
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalently crosslinked coacervates: immobilization and stabilization of proteins with enhanced enzymatic activity†
Mengmeng
Zhao
a,
Szu-Hao
Cho
a,
Xinchi
Wu
a,
Jingyi
Mao
a,
Bryan D.
Vogt
 *ab and
Nicole S.
Zacharia‡
*a
*ab and
Nicole S.
Zacharia‡
*a
aDepartment of Polymer Engineering, University of Akron, Akron OH 44325, USA
bDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: bdv5051@psu.edu
First published on 9th September 2024
Abstract
Coacervates represent models for membrane-free protocells and thus provide a simple route to synthetic cellular-like systems that provide selective encapsulation of solutes. Here, we demonstrate a simple and versatile post-coacervation crosslink method using the thiol–ene click reaction in aqueous media to prepare covalently crosslinked coacervates. The crosslinking of the coacervate enables stability at extreme pH where the uncrosslinked coacervate fully disassembles. The crosslinking also enhances the hydrophobicity within the coacervate environment to increase the encapsulation efficiency of bovine serum albumin (BSA), as compared to the uncrosslinked coacervate. Additionally, the crosslinked coacervate increases the stabilization of BSA at low pH. These crosslinked coacervates can act as carriers for enzymes. The enzymatic activity of alkaline phosphatase (ALP) is enhanced within the crosslinked coacervate compared to the ALP in aqueous solution. The post-coacervation crosslink approach allows the utilization of coacervates for encapsulation of biologicals under conditions where the coacervate would generally disassemble. We demonstrate that these crosslinked coacervates enable the protection of encapsulated protein against denaturation at extreme pH and enhance the enzymatic activity with encapsulation. This click approach to stabilization of coacervates should be broadly applicable to other systems for a variety of biologics and environmentally sensitive molecules.
Introduction
Compartmentalization of biomolecules within microscale aqueous environments is a promising route to synthetically mimic cellular systems with potential applications in biocatalysis,1–3 biosensing,4 therapeutics,5 and energy conversion.6 The design and construction of synthetic cellular systems have typically relied on membrane-bounded microcompartments, such as self-assembled bilayer vesicles,7–9 polymer-based capsules,10–12 inorganic vesicles,13,14 and water-in-oil emulsions.15,16 However, the low permeability of these membranes limits the mass transfer and reduces the possibility of continuous activity within the synthetic cellular systems.17 Membrane-free compartmentalization through complex coacervation avoids these limitations.3,17,18 Complex coacervation from the spontaneous phase separation of oppositely charged polyelectrolytes produces polymer-rich liquid microdroplets that provide a simple alternative for compartmentalization without a membrane, while also enabling passing of solutes due to the low interfacial tension of coacervates with water.19 Coacervates have been envisioned as molecularly crowded, membrane-free protocells due to their selective encapsulation of solutes, including small organic molecules,20–25 biomacromolecules3,26–28 and inorganic nanoparticles.24,25 The partitioning is driven by the relative affinities of solute–water and solute–polymer pairs; the relative partitioning of reactants and products between phases can act to facilitate reactions in aqueous environments.29A great challenge for many synthetic microreactors is constant intake of reagents and release of products for continuous reactions.12 The preferential segregation of solutes within the coacervate phase and their low interfacial tension make complex coacervates promising for synthetic microreactors. For example, complex coacervate droplets containing TiO2 nanosheets act as microreactors to photocatalytically degrade organic dyes with selectivity driven by equilibrium partitioning of small molecule dyes into the coacervate phase.24 Moreover, enzymatic reactions within coacervate droplets can exhibit increased reaction rates and yields due to preferential partitioning of the substrates within the coacervate phase.3 Encapsulation of proteins within complex coacervates can protect the proteins against the denaturation at extremes of pH,26 high temperature,26 and urea solutions.26,27 Proteins within coacervates retain their biological activity against heavy metal contamination.21,26 Moreover, coacervate droplets support the refolding of denatured proteins and facilitate the recovery of their secondary structure and biological activity.27 Together, these observations suggest that complex coacervation provides self-assembled microcompartments capable of stabilizing proteins under extreme conditions.
However, despite these favorable attributes, there are several shortcomings of these coacervate droplets that limit their use. First even in the absence of surface tension, the coacervate droplets tend to coalesce over time, leading to increasing diffusion lengths within the polymer-rich coacervate. The pH, ionic strength, and temperature requirements for stability of coacervate droplets may limit their use in certain applications.28,30 Therefore, the construction of robust, stable coacervate droplets with high permeability would enhance the working range for coacervates in various applications and enable engineering reactions that operate outside the conditions typically associated with protocells.
Towards the goal of improving the stability of coacervates, covalent crosslinking of complex coacervate core micelles has been demonstrated to enhance the stability of the micelles at high ionic strength.31 Application of crosslinking to coacervate droplets with tannic acid led to improved thermal stability, a significant increase in the droplet size and modification of the gelatin secondary structure.32 Aldehyde-based crosslinking of coacervates has been demonstrated to enhance the stability of protein–polysaccharide coacervates,33 but this chemistry has potential negative health effects; the physical transformation of the protein to amyloids provides an alternative approach to enhance the stability of the coacervates.34 These crosslinking approaches provide added stability to the coacervates but at costs to the functionality or droplet size control.
Here, we demonstrate a post-coacervation crosslinking approach for the stabilization of coacervate droplets that is generalizable. Thiol–ene click chemistry enables the formation of covalent crosslinks in aqueous media, without exposure to heat or organic solvents. An allyl modified (meth)acrylic acid was complexed with branched polyethylenimine (BPEI) and subsequently crosslinked using butanedithiol. The crosslinked coacervate droplets exhibit stability at low pH where the uncrosslinked coacervate droplets fully disassemble. The enhanced hydrophobicity from the crosslinking increases the encapsulation efficiency of various solutes, such as bovine serum albumin (BSA). The secondary structure of BSA is maintained when encapsulated into both uncrosslinked and crosslinked coacervates at neutral pH, while only the crosslinked coacervate can prevent denaturation of BSA at pH 2.0. In addition to stabilization of protein, the crosslinked coacervate can enhance the enzymatic activity of alkaline phosphatase (ALP). Changing to methacrylic acid from acrylic acid for the polyanion also enhanced the enzymatic activity of ALP, illustrating how tuning the hydrophobicity of the coacervate environment can be used to optimize these crosslinked droplets as microreactors.
Experimental
Materials
Poly(acrylic acid) (PAA, nominal Mw = 50 kg mol−1) and poly(methacrylic acid) (PMAA, nominal Mw = 150 kg mol−1) were purchased from Polysciences, Inc. Branched polyethylenimine (BPEI, nominal Mw = 25 kg mol−1) was purchased from Sigma-Aldrich. Allylamine, N,N′-dicyclohexylcarbodiimide (DCC), N-methyl-2-pyrrolidinone (NMP), 1,4-butanedithiol, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I-2959), 8-anilino-1-naphthalenesulfonic acid (ammonium salt, ANS), bovine serum albumin (BSA), alkaline phosphatase (ALP), fluorescein isothiocyanate (FITC, isomer I), 4-nitrophenyl phosphate disodium salt hexahydrate (4-NPP), tris(hydroxymethyl)aminomethane (Tris base), sodium acetate, acetic acid, and urea were obtained from Sigma-Aldrich. All these materials were used as received without further purification. All water was dispensed from a Milli-Q water system at a resistivity of 18.2 MΩ cm.Synthesis of poly(acrylic acid-co-allyl acrylamide) (allyl-PAA)
Amidization of PAA (1.0 g) dissolved in NMP (50 mL) was performed in a 100 mL three-neck flask equipped with a magnetic stir bar and condenser under a N2 atmosphere. To ensure complete dissolution, the flask was heated to 57 °C for 8 h. Subsequently, allylamine (0.176 g, 3.08 × 10−3 mol) was added rapidly to the flask, followed by the addition of 5 mL of 0.1656 g mL−1 DCC (0.828 g, 4.01 × 10−3 mol) solution in NMP. The reaction proceeded at 57 °C for 24 h with stirring and then terminated by rapidly cooling in an ice bath. Concentrated sodium hydroxide solution (10 M) was added dropwise to the reaction mixture to precipitate the allyl-PAA. The precipitated polymer was then redissolved in NMP at 60 °C and precipitated in cold methanol. The product was isolated by centrifugation and dried overnight at 60 °C. The polymer was dissolved in 10 mL of Milli-Q water and reprecipitated in cold methanol. The product was obtained by centrifugation and dried overnight at 60 °C. The refined allyl-PAA was ground into powder and dried in a vacuum. The allyl functionalization of the PAA was quantified using 1H NMR spectroscopy and determined to be approximately 16–18 mol% (Fig. S1, ESI†).Synthesis of poly(methacrylic acid-co-allyl methacrylamide) (allyl-PMAA)
Allyl-PMAA was synthesized via the amidization reaction using a similar approach as described for allyl-PAA except for the following changes. The original solution was 1.0 g PMAA and 50 mL NMP-DMSO cosolvent (volume ratio of NMP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO = 1:1). The amount of allylamine was reduced (0.0664 g, 1.16 × 10−3 mol). The initially precipitated allyl-PMAA was redissolved in NMP-DMSO at 60 °C and precipitated in cold methanol. The crude product was isolated by centrifugation and dried overnight in an oven (60 °C). The allyl functional group, as determined from 1H NMR spectroscopy, was calculated to be approximately 9–11 mol% (Fig. S2, ESI†) in the PMAA.
