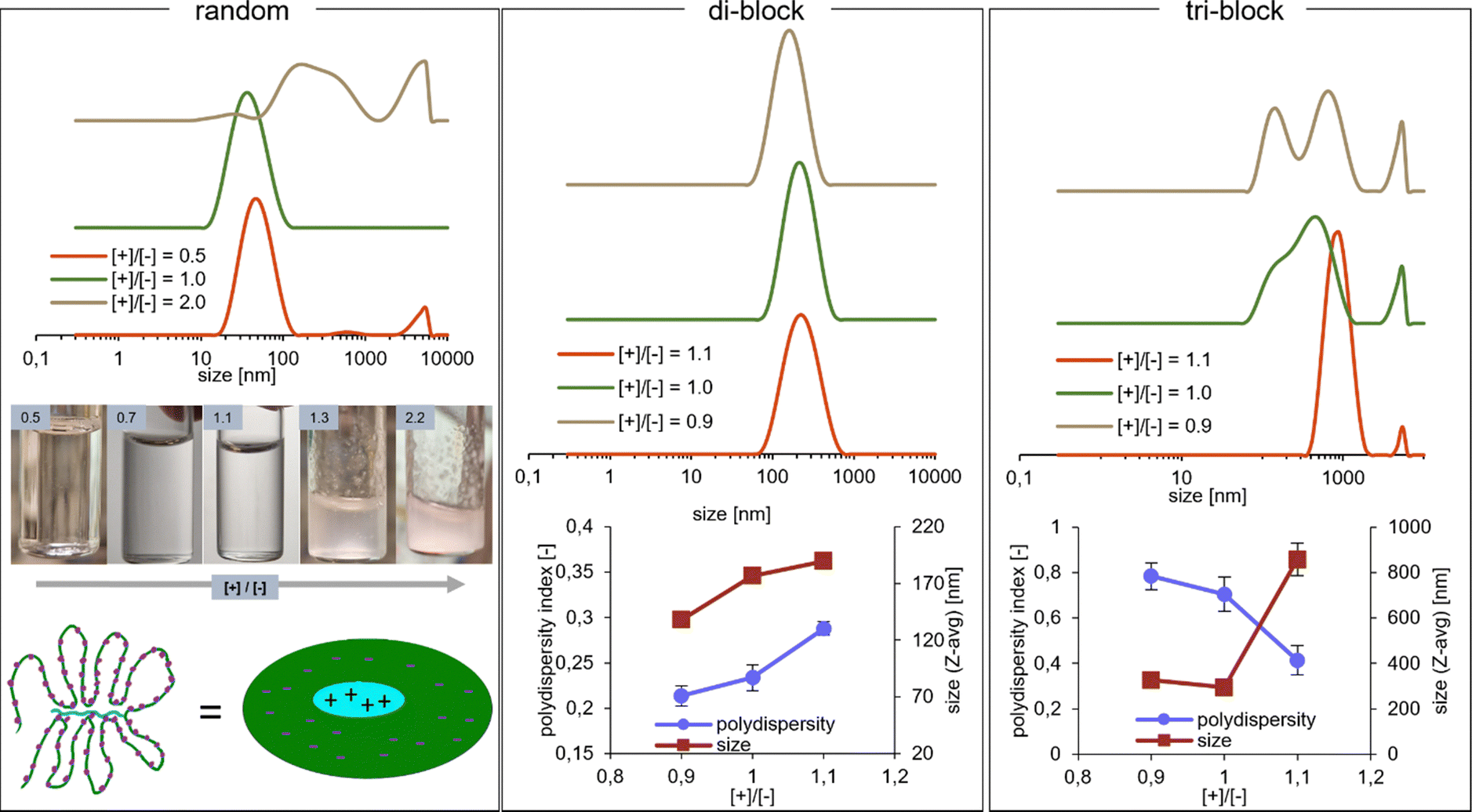 | ||
| Fig. 3 Influence of charge ratio on the coacervate dispersions, studied with DLS. Random: photos – appearance of the coacervate solutions at varying charge ratios. Tri-block: the Z-average mean size values and polydispersity indices are not representative of the object sizes in the multi-modal dispersions, but are provided to visualize the change happening in the system. | ||
The investigations of the random copolymer system were not pursued further within this study, concluding that the described effects disqualify it from applications as an efficient rheology modifier (see the following rheology section). However, it is a system worthy of further exploration, where the exact structure and charge distribution in such frustrated solutions would need to be thoroughly examined by zeta potential, SAXS, detailed DLS titrations and possibly simulations. The hypothetical presence of strong electric field gradients within the material even at the 1:1 charge ratio, caused by the introduction of molecular weight and charge density mismatches, could be an interesting way of providing building blocks for applications such as field-driven soft actuators.39
The charge stoichiometry was also found to influence the block copolymer PEC micelles. Preliminary experiments involving DLS measurements at [+]/[−] ratios of 0.9, 1.0, and 1.1 have been performed. For the di-block micelles, increasing the [+]/[−] ratio results in a slight increase in size and polydispersity, which could potentially be explained by coacervate core swelling due to excess charge (Fig. 3, di-block). Micelle aggregation number Nagg is not expected to increase, as it would contribute to unfavorable deformation of the corona poly(acrylamide) chains in a star-like micelle formed by very asymmetric block copolymers.40 In contrast, the tri-block micelles are affected much more profoundly by the charge ratio. When excess anionic component is present, multiple size populations are seen upon DLS, which are attributed to the co-existence of flowerlike micelles and network particles described before. However, at [+]/[−] = 1.1, it seems that only (nano)gel particles are present, with a single size population having a mean size of almost 1 μm (Fig. 3, tri-block). This is a surprising outcome, as deviation from charge balance generally results in decreased stability, partial disruption of the micelles and coexistence of soluble ion pairs,30,41 which should favour the disappearance of the triblock copolymer network. Furthermore, the observed effects seem to counter previous observations for the triblock + homopolymer PEC system, where the excess of the charged homopolymer component leads to micelle disruption,38 with an important remark that the charge imbalance applied here might not have been large enough to observe these effects. Hypothetically, if the addition of extra cationic charge weakens the micelle cores, the entropic gain due to the formation of bridges over loops could lead to stronger preference for network formation. In that case, the absence of the same effect at [+]/[−] = 0.9 could be explained by the lack of core weakening due to a mechanism proposed in the previous work,38 involving excess charge localization in the corona. Overall however, the behaviour of PEC micelle systems at deviation from 1:1 charge stoichiometry is still not fully understood,42 thus the observed effects, both for the di-block as well as the tri-block are at present difficult to explain satisfactorily without a more detailed experimental investigation.
