
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanochemical Cu(II) complexes and propargylamine synthetic adventures†
Ghadah Abdullah S.
Al Jomeh
a,
Andrew
McGown
a,
Emma
Richards
 b,
Graham J.
Tizzard
c,
Simon J.
Coles
c,
Ramón
González-Méndez
a,
Chris
Dadswell
a,
John
Spencer
a and
George E.
Kostakis
*a
b,
Graham J.
Tizzard
c,
Simon J.
Coles
c,
Ramón
González-Méndez
a,
Chris
Dadswell
a,
John
Spencer
a and
George E.
Kostakis
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: G.Kostakis@sussex.ac.uk
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, Wales, UK
cUK National Crystallography Service, Chemistry, University of Southampton, SO1 71BJ, UK
First published on 9th January 2024
Abstract
We synthesized and characterized known and new Cu(II)–salen complexes and investigated their efficacy in yielding propargylamines (PAs) via an A3 coupling reaction mechanochemically. Although the method appears simple, we explicitly describe synthetic obstacles that urge unavoidable solvent use to isolate the molecular entities in high purity. The recovered complexes retain structure and catalytic efficacy, and a library of twenty-four known and unknown PAs are isolated in very good to excellent yields. The scope and limitations of this method are presented.
Sustainability spotlightThe present portable methodology can be used in any developing country, escapes from traditional catalytic methods, and uses minimal solvent and abundant elements to produce heterobifunctional molecules that could remove specific unwanted proteins. This method aligns with UN SDG(s) 3, 9 and 12 and explicitly with targets 3.4, 9.4, 12.1 and 12.5. |
Introduction
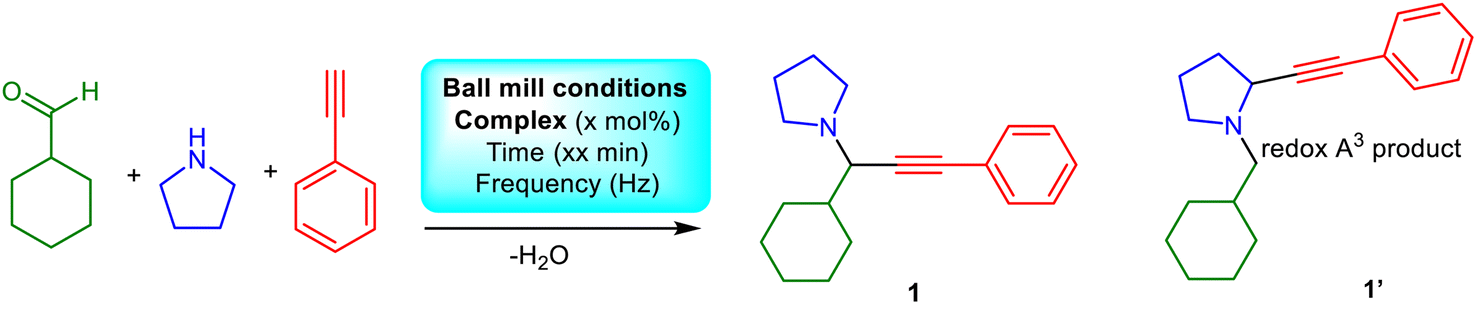
Traditional chemical processes often use toxic reagents and fossil fuels and generate harmful byproducts, significantly contributing to environmental pollution.1,2 Alternative, sustainable methods have been sought in the last two decades to minimize waste, decrease or avoid toxic materials, and incorporate renewable resources.3–5 Mechanochemistry uses mechanical forces, such as grinding or milling, to initiate or drive chemical reactions rather than relying on traditional solvent-based methods.6–11 Work by Toda et al. evidenced the reproducibility of several organic reactions, occurring in solution, in the solid state by mixing the ingredients into finely powdered form.12,13 Ball-milling represents a sophisticated upgrade of traditional grinding, minimizes waste, reduces energy consumption and can be easily scaled up for industrial production and thus can be considered a sustainable synthetic method, although its applicability can be limited in terms of capital expense (equipment costs), efficiency, and selectivity.8Propargylamines (PAs) are valuable precursors for organic scaffolds used for neurodegenerative diseases or as anti-depressants, anti-platelets and anticonvulsants.14,15 They can be synthesized via traditional catalytic protocols, based on alkylation or reductive amination paths, that incorporate Pd-based or Zn alkynyl reagents or sodium borohydride in significant amounts.16 The A3 coupling reaction is a popular organic pathway for synthesizing PAs in the presence of late (Cu, Pd, or Au) transition metals.17–29 This process involves three substrates, amine–alkyne–aldehyde and the in situ formation of a π complex (metal-alkyne), which weakens the C–H sp bond with subsequent proton elimination by a weak base, resulting in σ activation and formation of the acetylide, which can be subsequently added to the imine (product of the amine–aldehyde condensation), yielding the PA.14 The A3 coupling reaction has been a desirable target for mechanochemical synthesis, as it allows for the rapid and efficient construction of complex molecules with high structural diversity. In 2015, Su et al. demonstrated a solvent-free enantioselective PAs synthesis via a Cu(OTf)2/Ph-pybox system by ball-milling within 60 minutes. The in situ generated catalytic system can be reused several times without losing activity and applies to a broad scope of substrates, aromatic aldehydes and amines.23 In 2018, the mechanochemical activation of aldehydes, amines, and calcium carbide in the presence of CuI enabled the unprecedented formation of 1,4-diamino-2-butynes under solventless conditions.30
Our group have investigated the peculiar efficiency of Cu(II) complexes as vehicles to promote the A3 coupling reaction, in the absence of additives, at low loadings and in non-coordinating solvents.31,32 We identified the unprecedented efficacy of the neutral Cu(II)–N2O2 salen complex (Scheme 1), facilitating the A3 coupling at room temperature within 72 h.31 The reaction proceeds via a radical pathway, and the (N or O) heteroatoms of the salen frame act as a weak base to eliminate the proton from the alkyne while the reaction rate increases with temperature. The recovered complex retains its structure and catalytic efficacy after 10 cycles. The complex can be synthesized in two simple high-yielding steps from commercially available starting materials. The first step involves a condensation reaction, reflux for three hours, of the parent diamine and the 3,5-di-tert-butyl salicylaldehyde in MeOH, while the second step involves the complexation reaction of the resulting Schiff base and Cu(OTf)2. Inspired by the aforementioned solventless A3 reports and given the synthetic simplicity of these two steps, we envisioned the solventless ligand, Cu(II)–N2O2 salen complex, and PAs synthesis as feasible. To the best of our knowledge, a few studies have already reported the solventless synthesis of salen based complexes in one-pot33 or two steps,34–37 but no report identifies the synthesis of the ligand, complex and PAs mechanochemically. The results of this effort and the scope and limitations of this approach are described herein.