:DMSO = 1:1). The amount of allylamine was reduced (0.0664 g, 1.16 × 10−3 mol). The initially precipitated allyl-PMAA was redissolved in NMP-DMSO at 60 °C and precipitated in cold methanol. The crude product was isolated by centrifugation and dried overnight in an oven (60 °C). The allyl functional group, as determined from 1H NMR spectroscopy, was calculated to be approximately 9–11 mol% (Fig. S2, ESI†) in the PMAA.
Conjugation of BPEI, BSA and ALP with FITC
400 mM BPEI solution was prepared in 0.1 M carbonate buffer at pH 9.0. FITC was dissolved in DMSO at 5 mg mL−1. 175 μL FITC solution was slowly added to 1 mL of BPEI solution. These were reacted in the dark for 4 h to prepare FITC-labelled BPEI (FITC–BPEI). After the reaction, the FITC–BPEI solution was dialysed using the Spectra/Por dialysis membrane (3.5–5 kDa) against 1 L Milli-Q water for 1 day in the dark at room temperature to remove uncoupled FITC. The dialysis was repeated 3 times or until the absorption at 495 nm was <0.003 for the extracted solution. The dialysis membrane was washed prior to use in DI water.10 mg mL−1 BSA or 2 mg mL−1 ALP solution was prepared in 0.1 M carbonate buffer at pH 9.0. FITC was dissolved in DMSO at a concentration of 5 mg mL−1. For each 1 mL of protein solution, 50 μL of FITC solution was added slowly while gently and continuously stirring the protein solution. After the FITC solution was added, the reaction was incubated at room temperature in the dark for 4 h. The solutions were dialysed using the same procedure as described for the FITC–BPEI to obtain proteins without free FITC.
NMR measurements
1H NMR measurements of the synthesized allyl-PAA and allyl-PMAA were performed in D2O at approximately 15 mg mL−1. A Mercury 300 spectrometer with a proton resonance frequency of 300 MHz at 30 °C was used to determine the modification ratio of the allyl functional group on PAA and PMAA.Preparation of crosslinked complex coacervates
Stock solutions of 20 mM allyl-PAA and 20 mM BPEI (with respect to repeat unit) were prepared with pH adjusted to 6.5 using 1 M or 0.1 M NaOH and HCl solutions. These pH adjustments involved small quantities of acid or base, at most a handful of drops, which did not statistically alter the reported concentration of 20 mM. 1,4-Butanedithiol and I-2959 were added to the stock solution of allyl-PAA to produce 2.5 mM 1,4-butanedithiol and 1.25 mM I-2959 and stirred for 30 min. This allyl-PAA solution was then added dropwise to an equimolar concentration of BPEI stock solution under continuous stirring to form uncrosslinked complex coacervates as illustrated in Scheme 1 and stirred for 1 h. The complex coacervate was crosslinked by exposure to 365 nm light using a UV lamp (48 W) for 1 h under continuous stirring.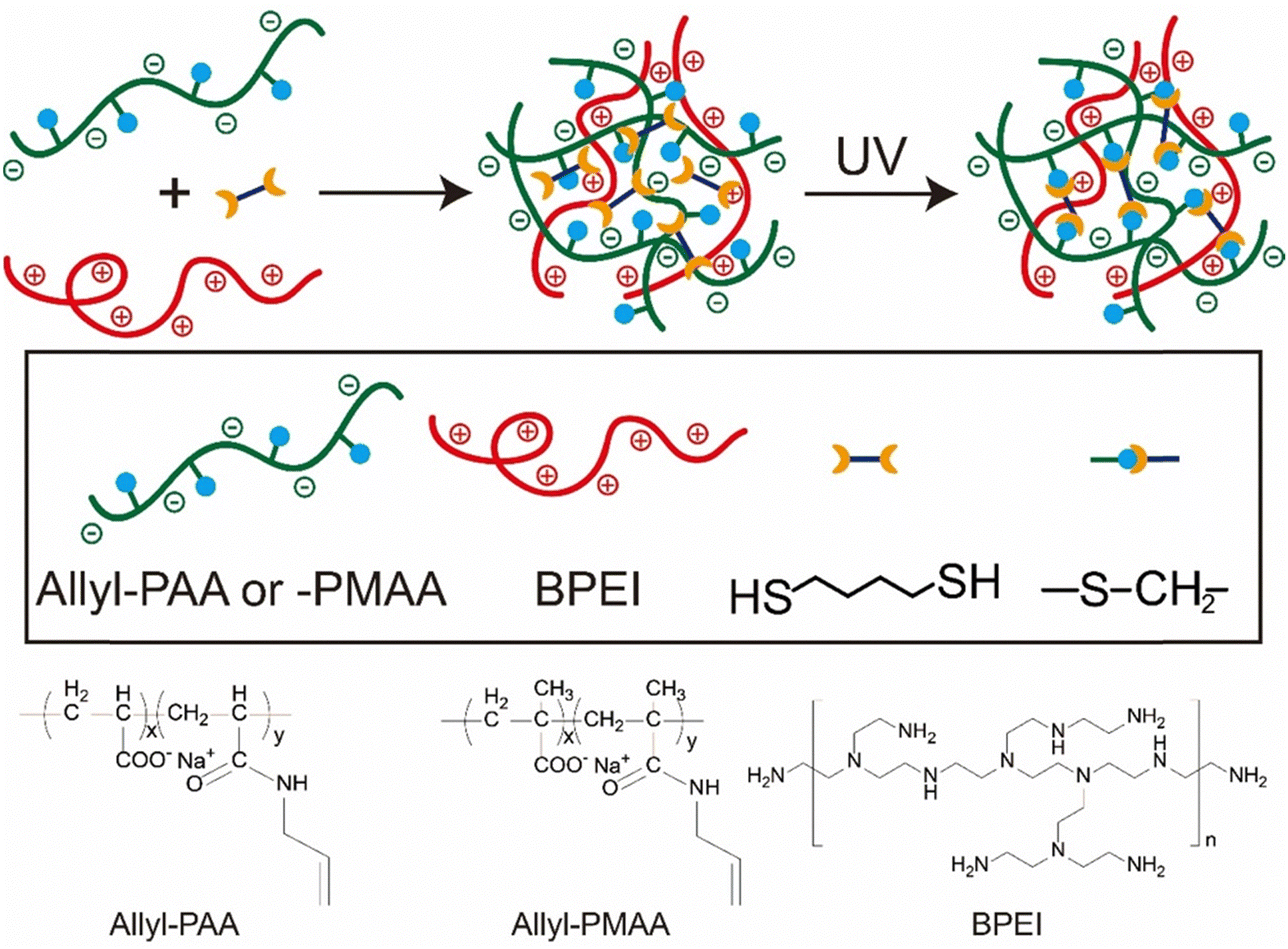 | ||
| Scheme 1 Strategy of post-coacervation crosslinking for the preparation of covalently crosslinked coacervate droplets and chemical structures of allyl-PAA, allyl-PMAA and BPEI. | ||
Stock solutions of 40 mg mL−1 BSA and 2 mg mL−1 ALP were used for the preparation of protein encapsulated coacervates. The stock solution of BSA or ALP was added dropwise to the BPEI stock solution to achieve final concentrations of 0.5 mg mL−1 for BSA and 0.08 mg mL−1 for ALP and stirred for 30 min to form intermediate BPEI–BSA or BPEI–ALP complexes. The BPEI–protein intermediate complexes were added to the stock allyl-PAA solution containing 2.5 mM 1,4-butanedithiol and 1.25 mM I-2959. A molar ratio of 1:1 for amine to acid was used for coacervate production. The mixture was stirred for 1 h to produce uncrosslinked complex coacervates. These coacervates were crosslinked using 365 nm radiation (48 W) for 1 h under continuous stirring.
