
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Open-source Python module to automate GC-MS data analysis developed in the context of bio-oil analyses
Matteo
Pecchi
 *ab and
Jillian L.
Goldfarb
ab
*ab and
Jillian L.
Goldfarb
ab
aDepartment of Biological & Environmental Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: mp933@cornell.edu
bSmith School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA
First published on 9th April 2024
Abstract
GC-MS (Gas Chromatography-Mass Spectrometry) is widely used to measure the composition of biofuels and complex organic mixtures. However, the proprietary GC-MS software associated with each instrument is often clunky and cannot quantify compounds based on similarity indices. Beyond slowing individual research group's efforts, the lack of universal free software to automatically process GC-MS data hampers field-wide efforts to improve bio-oil processes as data are often not comparable across research groups. We developed “gcms_data_analysis,” an open-source Python tool that automatically: (1) handles multiple GCMS semi-quantitative data tables (whether derivatized or not), (2) builds a database of all identified compounds and relevant properties using PubChemPy, (3) splits each compound into its functional groups using a published fragmentation algorithm, (4) applies calibrations and/or semi-calibration using Tanimoto and molecular weight similarities, and (5) produces multiple different reports, including one based on functional group mass fractions in the samples. The module is available on PyPI (https://pypi.org/project/gcms-data-analysis/) and on GitHub (https://github.com/mpecchi/gcms_data_analysis).
1 Introduction
Gas Chromatography-Mass Spectrometry (GC-MS) is routinely used to identify and quantify organic mixtures. In particular, GC-MS analysis of liquid biofuels enables assessment of the composition and therefore quality and value of these sustainable fuels.1–3 The detailed compositional analysis from GC-MS of bio-oil can reveal the chemical reactions underpinning biofuel production processes, thus enabling the optimization of process conditions based on feedstock composition.4–6Performing qualitative GC-MS analysis of bio-oil samples is relatively straightforward; bio-oil is routinely extracted using an organic solvent and compounds are identified by spectral library matches.7–9 In theory, the quantification of compounds via GC-MS is rather simple; chromatogram area is linearly related to the concentration of each analyte. In practice, quantifying the vast numbers of compounds detected in heterogeneous bio-oils requires the construction of a large calibration dataset.10–12 Depending on the GC-MS column, compounds present, and their concentrations, derivatization may be necessary to improve identification, reproducibility and therefore final quantification of species present.6,13,14 Derivatization requires its own calibration to quantify derivatized peaks, sometimes at different operating conditions than the non-derivatized analysis.
These experimental challenges are compounded by the handling and analysis of the large amount of data that GC-MS analyses produce. A single bio-oil sample can contain hundreds of compounds.15,16 A typical experimental campaign involves dozens of samples (hopefully with at least a modicum of replication). Thus, the analysis of such datasets is onerous; the application of a calibration curve to quantify each individual component in a bio-oil, and the translation from derivatized results to the original bio-oil components, both add downstream computational tasks. Manual handling of such large datasets introduces the risk of human error.
In general, most GC-MS instruments provide solutions for the identification of compounds through fragmentation pattern matching by means of proprietary software, and several open-source options are available for the task. One example is PyMS17 and its spin-off project, PyMassSpec.18 Using these open-source tools or the proprietary software available on GC-MS sample it is possible, for each sample, to produce a semi-quantitative table with the identified compounds and their measured chromatogram area, which indicates the intensity of the signal for each compound. If a user has developed a calibration curve for a particular compound, the instrument's software will (usually) report the concentration of the component in the sample vial. For derivatized samples, the derivatized version of the compound is usually returned (since the derivatized molecule enters the machine), which requires an additional step to translate the derivatized versions into their non-derivatized (original) compound. Most instrument companies' proprietary software handle calibrations, but their use can be counterintuitive and/or difficult to implement in additional automatic data analysis routines. Both from our own experience and conversations with researchers around the world, calibrations are often applied manually by researchers, especially when a semi-calibration mode is adopted to estimate the concentration of non-calibrated compounds based on similar compounds' calibration curves.
Another bottleneck in the analysis of GC-MS data is the classification of identified compounds based on their functional group(s). This is especially useful for heterogeneous mixtures such as bio-oils where aggregated compositional results (e.g., the fraction of compounds having a given functional group present in each sample) can be used to derive mechanistic conclusions.5,7,9,19 While performing these tasks manually is possible and has led to valuable results, automation could save time and minimize human error, while also increasing transparency in the methodological approach, specifically in how functional groups are attributed to each compound. Instruments' proprietary software is not always user-friendly or intuitive, and often requires extensive training, especially when used for quantitation of non-calibrated compounds. Such proprietary software does not allow for automation of the full GC-MS data analysis pipeline, usually limiting the automation at the single sample analysis. As such, a new tool for researchers that is open-access and incorporates multiple automated analysis pathways could save researchers' time and promote a higher level of standard best practices across GC-MS users, especially in the biofuels field.
To address this need, we designed an open-source Python tool to fully automate the handling of multiple GC-MS datasets simultaneously (a note, our tool does not perform the analysis of raw GC-MS spectral data, for that, please refer to 18). The tool relies on the Python package PubChemPy20 to access the PubChem website21 and build its own database with all identified chemicals and their relevant chemical properties. It also performs functional group fragmentation using each compound's SMILES22 retrieved from PubChem, employing the automatic fragmentation algorithm developed by ref. 23 to split each molecule into its functional groups and then assign to each group its mass fraction. Functional groups can also be specified by the user using their SMARTS codes.24 The code can apply calibrations for quantifying components present in a sample, with an semi-calibration option based on Tanimoto similarity with tunable thresholds.25,26 For derivatized samples, the procedure is unchanged except that the non-derivatized form of each identified compound is used to retrieve chemical information so that non-derivatized and derivatized samples can be directly compared. The tool groups replicate “files” (intended for single analyses) into the same “sample” (intended for the average of multiple replicates of single analyses of the same sample) to evaluate reproducibility; the user only needs to ensure the proper naming convention is adopted.
Besides producing single sample (or individual files) reports, the tool produces comprehensive reports that include all compounds across a series of samples (files) as well as aggregated reports for all samples (files) based on functional group mass fractions. The code also provides plotting functions for the aggregated reports to visualize the results and enable a preliminary investigation of their statistical significance. The module can be easily installed from PyPI (https://pypi.org/project/gcms-data-analysis/) using pip or downloaded from GitHub (https://github.com/mpecchi/gcms_data_analysis). A simplified example is available on GitHub as a tutorial for new users (with an artificial dataset that was simplified to make testing and maintenance feasible).