Rheology of the polyelectrolyte complex micelle solutions
The block copolymer coacervates are very effective thickeners and gelators. Both the di-block and the tri-block copolymers significantly raise the viscosity of the aqueous solution upon complexation with cationic poly(DMAPMA) and lead to the formation of gels. In Fig. 4A and B, the oscillatory frequency sweeps of the solutions are presented. All samples were measured within the linear viscoelastic regime (Fig. S11, ESI†). The high molecular weight of the neutral poly(acrylamide) block achieved by redox-RAFT polymerization resulted in unprecedentedly low gelation concentrations. The tri-block PEC micelles form an elastic gel already at 1 wt% due to the creation of a percolated network. The elastic modulus G′ clearly exceeds the loss modulus G′′, which suggests that the precise gelation concentration is even lower than 1 wt%. The di-block PEC micelles at 2 wt% display characteristics of a critical gel, with evident gels being formed between 2 and 3 wt% polymer, resulting from a jammed system of micelles and potential interdigitation of the non-charged poly(acrylamide) chains. At 1 wt%, the di-block PEC micelle solution is free-flowing, but the exact frequency behaviour cannot be measured reliably due to the low modulus mostly outside of the rheometer measurement window. Concentrations up to 20% were tested, resulting in gels of differing softness, ranging from extremely soft gels with moduli of approximately 0.1 Pa to moderately rigid gels at 10 kPa. This range conveniently overlaps with the rigidity of animal soft tissue,43 predisposing such materials for applications such as injectable gels and tissue engineering. At a high concentration of 20 wt%, the tri-block coacervate exhibits a hard gel scaling behaviour with the moduli almost independent of the frequency (G′ ∝ ω0.13), while the di-block can be described as a frequency-dependent glass (G′ ∝ ω0.32).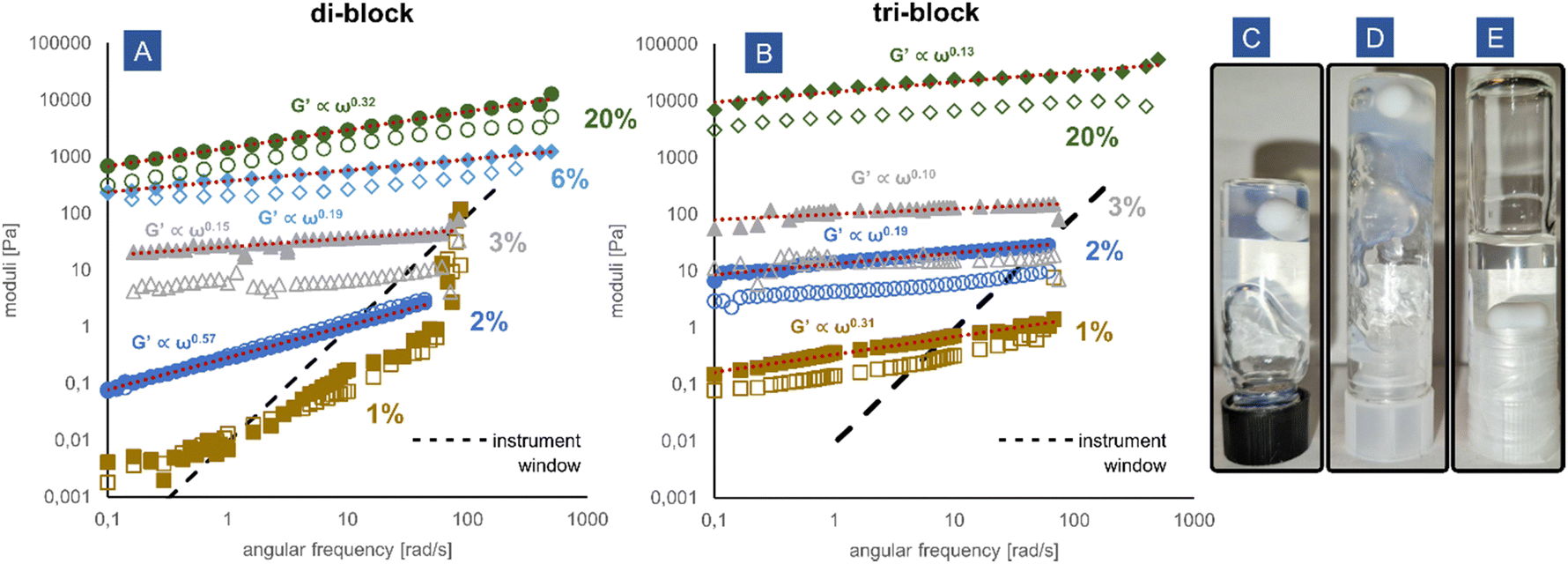 | ||
| Fig. 4 Characterization of the gels formed by the block copolymer coacervates. (A) and (B) Frequency sweeps at varying concentrations of polymer, (C) di-block coacervate at 3 wt%, (D) tri-block coacervate at 3 wt%, (E) random coacervate at 10 wt%. The open symbols correspond to loss modulus G′′, the filled symbols correspond to elastic modulus G′, and the dashed line denotes the instrument window. | ||
The gel concentrations obtained for the tested materials are significantly lower than in other attempts to make coacervate-driven gels found in the literature, where the concentrations at which the systems displayed a gel behaviour were 15 wt%,14 8–9 wt%,44,45 and 4%.46 The obtained moduli are also significantly higher at the respective concentrations. It is noteworthy that the presented system is a mixture of a block and homopolymers, while the mentioned examples from the literature typically use the more efficient14 block + block coacervate micelles, which makes our system even more remarkable in comparison. Lower gelation concentrations (although still higher than described herein) such as 2.5 wt% were possible for other network-forming strategies, such as polyampholytic block copolymers.47 However, due to their narrow pH stability window and the absence of a neutral hydrophilic block stabilizing controlled structures, the applicability of such systems is limited. To date, we believe that our system displays the lowest gelation concentration obtained in a synthetic charge-complexation-driven system. Further optimization is clearly possible by targeting even higher molecular weights (of the neutral, as well as the charged blocks), as new strategies for RAFT synthesis permit molecular weights well into the range of MDa.48 Additionally, it is expected that architectures involving multiple ionic blocks should be more effective than di-blocks and tri-blocks.49
In contrast, the random copolymer proved to be completely ineffective in the modification of rheology. Even at a concentration of 10 wt%, very little change was observed upon coacervation (Fig. 4E). The solution was a completely free flowing and Newtonian fluid with a viscosity of approximately 440 mPa s (Fig. S12, ESI†). The mixture displayed a remarkable lack of macroscopic phase separation and remained optically clear, as discussed in the previous section.