Characterization of coacervates
Fourier transform infrared spectroscopy (FTIR, Thermo Scientific Nicolet iS50 FTIR Spectrometer) was used to confirm the incorporation of both allyl-PAA and BPEI in the coacervate as well as the thiol–ene click reaction for the crosslinked coacervate. For FTIR analysis, samples were centrifuged for 1 h at 8000 rpm (9600 × g, Allegra X-30R Centrifuge, Beckman Coulter) to collect the coacervate. The coacervates obtained from centrifugation were dried for 12 h in a vacuum at 40 °C before FTIR measurements.An optical microscope (Carl Zeiss MicroImaging Axio Imager M2M) was used for bright field and fluorescence imaging of coacervate droplets in reflected light mode. The as-prepared uncrosslinked and crosslinked coacervate samples with pH adjusted to either 6.5 or 2.0 were centrifuged for 10 min at 4000 rpm (6800 × g, Allegra X-30R Centrifuge, Beckman Coulter) to achieve rapid sedimentation. After centrifugation, the supernatant was carefully removed using a micro-pipette, while the dense coacervate phase was transferred to a silica slide to obtain optical microscopy images.
Dynamic light scattering (DLS) analysis of uncrosslinked (pH 6.5) and crosslinked BPEI-allyl-PAA coacervates (pH 6.5 and 2.0) was performed using a Zeta PALS instrument (Brookhaven, USA). The volume-averaged distribution of hydrodynamic diameter was determined. Zeta potential measurements of uncrosslinked and crosslinked BPEI-allyl PAA coacervates at different pH (2.0–6.5) also used the Zeta PALS instrument (Brookhaven, USA). To study the release of BPEI from the crosslinked BPEI-allyl-PAA coacervate when PAA is fully uncharged, the zeta potential of the crosslinked BPEI-allyl-PAA coacervate at pH 2.0 was measured as a function of time. Each measurement was repeated at least 3 times. The electrophoretic mobility of the complex coacervate was converted into zeta potential using the Smoluchowski equation.
Turbidity measurements were performed using an Agilent 8453 UV-vis spectrometer at 500 nm to study the effect of pH and salt concentration on complex coacervation. Uncrosslinked and crosslinked coacervates were prepared as previously described and the light intensity was assessed. The turbidity is defined as eqn (1),35
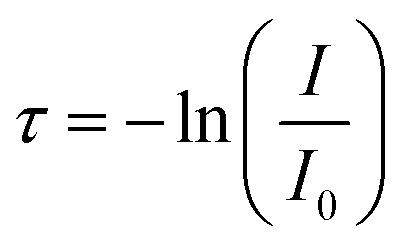 | (1) |
Determination of BSA and ALP encapsulation in coacervates
FITC–BSA and FITC–ALP were used to quantify the encapsulation of proteins in both uncrosslinked and crosslinked coacervates. All coacervate samples containing proteins were first prepared at pH 6.5. In some cases, the pH was reduced to 4.5 to assess partitioning at lower pH with an additional 1 h of stirring as the pH was adjusted. The coacervate samples were centrifuged for 1 h at 8000 rpm (Allegra X-30R Centrifuge, Beckman Coulter) to separate the supernatant. After centrifugation, the supernatant was removed using a micropipette. UV-vis measurement (Agilent 8453 spectrophotometer) was used to determine the BSA content of the supernatant. The encapsulation of BSA (E) into PAA-PAH coacervate phase was calculated using eqn (2).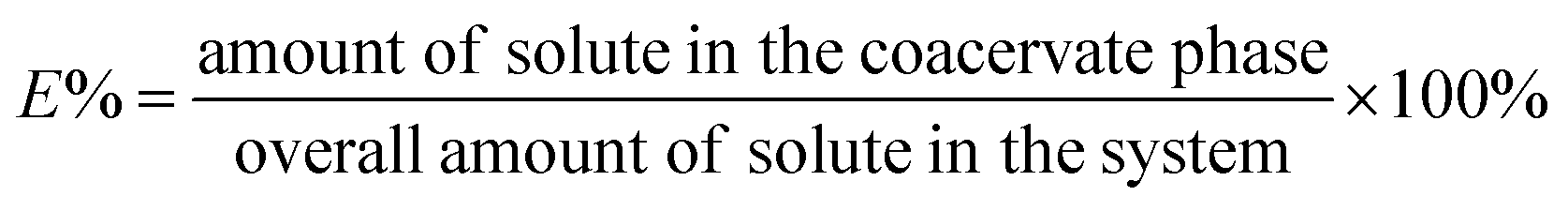 | (2) |
The concentration of the FITC–BSA and FITC–ALP was determined using a standard calibration curve.
Circular dichroism (CD) spectroscopy
To determine the secondary structure of free versus encapsulated BSA within the coacervates at pH 2.0 and 6.5, circular dichroism (CD) was performed at room temperature using a J-1500 circular dichroism spectrophotometer (Jasco Inc., Japan) with a quartz cuvette of 1 mm path length. The BSA encapsulated coacervate samples were prepared as previously described, followed by pH adjustment to either 2.0 or 6.5. CD measurements were performed from 280 nm to 200 nm. The residual molar ellipticity [θ] was calculated according to eqn (3), | (3) |
Determination of hydrophobicity within coacervates
Fluorescence spectroscopy was used to elucidate the polarity of the microenvironment within the BPEI-allyl-PAA and BPEI-allyl-PMAA coacervates through the addition of 0.2 mM ANS during their preparation. Steady state emission spectra of ANS within uncrosslinked and crosslinked coacervates were obtained using a Horiba FluoroMax 4 spectrofluorometer with an excitation wavelength of 350 nm. Fluorescence emission was measured from 400 to 675 nm in quartz cuvettes for microdroplet dispersions of coacervates. All fluorescence was attributed to ANS within the microdroplets as ANS fluorescence was quenched in water.Determination of enzymatic activity using UV-vis spectroscopy
UV-vis spectroscopy was used to monitor the enzymatic activity of alkaline phosphatase (ALP) at 25 °C. Alkaline phosphatase catalyzes the hydrolysis of primary phosphate esters. 4-Nitrophenyl phosphate (4-NPP) was used as the substrate, which is hydrolyzed to produce phosphate and 4-nitrophenolate (4-NP). 10 mM acetate buffer and tris buffer were added at pH 4.5 and 6.5, respectively, to maintain a constant pH during the reaction. 4-NP exhibits strong absorbance at 405 nm. A calibration curve was developed to correlate 4-NP concentration with absorbance at 405 nm. Spectra were collected as a function of time to track the reaction.Results and discussion
Coacervation occurs spontaneously on mixing of the aqueous allyl-PAA and BPEI solutions. The allyl functionalization of PAA does slightly alter the phase diagram, but coacervation based on the association between the carboxylic acid groups in PAA and amines in BPEI occurs readily. The inclusion of the allyl group enables covalent crosslinking of the coacervate droplets as shown in Scheme 1. The BPEI-allyl-PAA coacervates were characterized using FTIR spectroscopy as shown in Fig. S3 (ESI†). Characteristic peaks associated with BPEI and allyl-PAA are present in the spectra between 1500 and 1850 cm−1. Gaussian fits were used to deconvolute the peaks with absorption bands at ∼1554 cm−1 and 1710 cm−1 attributed to the asymmetric stretching of the carboxylate group and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxyl group, respectively,36 while bands at ∼1660 cm−1 and 1600 cm−1 were assigned to the CO stretching of amide and NH3+ of BPEI, respectively.36,37 Additionally, the bands at 1638 cm−1 and ∼3082 cm−1, associated with CC stretching and C–H stretching for unsaturated bonds, respectively, were reduced dramatically after the crosslinking reaction (Fig. S3c, ESI†). These indicate the successful thiol–ene click reaction that should crosslink the coacervate. These thiol–ene reactions have been demonstrated extensively for the fabrication of hydrogels with the hydrolytic stability of these bonds being greater than those within the polymer.38,39
O stretching of the carboxyl group, respectively,36 while bands at ∼1660 cm−1 and 1600 cm−1 were assigned to the CO stretching of amide and NH3+ of BPEI, respectively.36,37 Additionally, the bands at 1638 cm−1 and ∼3082 cm−1, associated with CC stretching and C–H stretching for unsaturated bonds, respectively, were reduced dramatically after the crosslinking reaction (Fig. S3c, ESI†). These indicate the successful thiol–ene click reaction that should crosslink the coacervate. These thiol–ene reactions have been demonstrated extensively for the fabrication of hydrogels with the hydrolytic stability of these bonds being greater than those within the polymer.38,39