In this paper, we demonstrate the tool's capability using a set of three real non-derivatized and derivatized bio-oil samples (analyzed in triplicate); the real data used in this manuscript (found in the subfolder “RCSdata” in the GitHub repository) differ from the example case on GitHub, which is simplified to ensure that no user accidentally employs our calibration curves for their instruments.
2 Algorithm
This section describes the conceptual principles used to design the algorithm. For details about function, naming conventions, and detailed documentation, please refer to the GitHub repository (https://github.com/mpecchi/gcms_data_analysis). For examples of file formatting, the reader should refer directly to the “RCS/data” folder in said GitHub repository.An overview of the algorithm is provided in Table 1. The algorithm on the left is divided into steps that perform sequential tasks, highlighting the inputs and outputs the code requires or produces in each part. Main libraries and functions used by the algorithm are described in the relevant sections. Details of each part are given in the following subsections.
| Part | Action | Input | Output | Methods |
|---|---|---|---|---|
| 1 | Load input files | “files_info.xlsx” Files (GC results), calibration files “compounds_properties” | Data is stored in the project class | Pandas |
| 2 | Create compounds properties (if not available) | “classifications_codes_fractions” | “compounds_properties” | pubchempy, rdikit, frag. Algoritm23 |
| 3 | Apply calibration to files | Files, calibrations | Updated files with concentrations | Pandas, numpy |
| 4 | Create samples from files | Files | Samples (average), samples (standard deviation) | Pandas, numpy |
| 5 | Create multi-file (or sample) reports | Files (or samples) | Report: one parameter (conc, area) for all files or samples | Pandas |
| 6 | Create multi-file (or sample) aggregated reports (by functional groups) | Files (or samples), compounds properties with functional group splitting | Report: one aggregated parameter (conc, area) for all files or samples | Pandas |
| 7 | Plot reports or aggregated reports | Report or aggregated report | Figures | Matplotlib, pandas |
2.1 Part 1: load (or create) input files
The user needs to firstly specify the path to the project folder where the input files are located and where the output files will be created.The algorithm can then load (or create) the following input files:
(1) “files_info.xlsx” file: a table that contains, for each “file” (intended as a single experiment or replicate), the information provided below as columns.
• “filename”: the name of the GC-MS semi-quantitative data table file (the report of the GC-MS analysis of the sample that contains the identified compounds' names and their peak area).
• “derivatized”: Boolean indicating whether the sample was derivatized or not.
• “dilution_factor”: a int or float that indicates the dilution factor of the sample (in x-folds, e.g., a 10-fold dilution would only report “10”). If no calibration is used, the only available comparison is between absolute area for the same identified compound across samples (since area is directly proportional to concentration but with an unknown factor for each compound). If different samples are obtained with the same methodology but need to be analyzed at different dilutions (for example to protect the GC-MS detector from saturation), specifying such dilution factor allows the code to compute the “area_if_undiluted” (measured area multiplied by the dilution factor) of compounds in each sample that can then be used to compare “relative concentrations” (not absolute ones). If unspecified, the default value is 1. The same approach is used to compute “conc_vial_if_undiluted_mg_L′′ if a calibration is available.
• “calibration_file”: The name of the Calibration file to use for the sample, if available (in the example “calibration” and “deriv_calibration”. This is used to load the right calibration file to compute concentration for the sample.
• “total_sample_conc_in_vial_mg_L”: The measured sample concentration in mgsample per Lsolvent where mgsample indicates the mg of undiluted sample (without any solvent) in the GC-MS vial (must be obtained either gravimetrically or via mass balances). If a calibration is available, the code computes the concentration of each identified compound in the sample. The sum of the identified fractions gives the overall sample fraction; it is compared with the measured sample concentration to calculate the detected sample fraction, an indication of how much of the sample was identifiable through GC-MS analysis. If not available, default is set to 1.
• “sample_yield_on_feedstock_basis_fr”: If the sample is produced from a feedstock with a known yield, the user can include such yield (ex.: the bio-oil yield on feedstock basis). This is used to compute single compound yields on feedstock basis. If no yield is specified, 1 is used as default and the results can be ignored.
(2) One semi-quantitative data table for each sample, usually as given by the GC-MS. Files must be named “samplename_replicatenumber” (with samplename a string, and replicatenumber an integer). To make this software agnostic to the type of GC-MS used, the user must specify the number of rows at the top of the file that must be skipped (typically if the file contains a summary of the GCMS method before the peak table) and the exact name for the columns that, for each identified peak, contain information about:
• Retention time of peaks.
• Names of the identified compounds (not necessarily in the IUPAC form).
• Area of each peak.
• Height of each peak.
(3) Calibration files, each sample can refer to a different calibration file by specifying it in the “files_info” table, that contains:
• The name of each calibrated compound.
• The concentrations (in our example, we use ppm on a mass per volume basis, mg L−1) and of intensity (area) for each calibrated compound. Columns must be named “PPM #” and “Area #” (# = incremental natural number) for each investigated concentration. The calculated results will match the provided concentrations (ppm on mass or volume basis, or on mass/volume, depending on the users' calibration units).
(4) “classification_codes_fractions.xlsx”: the list of Functional Group Classification Code Fractions: a list of names of the functional group, the SMARTS codes (SMILES arbitrary target specification) associated, and their molecular weight. This information is used to automatically fragment each identified compound into its functional groups also calculating the mass fraction of said functional groups in the identified compound. The file can be modified by the user depending on the functional groups that are of interest (details are given in Section 2.2.1).
(5) “compounds_properties.xlsx” or “deriv_compounds_properties.xlsx”: see next subsection.
2.2 Part 2: build compound's property file
The code can create (or load, if available) a database that contains the properties of all compounds identified in all files and in all calibrations, the “compounds_properties”. These files contain information about IUPAC name, molecular formula, canonical SMILES, molecular weight, elemental composition, etc. for every compound. Data are retrieved from the PubChem website21 using PubChemPy,20 a Python package to automatically access the PubChem database. PubChemPy needs internet access to perform its operations and can retrieve the full set of properties of a chemical compound using any of its synonyms.If a file is derivatized, identified compounds in the semi-quantitative data table should be in their derivatized form (ex.: “phenol, TMS derivative”); the non-derivatized form is obtained splitting the name at the last comma (ex.: “phenol”). Then, the code queries the PubChem database through PubChemPy using the non-derivatized form to retrieve chemical information. If at least one sample is derivatized, a second database (named “deriv_compounds_properties”) with only compounds found in derivatized samples is created or loaded. If a derivatized calibration file is provided (in the example “deriv_calibration”), compound names must be given in their underivatized form.
After these chemical properties are retrieved, each compound is categorized by its functional groups and corresponding their mass fraction (see Section 2.2.1 for details). This operation relies on an heuristic algorithm developed in Python;23 its code is available at ref. 27. It also requires a list of the functional groups to be searched, the “classification_codes_fractions.xlsx” file, specified in Section 2. In the present work, the simple fragmentation option of the heuristic algorithm is used.