Responsiveness of the polyelectrolye complex micelles
The salt stability of the block copolymer micelles was tested by performing DLS measurements at varying concentrations of NaCl (Fig. 5A and B). A disruption of the micelles caused by the screening of the ionic interactions is clearly detected by the sharp decrease in the measured scattering intensity and hydrodynamic size. For both di-blocks and tri-blocks, this critical salt concentration is around 1.1 M NaCl, which is higher than the value observed for the other systems in the literature.50 The increased static stability of the micelles could be a result of both polyelectrolytes involved having higher than usually reported molecular weights, increasing the thermodynamic driving force for coacervation, as well as due to possible non-ionic (e.g. hydrophobic) interactions between AMPS and DMAPMA monomers, both of which possess relatively highly hydrophobic alkyl units in their structures.51 It is also known that the homopolymer + block copolymer PEC micelle configuration displays higher stability than the block + block.52 Additionally, the large-sized populations (observed by DLS) in the tri-block micelles attributed before to (nano)gel network particles seem to disappear upon the addition of small amounts of salt, lower than the critical concentration (Fig. S13, ESI†), potentially signifying that the fragments of the percolated network display less stability than the isolated flowerlike micelles.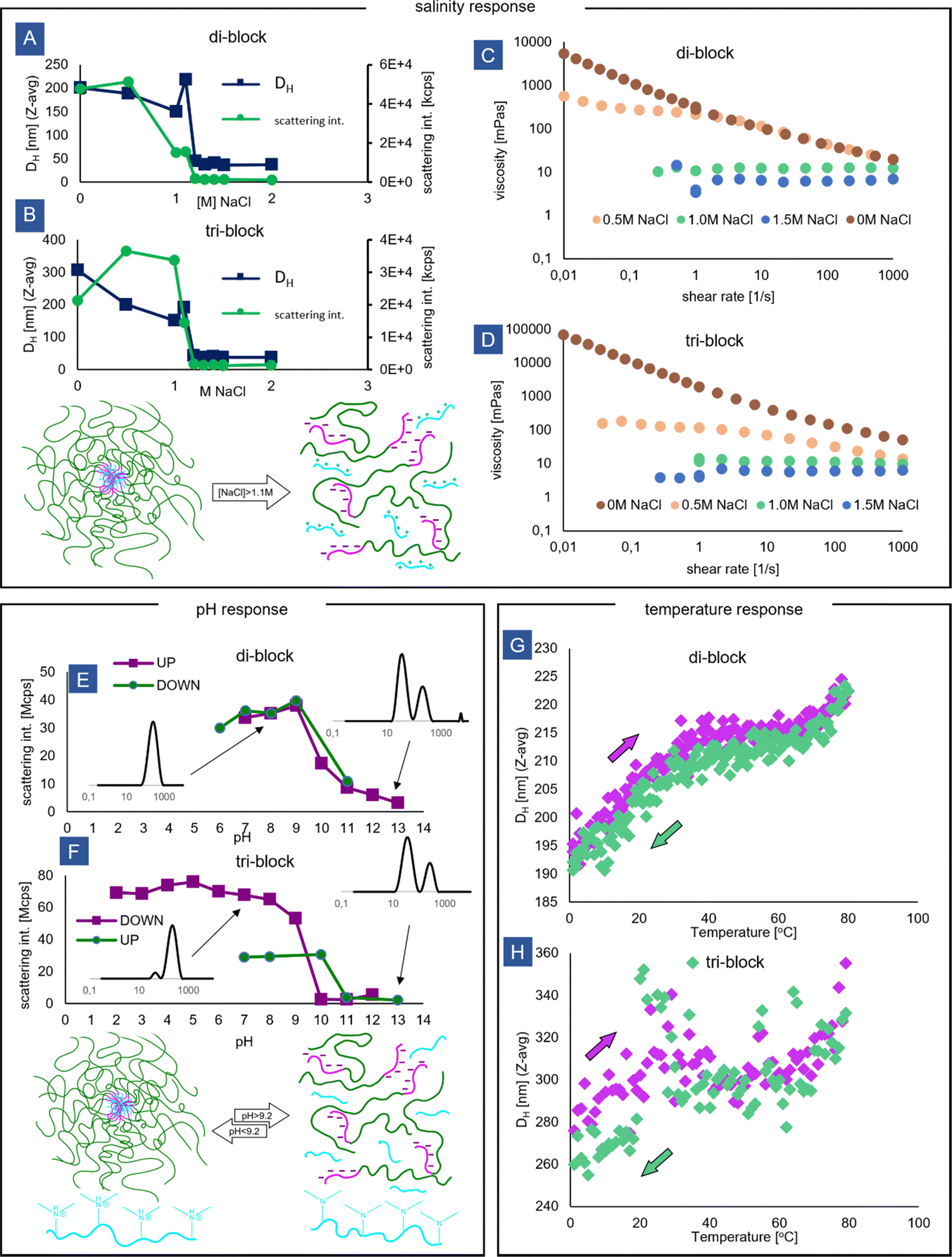 | ||
| Fig. 5 Responsiveness of the complex coacervate core micelles to various stimuli. (A) and (B) Salinity tolerance of the PEC micelles studied by DLS at the concentration of 0.05 wt%, (C) and (D) influence of salinity on the shear viscosity of the 2 wt% solutions of PEC micelles – data points outside of the instrument window are not shown, (E) and (F) pH responsiveness of aggregates studied by DLS (concentration approx. 0.05 wt%), (G) and (H) temperature effect on aggregate size determined by DLS at a micelle concentration of 0.05 wt%. | ||
The salt concentration also has an effect on the micelle size measured by DLS. For both di-block and tri-block micelles, a gradual size decrease is seen, followed by a sharp increase just before reaching the critical salt concentration, which is accompanied by a change in light scattering (Fig. S14, ESI†). The size decrease could be driven by a reduction in Nagg following a change in the preferred micelle curvature, resulting from the expansion and increased crowding of polyacrylamide chains, which are better hydrated in salt solution than in water.53 In general, the addition of salt causes a reduction in surface energy (interfacial tension) of the PEC core; however, in this case, due to the extreme asymmetry between the blocks, the size of the micelles is expected to be completely controlled by the entropic effects arising from crowding within the corona polymer brush according to the star-like micelle model,30 rather than by the core surface energy.
The brief spike in size at high salt concentration has been observed for other PEC systems and could be linked to strong core swelling and loosening of the aggregates resulting from severely weakened ionic attractive interactions and the vanishing difference between the coacervate and solution phase. As the interfacial tension approaches zero, large, loose and non-spherical aggregates could appear,31 accounting for the increased size.
In the random copolymer coacervate, the addition of NaCl seems to reverse the effects observed earlier upon coacervation, with a small decrease in the scattering intensity and little change in the measured hydrodynamic size (Fig. S15, ESI†). The reversal seems to be complete already at 0.5 M NaCl, which is much lower than for the block copolymers, which is in line with previous observations that coacervates with less degree of blockiness of the charged co-monomers exhibit lower entropic driving force for coacervation and are therefore less stable.54