pH and ionic strength stability
The charge density of weak polyelectrolytes can be tuned with pH, which impacts complexation.40 The stability of the equimolar BPEI-allyl-PAA coacervate droplets prepared at pH 6.5 to changes in pH is elucidated from changes in turbidity as shown in Fig. 1a. Irrespective of crosslinking in coacervates, the maximum turbidity was observed at pH 5.0. This maximum represents the maximum extent of coacervate formation for the polyelectrolyte mixture. This pH deviates from the expected 1:1 charge ratio at approximately pH 5.8 as determined from the potentiometric titration for allyl-PAA and BPEI in aqueous solutions (Fig. S4, ESI†). The change in charge of the weak polyelectrolytes with pH is illustrated pictorially in Scheme 2. The deviation from the 1:1 charge ratio for maximum coacervate formation is not surprising, as the pKa of weak polyelectrolytes shifts upon complexation due to charge–charge interactions.41 The turbidity of the crosslinked coacervates tends to be lower than that of the uncrosslinked coacervate at pH > 3.0, which could be associated with the fixed size of the droplets after crosslinking and the scaling of scattering with particle size.42
 | ||
Fig. 1 (a) Turbidity of uncrosslinked (□) and crosslinked ( ) BPEI-allyl-PAA coacervate droplets as a function of decreasing pH from 6.5 to 2.0. Optical micrographs of (b) uncrosslinked and (c) crosslinked coacervate droplets at pH 6.5 as well as (d) uncrosslinked and (e) crosslinked coacervate droplets at pH 2.0. Error bars represent the standard deviations from three measurements. ) BPEI-allyl-PAA coacervate droplets as a function of decreasing pH from 6.5 to 2.0. Optical micrographs of (b) uncrosslinked and (c) crosslinked coacervate droplets at pH 6.5 as well as (d) uncrosslinked and (e) crosslinked coacervate droplets at pH 2.0. Error bars represent the standard deviations from three measurements. | ||
 | ||
| Scheme 2 Pictorial illustration of charge density on allyl-PAA and BPEI as a function of pH. Coacervation is favored at intermediate pH where both weak polyelectrolytes are highly charged. | ||
However, with a further decrease in pH to <3.0, the uncrosslinked coacervate dissolves to a single-phase solution with effective measurement of zero turbidity. At low pH, the allyl-PAA is fully protonated and neutralized as illustrated in Scheme 2, so it cannot effectively complex with the BPEI. However, the crosslinked coacervate droplets are stable at low pH as the covalent crosslinks prevent dissociation of the drops even when the driving force for coacervation is removed. The turbidity of the crosslinked coacervate at pH <3.0 is similar to the initial turbidity of the crosslinked coacervate (pH 6.5). This limited change in turbidity indicates that the thiol–ene click reaction sufficiently crosslinks the coacervate droplets to minimize changes, but there appears to be a small decrease in turbidity as the pH is further decreased, which can be associated with droplet number density, size and refractive index. The pH stability of the coacervates was also confirmed visually using optical microscopy (Fig. 1b–e), where droplets are clearly present in both crosslinked and uncrosslinked coacervates at pH 6.5, but droplets were only observed in the crosslinked coacervates at pH 2.0. Additionally, the diameter of the crosslinked coacervate droplets increases as the pH drops from 6.5 to 2.0, which was also confirmed by dynamic light scattering measurement (Fig. S5, ESI†). This increase in size is attributed to the loss of ionic crosslinks between allyl-PAA and BPEI to allow additional swelling as well as the increasing electrostatic repulsion between positively charged BPEI chains at pH 2.0.
This click chemistry crosslinking approach can be equally applied to PMAA-BPEI coacervates. However, PMAA exhibits lower water solubility than PAA at low pH due to a hypercoiling transition where a globular collapse is associated with the methyl groups on the backbone.43,44 As such, the stability of uncrosslinked and crosslinked BPEI-allyl-PMAA coacervates was probed by increasing the pH to deprotonate the amine to avoid complications in interpreting the turbidity from insoluble PMAA. As shown in Fig. S6 (ESI†) based on turbidity measurements, the uncrosslinked BPEI-allyl-PMAA coacervates dissolve at pH > 8.0, while the turbidity signal from the crosslinked BPEI-allyl-PAA coacervate remains stable on increasing pH. In addition to pH, the ionic strength of the media can act to destabilize coacervates. Fig. 2a illustrates the stability of the BPEI-allyl-PAA coacervates at high salt concentrations up to 1 M NaCl. The turbidity of the uncrosslinked coacervate disappears for NaCl concentrations > 500 mM, but the turbidity only decreases slightly at high ionic strength for the crosslinked coacervates. These results indicate that the thiol–ene click reaction is effective and prevents dissolution of the coacervates.
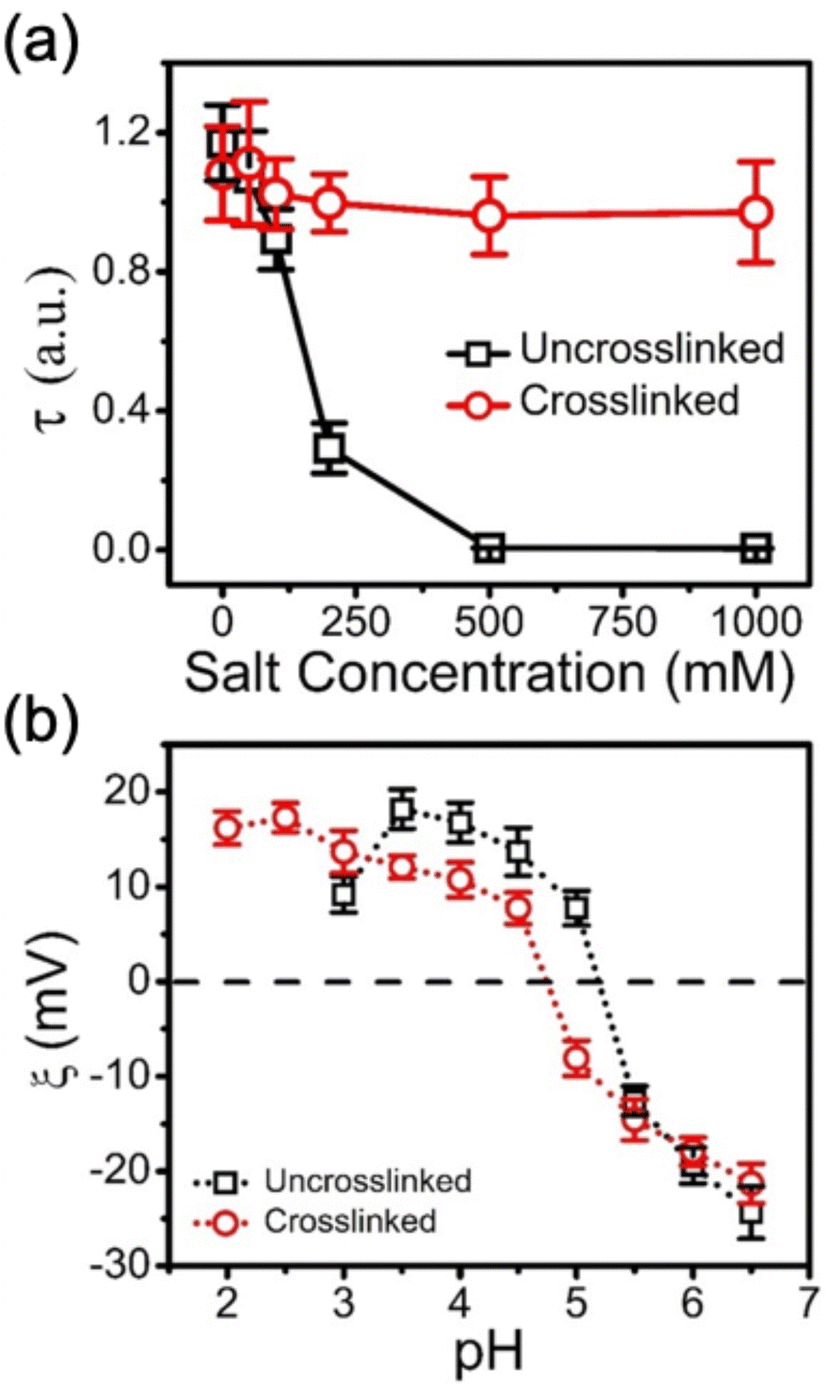 | ||
Fig. 2 (a) Turbidity of uncrosslinked (□) and crosslinked ( ) BPEI-allyl-PAA coacervate droplets as a function of increasing NaCl concentration from 0 to 1000 mM. (b) Zeta potential of BPEI-allyl-PAA coacervates as a function of pH. Error bars represent the standard deviations of the three measurements. ) BPEI-allyl-PAA coacervate droplets as a function of increasing NaCl concentration from 0 to 1000 mM. (b) Zeta potential of BPEI-allyl-PAA coacervates as a function of pH. Error bars represent the standard deviations of the three measurements. | ||
The crosslinking of the coacervates does not dramatically impact their charge as determined by zeta potential at decreasing pH (Fig. 2b). To ensure that these measurements correspond to the coacervate, electrophoretic measurements of the individual polyelectrolyte solutions at their highest charge densities, BPEI aqueous solution at pH 2.0 and allyl-PAA aqueous solution at pH 6.5, were performed as controls. The Zeta PALS data for PAA and BPEI aqueous solutions confirm that the data in Fig. 2b are related to the complex coacervate. The coacervate samples formed at pH 6.5 are negatively charged. Decreasing pH leads to the zeta potential increasing. These changes in zeta potential are consistent with the protonation of PAA and BPEI at low pH. The zeta potential decrease at pH 3.0 for the uncrosslinked coacervate is consistent with the partial disassembly of coacervates to reduce the total surface change. At lower pH, the zeta potential was consistent with polyelectrolyte solutions for the uncrosslinked coacervates, while the zeta potential for the crosslinked coacervate increased as pH was decreased to 2.0, indicating further protonation of the coacervate and stability of the crosslinked coacervate against extremes in pH.