An alternative approach (though extremely time consuming if performed manually) is to calculate the mass fraction of each functional group present in the compound and use these mass fractions to split the area or the concentration of each molecule into weighted averages of the different functional groups present, and then compute the aggregated value for all compounds present in the sample. This has the advantage of fully accounting for all functional groups in each sample. This approach uses eqn (1) to compute the aggregated concentration of each functional group (Cfg) in the sample, obtained as the sum over all n identified compounds in the sample of the concentration of the compound (Ci) multiplied by the mass fraction of the considered functional group in the compound itself (mffg,i). This equally applies to area, concentration, and yield, though of course only concentration and yield data would be comparable across different laboratories.
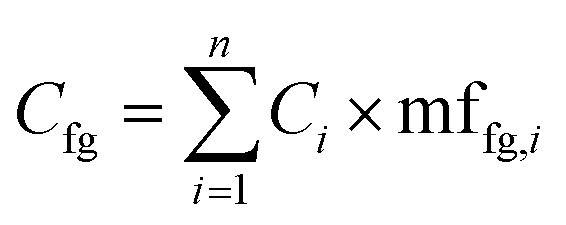 | (1) |
This latter approach is automated in the present tool. The fragmentation into functional groups relies on the fragmentation algorithm developed by ref. 23, available at ref. 27. This heuristic algorithm performs the automatic fragmentation of molecules (relying on their canonical SMILES) into subunits (patterns), specified by their SMARTS code, (SMILES arbitrary target specification). This is done via heuristic group prioritization; heuristically determined descriptors are used to sort the patterns of the fragmentation scheme, parent–child group prioritization (patterns contained in other larger patterns are searched last), and adjacent group search (subsequent matches must be adjacent to the already fragmented structure to avoid incomplete group assignment). The algorithm provides two options: a simple fragmentation and a complete fragmentation.
In the present paper, the code loads the “classification_codes_fractions.xlsx” file described in Section 2.1, that contains a list of names of the functional group, their SMARTS codes, and their molecular weights. The smarts codes are used by the automatic fragmentation algorithm developed by ref. 23 to count the instances of each functional group, and their molecular weights are used to compute their mass fractions. A brief example of functional group SMARTS and mass fractions is provided in Table 2; when more variants (ex. ester and ester_1) of the same functional group are available (with slightly different molecular mass, mostly due to H atoms), the code considers them to be of the same functional group (they are named as the general name of the functional group followed by “_#”, with # = incremental natural number). An example of automatic fragmentation for selected chemical compounds with different functional groups is shown in Table 3.
| Functional group code | SMARTS code | Mass fraction | Figure code | |
|---|---|---|---|---|
| Ester | [CH0](![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O[CH3] O)O[CH3] |
59.044 | a |
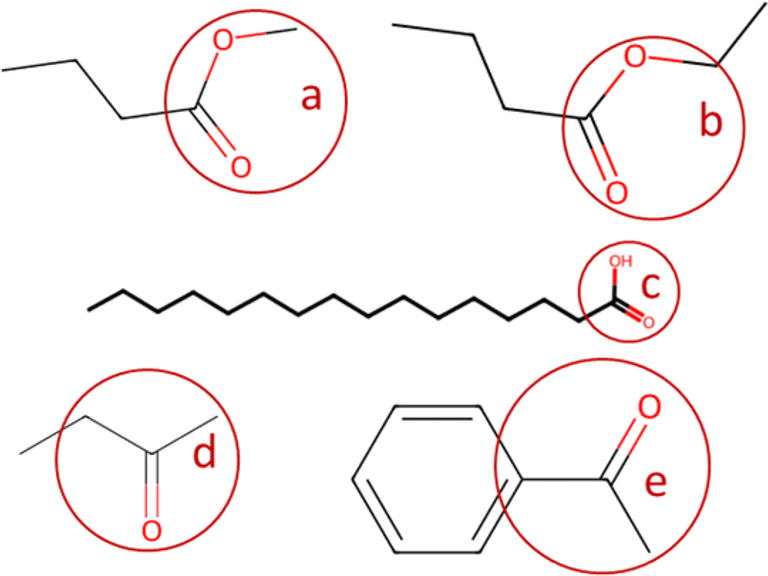
|
| Ester_1 | [CH0](O)O[CH2] |
58.036 | b | |
| Carboxyl | [CH0](O)O |
45.017 | c | |
| Ketone | [CH2]C(O)[CH3] |
58.08 | d | |
| Ketone_1 | [CH3]C(O)[C] |
55.056 | e |
| IUPAC name | C-aliph | C-arom | N-arom | O-arom | Alcohol | Aldehyde | Carboxyl | Ester | Ether | Ketone |
|---|---|---|---|---|---|---|---|---|---|---|
| Tetradecanoic acid | 0.80 | — | — | — | — | — | 0.20 | — | — | — |
| Benzene-1,4-diol | — | 0.69 | — | — | 0.31 | — | — | — | — | — |
| 3-Hydroxybenzaldehyde | — | 0.62 | — | — | 0.14 | 0.24 | — | — | — | — |
| Ethenyl hexanoate | 0.60 | — | — | — | — | — | — | 0.40 | — | — |
| 1-(3-Hydroxyphenyl)ethanone | — | 0.47 | — | — | 0.12 | — | — | — | — | 0.40 |
| 4-Butoxyphenol | 0.26 | 0.39 | — | — | 0.10 | — | — | — | 0.25 | — |
| 2-Methyl-1-benzofuran-5-ol | 0.10 | 0.68 | — | 0.11 | 0.11 | — | — | — | — | — |
| Phenol | — | 0.82 | — | — | 0.18 | — | — | — | — | — |
| 2-Methylpyrazine | 0.16 | 0.54 | 0.30 | — | — | — | — | — | — | — |
| Benzoic acid | — | 0.63 | — | — | — | — | 0.37 | — | — | — |
2.3 Part 3: apply calibrations to files
For each file specified in the “files_info” table, its GC-MS semi-quantitative data table is loaded, together with the relevant calibration file (also specified in the “files_info” table). The semi-quantitative table is updated – the original information is maintained, except all unidentified peaks are grouped under one peak – with all the results calculated by the algorithm.The updated semi-quantitative table for files contain, for each identified compound:
• Its IUPAC name, molecular weight, and functional group mass fractions.
• The original peak height and peak area as measured by the GC-MS.
• The computed “area_if_undiluted” (using the dilution factor, if provided, see Section 2.1).