Beyond the critical salt concentration at 1.5 M NaCl, both the di-block and tri-block solutions behave as completely Newtonian fluids, signifying that only unimers (or neutral anionic/cationic soluble pairs) are present in the solution (Fig. 5C and D). However, interestingly, the effects of salt on the shear viscosity of the solutions are also seen before the critical salt concentration is reached. For the di-block, at 0.5 M NaCl, the flow curve remains unchanged in the shear-thinning region, but in the low-shear region, the behaviour changes from a clear yield point to a Newtonian plateau (Fig. 5C). Potentially, this effect is caused by increased intermicellar chain exchange55 and micelle core softening (increased deformability) observed for coacervates in solutions of electrolytes, which would lead to the mobilization of the jammed micelle system responsible for the gelation of the di-block solution.
A particularly strong effect of salt is seen for the tri-block (Fig. 5D). At just 0.5 M NaCl, the viscosity is severely decreased and the solution becomes Newtonian over a wide range of shear rates. Here, the micellar bridges in the network could be preferentially affected by the increased mobility within the coacervate cores. Since the rheology of the tri-block solution is primarily controlled by the presence of the network and by the relaxation rate of the linkages,56,57 these changes would lead to a drastic decrease in viscosity even at salt concentrations that are not high enough to disrupt the micelle cores completely. This explanation coincides with the prior observation of disappearance of the larger-sized population in the tri-block micelles at 0.5 M NaCl. Our observations are in line with other experimental work where lower-than-critical salt concentrations have been shown to weaken the triblock network by reducing the number of elastically active links and causing the micelle arrangement to change, resulting in a weaker gel.14 However, it has also been suggested before that the number of linkages does not change upon salt addition, which would imply that the elasticity of the solutions is insensitive to salt concentration.35 According to that model, the observed drop in viscosity would be only connected to the decreased loss modulus, affected by the decreased lifetime of the linkages.
The aggregates were found to be pH-responsive due to the weak base nature of the cationic amine block (Fig. 5E and F). At high pH, coinciding with the pKa of the DMAPMA monomer (9.2), a breakdown of the aggregates is observed, accompanied by a drastic drop in the scattering intensity and a drop in size measured by DLS. The aggregates are stable under acidic pH conditions, as AMPS is a relatively strong acid. Based on the DLS size distributions, a small amount of the micelles persists both for the di-block as well as for the tri-block in the strongly basic regime (Fig. 5E and F insets). As DLS measurements were carried out immediately after adding a portion of the base to prevent polyacrylamide hydrolysis under the basic conditions, this may reflect a kinetic effect related to micelle reorganization through fission before reaching equilibrium which for some PEC systems can take tens of minutes36,58 or even hours, especially if polydisperse chains are present.59 Alternatively, other interactions, such as hydrophobic ones, can still lead to some aggregation even if one of the polymers is not charged.50
The pH effect is fully reversible for the diblock, with similar sized micelle population re-appearing upon adding acid. However, for the triblock, the larger-sized population attributed earlier to nanogel particles is not re-formed, and only a population related to single micelles is present in the distribution after a pH cycle (Fig. S16, ESI†). It is known that for PEC micelles, the self-assembly pathway can affect the morphology of the created nanoobjects,60 and micelle re-formation in conditions of uniform molecular mixing of anionic and cationic chains could potentially favor a more uniform micelle distribution. Alternatively, the small amount of salt that is created during a NaOH → HCl pH cycle could disrupt the nanogel particles, which would be in line with the above observations regarding the influence of salinity.