Release of BPEI from crosslinked coacervates at low pH
As the effective electrostatic interaction between allyl-PAA and BPEI to produce a coacervate is lost at pH 2.0, the BPEI and allyl-PAA within the coacervate can only be held in place by the crosslinks induced by click chemistry. The gel fraction in the crosslinked coacervates can be qualitatively accessed by the examination of the temporal evolution zeta potential at pH 2.0. Zeta potential measurements of the crosslinked coacervate droplets at pH 2.0 were recorded as a function of time (Fig. 3). There is a slow decay in the zeta potential from approximately 16 to 13 mV over 5 days, which suggests a reduction in the protonated PBEI in the coacervate. This release of BPEI was quantified using FITC-labeled BPEI in the coacervate. Initially, the distribution coefficient of BPEI at pH 2.0, defined as the concentration ratio of the BPEI in the crosslinked coacervate to the BPEI in the supernatant, was approximately 400 based on UV-vis measurements of the phases. The intra- to extra-coacervate concentration gradient of BPEI is high, thus favoring the release of free BPEI. The concentration of FITC-labeled BPEI in the supernatant was assessed periodically through UV-vis measurements after centrifugation. The fraction of BPEI in the coacervate was calculated from a mass balance based on the increase in FITC concentration in the supernatant. Fig. 3 illustrates that only 3.5% of the FITC–BPEI was released from the crosslinked coacervate over 5 days. This corresponds to 96.5% gel fraction for the coacervate, which is consistent with other reports for crosslinking with click chemistries for network formation.45,46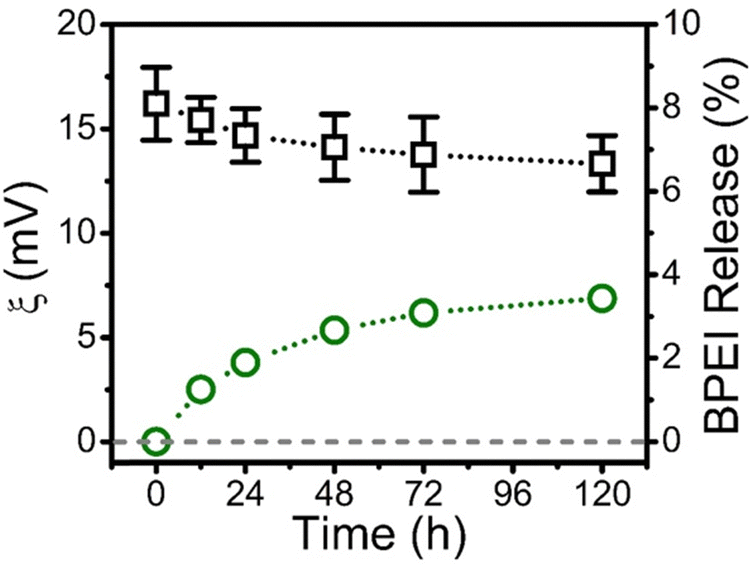 | ||
Fig. 3 Zeta potential (□) and release of BPEI ( ) of crosslinked BPEI-allyl-PAA coacervates at pH 2.0 as a function of time. Both the zeta potential results and release of BPEI of crosslinked coacervates at pH 2.0 indicate that the covalent crosslinks in the BPEI-allyl-PAA coacervate inhibit the release of BPEI. ) of crosslinked BPEI-allyl-PAA coacervates at pH 2.0 as a function of time. Both the zeta potential results and release of BPEI of crosslinked coacervates at pH 2.0 indicate that the covalent crosslinks in the BPEI-allyl-PAA coacervate inhibit the release of BPEI. | ||
Encapsulation of BSA and ALP within complex coacervates
Bovine serum albumin (BSA) and alkaline phosphatase (ALP) at pH 6.5 were encapsulated within BPEI-allyl-PAA during coacervation. As both FITC-labeled BSA and ALP are negatively charged, an intermediate complex was first formed by mixing with BPEI. Subsequently, the intermediate was mixed with allyl-PAA to form protein-encapsulated complex coacervate droplets. The successful encapsulation of FITC-labeled BSA was confirmed visually using optical microscopy (Fig. S7, ESI†). The complexes are reversible during complexation and there will be competition for the amines in the BPEI between the components. However, there is also partitioning of BSA and ALP due to the environmental differences in the coacervate and solution phase. Thus, the composition of components within the coacervate phase may be altered by the crosslinking. The efficiency of protein encapsulation was determined from UV-vis measurements of the supernatant after coacervation. The crosslinked coacervate shows a higher efficiency of encapsulation for BSA and ALP than the uncrosslinked coacervate (Fig. 4). This is attributed to the more hydrophobic environment in the crosslinked coacervate that can interact with the globular proteins.47–49 This more hydrophobic environment promotes the uptake of BSA and ALP from the solution phase into the coacervate after crosslinking.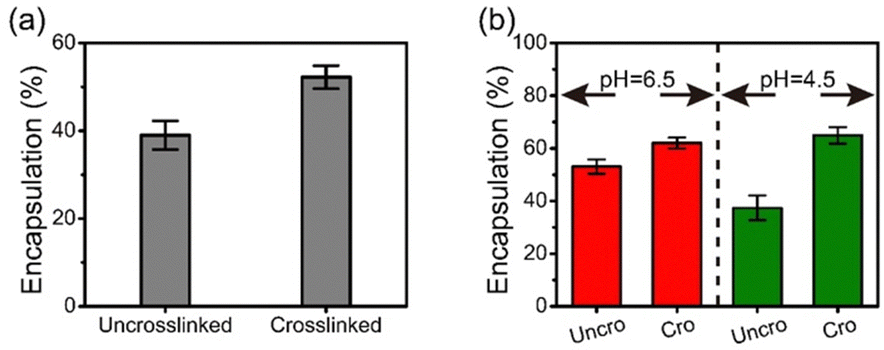 | ||
| Fig. 4 (a) BSA encapsulation efficiency of uncrosslinked and crosslinked coacervates at pH 6.5, and (b) ALP encapsulation efficiency of uncrosslinked (Uncro) and crosslinked (Cro) coacervates at pH 6.5 and 4.5. The error bars represent the standard deviations of the three measurements. | ||
The hydrophobicity of the coacervates was probed with ANS within uncrosslinked and crosslinked BPEI-allyl-PAA coacervates. The fluorescence spectra confirmed increased hydrophobicity after crosslinking (Fig. S8, ESI†). Changes in the water content of the BPEI-PAA coacervate, uncrosslinked and crosslinked BPEI-allyl PAA coacervates shown in Fig. S9 (ESI†) are also consistent with the increased hydrophobicity with the addition of the allyl function group and upon crosslinking. The decrease in water content is indirect proof that the hydrophobicity of complex coacervates increases upon allyl modification and crosslinking.20 The crosslinking of the coacervate prevents the release of BSA at pH 2.0. No significant fluorescence was detected in the pH 2.0 supernatant after 1 week.