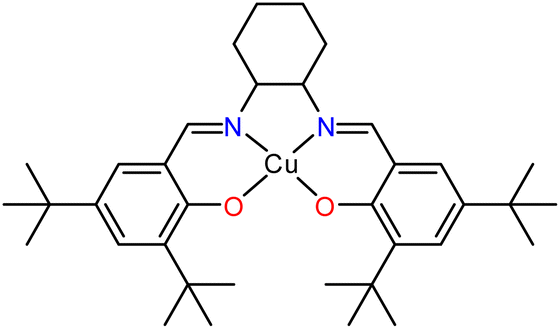 | ||
| Scheme 1 A schematic representation of the Cu(II)–N2O2 salen complex used in previous studies.31 | ||
Results
Ligand synthesis
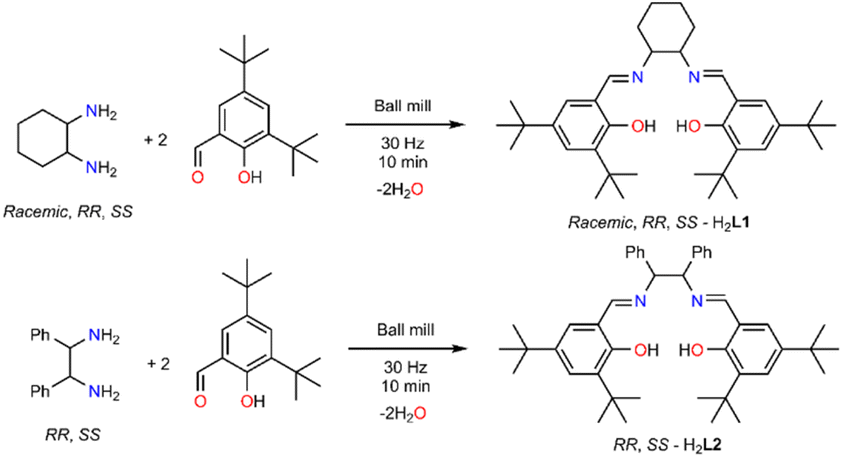
The solventless ligand synthesis, as anticipated, is a straightforward procedure. Initial screenings, varying the frequency and the milling time, identified the optimum conditions (Table 1, entry 6). A detailed optimisation table can be found in the ESI (Table S1†). The ligand purity was identified by 1H and 13C NMR, IR, and ESI-MS, although two important observations should be considered for future reference. Using steel-based balls and jars and/or prolonged reactions (40 or 80 minutes) yielded yellow solids for which the 1H NMR identified the presence of a different species (results not shown in Table 1), possibly the ketonic form of the targeted species, in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S1 and S2†). The presence of additional peaks in the aromatic and aliphatic regions and integration comparison could support this observation and the tert-butyl groups act as convenient fingerprints in identifying a 4:1 ratio. Moreover, the IR data (Fig. S3†) of the yellow solids indicate the absence of the hydroxyl group and H-bonding interactions (3500–3000 cm−1) and the presence of a ketone (C
:1 ratio (Fig. S1 and S2†). The presence of additional peaks in the aromatic and aliphatic regions and integration comparison could support this observation and the tert-butyl groups act as convenient fingerprints in identifying a 4:1 ratio. Moreover, the IR data (Fig. S3†) of the yellow solids indicate the absence of the hydroxyl group and H-bonding interactions (3500–3000 cm−1) and the presence of a ketone (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond at 1700 cm−1. In contrast, the use of Teflon-based jars and ceramic balls eliminates the formation of the other species. After overcoming these obstacles, we could successfully synthesise the chiral versions (Table 1, entries 7 and 8, H2L1). All ligands were characterised by 1H and 13C NMR, IR, UV-Vis, ESI-MS, while their chiral nature was confirmed by circular dichroism (Fig. S4 and S5†). Thereafter, we extended the study to a different chiral diamine (Table 1, entries 9 and 10, H2L2). As before, we performed several reactions; the use of a steel-based jar and balls for 80 minutes yielded a mixture of diol–ketone forms in a 4:1 ratio, as previously, thus supporting our choice to use only Teflon based jars and balls (see 1H NMR data Fig. S6†). Single crystal X-ray diffraction studies of a recrystallised material from MeOH for the prolonged experiment identified loss of chirality (Table S1 and Fig. S7†); a notion that should be considered for future investigations for us and others. However, when the reactions are performed for a short period (i.e. 10 minutes) with Teflon and ceramic components, chirality is retained. The single-crystal X-ray diffraction results are similar to the recently reported components (Table S2†).38,39 As for the first family of ligands, SS-H2L2 and RR-H2L2 were characterised by 1H and 13C NMR, IR, UV-Vis, ESI-MS, while their chirality was determined by circular dichroism (Fig. S4†). After reaction completion, the solids were placed in the oven (60 °C) overnight to dry. This process is convenient compared with reactions performed in the presence of molecular sieves, which eliminates water molecules as byproducts, but requires treatment with solvent to obtain the targeted species.