• The computed concentrations (“conc_vial _mg_L” and “conc_vial_if_undiluted_mg_L”) obtained by applying the calibration to the measured areas. This step requires a calibration file, if not available, nan values (not a number) are returned instead. The code also has the possibility of using a semi-quantitative calibration based on Tanimoto and molecular weight similarity to apply a similar compound's calibration to compounds that are not calibrated; details are provided in Section 2.3.1.
• The “compound_used_for_calibration” column contains information about the calibration: if a calibration for the compound was available, the column indicates “self” (meaning the compound was calibrated using “itself”). If a calibration based on a similarity index is applied, the name of the compound used as calibration surrogate, its similarity index with the compound that is semi-calibrated, and their difference in molecular weight are instead reported in the column.
• The computed mass fraction of compounds on a sample basis (“fraction_of_sample_fr”). The “fraction_of_sample_fr” is obtained by diving the concentration of the compound in the GC-MS vial (obtained by applying the calibration) by the “total_sample_conc_in_vial_mg_L” (that must be computed by the operator and included in the “files_info” table, see Section 2.1). This requires a calibration file and the sample concentration in the vial. The sum of all compound fractions gives the total amount of sample that can be identified by GC-MS.
• The computed mass fraction of compounds on a feedstock basis (“fraction_of_feedstock_fr”). The yield of the compound on feedstock basis (if provided). This requires a calibration file, the net amount of sample in the vial, and the yield of sample on feedstock basis (“sample_yield_on_feedstock_basis_fr”).
The present code allows for semi-calibration (if the option is selected); for compounds without a calibration curve, their Tanimoto similarity with all calibrated compounds is evaluated. The calibrated compound with the highest similarity is selected (if more compounds share the same similarity, as happens for compounds of the same class, the compound with the closest molecular weight is selected). The Python package rdkit35 is used to convert canonical SMILES into molecular fingerprints and to compute the Tanimoto similarity among compounds. The user can change the minimum similarity threshold and maximum molecular weight difference to apply the semi-calibration to each non-calibrated compound. If no calibrated compound meets the criteria or if the user sets the semi-calibration option to false, the compounds for which no calibration compounds are available will have all concentrations set to not-a-number (though its areas are persevered). An example of similarity table for tetradecanoic acid as compared to several compounds is provided in Table 4.
O)O) and several compounds
| Compound | SMILES | Tanimoto similarity | Molecular weight diff. [g mol−1] |
|---|---|---|---|
| Hexadecanoic acid | CCCCCCCCCCCCCCCC(O)O |
1 | 28.05 |
| Octadecanoic acid | CCCCCCCCCCCCCCCCCC(O)O |
1 | 56.13 |
| (z)-Octadec-9-enoic acid | CCCCCCCCCCCCCCCCCC(O)O |
0.74 | 54.13 |
| (9z,12z)-Octadeca-9,12-dienoic acid | CCCCCCCCCCCCCCCCCC(O)O |
0.65 | 52.03 |
| Oxacycloheptadecan-2-one | C1CCCCCCCC(O)OCCCCCCC1 |
0.03 | 26.04 |
 | (2) |
This error has been assessed for all combinations of calibrated compounds in our calibration dataset in our GC-MS (details are provided in Section 3); this dataset is not the one provided in the GitHub repository in the “example” subdirectory as its size is not appropriate for a tutorial. The dataset is included in the RCSdata subfolder in the GitHub repo. The data set comprises 89 compounds for which a ≥4 points calibration (up to 6 points) is available in the form of mg L−1vs. detected area; this results in 3827 combinations. For each combination of compounds, the Tanimoto similarity and the molecular weight difference is also computed. Fig. 1 plots the average error as a function of the Tanimoto similarity of compounds and their molecular weight difference. The error decreases with the increasing Tanimoto similarity, while there seems to be no marked effect of molecular weight difference. Selecting a Tanimoto similarity threshold of 0.4 minimizes the risk of errors that are above one order of magnitude, while a similarity of 0.7 avoids this almost entirely (at least in our dataset). The percentage error threshold of 100% may seem unreasonably high, however the alternative would be to simply ignore all the compounds for which no calibration is available which also implies a large data loss. The selection of a Tanimoto similarity threshold that avoids unrealistic overestimation of concentrations (underestimations are less of a concern, since the alternative would be to ignore the compound entirely) improves the quality of GC-MS results. A similarity threshold of 0.4 was arbitrarily chosen as the default threshold in the code based on these results, with a molecular weight difference a threshold of 100 atomic mass units set as the default.
 | ||
| Fig. 1 Average error between calibration curves of combination of compounds as a function of their Tanimoto similarity. The molecular weight difference of the compounds is reported in the colormap. The horizontal like indicates 100% error, the vertical line reports the default similarity threshold adopted in the code. | ||
The user is encouraged to run a similar analysis on their calibration database to assess what thresholds are most appropriate for their use case.
2.4 Create samples from files (replicates)
After each file is processed, those files that belong to the same sample are used to compute average values and standard deviations for each sample (in the example, files “A_1”, “A_2”, and “A_3” are replicates of sample “A”).2.5 Part 4: build multi-file and sample reports
The code can create multi-file and multi-sample report tables with a list of all identified compounds as the rows, and the results for each sample and for each parameter (area, concentrations, and yield) as the columns.2.6 Part 6: build multi-file and sample aggregated reports
The tool can also produce aggregated reports by functional group for each sample. Aggregated reports have samples as rows and aggregated results by functional group as columns, obtained as the weighted sum of each parameter, (area, concentration) where the weights are the functional group mass fractions of each compound. Details of the aggregation by functional group procedure are given in Section 2.2.1.2.7 Part 7: saving and displaying results as plots
The module automatically saves all tabulated outputs by default in dedicated subfolders in a “output” subdirectory. It also provides a straightforward way to directly plot the content of multi-file and multi-sample reports and aggregated reports. With files (as opposed to samples), plots do not show error bars (as each file is analyzed alone). For sample, each value is reported as the average and standard deviation of the files that constitute the sample. For details, we refer the reader to the documentation and to the example in the GitHub repository.3 Test cases of three bio-oil samples
This case study describes real results for 3 bio-oil samples that belong to a larger experimental campaign that will be discussed in a separate publication. The context of the experiments and the implications of the results are only discussed here to showcase the capabilities of the proposed tool. The code and the data required to obtain the results here present are publicly available in the GitHub subfolder “RCSdata” (of course the “gcms_data_analysis” package must be installed). For conciseness, only a small subset of the results is discussed here.This case differs from the main example/tutorial in the GitHub repository, as the data presented here is complete and accurate. The GitHub example has the task of showing how to run the module and to perform automatic tests on the code to ensure consistency and thus contains a subset of the data presented here that has been manipulated to make it small and manageable.