Preservation of secondary structure of encapsulated BSA
Although the crosslinking of the coacervate prevents leaching of the BSA into the solution, this is only one criterion for applications. Preservation of the secondary structure is critical for protein encapsulation, as it directly relates to activity and function of protein. Circular dichroism (CD) spectra for native free BSA consist of minima at 222 and 208 nm, which are characteristic of an α-helical secondary structure.26,28,50Fig. 5a illustrates this spectrum of BSA in aqueous solution at pH 6.5. Exposure to UV irradiation used for crosslinking has a negligible impact on the secondary structure of BSA. Decreasing the pH to 2.0 or including high concentration of urea leads to a change in CD (Fig. 5a). BSA encapsulated in the coacervate exhibits CD spectra similar to the BSA in solution, irrespective of crosslinking, as shown in Fig. 5b. This CD confirms the retention of the secondary structure of BSA during the complex coacervation and photo-crosslinking processes. There is a small decrease in intensity at low wavelengths with the encapsulated BSA, which is likely associated with scattering from coacervate droplets.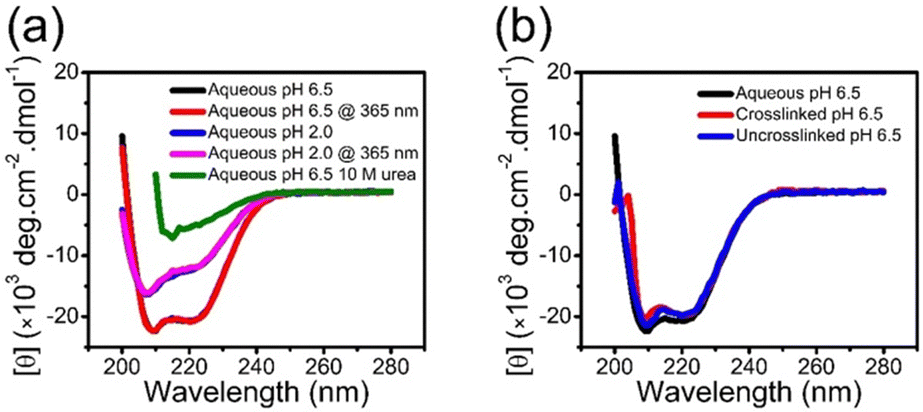 | ||
| Fig. 5 (a) CD spectra of BSA in aqueous solution at pH 6.5 and 2.0 with and without 1 h of UV (365 nm) irradiation. (b) CD spectra of BSA in aqueous solution, and in uncrosslinked and crosslinked coacervates at pH 6.5. The encapsulation of BSA in uncrosslinked and crosslinked coacervates has a negligible impact on the secondary structure of BSA. | ||
Stabilization of encapsulated BSA against extremely low pH
Low pH is known to destabilize BSA as shown through CD spectroscopy in Fig. 5a and Fig. 6. Low pH destabilizes proteins through unfavorable electrostatic repulsion from added positive charges on the protein.51 As shown in Fig. 6a, the CD spectra for BSA in aqueous solution or in the uncrosslinked coacervate, which dissolves, at pH 2.0 are similar. However, the BSA in the crosslinked coacervate maintains substantially more of the secondary structure (Fig. 6a). From CD spectra, the unfolded fraction of BSA (Σ) can be calculated from eqn (4)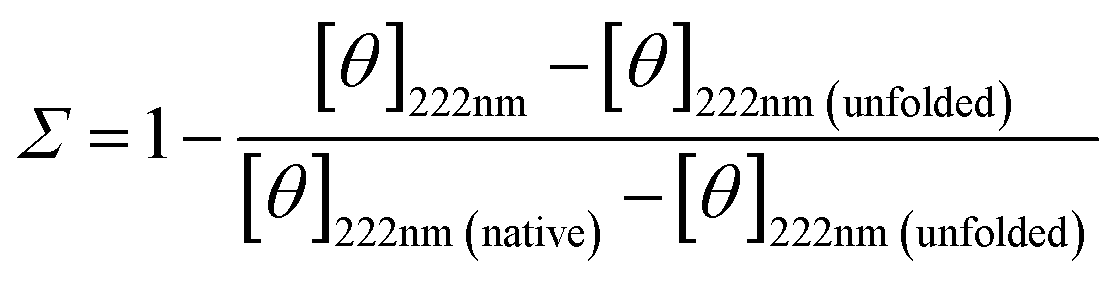 | (4) |
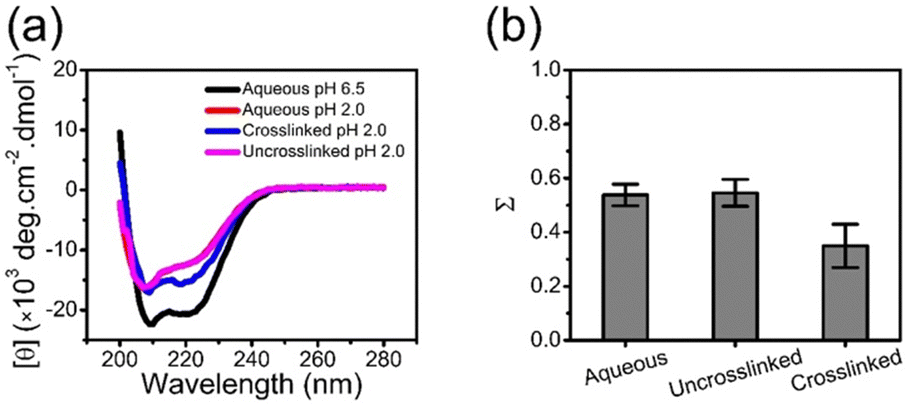 | ||
| Fig. 6 (a) CD spectra of BSA in aqueous solution at pH 6.5 (black) and 2.0 (red), and in uncrosslinked (magenta) and crosslinked (blue) coacervates at pH 2.0. (b) Fraction of unfolded BSA at pH 2.0, in aqueous solution, and in uncrosslinked and crosslinked coacervates. The error bars represent the standard deviation of the 3 measurements. | ||
Enhanced enzymatic activity of ALP within coacervates
Stabilization of the secondary structure within the crosslinked coacervate at low pH suggests that the activity of the protein will be maintained. To demonstrate sustained activity within the crosslinked coacervates, alkaline phosphatase (ALP) was used as a model enzyme to catalyze the hydrolysis of 4-nitrophenylphosphate (4-NPP). This reaction leads to the formation of 4-nitrophenol (4-NP) and phosphate. Measurement of the increase in 4-NP concentration using UV-vis spectroscopy provides a measure of the reaction and activity of the ALP. The absorption peak of 4-NP at 405 nm was used to determine the evolution in the concentration of 4-NP as shown in Fig. 7a. The initial rate of 4-NP production is strongly dependent on the environment. The activity of the ALP is reduced at lower pH in solution. However, the impact of the coacervate on the activity depends on the pH of the media. At pH 6.5, the uncrosslinked BPEI-allyl-PAA coacervate provides a faster reaction rate than the crosslinked analog, but both show a significant improvement over the ALP in solution. At pH 4.5, the fastest rate is for the crosslinked coacervate, illustrating the potential for the crosslinked coacervate to improve the performance of biologics when operating in non-ideal environments.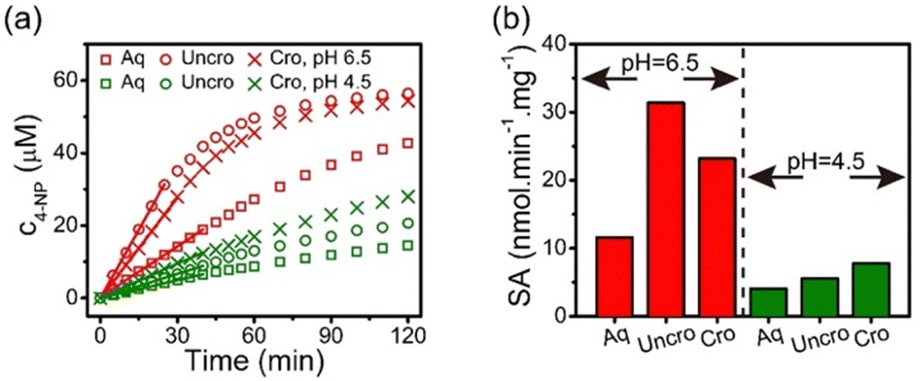 | ||
| Fig. 7 (a) Enzymatic activity of ALP in (□) aqueous solution, (○) uncrosslinked and (×) crosslinked coacervates at pH 6.5 (red) and 4.5 (green). (b) Specific activity (SA) of ALP determined at 25 °C. Encapsulating ALP in coacervates improved the SA. | ||
The changes in the performance of the ALP are quantified through the specific activity (SA) of ALP as shown in eqn (5),
 | (5) |
At pH 4.5, the activity of ALP remains higher in the coacervates than in aqueous solution. However at pH 4.5, the ALP loaded within the crosslinked coacervate shows a higher activity than the ALP within the uncrosslinked coacervate. To explain this phenomenon, reorganization within the uncrosslinked coacervate can occur as the enzyme ALP becomes positively charged at pH 4.5 with its isoelectric point at 4.8. This change in the charge of the ALP enables its release from the coacervate due to the strength of the electrostatic interactions between components as shown in Fig. 4b. The change in pH from 6.5 to 4.5 leads to a small increase in the efficacy of encapsulation of ALP in crosslinked coacervates, while the encapsulation of ALP in uncrosslinked coacervates decreases from approximately 53% to 37% as pH decreases from 6.5 to 4.5.