O) bond at 1700 cm−1. In contrast, the use of Teflon-based jars and ceramic balls eliminates the formation of the other species. After overcoming these obstacles, we could successfully synthesise the chiral versions (Table 1, entries 7 and 8, H2L1). All ligands were characterised by 1H and 13C NMR, IR, UV-Vis, ESI-MS, while their chiral nature was confirmed by circular dichroism (Fig. S4 and S5†). Thereafter, we extended the study to a different chiral diamine (Table 1, entries 9 and 10, H2L2). As before, we performed several reactions; the use of a steel-based jar and balls for 80 minutes yielded a mixture of diol–ketone forms in a 4:1 ratio, as previously, thus supporting our choice to use only Teflon based jars and balls (see 1H NMR data Fig. S6†). Single crystal X-ray diffraction studies of a recrystallised material from MeOH for the prolonged experiment identified loss of chirality (Table S1 and Fig. S7†); a notion that should be considered for future investigations for us and others. However, when the reactions are performed for a short period (i.e. 10 minutes) with Teflon and ceramic components, chirality is retained. The single-crystal X-ray diffraction results are similar to the recently reported components (Table S2†).38,39 As for the first family of ligands, SS-H2L2 and RR-H2L2 were characterised by 1H and 13C NMR, IR, UV-Vis, ESI-MS, while their chirality was determined by circular dichroism (Fig. S4†). After reaction completion, the solids were placed in the oven (60 °C) overnight to dry. This process is convenient compared with reactions performed in the presence of molecular sieves, which eliminates water molecules as byproducts, but requires treatment with solvent to obtain the targeted species.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Amine–aldehyde ratio | Time (min) | Frequency (Hz) | Yield (%) | Ligand |
| 1 | 1:2 |
5 | 3 | — | Racemic-H2L1 |
| 2 | 1:2 |
5 | 5 | — | Racemic-H2L1 |
| 3 | 1:2 |
5 | 10 | — | Racemic-H2L1 |
| 4 | 1:2 |
5 | 15 | 50 | Racemic-H2L1 |
| 5 | 1:2 |
5 | 20 | 70 | Racemic-H2L1 |
| 6 | 1:2 |
10 | 25 | >99 | Racemic-H2L1 |
| 7 | 1:2 |
10 | 25 | >99 | RR-H2L1 |
| 8 | 1:2 |
10 | 25 | >99 | SS-H2L1 |
| 9 | 1:2 |
10 | 25 | >99 | RR-H2L2 |
| 10 | 1:2 |
10 | 25 | >99 | SS-H2L2 |
Complex synthesis
With the ligands in hand, we then focused on the synthesis of the corresponding complexes. The crystalline complex made under reflux conditions with lack of lattice solvent molecules and decomposition starting at 282 °C was used as the benchmark, and we employed thermogravimetric analysis (TGA) as a tool to identify impurities and to determine the optimal reaction conditions (Fig. 1).31 We screened several parameters, altering (a) slightly the metal salt:ligand ratio (0.95:1, 1:1, 1:1.05); (b) the metal salt, incorporating weakly (Cl, Br, OTf, BF4) or strongly coordinating (NO3, OAc) anions; (c) the frequency; and (d) the time of the reaction (Table S3†). Also, we incorporated the ligand(s) with(out) drying overnight, ligand(s) made under reflux solvent conditions. The optimal conditions were determined to be as follows: ceramic balls, Teflon-based jars, metal salt:ligand ratio 1:1, CuCl2 as the Cu(II) source, 180 minutes and ball-milling frequency 25 Hz.
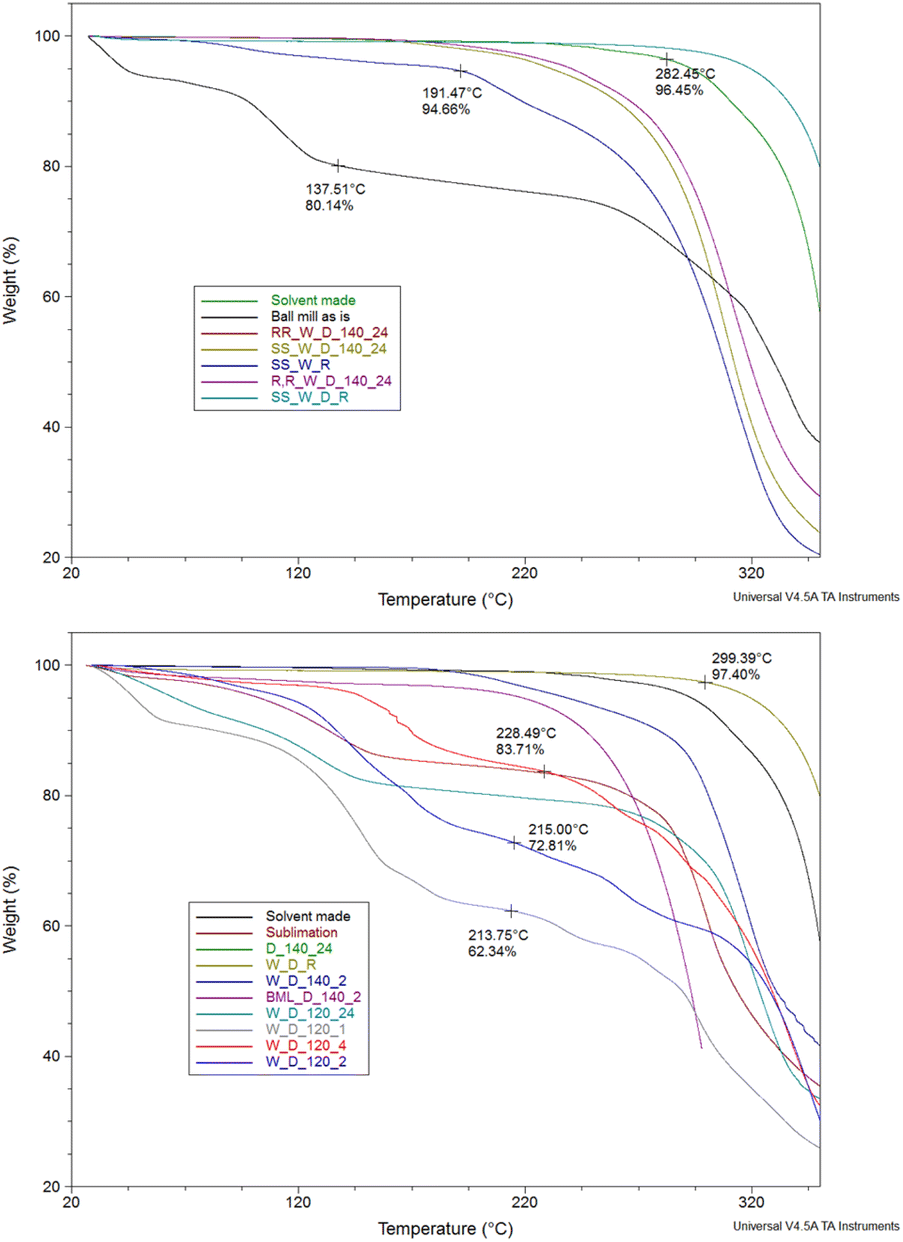 | ||
| Fig. 1 Thermogravimetric analysis of different trials; washed (W), dried (D), recrystallised (R) drying temperatures (120 or 140 °C), drying time (1, 2, 4 or 24 hours), chiral versions (RR/SS). | ||
We chose CuCl2 as the metal salt because it bears a weakly coordinating anion and no lattice solvent molecules. The reaction of the ligand racemic-H2L1, without drying, with CuCl2 in 1:1 molar ratio resulted in a brownish material possessing a significant amount of lattice molecules (at 138 °C presents 20% weight loss). It is worth noting that two hydrochloric acid molecules (Scheme 2) are produced during the complexation reaction.