Bio-oil samples were produced by dichloromethane extraction of hydrochars, which were in turn produced via hydrothermal carbonization at 250 °C for holding time of 1 hour using food waste (named A and Ader in the underivatized and derivatized form, respectively) and cellulose (named B) as feedstock with a 15 and 20% biomass to water ratio. The preparation method for hydrochar production is described in ref. 36. The solvent extraction – performed using an accelerated solvent extractor (ASE Dionex 350) – uses 0.5 g hydrochar and ∼25 mL of dichloromethane per sample, with an extraction temperature of 100 °C at ∼100 bar. While samples A and B are analyzed after simple dilution, Ader is firstly dried under vacuum, mixed with excess BSTFA, heated at 70 °C for 1 hour, vacuum dried a second time to remove BSTFA, and then reconstituted with dichloromethane before the analysis. The sample are analyzed on a Shimadzu GC 2010 Plus Gas Chromatograph-Mass Spectrometer (GC-MS), equipped with a Restek Rtx-5Sil MS 30 m fused silica column and the semi-quantitative data tables for each sample replicate, the “files_info.xlsx” table (with dilution factor, sample mass in the vial, and name of calibration files), a non-derivatized and a derivatized calibration files, and the “classifications_codes_fractions.xlsx” files are provided in the “RCSdata” folder.
Table 5 shows a part of the updated semi-quantitative data table results for a single sample, A_1. The “area_if_undiluted” is computed considering the sample dilution of 25× reported in the “files_info” table. The “conc_vial_mg_L” column contains the concentration of each compound in the GC vial, calculated by applying the calibration (and a semi-calibration with Tanimoto similarity threshold of 0.4 and molecular weight difference threshold of 100). The total concentration of sample in the vial (provided in the “files_info” table) is used to compute the fraction of each compound in the bio-oil sample, shown in the “fraction_of_sample_fr” column in g g−1. The “fraction_of_feedstock_fr” column in g g−1 contains the yield of each compound on a feedstock basis, computed using the sample yield vial (provided in the “files_info” table). In this example, the “fraction_of_feedstock_fr” contains the yield of each compound per unit mass of initial biomass.
| A_1 | iupac_name | retention_time | Area | area_if_undiluted | conc_vial_if_undiluted_mg_L | fraction_of_sample_fr | fraction_of_feedstock_fr | compound_used_for_calibration |
|---|---|---|---|---|---|---|---|---|
| Unidentified | Unidentified | 6.025 | 373![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 897 897 |
9347425 |
n.a. | |||
| Tetradecanoic acid | Tetradecanoic acid | 36.163 | 44389 |
1109725 |
589.5376 | 0.04211 | 0.021055 | Self |
| Oxacycloheptadecan-2-one | Oxacycloheptadecan-2-one | 40.052 | 15068 |
376700 |
n.a. | |||
| n-Hexadecanoic acid | Hexadecanoic acid | 40.492 | 1878180 |
46954500 |
1651.359 | 0.117954 | 0.058977 | Self |
| 9,12-Octadecadienoic acid (z,z)- | (9z,12z)-Octadeca-9,12-dienoic acid | 43.847 | 1456119 |
36402975 |
3279.7 | 0.234264 | 0.117132 | Self |
| Oleic acid | (z)-Octadec-9-enoic acid | 43.986 | 6379752 |
1.59 × 108 | 2840.463 | 0.20289 | 0.101445 | (e)-Octadec-9-enoic acid (sim = 1.0; dwt = 0) |
| Octadecanoic acid | Octadecanoic acid | 44.402 | 1635947 |
40898675 |
1648.929 | 0.117781 | 0.05889 | Self |
| 9-Octadecenamide, (z)- | (z)-Octadec-9-enamide | 44.733 | 62240 |
1556000 |
635.495 | 0.045393 | 0.022696 | (e)-Octadec-9-enoic acid (sim = 0.7; dwt = 1) |
| 13-Docosenamide, (z)- | (z)-Docos-13-enamide | 47.947 | 21557 |
538925 |
621.2956 | 0.044378 | 0.022189 | (e)-Octadec-9-enoic acid (sim = 0.7; dwt = 55) |
For each file, an updated report is produced. These results are all collected and summarized into report files for each parameter: Table 6 that shows the first 10 rows of the file report with “conc_vial_mg_L” as the parameter. The algorithm aligns derivatized and non-derivatized instances of the same compound to simplify comparison. Please note that if a non-derivatized and a derivatized calibration are available, the results can be compared, but areas should not be used to describe relative concentrations of a derivatized and underivatized compound. Additional reports with all other parameters can be produced and saved in the “files report” subfolder.
| conc_vial_mg_L | A_1 | A_2 | A_3 | Ader_1 | Ader_2 | Ader_3 | B_1 | B_2 | B_3 |
|---|---|---|---|---|---|---|---|---|---|
| 4-Oxopentanoic acid | 0 | 0 | 0 | 0 | 0 | 0 | 683.4494 | 776.1829 | 768.8858 |
| (9z,12z)-Octadeca-9,12-dienoic acid | 131.188 | 125.3808 | 86.76279 | 31.36777 | 36.81299 | 27.92726 | 0 | 0 | 0 |
| (z)-Octadec-9-enoic acid | 113.6185 | 113.8273 | 84.92259 | 21.66908 | 24.27344 | 19.93824 | 0 | 0 | 0 |
| Furan-2-carbaldehyde | 0 | 0 | 0 | 0 | 0 | 0 | 72.39856 | 0 | 0 |
| Hexadecanoic acid | 66.05436 | 61.11673 | 47.13392 | 27.62319 | 27.3815 | 19.56924 | 29.1569 | 31.45626 | 32.20709 |
| Octadecanoic acid | 65.95717 | 59.31563 | 53.60064 | 14.2637 | 17.55913 | 11.2483 | 35.62859 | 37.75122 | 37.75725 |
| 3-Methylcyclopentane-1,2-dione | 0 | 0 | 0 | 0 | 0 | 0 | 36.33573 | 7.318511 | 7.234667 |
| 5-(Hydroxymethyl)furan-2-carbaldehyde | 0 | 0 | 0 | 0 | 0 | 0 | 31.55445 | 34.74618 | 35.66275 |
| Ethenyl hexanoate | 0 | 0 | 0 | 0 | 0 | 0 | 33.06538 | 0 | 0 |
| 5-Methylfuran-2-carbaldehyde | 0 | 0 | 0 | 0 | 0 | 0 | 25.57821 | 27.62023 | 27.29056 |
The same reports can be produced using samples instead of files (average and standard deviation of replicates instead of single replicates). In this case, two reports are produced, one with average and one with standard deviation results. Table 7 contains the first 10 rows for average and standard deviation reports for samples using “conc_vial_mg_L” as the parameters (they are directly obtained from Table 5).