Finally, the effect of temperature on the PEC micelles was tested. For both the tri-block and di-block, the micelles exist in the entire temperature range tested (0–80 °C), and the system does not display an LCST nor UCST behaviour reported for some other PEC micelle systems.61,62 For the di-block, the micelles grow slightly in size, reversibly with temperature. It may be related to the hydration and slight expansion of the polyacrylamide corona, or a growth of the micelle cores in response to decreased Bjerrum length.61 For the tri-block, a size increase with temperature is observed as well. However, due to the complex multi-modal size distribution and the existence of very large particles, the size readings are very noisy, making identification of a clear trend impossible.
Conclusions
We successfully demonstrated a PEC micelle system capable of efficient viscosification of aqueous solutions and forming a viscoelastic gel at below 1 wt% concentration of solids, which we believe to date is the lowest gelation concentration reported for a synthetic ionically self-assembled block copolymer system and is comparable to some hydrophobically modified systems used as thickeners and gelators. With this, we open the possibility to consider broader industrial applications of coacervate-driven self-assembled systems such as in the petroleum industry, paints, and cosmetics, where up to now they remained inaccessible due to high polymer concentrations required. The gels exhibited a range of moduli depending on polymer concentration, ranging from extremely soft (0.1 Pa) to moderately rigid (10 kPa), further predisposing these systems for applications in injectable gels and tissue engineering.The high viscosification capacity was achieved by targeting a high molecular weight of the synthesized block copolymers, which was made possible by utilizing redox-initiated RAFT polymerization in water at low (RT-45 °C) temperatures. Poly(NaAMPS)-block-poly(acrylamide)-block-poly(NaAMPS) tri-block, poly(NaAMPS)-block-poly(acrylamide) di-block, and poly(acrylamide)-co-poly(NaAMPS) random copolymers with respective theoretical molecular weights of 289 kDa, 237 kDa, and 230 kDa, respectively, were synthesized. A synthesis protocol was developed allowing such copolymers at high conversions and moderate dispersities to be obtained.
Upon mixing with cationic homopolymer poly(DMAPMA), large (∼200 nm) PEC micelles were created, which was confirmed by DLS and cryoTEM observations. Responsiveness of the micelles was preliminarily tested, finding the critical salt concentration to be 1.1 M NaCl, and confirming a pH-responsive behaviour leading to a reversible breakdown of micelles under alkaline conditions. The micelles formed from the di-block copolymer were well defined and spherical, while the tri-block formed a mixture of flower-like micelles and (nano)gel particles created from fragments of a percolated network. The (nano)gel particles were observed even under strong dilution and were found to be more sensitive to ionic conditions than the isolated micelles: they vanished at salinities well below the critical NaCl concentration, as well as during a NaOH → HCl pH cycle. Surprisingly, changing the charge ratio in favour of the cationic component seemed to favour (nano)gel formation and diminish the amount of isolated triblock micelles.
The rheology of solutions of tri-block copolymer PEC micelles was more sensitive to salt than the di-block, with a drastic reduction in viscosity observed already for salt concentrations well below the critical concentration, which was attributed to the preferential breaking of the inter-micellar bridges, combined with high sensitivity of the solution to the presence and dynamics of the transient network. On the other hand, the di-block PEC micelle solution was affected only in the low-shear-rate region, which is attributed to the un-jamming of the micelles. In this regard, the di-block system presents an interesting new way of obtaining coacervation-driven rheology modifiers with increased salt resistance and well-defined aggregates, in contrast to typically applied network-forming copolymers.
On the other hand, the random poly(acrylamide)-co-poly(NaAMPS) copolymer proved to be completely ineffective as a rheology modifier, with concentrations as high as 10 wt% still exhibiting a free flowing liquid behaviour. Interestingly, no macroscopic phase separation was observed at the 1:1 charge ratio. The system only phase-separated when a significant deviation towards excess positive charge ([+]/[−] = 1.3) was reached, while it did not phase-separate upon excess negative charge. These effects are speculated to arise from the frustration brought about by the extreme mismatch between molecular weights and charge densities of the anionic and cationic components, leading to the separation of charged domains and this phenomenon could be relevant to other applications of random co-polymer coacervates.
Data availability
All data connected with this work are available upon request to the corresponding author.Conflicts of interest
There are no conflicts to declareAcknowledgements
This work is part of the research programme of DPI, project #821, which is kindly acknowledged for funding.References
- M. A. Cohen Stuart, B. Hofs, I. K. Voets and A. de Keizer, Assembly of Polyelectrolyte-Containing Block Copolymers in Aqueous Media, Curr. Opin. Colloid Interface Sci., 2005, 10(1), 30–36, DOI:10.1016/j.cocis.2005.04.004.
- X. Xin, Y. Wang and W. Liu, Synthesis of Zwitterionic Block Copolymers via RAFT Polymerization, Eur. Polym. J., 2005, 41(7), 1539–1545, DOI:10.1016/j.eurpolymj.2005.01.015.
- C. C. M. Sproncken, J. R. Magana and I. K. Voets, 100th Anniversary of Macromolecular Science Viewpoint: Attractive Soft Matter: Association Kinetics, Dynamics, and Pathway Complexity in Electrostatically Coassembled Micelles, ACS Macro Lett., 2021, 10(2), 167–179, DOI:10.1021/acsmacrolett.0c00787.