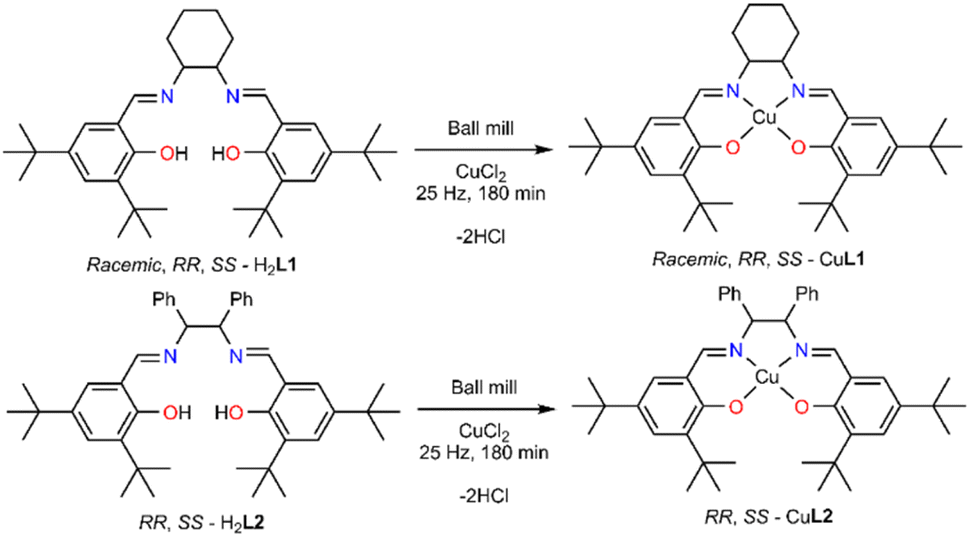 | ||
| Scheme 2 Solventless synthesis of CuL1 and CuL2. | ||
Given this notion, we washed the final product with five drops (∼0.25 ml) of water to remove hydrochloric acid and dried it for different times (1, 2, 24 h) or temperatures (120 or 140 °C), aiming to identify the least energy consuming conditions. However, none of these efforts delivered a solid product with similar thermal behaviour to the benchmark material. Next, we attempted to sublime the produced material, but again we obtained material containing a significant amount of lattice molecules. After all these trials, we identified that recrystallisation from a minimal amount of non-coordinating and volatile acetone provides a solid with identical thermal behaviour to the parent molecule. PXRD studies of the products, in bulk, strongly deviate from the calculated pattern, possibly indicating crystallisation in a different space group (Fig. S8†). Last, we adopted the previously described one-pot synthesis,33 (Table S3,† entry 28); however, the yield of the targeted material was only 30% after washing, drying and recrystallisation. After optimising this process, we applied this synthetic protocol to obtain all the Cu(II) complexes; the yield for all five complexes is over 99%. The synthesis of the final molecules in the solid state was further validated by recording elemental analysis and supplementary EPR studies. The EPR spectrum of solid dehydrated SS-CuL2 is shown in Fig. S13,† indicating an approximately pseudo-axial profile but with no resolvable copper hyperfine splitting, as would be expected for the spin active metal centre I(63,65Cu) = 3/2. Further weak signals at half-field (B ∼ 150 mT) are typical of high-spin (i.e. S > ½) spin systems and suggests magnetic interaction between neighbouring metal centres in the solid state (Table S5†). Therefore, for improved resolution of the spin Hamiltonian parameters characterizing the paramagnetic centre, further measurements were performed in a toluene:dichloromethane solvent system in the presence of the A3 substrates (Fig. S13–S18†). SS-CuL2 is characterized by a predominantly axial g-tensor (g‖ = 2.150, g⊥ = 2.010; giso = 2.056) and dominated by copper hyperfine coupling (A‖ = 590, A⊥ = 90 MHz; aiso = 256.7 MHz), in which the g-tensor and A-tensor are coincident with the molecular frame, as reported previously for a closely analogous derivative of CuL1 in which the cyclohexyl backbone is replaced by a phenyl ring.40 The spin Hamiltonian parameters characterizing the EPR spectra change upon addition of the A3 coupling components, notably an increase in the g‖ value accompanied by a decrease in A‖ (Fig. S13–S18†), indicating transfer of electron spin density away from the copper centre through weak coordination of the substrate to an axial coordination site. This is analogous to previous reports for binding of chiral amines to Schiff-base complexes.41,42
To determine the absolute structure of the resulting complexes, we recorded single crystal X-ray diffraction data for compounds SS-CuL2 and RR-CuL2 (Table S2†), and identified the unit cell data (data not presented) for compounds SS-CuL1, RR-CuL1 and racemic-CuL1.43 For compounds SS-CuL2 and RR-CuL2 (Fig. 2 and Table S4†), the structures are solved in chiral space groups, in contrast to the already known data.44 To our surprise, one of the screening reactions with steel-based jars and balls, RR-H2L2 and CuCl2, yielded a crystalline byproduct material formulated as RR-FeClL2 (Fig. 2, bottom) that was not further characterised but intimated that metal leaching from either the jar or balls could occur during the milling process. All these studies identify the technical obstacles in our efforts to obtain the targeted complexes in high purity, despite the simplicity of the system and previous reports for one or two-step procedures.33–37 Furthermore, the produced complexes are susceptible to absorbing moisture from the environment as determined by elemental analysis (ESI†); therefore, the final complexes should be stored in the oven at 60 °C or be “activated” before use in any catalytic reaction. All these factors are essential when applying these chiral complexes in the asymmetric A3 coupling reactions or for redox purposes.45 In solution, the complexes retain their chiral character (Fig S9†), and showcase a typical UV-Vis spectrum corresponding to a tetradentate N2O2 chromophore (Fig S10†), while the ESI-MS data provide structures of the absolute formulae (Fig S11 and S12†).