Hydrophobicity is known to be important to the encapsulation of organic small molecules. For example, an increase in the hydrophobicity of the coacervate phase increases the sequestration efficiency of organic dyes.22 The increased hydrophobicity from the α-methyl group in PMAA provides a simple comparison with PAA. The role of hydrophobicity in complexes has been a topic of interest for more than 25 years.64,65 Hydrophobicity may contribute to the preferential uptake of substrates, and therefore enhance the rates of reaction confined within coacervate droplets. Here, allyl-PMAA was compared to allyl-PAA for the encapsulation of ALP in coacervates to more directly probe the impact of hydrophobicity on the enzymatic activity of ALP. The increase in hydrophobicity within the coacervate droplets with allyl-PMAA as the polyanion is confirmed by fluorescence spectra using ANS (Fig. S8, ESI†). Fig. 8 illustrates the enhanced enzymatic activity for the encapsulated ALP with the BPEI-allyl-PMAA coacervate. The partition coefficient of 4-NPP is greater for the BPEI-allyl-PMAA coacervate than for the BPEI-allyl-PAA coacervate (Fig. S10, ESI†). These results illustrate the importance of the partition coefficient of the substrate for enzymatic activity in encapsulated coacervates.
 | ||
| Fig. 8 Enzymatic activity of ALP in (□) aqueous solution and (○) uncrosslinked and (×) crosslinked coacervates at pH 6.5. Both (red) BPEI-allyl-PAA and (green) BPEI-allyl-PMAA coacervates are compared with the allyl functional group representing 9–11% of the repeat unit for both PAA and PMAA. | ||
The click chemistry approach66 described here requires functionality on at least one of the polyelectrolytes in the coacervate and a bifunctional crosslinker that can be light activated after the coacervate is formed. Bowman and coworkers have described the chemistry requirements for effective photoclick reactions and a variety of complementary pairs.67 The key to this approach is including one functionality on a polyelectrolyte, but this has been overcome in other bio-related fields.68 The crosslinker can be selected to augment the properties of the coacervate. For example, the chemistry between the functional ends, such as the thiols used here, provides a simple handle to further modify hydrophobicity with the crosslinker without impacting the structure of biologics in coacervates and stabilizing the coacervate environment under conditions where disassembly would typically occur.
Conclusions
Allyl functionalization of polyelectrolytes provides a facile handle for crosslinking through click chemistry. A post-coacervation crosslinking approach under benign, aqueous conditions using the photoinitiated thiol–ene click reaction provides stability to the coacervate droplets at extremes of pH and high ionic strength where the uncrosslinked coacervate dissociates into solution. Beyond the increased stability of the coacervate, crosslinking also offers advantages in partitioning of proteins into coacervates due to the increased hydrophobicity of the coacervate environment. The crosslinking reaction does not alter the structure of BSA within the coacervate and improves stability of BSA at low pH. Similarly, the crosslinked coacervate acts as a carrier for the enzyme ALP. The enzymatic activity is found to effectively increase due to the partitioning of the substrate within the coacervate. This partitioning is enhanced by switching the polyanion from allyl-PAA to allyl-PMAA, which leads to a significantly enhanced enzymatic activity for the encapsulated ALP.Coacervate droplets provide an attractive route to enzyme encapsulation, stabilization, and possibly improvement in the activity of the sequestered enzymes. The encapsulation of proteins within coacervate drops may have several protective effects on the protein structure in harsh environments including extremes of pH, change in ionic strength, high temperature, and the presence of chemicals including urea and heavy metals.26 The crosslinking of the coacervate should enable a broader window to be explored as well as the density of the coacervate itself through the conditions under which the crosslinking is applied. A system like this crosslinked coacervate serving as a carrier for enzymes could address challenges with the instability of polyelectrolyte coacervates to protect encapsulated proteins against extreme environments while enhancing the enzymatic activity of the sequestered enzymes. The flexibility of this post coacervation crosslinking should be broadly applicable to extend the potential operating environments for coacervates to protect components, especially in emerging areas of oncology,69 vaccines,70,71 and catalysis.72 Additionally, the increased stability of the crosslinked coacervate droplets could have significant implications towards industrial applications.73
Author contributions
Mengmeng Zhao: methodology, investigation, data curation, writing – original draft, validation, and visualization. Szu-Hao Cho: investigation and visualization. Xinchi Wu: investigation. Jingyi Mao: investigation. Bryan D. Vogt: writing – review & editing. Nicole S. Zacharia: conceptualization, methodology, resources, supervision, funding acquisition, and writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF) under grant no. CBET-1744459. The authors would like to thank Prof. Jie Zheng and Dr Baiping Ren at the University of Akron (Department of Chemical Engineering) for help with circular dichroism measurement.Notes and references
- C. Schmidt-Dannert and F. Lopez-Gallego, Microb. Biotechnol., 2016, 9, 601–609 CrossRef PubMed.
- B. Li, Z. Yuan, P. Zhang, A. Sinclair, P. Jain, K. Wu, C. Tsao, J. Xie, H. C. Hung, X. Lin, T. Bai and S. Jiang, Adv. Mater., 2018, 1705728 CrossRef PubMed.
- B. V. V. S. Pavan Kumar, J. Fothergill, J. Bretherton, L. Tian, A. J. Patil, S. A. Davis and S. Mann, Chem. Commun., 2018, 54, 3594–3597 RSC.
- L. Tian, N. Martin, P. G. Bassindale, A. J. Patil, M. Li, A. Barnes, B. W. Drinkwater and S. Mann, Nat. Commun., 2016, 7, 13068 CrossRef CAS PubMed.
- G. Carturan, R. Dal Toso, S. Boninsegna and R. Dal Monte, J. Mater. Chem., 2004, 14, 2087–2098 RSC.
- C. F. Meunier, J. C. Rooke, A. Léonard, H. Xie and B.-L. Su, Chem. Commun., 2010, 46, 3843–3859 RSC.
- K. Kurihara, M. Tamura, K. Shohda, T. Toyota, K. Suzuki and T. Sugawara, Nat. Chem., 2011, 3, 775–781 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122–125 CrossRef CAS PubMed.
- P. Stano and P. L. Luisi, Chem. Commun., 2010, 46, 3639–3653 RSC.
- B. Städler, A. D. Price, R. Chandrawati, L. Hosta-Rigau, A. N. Zelikin and F. Caruso, Nanoscale, 2009, 1, 68–73 RSC.
- S. F. M. van Dongen, H.-P. M. de Hoog, R. J. R. W. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS PubMed.
- A. D. Price, A. N. Zelikin, Y. Wang and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 329–332 CrossRef CAS.
- C. Wu, S. Bai, M. B. Ansorge-Schumacher and D. Wang, Adv. Mater., 2011, 23, 5694–5699 CrossRef CAS PubMed.
- A. B. Subramaniam, J. Wan, A. Gopinath and H. A. Stone, Soft Matter, 2011, 7, 2600–2612 RSC.
- F. J. Ghadessy, J. L. Ong and P. Holliger, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4552–4557 CrossRef CAS.
- B. T. Kelly, J.-C. Baret, V. Taly and A. D. Griffiths, Chem. Commun., 2007, 1773 RSC.
- M. Li, X. Huang, T. Y. D. Tang and S. Mann, Curr. Opin. Chem. Biol., 2014, 22, 1–11 CrossRef CAS.
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 CrossRef CAS PubMed.