| conc_vial_mg_L | A | Ader | B | A | Ader | B |
|---|---|---|---|---|---|---|
| Average | Standard deviation | |||||
| 4-Oxopentanoic acid | 0 | 0 | 742.8394 | 51.56245 | ||
| (9z,12z)-Octadeca-9,12-dienoic acid | 114.4439 | 32.03601 | 0 | 24.14771 | 4.480395 | |
| (z)-Octadec-9-enoic acid | 104.1228 | 21.96026 | 0 | 16.6282 | 2.182219 | |
| Octadecanoic acid | 59.62448 | 14.35704 | 37.04568 | 6.184055 | 3.156452 | 1.227244 |
| Hexadecanoic acid | 58.10167 | 24.85798 | 30.94008 | 9.813955 | 4.581773 | 1.589258 |
| 5-(Hydroxymethyl)furan-2-carbaldehyde | 0 | 0 | 33.98779 | 2.156596 | ||
| Tridecane-5,8-dione | 0 | 0 | 26.88192 | |||
| 5-Methylfuran-2-carbaldehyde | 0 | 0 | 26.82967 | 1.096256 | ||
| Furan-2-carbaldehyde | 0 | 0 | 24.13285 | 41.79933 | ||
The concentrations for each compound in Tables 5 and 6 can be used, together with the mass fraction of each functional group in such compound, to compute aggregated results for each functional group in each sample (using eqn (1), described in Section 2.2.1). Table 8 shows the aggregated results for each functional group in each sample in terms of for sample fraction [g g−1]; additional aggregated reports for the other parameters (area, injection concentration, yield) are also produced by the code but are not reported here.
| conc_vial_mg_L | A | Ader | B | A | Ader | B |
|---|---|---|---|---|---|---|
| Average | Standard deviation | |||||
| C-aliph | 0.615577 | 0.827408 | 0.078768 | 0.136911 | 0.118395 | 0.012261 |
| Ketone | 0 | 0 | 0.168349 | 0 | 0 | 0.016044 |
| Carboxyl | 0.116397 | 0.140476 | 0.113552 | 0.016592 | 0.021211 | 0.007693 |
| C-arom | 0 | 0 | 0.039598 | 0 | 0 | 0.01276 |
| Ester | 0.002009 | 0.025475 | 0.006185 | 0.001229 | 0.002409 | 0.003682 |
| Alcohol | 0.001184 | 0.014931 | 0.00717 | 0.000726 | 0.001412 | 0.000767 |
| Aldehyde | 0 | 0 | 0.008519 | 0 | 0 | 0.005208 |
| O-arom | 0 | 0 | 0.006789 | 0 | 0 | 0.003154 |
| N-aliph | 0.002403 | 0 | 7.73 × 10−5 | 0.002689 | 0 | 0 |
| O-aliph | 0.002399 | 0 | 0.001148 | 0.002685 | 0 | 0.001031 |
| Ether | 0 | 0 | 0.001016 | 0 | 0 | 0.000228 |
Each report (an example is given by Tables 6–8) can be plotted by a dedicated function. Examples are shown in Fig. 2.
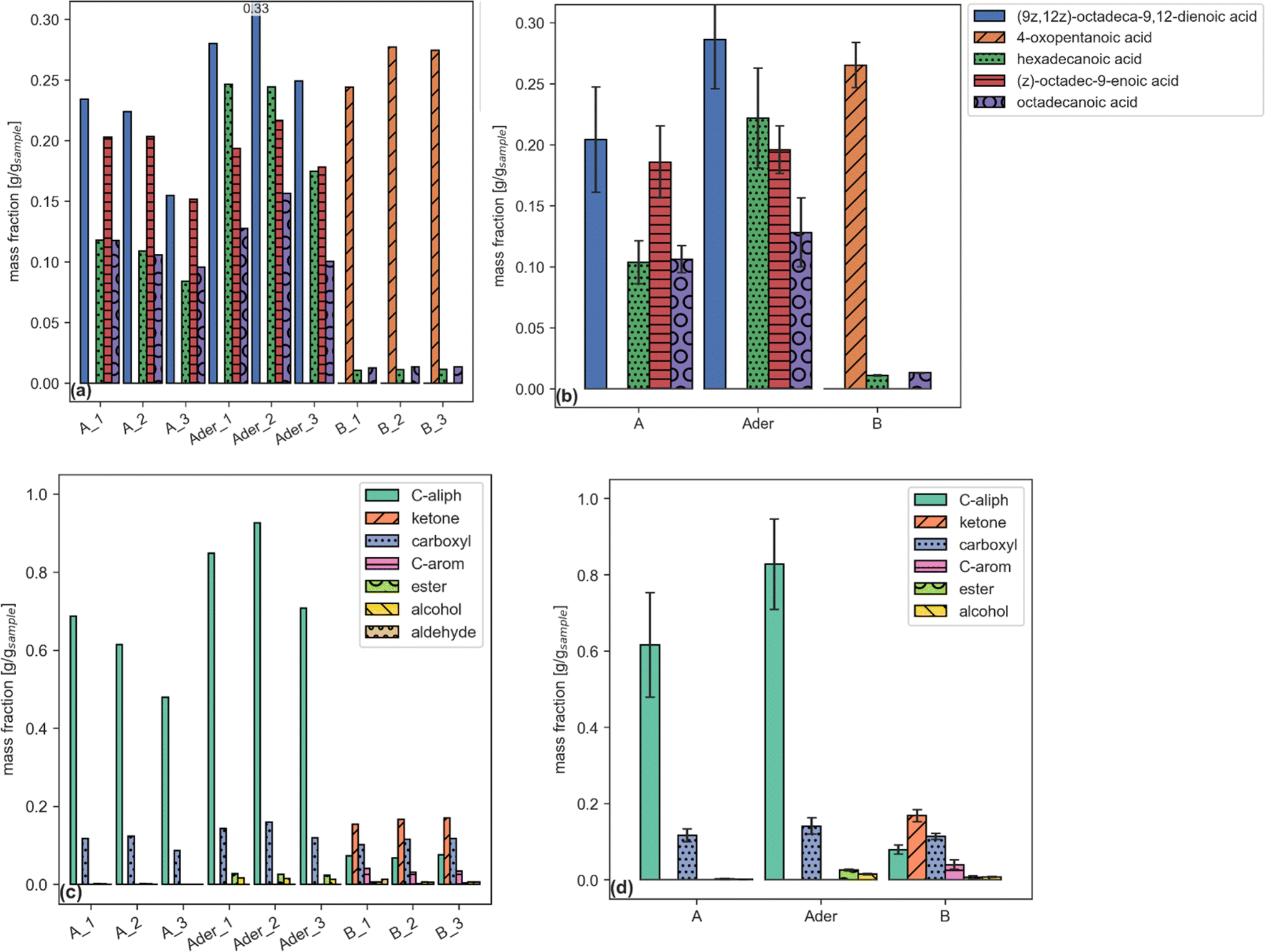 | ||
| Fig. 2 Single compound (a, b) and functional group aggregated (c, d) plots reporting sample fraction for files (a, c) and samples (b, d). (a and b) share the same legend. | ||
4 Conclusions
The lack of open-source tools to automate the analysis of large datasets from Gas Chromatography coupled with Mass Spectrometry (GC-MS) of biofuels results in time-consuming manual analyses of such data that employ sub-optimal methodologies, are difficult to replicate, and present an increased risk of error. We developed a Python package to automate GC-MS data analysis of complex, heterogeneous organic mixtures. The code retrieves properties for each identified chemical from PubChem, applies calibration and semi-calibration using Tanimoto similarity index and molecular weight similarity, splits each identified compound into its functional groups, and handles both derivatized and non-derivatized samples. Replicates of the same samples are automatically combined to compute averages and standard deviations. The outputs include single and multiple sample reports with area, concentrations and yield on feedstock basis and aggregated reports based on the cumulative fraction of each functional group in each analyzed sample. This tool reduces GC-MS analysis time from hours days to minutes, avoiding human errors and promoting standardization and best practices for GC-MS data handling. The module is publicly available on PyPI at https://pypi.org/project/gcms-data-analysis/and on GitHub at https://github.com/mpecchi/PyGCMS with MIT license.Data availability
The code and the data required to obtain the results here present are publicly available in the GitHub subfolder “RCSdata” at https://github.com/mpecchi/gcms_data_analysis (the “gcms_data_analysis” package must be installed https://pypi.org/project/gcms-data-analysis/).Conflicts of interest