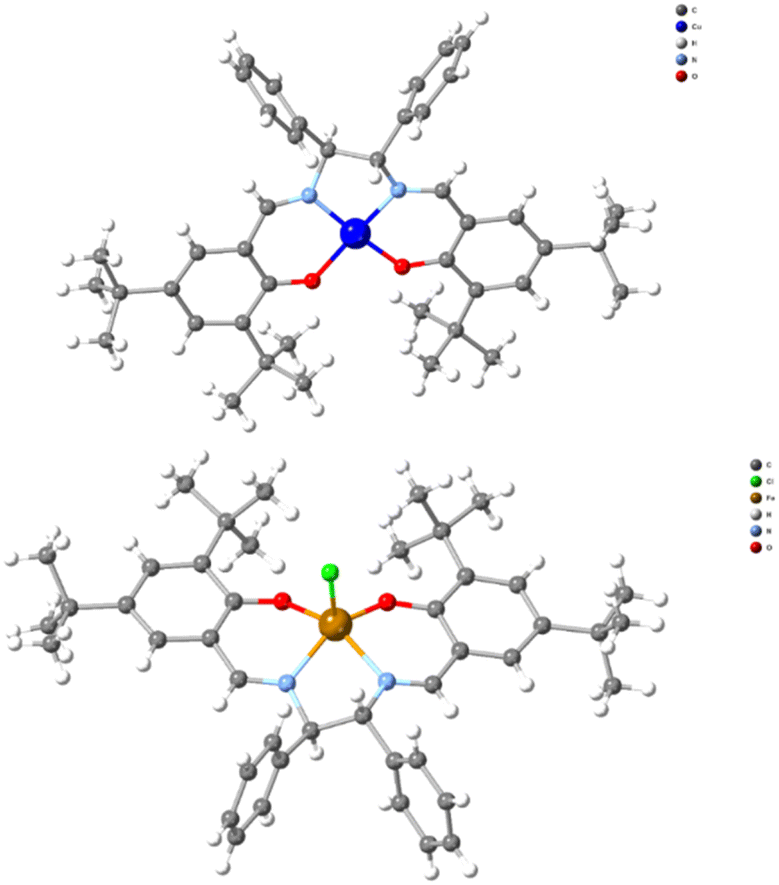 | ||
| Fig. 2 A projection of compound SS-CuL2 and the unprecedented byproduct (RR-FeClL2) was observed when steel-based jars and balls were used. | ||
Propargylamine synthesis
We aimed to identify the optimum conditions for the A3 coupling reaction with the high-purity complexes in our hands.23,30 As previously, we chose cyclohexanecarboxaldehyde, pyrrolidine, and phenylacetylene as model substrates to evaluate the title reaction,31 and incorporated the racemic-CuL1 as the catalyst. Table 2 and Fig. 3 provide the average yield and calculated standard deviation out of at least three trials. Our first control experiments focused on determining the impact of the type of jar and/or number of balls; however, a reaction using Teflon-based jars and balls failed to produce the desired product (Table 2, entry 1 and Fig S19†). Given that reactions with steel-based jars led to the production of unwanted (RR-FeClL2) impurities vide supra, we discarded their use for catalytic reactions. Hence, we instead attempted the reaction using already known conditions,23,30 or modified jars and balls and the substrate molar ratio to optimise our protocol (Table 2, entries 2–9). The reaction in the presence of silica gel23 does not improve performance (entry 3, Table 2). We identified that the optimum substrate ratio is cyclohexanecarboxaldehyde (1.5 mmol), pyrrolidine (1.5 mmol), and phenylacetylene (1 mmol).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Aldehyde (mmol) | Amine (mmol) | Alkyne (mmol) | Cu source (% mol) | Time (h) | Frequency (Hz) | Conversiona (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Calculated conversion from crude 1H NMR. Number in parenthesis represents standard deviation while those in bold represent formation of different product. b NR symbolises no reaction. c Silica gel present. d Stainless steel jar & stainless steel ball. e Stainless steel jar & ceramic ball. f Plastic jar & ceramic ball. g Stainless steel jar & ceramic ball. h Plastic jar & ceramic ball. i In the presence of molecular sieves. j Complex stored in the oven (60 °C). k Recovered. l Recovered dried (10th time). m We used complex named “Ball mill as is” (Fig. 1, left). n We used complex named “W_D_120_1” (Fig. 1, right). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1.5 | 1.5 | 2 | — | 3 | 25 | NRb | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1.2 | 1.1 | 1 | rac-CuL1 (5) | 2 | 25 | 32 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1.0 | 1.2 | 1.5 | rac-CuL1 (7) | 2 | 25 | NRc | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 1 | 1.1 | 1.2 | rac-CuL1 (2) | 2 | 25 | 34d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1 | 1.1 | 1.2 | rac-CuL1 (5) | 3 | 25 | 52e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1 | 1.1 | 1.2 | rac-CuL1 (5) | 3 | 25 | 59f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 1.5 | 1.5 | 1.0 | rac-CuL1 (5) | 3 | 25 | 81g | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 1.5 | 1.5 | 1.0 | rac-CuL1 (5) | 3 | 25 | 89h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 1.5 | 1.5 | 1.0 | rac-CuL1 (2) | 3 | 25 | 92 (3)h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1.5 | 1.5 | 1.0 | rac-CuL1 (2) | 3 | 25 | 99 (3)h,i | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 1.5 | 1.5 | 1.0 | rac-CuL1 (2) | 3 | 25 | 99 (1)h,j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 1.5 | 1.5 | 1.0 | CuCl2 (2) | 3 | 25 | NRh | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 1.5 | 1.5 | 1.0 | CuCl2 (10) | 3 | 25 | NRh | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 1.5 | 1.5 | 1.0 | CuCl2 (20) | 3 | 25 | 