- D. Priftis, R. Farina and M. Tirrell, Langmuir, 2012, 28, 8721–8729 CrossRef CAS.
- S. Huang, M. Zhao, M. B. Dawadi, Y. Cai, Y. Lapitsky, D. A. Modarelli and N. S. Zacharia, J. Colloid Interface Sci., 2018, 518, 216–224 CrossRef CAS PubMed.
- M. Zhao, X. Xia, J. Mao, C. Wang, M. B. Dawadi, D. A. Modarelli and N. S. Zacharia, Mol. Syst. Des. Eng., 2019, 4, 110–121 RSC.
- M. Zhao, S. A. Eghtesadi, M. B. Dawadi, C. Wang, S. Huang, A. E. Seymore, B. D. Vogt, D. A. Modarelli, T. Liu and N. S. Zacharia, Macromolecules, 2017, 50, 3818–3830 CrossRef CAS.
- M. Zhao and N. S. Zacharia, Macromol. Rapid Commun., 2016, 37, 1249–1255 CrossRef CAS PubMed.
- K. Lv, A. W. Perriman and S. Mann, Chem. Commun., 2015, 51, 8600–8602 RSC.
- D. S. Williams, S. Koga, C. R. C. Hak, A. Majrekar, A. J. Patil, A. W. Perriman and S. Mann, Soft Matter, 2012, 8, 6004–6014 RSC.
- M. Zhao and N. S. Zacharia, J. Chem. Phys., 2018, 149, 163326 CrossRef.
- N. Martin, M. Li and S. Mann, Langmuir, 2016, 32, 5881–5889 CrossRef CAS PubMed.
- K. A. Black, D. Priftis, S. L. Perry, J. Yip, W. Y. Byun and M. Tirrell, ACS Macro Lett., 2014, 3, 1088–1091 CrossRef CAS PubMed.
- I. B. A. Smokers, B. S. Visser, A. D. Slootbeek, W. T. S. Huck and E. Spruijt, Acc. Chem. Res., 2024, 57, 1885–1895 CrossRef CAS PubMed.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- R. Kembaren, J. M. Kleijn, J. W. Borst, M. Kamperman and A. H. Hofman, Soft Matter, 2022, 18, 3052–3062 RSC.
- B. Muhoza, S. Xia and X. Zhang, Food Hydrocolloids, 2019, 97, 105174 CrossRef CAS.
- E. J. Bealer, S. Onissema-Karimu, A. Rivera-Galletti, M. Francis, J. Wilkowski, D. S. de la Cruz and X. Hu, Polymers, 2020, 12, 464 CrossRef CAS.
- M. Peydayesh, S. Kistler, J. Zhou, V. Lutz-Bueno, F. D. Victorelli, A. B. Meneguin, L. Spósito, T. M. Bauab, M. Chorilli and R. Mezzenga, Nat. Commun., 2023, 14, 1848 CrossRef CAS PubMed.
- R. Chollakup, J. B. Beck, K. Dirnberger, M. Tirrell and C. D. Eisenbach, Macromolecules, 2013, 46, 2376–2390 CrossRef CAS.
- N. S. Zacharia, M. Modestino and P. T. Hammond, Macromolecules, 2007, 40, 9523–9528 CrossRef CAS.
- A. Roguska, M. Pisarek, M. Andrzejczuk, M. Dolata, M. Lewandowska and M. Janik-Czachor, Mater. Sci. Eng. C, 2011, 31, 906–914 CrossRef CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2006, 7, 2827–2836 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- D. Priftis, X. Xia, K. O. Margossian, S. L. Perry, L. Leon, J. Qin, J. J. De Pablo and M. Tirrell, Macromolecules, 2014, 47, 3076–3085 CrossRef CAS.
- Y. Zhang, F. Li, L. D. Valenzuela, M. Sammalkorpi and J. L. Lutkenhaus, Macromolecules, 2016, 49, 7563–7570 CrossRef CAS.
- A. R. Jones, Prog. Energy Combust. Sci., 1999, 25, 1–53 CrossRef.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- L. Zhang, Y. Yang, H. Ni, J. Liu, M. Wu and G. Chen, Colloid Polym. Sci., 2015, 293, 2035–2044 CrossRef.
- A. K. Zhang, J. Ling, K. Li, G. D. Fu, T. Nakajima, T. Nonoyama, T. Kurokawa and J. P. Gong, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1227–1236 CrossRef CAS.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- E. Brynda, N. A. Cepalova and M. Stol, J. Biomed. Mater. Res., 1984, 18, 685–694 CrossRef CAS PubMed.
- K. Al-Malah, J. McGuire and R. Sproull, J. Colloid Interface Sci., 1995, 170, 261–268 CrossRef CAS.
- P. Quellec, R. Gref, L. Perrin, E. Dellacherie, F. Sommer, J. M. Verbavatz and M. J. Alonso, J. Biomed. Mater. Res., 1998, 42, 45–54 CrossRef CAS.
- K. Takeda, A. Wada, K. Yamamoto, Y. Moriyama and K. Aoki, J. Protein Chem., 1989, 8, 653–659 CrossRef CAS PubMed.
- D. E. Anderson, W. J. Becktel and F. W. Dahlquist, Biochemistry, 1990, 29, 2403–2408 CrossRef CAS.
- Y. H. Kim and W. E. Stites, Biochem, 2008, 47, 8804–8814 CrossRef CAS PubMed.
- H.-X. Zhou, Arch. Biochem. Biophys., 2008, 469, 76–82 CrossRef CAS PubMed.
- E. V. Musin, A. V. Dubrovskii, A. L. Kim and S. A. Tikhonenko, Int. J. Mol. Sci., 2022, 23, 9917 CrossRef CAS.
- J. Choi and M. F. Rubner, Macromolecules, 2005, 38, 116–124 CrossRef CAS.
- C. Cho and N. S. Zacharia, Langmuir, 2012, 28, 841–848 CrossRef CAS PubMed.
- M. Lund and B. Jönsson, Q. Rev. Biophys., 2013, 46, 265–281 CrossRef CAS PubMed.
- D. Srivastava, E. Santiso, K. Gubbins and F. L. Barroso Da Silva, Langmuir, 2017, 33, 11417–11428 CrossRef CAS PubMed.
- F. L. B. da Silva and B. Jönsson, Soft Matter, 2009, 5, 2862 RSC.
- D. Goula, C. Benoist, S. Mantero, G. Merlo, G. Levi and B. Demeneix, Gene Ther., 1998, 5, 1291–1295 CrossRef CAS PubMed.
- K. A. Curtis, D. Miller, P. Millard, S. Basu, F. Horkay and P. L. Chandran, PLoS One, 2016, 11, 1–20 Search PubMed.
- H. Fernley and P. Walker, Biochem. J., 1967, 104, 1011–1018 CrossRef CAS PubMed.
- H. Tokuyama, Y. Nakahata and T. Ban, J. Membr. Sci., 2020, 595, 117533 CrossRef CAS.
- T. Ikawa, K. Abe, K. Honda and E. Tsuchida, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1505–1514 CrossRef CAS.
- H. T. Oyama, W. T. Tang and C. W. Frank, Macromolecules, 1987, 20, 1839–1847 CrossRef CAS.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS.
- A. K. Agrahari, P. Bose, M. K. Jaiswal, S. Rajkhowa, A. S. Singh, S. Hotha, N. Mishra and V. K. Tiwari, Chem. Rev., 2021, 121, 7638–7956 CrossRef CAS.
- Y. Tian, Q. Hu, Z. Sun, Y. Yu, X. Li, T. Tian, X. Bi, Y. Li, B. Niu and Z. Zhang, Small, 2024, 20, 2311890 CrossRef CAS PubMed.
- C. Forenzo and J. Larsen, Mol. Pharm., 2023, 20, 4387–4403 CrossRef CAS PubMed.
- P. U. Joshi, C. Decker, X. Zeng, A. Sathyavageeswaran, S. L. Perry and C. L. Heldt, Biomacromolecules, 2024, 25, 741–753 CrossRef CAS PubMed.
- S. Cao, T. Ivanov, J. Heuer, C. T. J. Ferguson, K. Landfester and L. Caire da Silva, Nat. Commun., 2024, 15, 39 CrossRef CAS PubMed.
- W. Chen, S. Zheng, F. Zhou, Y. Huang, M. Tu and L. Jiang, Chem. Eng. J., 2023, 462, 142165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00765d |
| ‡ Present address: University of Wisconsin-Madison, Madison WI 53706 USA. E-mail: nzacharia@wisc.edu |
| This journal is © The Royal Society of Chemistry 2024 |