- S. Shah and L. Leon, Structural Dynamics, Phase Behavior, and Applications of Polyelectrolyte Complex Micelles, Curr. Opin. Colloid Interface Sci., 2021, 53, 101424, DOI:10.1016/j.cocis.2021.101424.
- S. Gao, A. Holkar and S. Srivastava, Protein–Polyelectrolyte Complexes and Micellar Assemblies, Polymers, 2019, 11(7), 1097, DOI:10.3390/polym11071097.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev., 2018, 118(14), 6844–6892, DOI:10.1021/acs.chemrev.8b00199.
- J. M. Fay and A. V. Kabanov, Interpolyelectrolyte Complexes as an Emerging Technology for Pharmaceutical Delivery of Polypeptides, Rev. Adv. Chem., 2022, 12(3), 137–162, DOI:10.1134/S2634827622600177.
- C. Forenzo and J. Larsen, Complex Coacervates as a Promising Vehicle for mRNA Delivery: A Comprehensive Review of Recent Advances and Challenges, Mol. Pharmaceutics, 2023, 20(9), 4387–4403, DOI:10.1021/acs.molpharmaceut.3c00439.
- V. T. A. Nguyen, M.-C. De Pauw-Gillet, M. Gauthier and O. Sandre, Magnetic Polyion Complex Micelles for Cell Toxicity Induced by Radiofrequency Magnetic Field Hyperthermia, Nanomaterials, 2018, 8(12), 1014, DOI:10.3390/nano8121014.
- A. M. Brzozowska, E. Spruijt, A. de Keizer, M. A. Cohen Stuart and W. Norde, On the Stability of the Polymer Brushes Formed by Adsorption of Ionomer Complexes on Hydrophilic and Hydrophobic Surfaces, J. Colloid Interface Sci., 2011, 353(2), 380–391, DOI:10.1016/j.jcis.2010.09.074.
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, On Complex Coacervate Core Micelles: Structure-Function Perspectives, Polymers, 2020, 12(9), 1953, DOI:10.3390/polym12091953.
- A. E. Marras, J. M. Ting, K. C. Stevens and M. V. Tirrell, Advances in the Structural Design of Polyelectrolyte Complex Micelles, J. Phys. Chem. B, 2021, 125(26), 7076–7089, DOI:10.1021/acs.jpcb.1c01258.
- A. L. Z. Lee, Z. X. Voo, W. Chin, R. J. Ono, C. Yang, S. Gao, J. L. Hedrick and Y. Y. Yang, Injectable Coacervate Hydrogel for Delivery of Anticancer Drug-Loaded Nanoparticles in Vivo, ACS Appl. Mater. Interfaces, 2018, 10(16), 13274–13282, DOI:10.1021/acsami.7b14319.
- D. V. Krogstad, N. A. Lynd, S.-H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Effects of Polymer and Salt Concentration on the Structure and Properties of Triblock Copolymer Coacervate Hydrogels, Macromolecules, 2013, 46(4), 1512–1518, DOI:10.1021/ma302299r.
- D. Li, T. Göckler, U. Schepers and S. Srivastava, Polyelectrolyte Complex-Covalent Interpenetrating Polymer Network Hydrogels, Macromolecules, 2022, 55(11), 4481–4491, DOI:10.1021/acs.macromol.2c00590.
- C. M. Papadakis and C. Tsitsilianis, Responsive Hydrogels from Associative Block Copolymers: Physical Gelling through Polyion Complexation, Gels, 2017, 3(1), 3, DOI:10.3390/gels3010003.
- I. Bos, M. Timmerman and J. Sprakel, FRET-Based Determination of the Exchange Dynamics of Complex Coacervate Core Micelles, Macromolecules, 2021, 54(1), 398–411, DOI:10.1021/acs.macromol.0c02387.
- C. E. Mills, A. Obermeyer, X. Dong, J. Walker and B. D. Olsen, Complex Coacervate Core Micelles for the Dispersion and Stabilization of Organophosphate Hydrolase in Organic Solvents, Langmuir, 2016, 32(50), 13367–13376, DOI:10.1021/acs.langmuir.6b02350.
- J. Es Sayed, H. Brummer, M. C. A. Stuart, N. Sanson, P. Perrin and M. Kamperman, Responsive Pickering Emulsions Stabilized by Frozen Complex Coacervate Core Micelles, ACS Macro Lett., 2022, 11(1), 20–25, DOI:10.1021/acsmacrolett.1c00647.
- H. Senebandith, D. Li and S. Srivastava, Advances, Applications, and Emerging Opportunities in Electrostatic Hydrogels, Langmuir, 2023, 39(48), 16965–16974, DOI:10.1021/acs.langmuir.3c02255.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Ultra-High Molecular Weights via Aqueous Reversible-Deactivation Radical Polymerization, Chem, 2017, 2(1), 93–101, DOI:10.1016/j.chempr.2016.12.007.
- J. Rzayev and J. Penelle, HP-RAFT: A Free-Radical Polymerization Technique for Obtaining Living Polymers of Ultrahigh Molecular Weights, Angew. Chem., Int. Ed., 2004, 43(13), 1691–1694, DOI:10.1002/anie.200353025.
- E. Read, A. Guinaudeau, D. James Wilson, A. Cadix, F. Violleau and M. Destarac, Low Temperature RAFT/MADIX Gel Polymerisation: Access to Controlled Ultra-High Molar Mass Polyacrylamides, Polym. Chem., 2014, 5(7), 2202, 10.1039/c3py01750h.
- L. Martin, G. Gody and S. Perrier, Preparation of Complex Multiblock Copolymers via Aqueous RAFT Polymerization at Room Temperature, Polym. Chem., 2015, 6(27), 4875–4886, 10.1039/C5PY00478K.