90 (1)/9 (1′)h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 1.5 | 1.5 | 1.0 | CuCl2 (2) + H2L1 (2) | 3 | 25 | NRh | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 1.5 | 1.5 | 1.0 | CuCl2 (20) + H2L1 (20) | 3 | 25 | 90 (1)/9 (1′)h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 1.5 | 1.5 | 1.0 | rac-CuL1 (2) | 3 | 25 | 93 (3)h,k | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 1.5 | 1.5 | 1.0 | rac-CuL1 (2) | 3 | 25 | 99 (1)h,j,l | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | 1.5 | 1.5 | 1.0 | rac-CuL1m | 3 | 25 | 20h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20 | 1.5 | 1.5 | 1.0 | rac-CuL1n | 3 | 25 | 31h | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21 | 1.5 | 1.5 | 1.0 | SS-CuL1 | 3 | 25 | 99 (1)h,j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 22 | 1.5 | 1.5 | 1.0 | RR-CuL1 | 3 | 25 | 99 (1)h,j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 23 | 1.5 | 1.5 | 1.0 | SS-CuL2 | 3 | 25 | 99 (1)h,j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 24 | 1.5 | 1.5 | 1.0 | RR-CuL2 | 3 | 25 | 99 (1)h,j | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
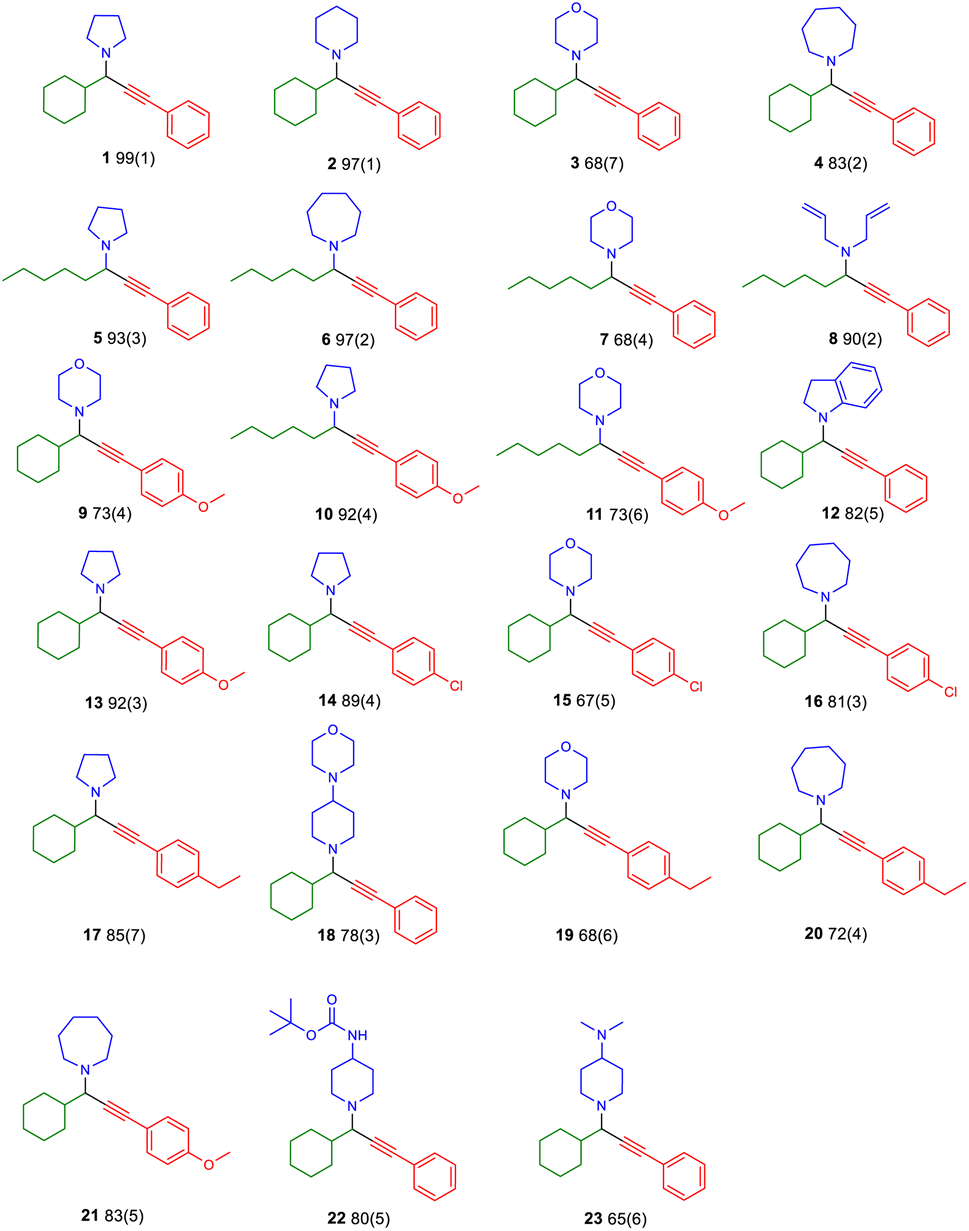 | ||
| Fig. 3 Substrate scope of the reaction with aldehydes, secondary amines and alkynes. Isolated yields, after purification. Reaction conditions; catalyst (2 mol%), 1.5 mmol aldehyde, 1.5 mmol amine, 1.0 mmol alkyne, time 3 h, frequency 25 Hz. Numbers in parenthesis represent the standard deviation (n = 3). | ||
Using molecular sieves to capture the in situ generated water molecules improves performance (entry 10, Table 2); however, incorporation of the drying agents complicates the recovery process, requiring additional solvent treatment, thus we decided to proceed without their use. These salen complexes are susceptible to absorbing moisture upon storage, which significantly impacts performance (entry 9, Table 2), although storing the complex at 60 °C or activation prior to catalysis is required for optimum performance (entry 11, Table 2). To our delight, the reaction employing the same substrate ratio of CuCl2 instead of the racemic-CuL1 complex, does not promote the A3 coupling. Higher copper salt loadings (10%, 20%) yield the anticipated product and its redox analogue (entries 12–14, Table 2, Fig. S20 and S21†), which, according to the literature, corresponds to the redox A3 coupling product.46,47 Our efforts to in situ generate the complex from CuCl2 and racemic-H2L1 in loadings 2% and 20% either did not yield the expected product (entry 15, Table 2), or yielded the expected compounds in addition to the byproduct 1′ (entry 16, Table 2), signifying the importance of isolating the well-characterised complexes prior to catalysis.
Treating the reaction with isopropanol,31 allows for recovery of the complex and single crystal X-ray diffraction studies (Table S4†) and catalytic trials of the recovered material identify that it retains its structure and catalytic efficacy after ten cycles (entries 17 and 18, Table 2). Also, we tested the catalytic efficacy of the “impure” complexes (entries 19 and 20, Table 2), and, to our delight, identified moderate performance, justifying our persistence and choice to use well-characterised complexes. After optimising the conditions, we expanded our study to the other complexes, which outperformed the others (entries 21–24, Table 2). Finally, we attempted to expand the scope of the method to other substrates (Fig. 3). The method applies to aliphatic aldehydes, secondary amines, and various alkynes and provides twenty PAs in very good to excellent yields, demonstrating the excellent utility of this approach.