There are no conflicts of interest to declare.Biofuels play a pivotal role in reducing our carbon footprint and transitioning towards a more sustainable energy future. However, the reliance on outdated proprietary software for GC-MS (Gas Chromatography-Mass Spectrometry) analysis not only wastes researchers' time but also hinders comparability across research groups. Our open-source Python tool directly addresses these challenges, ensuring faster, more accurate, and standardized analysis. By democratizing access through open-source software, we also take a stand against knowledge inequality, aligning with the UN's Sustainable Development Goal 10 (Reduced Inequality). Furthermore, our tool contributes to Goal 7 (Affordable and Clean Energy) and Goal 9 (Industry, Innovation, and Infrastructure) as it promotes the adoption of sustainable biofuels and encourages innovation in clean energy research.
Acknowledgements
This work was supported by the National Science Foundation CBET under grant numbers 1933071 and 2031710. The authors thank James Li Adair for the constructive criticism on the manuscript; Alessandro Cascioli for the help on Python package structuring; and Alessandro Cascioli, James Li Adair, and Madeline Karod for being early testers of the code.References
- Y. Lu, G. S. Li, Y. C. Lu, X. Fan and X. Y. Wei, Analytical Strategies Involved in the Detailed Componential Characterization of Biooil Produced from Lignocellulosic Biomass, Int. J. Anal. Chem., 2017, 9298523 Search PubMed . Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5745679/.
- K. Sharma, T. H. Pedersen, S. S. Toor, Y. Schuurman and L. A. Rosendahl, Detailed Investigation of Compatibility of Hydrothermal Liquefaction Derived Biocrude Oil with Fossil Fuel for Corefining to Drop-in Biofuels through Structural and Compositional Analysis, ACS Sustainable Chem. Eng., 2020, 8(22), 8111–8123 CrossRef CAS . Available from: https://doi.org/10.1021/acssuschemeng.9b06253.
- V. Sugumaran, S. Prakash, E. Ramu, A. K. Arora, V. Bansal and V. Kagdiyal, et al., Detailed characterization of bio-oil from pyrolysis of non-edible seed-cakes by Fourier Transform Infrared Spectroscopy (FTIR) and gas chromatography mass spectrometry (GC–MS) techniques, J. Chromatogr. B, 2017, 1058, 47–56 CrossRef CAS PubMed . Available from: https://www.sciencedirect.com/science/article/pii/S157002321730079X.
- J. Grams, Chromatographic analysis of bio-oil formed in fast pyrolysis of lignocellulosic biomass, Rev. Anal. Chem., 2020, 39(1), 65–77 CrossRef CAS . Available from: https://www.degruyter.com/document/doi/10.1515/revac-2020-0108/html.
- H. Sudibyo, M. Pecchi and J. W. Tester, Experimental-based mechanistic study and optimization of hydrothermal liquefaction of anaerobic digestates, Sustain. Energy Fuels, 2022, 6(9), 2314–2329 RSC . Available from: http://xlink.rsc.org/?DOI=D2SE00206J.
- Y. Wang, Y. Han, W. Hu, D. Fu and G. Wang, Analytical strategies for chemical characterization of bio-oil, J. Sep. Sci., 2020, 43(1), 360–371 CrossRef CAS PubMed . Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jssc.201901014.
- E. Heracleous, M. Vassou, A. A. Lappas, J. K. Rodriguez, S. Chiaberge and D. Bianchi, Understanding the Upgrading of Sewage Sludge-Derived Hydrothermal Liquefaction Biocrude via Advanced Characterization, Energy Fuels, 2022, 36(19), 12010–12020 CrossRef CAS PubMed . Available from: https://doi.org/10.1021/acs.energyfuels.2c01746.
- Y. Kostyukevich, M. Vlaskin, A. Zherebker, A. Grigorenko, L. Borisova and E. Nikolaev, High-Resolution Mass Spectrometry Study of the Bio-Oil Samples Produced by Thermal Liquefaction of Microalgae in Different Solvents, J. Am. Soc. Mass Spectrom., 2019, 30(4), 605–614 CrossRef CAS PubMed . Available from: https://doi.org/10.1007/s13361-018-02128-9.
- Z. Zhu, X. Guo, L. Rosendahl, S. S. Toor, S. Zhang and Z. Sun, et al., Fast hydrothermal liquefaction of barley straw: reaction products and pathways, Biomass Bioenergy, 2022, 165, 106587 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0961953422002495.
- K. Kohansal, K. Sharma, M. S. Haider, S. S. Toor, D. Castello and L. A. Rosendahl, et al., Hydrotreating of bio-crude obtained from hydrothermal liquefaction of biopulp: effects of aqueous phase recirculation on the hydrotreated oil, Sustain. Energy Fuels, 2022, 6(11), 2805–2822 RSC . Available from: https://rsc.66557.net/en/content/articlelanding/2022/se/d2se00399f.