- A. Reyhani, S. Allison-Logan, H. Ranji-Burachaloo, T. G. McKenzie, G. Bryant and G. G. Qiao, Synthesis of Ultra-High Molecular Weight Polymers by Controlled Production of Initiating Radicals, J. Polym. Sci., Part A: Polym. Chem., 2019, 57(18), 1922–1930, DOI:10.1002/pola.29318.
- J. Y. Rho, A. B. Korpusik, M. Hoteit, J. B. Garrison and B. S. Sumerlin, Ultra-High Molecular Weight Complex Coacervates via Polymerization-Induced Electrostatic Self-Assembly, Polym. Chem., 2024, 15(18), 1821–1825, 10.1039/D4PY00273C.
- L. Despax, J. Fitremann, M. Destarac and S. Harrisson, Low Concentration Thermoresponsive Hydrogels from Readily Accessible Triblock Copolymers, Polym. Chem., 2016, 7(20), 3375–3377, 10.1039/C6PY00499G.
- C. Bray, R. Peltier, H. Kim, A. Mastrangelo and S. Perrier, Anionic Multiblock Core Cross-Linked Star Copolymers via RAFT Polymerization, Polym. Chem., 2017, 8(36), 5513–5524, 10.1039/C7PY01062A.
- J. Liang, G. Shan and P. Pan, Aqueous RAFT Polymerization of Acrylamide: A Convenient Method for Polyacrylamide with Narrow Molecular Weight Distribution, Chin. J. Polym. Sci., 2017, 35(1), 123–129, DOI:10.1007/s10118-017-1874-0.
- S. van der Burgh, A. de Keizer and M. A. Cohen Stuart, Complex Coacervation Core Micelles. Colloidal Stability and Aggregation Mechanism, Langmuir, 2004, 20(4), 1073–1084, DOI:10.1021/la035012n.
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, On the Stability and Morphology of Complex Coacervate Core Micelles: From Spherical to Wormlike Micelles, Langmuir, 2012, 28(40), 14180–14191, DOI:10.1021/la303211b.
- J. Wang, A. de Keizer, R. Fokkink, Y. Yan, M. A. Cohen Stuart and J. van der Gucht, Complex Coacervate Core Micelles from Iron-Based Coordination Polymers, J. Phys. Chem. B, 2010, 114(25), 8313–8319, DOI:10.1021/jp1003209.
- S. Srivastava, M. Andreev, A. E. Levi, D. J. Goldfeld, J. Mao, W. T. Heller, V. M. Prabhu, J. J. de Pablo and M. V. Tirrell, Gel Phase Formation in Dilute Triblock Copolyelectrolyte Complexes, Nat. Commun., 2017, 8(1), 14131, DOI:10.1038/ncomms14131.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, Block Copolymer Micelles: Preparation, Characterization and Application in Drug Delivery, J. Controlled Release, 2005, 109(1), 169–188, DOI:10.1016/j.jconrel.2005.09.034.
- M. Lemmers, K. I. Voets, M. A. C. Stuart and J. van der Gucht, Transient Network Topology of Interconnected Polyelectrolyte Complex Micelles, Soft Matter, 2011, 7(4), 1378–1389, 10.1039/C0SM00767F.
- M. Amann, J. S. Diget, J. Lyngsø, J. S. Pedersen, T. Narayanan and R. Lund, Kinetic Pathways for Polyelectrolyte Coacervate Micelle Formation Revealed by Time-Resolved Synchrotron SAXS, Macromolecules, 2019, 52(21), 8227–8237, DOI:10.1021/acs.macromol.9b01072.
- M. Mende, G. Petzold and H.-M. Buchhammer, Polyelectrolyte Complex Formation between Poly(Diallyldimethyl-Ammonium Chloride) and Copolymers of Acrylamide and Sodium-Acrylate, Colloid Polym. Sci., 2002, 280(4), 342–351, DOI:10.1007/s00396-001-0614-7.
- M. Lemmers, E. Spruijt, L. Beun, R. Fokkink, F. Leermakers, G. Portale, M. A. C. Stuart and J. van der Gucht, The Influence of Charge Ratio on Transient Networks of Polyelectrolyte Complex Micelles, Soft Matter, 2011, 8(1), 104–117, 10.1039/C1SM06281F.
- J. Ahn, J. Gu, J. Choi, C. Han, Y. Jeong, J. Park, S. Cho, Y. S. Oh, J.-H. Jeong, M. Amjadi and I. Park, A Review of Recent Advances in Electrically Driven Polymer-Based Flexible Actuators: Smart Materials, Structures, and Their Applications, Adv. Mater. Technol., 2022, 7(11), 2200041, DOI:10.1002/admt.202200041.
- O. V. Borisov and E. B. Zhulina, Effect of Salt on Self-Assembly in Charged Block Copolymer Micelles, Macromolecules, 2002, 35(11), 4472–4480, DOI:10.1021/ma010934n.
- D. J. Adams, S. H. Rogers and P. Schuetz, The Effect of PEO Block Lengths on the Size and Stability of Complex Coacervate Core Micelles, J. Colloid Interface Sci., 2008, 322(2), 448–456, DOI:10.1016/j.jcis.2008.03.010.
- J. B. Sabadini, C. L. P. Oliveira and W. Loh, Assessing the Structure and Equilibrium Conditions of Complex Coacervate Core Micelles by Varying Their Shell Composition and Medium Ionic Strength, Langmuir, 2024, 40(4), 2015–2027, DOI:10.1021/acs.langmuir.3c01606.
- J. Liu, H. Zheng, P. S. P. Poh, H.-G. Machens and A. F. Schilling, Hydrogels for Engineering of Perfusable Vascular Networks, Int. J. Mol. Sci., 2015, 16(7), 15997–16016, DOI:10.3390/ijms160715997.