To further expand and understand the limitations of our method, and given the chiral nature of the PA products, we attempted enantioselective determination for compound 1 (entries 21–24, Table 2). To our disappointment, analysis with a chiral column identified the formation of only racemic products (Fig. S22–S26†). Then, to evaluate copper metal leaching during the process, we performed a series of ICP-MS studies with(out) the use of a metal scavenger and the results are shown in Table 3. These data suggest that the amount of Cu found in the synthesised PAs using racemic-CuL1 is significantly below the acceptable limits with or without using a metal scavenger, which confirms that metal leaching from the active catalyst does not occur during the catalytic process.48 A comparison with a reaction using 20% CuCl2 loading (reactions with 2% or 10% loading do not proceed) suggests that using a metal scavenger is vital, greatly reducing the remaining Cu entity in the organic scaffold.
| Entry | Catalyst (loading%) | Metal scavenger | Yield (%) | ICP-MS Cu detection level (ppm) |
|---|---|---|---|---|
| 1 | rac-CuL1 (2) | Yes | 99 (1) | 0.25 |
| 2 | rac-CuL1 (2) | No | 99 (1) | 0.43 |
| 3 | rac-CuL1 (10) | Yes | 99 (1) | 0.61 |
| 4 | rac-CuL1 (10) | No | 99 (1) | 3.31 |
| 5 | CuCl2 (2) | Yes | No reaction | — |
| 6 | CuCl2 (2) | No | No reaction | — |
| 7 | CuCl2 (10) | Yes | No reaction | — |
| 8 | CuCl2 (10) | No | No reaction | — |
| 9 | CuCl2 (20) | Yes | 90 (1)/9 (1′) | 0.01 |
| 10 | CuCl2 (20) | No | 90 (1)/9 (1′) | 0.23 |
Lastly, to evaluate the applicability of our method and given the very good yield in isolating PA 22, we expanded our study to the synthesis of PA 24.
The chosen PA can be used as a template for synthesising heterobifunctional molecules. Compound 24 can be synthesised under microwave irradiation on a gram scale (method A; Scheme 3), however, using our protocol, 24 can be synthesized up to 500 mg scale (method B).
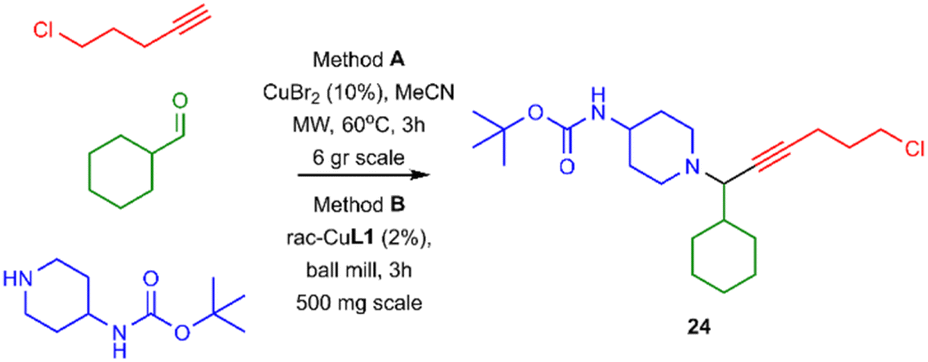 | ||
| Scheme 3 The application of our protocol and gram-scale synthesis of organic scaffold 24. | ||
Discussion
Building on our previous studies,31 this work investigated the solventless synthesis of Cu(II) salen complexes and propargylamines. Despite the simplicity of the task, we identified significant synthetic technical obstacles and the unavoidable use of a minimum amount of solvent to access the complexes in high purity. This notion is vital considering, for example, similar molecular scaffolds used in the electrochemical CO2 reduction and formation of C2+ products;45 the presence of impurities may affect catalytic performance significantly, as is the case in this study.The Cu(II) complexes catalyse the A3 coupling reaction yielding twenty-four PAs in very good to excellent yields, but most importantly, the recovered molecular complexes retain their structure and catalytic efficacy after ten reaction cycles. Control experiments applaud the use of well-characterised species over metal salts that retain structure and efficacy and are hence recoverable, requiring one order of magnitude less loading and resulting in Cu content (as impurities) within the final organic species that are significantly below the acceptable limits. The method reported herein (ligand, complex and PA synthesis) uses significantly less solvent than our previous study, proceeds to completion in a shorter period (Table 4), incorporates non-toxic, abundant, commercially available chemicals, and operates in the open air. It therefore, has a significant “green” character and is sustainable compared to traditional catalytic methods.
| Previous work31 | This work | |||||
|---|---|---|---|---|---|---|
| Solvent | Time | Yield (%) | Solvent | Time | Yield (%) | |
| Ligand synthesis | EtOH | 2 h | 97 | None | 15 min | >99 |
| Complex synthesis | MeOH | 1 h | 90 | None | 15 min | >99 |
| Complex purification | Not needed | Acetone/water | ||||
| Complex recovery post catalysis | Isopropanol | Isopropanol | ||||
| PAs synthesis | DCM | 72 h | 97 | None | 3 h | >99 |
| PAs purification | EtOAc/hexane | EtOAc/hexane | ||||
Conclusions
We encountered several synthetic/technical obstacles in our effort to transition from traditional catalytic protocols to greener methodologies and provide functional organic scaffolds conveniently. We entrust the reported exemplary method paves the way for modern and sustainable coordination and synthetic principles and will be inspirational for several research groups.Author contributions
GEK devised the project with critical input from GAJ. GAJ synthesised and characterised the ligands and complexes, performed the catalytic studies and purified the propargylamines. GAJ recorded the thermogravimetric analysis data. GAJ, GEK, GJT, SJC produced and processed crystallographic data. ER recorded the EPR data. RGM performed MS studies. CD performed ICP-MS studies. AM and JS assisted in the method optimisation and purification of organic products. All authors contributed to the preparation of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GAJ thanks Imam Muhammed Ibn Saud University for offering financial support for a PhD fellowship. GEK thanks the EPSRC UK National Crystallography Service at the University of Southampton for collecting the crystallographic data.49 GEK/ER acknowledge financial support from RSC Research Enablement Grant (E22-9127990267). AM, GEK, JS are grateful to the University of Sussex for HEIF Business Collaboration & Commercialisation 2023 funding. We thank Prof. Louise C. Serpell and Dr Youssra Al-Hilaly (Sussex Neuroscience) for accessing the Circular Dichroism apparatus. We acknowledge REACH Separations Ltd in Biocity Nottingham for chirality determination of the products.References
- P. T. Anastas, Crit. Rev. Anal. Chem., 2010, 29, 167–175 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- C. J. Elsevier, J. Reedijk, P. H. Walton and M. D. Ward, Dalton Trans., 2003, 1869–1880 RSC.