- E. Panisko, T. Wietsma, T. Lemmon, K. Albrecht and D. Howe, Characterization of the aqueous fractions from hydrotreatment and hydrothermal liquefaction of lignocellulosic feedstocks, Biomass Bioenergy, 2015, 74, 162–171 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0961953415000124.
- S. R. Villadsen, L. Dithmer, R. Forsberg, J. Becker, A. Rudolf and S. B. Iversen, et al., Development and Application of Chemical Analysis Methods for Investigation of Bio-Oils and Aqueous Phase from Hydrothermal Liquefaction of Biomass, Energy Fuels, 2012, 26(11), 6988–6998 CrossRef CAS . Available from: https://doi.org/10.1021/ef300954e.
- I. Leonardis, S. Chiaberge, T. Fiorani, S. Spera, E. Battistel and A. Bosetti, et al., Characterization of Bio-oil from Hydrothermal Liquefaction of Organic Waste by NMR Spectroscopy and FTICR Mass Spectrometry, ChemSusChem, 2013, 6(1), 160–167 CrossRef CAS PubMed . Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cssc.201200314.
- R. B. Madsen, M. M. Jensen, A. J. Mørup, K. Houlberg, P. S. Christensen and M. Klemmer, et al., Using design of experiments to optimize derivatization with methyl chloroformate for quantitative analysis of the aqueous phase from hydrothermal liquefaction of biomass, Anal. Bioanal. Chem., 2016, 408(8), 2171–2183 CrossRef CAS PubMed . Available from: https://doi.org/10.1007/s00216-016-9321-6.
- M. S. Haider, D. Castello and L. A. Rosendahl, Two-stage catalytic hydrotreatment of highly nitrogenous biocrude from continuous hydrothermal liquefaction: a rational design of the stabilization stage, Biomass Bioenergy, 2020, 139, 105658 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0961953420301926.
- J. Han, X. Li, S. Kong, G. Xian, H. Li and X. Li, et al., Characterization of column chromatography separated bio-oil obtained from hydrothermal liquefaction of Spirulina, Fuel, 2021, 297, 120695 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0016236121005718.
- S. O'Callaghan, D. P. De Souza, A. Isaac, Q. Wang, L. Hodkinson and M. Olshansky, et al., PyMS: a Python toolkit for processing of gas chromatography-mass spectrometry (GC-MS) data. Application and comparative study of selected tools, BMC Bioinf., 2012, 13(1), 115 CrossRef PubMed . Available from: https://doi.org/10.1186/1471-2105-13-115.
- D. Davis-Foster, PyMassSpec, PyMassSpec, 2024, available from: https://github.com/PyMassSpec/PyMassSpec.
- J. Yang, Q. He, K. Corscadden and H. Niu, The impact of downstream processing methods on the yield and physiochemical properties of hydrothermal liquefaction bio-oil, Fuel Process. Technol., 2018, 178, 353–361 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0378382018307574.
- Matt Swain, PubChemPy, 2017, available from: https://pubchempy.readthedocs.io/en/latest/guide/introduction.html Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He and S. He, et al., PubChem 2023 update, Nucleic Acids Res., 2023, 51(D1), D1373–D1380 CrossRef PubMed . Available from: https://doi.org/10.1093/nar/gkac956.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS . Available from: https://doi.org/10.1021/ci00057a005.
- S. Müller, Flexible heuristic algorithm for automatic molecule fragmentation: application to the UNIFAC group contribution model, J. Cheminf., 2019, 11(1), 57 Search PubMed . Available from: https://doi.org/10.1186/s13321-019-0382-3.
- Daylight Chemical Information Systems, Inc., SMARTS - A Language for Describing Molecular Patterns, 1997, available from: https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html.
- D. Bajusz, A. Rácz and K. Héberger, Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?, J. Cheminf., 2015, 7(1), 20 Search PubMed . Available from: https://doi.org/10.1186/s13321-015-0069-3.
- X. Chen and C. H. Reynolds, Performance of Similarity Measures in 2D Fragment-Based Similarity Searching: Comparison of Structural Descriptors and Similarity Coefficients, J. Chem. Inf. Comput. Sci., 2002, 42(6), 1407–1414 CrossRef CAS PubMed . Available from: https://doi.org/10.1021/ci025531g.
- S. Müller, Repository for: Flexible Heuristic Algorithm for Automatic Molecule Fragmentation: Application to the UNIFAC Group Contribution Model (New Version), 2022, available from: https://github.com/simonmb/fragmentation_algorithm Search PubMed.
- D. Castello, M. S. Haider and L. A. Rosendahl, Catalytic upgrading of hydrothermal liquefaction biocrudes: different challenges for different feedstocks, Renewable Energy, 2019, 141, 420–430 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0960148119304768.
- X. Liu, Organic Chemistry I, Kwantlen Polytechnic University, 2021, available from: https://kpu.pressbooks.pub/organicchemistry/ Search PubMed.
- J. W. Ahn, S. K. Pandey and K. H. Kim, Comparison of GC-MS Calibration Properties of Volatile Organic Compounds and Relative Quantification Without Calibration Standards, J. Chromatogr. Sci., 2011, 49(1), 19–28 CAS . Available from: https://doi.org/10.1093/chrsci/49.1.19.
- A. H. Hubble and J. L. Goldfarb, Synergistic effects of biomass building blocks on pyrolysis gas and bio-oil formation, J. Anal. Appl. Pyrolysis, 2021, 156, 105100 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0165237021000863.
- R. Olcese, V. Carré, F. Aubriet and A. Dufour, Selectivity of Bio-oils Catalytic Hydrotreatment Assessed by Petroleomic and GC*GC/MS-FID Analysis, Energy Fuels, 2013, 27(4), 2135–2145 CrossRef CAS . Available from: https://doi.org/10.1021/ef302145g.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, Understanding the Fast Pyrolysis of Lignin, ChemSusChem, 2011, 4(11), 1629–1636 CrossRef CAS PubMed . Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cssc.201100133.
- D. Butina, Unsupervised Data Base Clustering Based on Daylight's Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets, J. Chem. Inf. Comput. Sci., 1999, 39(4), 747–750 CrossRef CAS . Available from: https://doi.org/10.1021/ci9803381.
- G. Landrum, P. Tosco, B. Kelley, et al., RDKit: Open-source cheminformatics, available from: https://www.rdkit.org Search PubMed.
- M. Pecchi, M. Baratieri, J. L. Goldfarb and A. R. Maag, Effect of solvent and feedstock selection on primary and secondary chars produced via hydrothermal carbonization of food wastes, Bioresour. Technol., 2022, 348, 126799 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0960852422001286.
| This journal is © The Royal Society of Chemistry 2024 |