- J.-M. Kim, T.-Y. Heo and S.-H. Choi, Structure and Relaxation Dynamics for Complex Coacervate Hydrogels Formed by ABA Triblock Copolymers, Macromolecules, 2020, 53(21), 9234–9243, DOI:10.1021/acs.macromol.0c01600.
- S. Kim, J.-M. Kim, K. Wood and S.-H. Choi, Ionic Group-Dependent Structure of Complex Coacervate Hydrogels Formed by ABA Triblock Copolymers, Soft Matter, 2022, 18(21), 4146–4155, 10.1039/D2SM00255H.
- M. Lemmers, J. Sprakel, I. K. Voets, J. van der Gucht and M. A. Cohen Stuart, Multiresponsive Reversible Gels Based on Charge-Driven Assembly, Angew. Chem., Int. Ed., 2010, 49(4), 708–711, DOI:10.1002/anie.200905515.
- F. Bossard, V. Sfika and C. Tsitsilianis, Rheological Properties of Physical Gel Formed by Triblock Polyampholyte in Salt-Free Aqueous Solutions, Macromolecules, 2004, 37(10), 3899–3904, DOI:10.1021/ma0353890.
- S. Allison-Logan, F. Karimi, Y. Sun, T. G. McKenzie, M. D. Nothling, G. Bryant and G. G. Qiao, Highly Living Stars via Core-First Photo-RAFT Polymerization: Exploitation for Ultra-High Molecular Weight Star Synthesis, ACS Macro Lett., 2019, 8(10), 1291–1295, DOI:10.1021/acsmacrolett.9b00643.
- R. Staňo, P. Košovan, A. Tagliabue and C. Holm, Electrostatically Cross-Linked Reversible Gels—Effects of pH and Ionic Strength, Macromolecules, 2021, 54(10), 4769–4781, DOI:10.1021/acs.macromol.1c00470.
- Z. A. Digby, M. Yang, S. Lteif and J. B. Schlenoff, Salt Resistance as a Measure of the Strength of Polyelectrolyte Complexation, Macromolecules, 2022, 55(3), 978–988, DOI:10.1021/acs.macromol.1c02151.
- K. Sadman, Q. Wang, Y. Chen, B. Keshavarz, Z. Jiang and K. R. Shull, Influence of Hydrophobicity on Polyelectrolyte Complexation, Macromolecules, 2017, 50(23), 9417–9426, DOI:10.1021/acs.macromol.7b02031.
- B. Hofs, I. K. Voets, A. de Keizer and M. A. C. Stuart, Comparison of Complex Coacervate Core Micelles from Two Diblock Copolymers or a Single Diblock Copolymer with a Polyelectrolyte, Phys. Chem. Chem. Phys., 2006, 8(36), 4242–4251, 10.1039/B605695D.
- Y. D. Livney, I. Portnaya, B. Faupin, O. Ramon, Y. Cohen, U. Cogan and S. Mizrahi, Interactions between Inorganic Salts and Polyacrylamide in Aqueous Solutions and Gels, J. Polym. Sci., Part B: Polym. Phys., 2003, 41(5), 508–519, DOI:10.1002/polb.10406.
- A. M. Rumyantsev, N. E. Jackson, B. Yu, J. M. Ting, W. Chen, M. V. Tirrell and J. J. de Pablo, Controlling Complex Coacervation via Random Polyelectrolyte Sequences, ACS Macro Lett., 2019, 8(10), 1296–1302, DOI:10.1021/acsmacrolett.9b00494.
- I. Bos and J. Sprakel, Langevin Dynamics Simulations of the Exchange of Complex Coacervate Core Micelles: The Role of Nonelectrostatic Attraction and Polyelectrolyte Length, Macromolecules, 2019, 52(22), 8923–8931, DOI:10.1021/acs.macromol.9b01442.
- M. S. Green and A. V. Tobolsky, A New Approach to the Theory of Relaxing Polymeric Media, J. Chem. Phys., 1946, 14(2), 80–92, DOI:10.1063/1.1724109.
- F. Tanaka and S. F. Edwards, Viscoelastic Properties of Physically Crosslinked Networks. 1. Transient Network Theory, Macromolecules, 1992, 25(5), 1516–1523, DOI:10.1021/ma00031a024.
- H. Wu, J. M. Ting and M. V. Tirrell, Mechanism of Dissociation Kinetics in Polyelectrolyte Complex Micelles, Macromolecules, 2020, 53(1), 102–111, DOI:10.1021/acs.macromol.9b01814.
- D. Schaeffel, A. Kreyes, Y. Zhao, K. Landfester, H.-J. Butt, D. Crespy and K. Koynov, Molecular Exchange Kinetics of Diblock Copolymer Micelles Monitored by Fluorescence Correlation Spectroscopy, ACS Macro Lett., 2014, 3(5), 428–432, DOI:10.1021/mz500169n.
- S. Lindhoud, W. Norde and M. A. Cohen Stuart, Reversibility and Relaxation Behavior of Polyelectrolyte Complex Micelle Formation, J. Phys. Chem. B, 2009, 113(16), 5431–5439, DOI:10.1021/jp809489f.
- S. Ali, M. Bleuel and V. M. Prabhu, Lower Critical Solution Temperature in Polyelectrolyte Complex Coacervates, ACS Macro Lett., 2019, 8(3), 289–293, DOI:10.1021/acsmacrolett.8b00952.
- Z. Ye, S. Sun and P. Wu, Distinct Cation–Anion Interactions in the UCST and LCST Behavior of Polyelectrolyte Complex Aqueous Solutions, ACS Macro Lett., 2020, 9(7), 974–979, DOI:10.1021/acsmacrolett.0c00303.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00763h |
| This journal is © The Royal Society of Chemistry 2024 |