- A. Reichle and O. Reiser, Chem. Sci., 2023, 14, 4449–4462 RSC.
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef PubMed.
- J. Yu, X. Yang, C. Wu and W. Su, J. Org. Chem., 2020, 85, 1009–1021 CrossRef CAS PubMed.
- A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustain. Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- S. L. James and T. Frišcic, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- F. Toda, K. Tanaka and S. Iwata, J. Org. Chem., 1989, 54, 3007–3009 CrossRef CAS.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- J. Farhi, I. N. Lykakis and G. E. Kostakis, Catalysts, 2022, 12, 660 CrossRef CAS.
- K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS PubMed.
- A. Kumar, P. Sharma, N. Sharma, Y. Kumar and D. Mahajan, RSC Adv., 2021, 11, 25777–25787 RSC.
- B. V. Rokade, J. Barker and P. J. Guiry, Chem. Soc. Rev., 2019, 48, 4766–4790 RSC.
- V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861–3898 RSC.
- G. Abbiati and E. Rossi, Beilstein J. Org. Chem., 2014, 10, 481–513 CrossRef PubMed.
- S. Shah, B. G. Das and V. K. Singh, Tetrahedron, 2021, 93, 132238 CrossRef CAS.
- Q. Liu, H. Xu, Y. Li, Y. Yao, X. Zhang, Y. Guo and S. Ma, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- C. Koradin, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 2535–2538 CrossRef CAS PubMed.
- Z. Li, Z. Jiang and W. Su, Green Chem., 2015, 17, 2330–2334 RSC.
- C. Wetzel, P. C. Kunz, I. Thiel and B. Spingler, Inorg. Chem., 2011, 50, 7863–7870 CrossRef CAS PubMed.
- J. Dulle, K. Thirunavukkarasu, M. C. Mittelmeijer-Hazeleger, D. V Andreeva, N. R. Shiju and G. Rothenberg, Green Chem., 2013, 15, 1238–1243 RSC.
- A. Mariconda, M. Sirignano, C. Costabile and P. Longo, Mol. Catal., 2020, 480, 110570 CrossRef.
- M.-T. Chen, B. Landers and O. Navarro, Org. Biomol. Chem., 2012, 10, 2206–2208 RSC.
- I. Jesin and G. C. Nandi, Eur. J. Org Chem., 2019, 2704–2720 CrossRef CAS.
- K. Dong, M. Liu and X. Xu, Molecules, 2022, 27, 3088 CrossRef CAS PubMed.
- M. Turberg, K. J. Ardila-Fierro, C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2018, 57, 10718–10722 CrossRef CAS PubMed.
- S. I. Sampani, V. Zdorichenko, M. Danopoulou, M. C. Leech, K. Lam, A. Abdul-Sada, B. Cox, G. J. Tizzard, S. J. Coles, A. Tsipis and G. E. Kostakis, Dalton Trans., 2020, 49, 289–299 RSC.
- J. Devonport, L. Sully, A. K. Boudalis, S. Hassell-Hart, M. C. Leech, K. Lam, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, J. Spencer, A. Vargas and G. E. Kostakis, JACS Au, 2021, 1, 1937–1948 CrossRef CAS PubMed.
- M. Ferguson, N. Giri, X. Huang, D. Apperley and S. L. James, Green Chem., 2014, 16, 1374–1382 RSC.
- H. Katouah, A. M. Hameed, A. Alharbi, F. Alkhatib, R. Shah, S. Alzahrani, R. Zaky and N. M. El-Metwaly, ChemistrySelect, 2020, 5, 10256–10268 CrossRef CAS.
- L. Leoni, A. Carletta, L. Fusaro, J. Dubois, N. A. Tumanov, C. Aprile, J. Wouters and A. D. Cort, Molecules, 2019, 24, 2314 CrossRef CAS PubMed.
- V. K. Singh, A. Chamberlain-Clay, H. C. Ong, F. León, G. Hum, M. Y. Par, P. Daley-Dee and F. Garciá, ACS Sustain. Chem. Eng., 2021, 9, 1152–1160 CrossRef CAS.
- S. Zuo, S. Zheng, J. Liu and A. Zuo, Beilstein J. Org. Chem., 2022, 18, 1416–1423 CrossRef CAS PubMed.
- M. S. Maru, S. Barroso, P. Adão, L. G. Alves and A. M. Martins, J. Organomet. Chem., 2018, 870, 136–144 CrossRef CAS.
- W. Tan, J. Gao, J. Guan, X. Bi, Y. Tang, C. Zheng, T. Yan and C. Zhang, J. Mol. Struct., 2022, 1269, 133868 CrossRef CAS.
- I. Caretti, E. Carter, I. A. Fallis, D. M. Murphy and S. Van Doorslaer, Phys. Chem. Chem. Phys., 2011, 13, 20427–20434 RSC.
- D. M. Murphy, I. Caretti, E. Carter, I. A. Fallis, M. C. Göbel, J. Landon, S. Van Doorslaer and D. J. Willock, Inorg. Chem., 2011, 50, 6944–6955 CrossRef CAS PubMed.
- S. Zamani, E. Carter, D. M. Murphy and S. Van Doorslaer, Dalton Trans., 2012, 41, 6861–6870 RSC.
- S. Bunce, R. J. Cross, L. J. Farrugia, S. Kunchandy, L. L. Meason, K. W. Muir, M. O'Donnell, R. D. Peacock, D. Stirling and S. J. Teat, Polyhedron, 1998, 17, 4179–4187 CrossRef CAS.
- A. Sharma, K. Mejia, H. Ueno, W. Zhou and L. Chiang, Inorg. Chim. Acta, 2022, 542, 121106 CrossRef CAS.
- L. J. Zhu, D. H. Si, F. X. Ma, M. J. Sun, T. Zhang and R. Cao, ACS Catal., 2023, 13, 5114–5121 CrossRef CAS.
- D. Seidel, Org. Chem. Front., 2014, 1, 426–429 RSC.
- D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765–3769 CrossRef CAS PubMed.
- ICH, ICH Official web site: ICH, https://www.ich.org/page/quality-guidelines, accessed 13 June 2023 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, HRMS, TG, IR, EPR, UV-Vis, CD. CCDC 2284741–2284750. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3su00315a |
| This journal is © The Royal Society of Chemistry 2024 |