
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable synthesis of diethyl carbonate from carbon dioxide and ethanol featuring acetals as regenerable dehydrating agents†
Wahyu S.
Putro
 a,
Seiichiro
Ijima
a,
Seiji
Matsumoto
b,
Satoshi
Hamura
b,
Mizuho
Yabushita
c,
Keiichi
Tomishige
c,
Norihisa
Fukaya
*a and
Jun-Chul
Choi
*a
a,
Seiichiro
Ijima
a,
Seiji
Matsumoto
b,
Satoshi
Hamura
b,
Mizuho
Yabushita
c,
Keiichi
Tomishige
c,
Norihisa
Fukaya
*a and
Jun-Chul
Choi
*a
aNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: n.fukaya@aist.go.jp; junchul.choi@aist.go.jp
bTosoh Corporation, 3-8-2 Shiba, Minato-ku, Tokyo 105-8623, Japan
cDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07, Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8579, Japan
First published on 23rd April 2024
Abstract
CO2 valorization, which helps to offset the negative environmental impact of anthropogenic CO2 emission, is a key aspect of the circular economy and a promising carbon capture and utilization strategy. Among the products of this valorization, dialkyl carbonates such as diethyl carbonate (DEC) draw much attention because of their low toxicity, high polarity, and broad application scope, and the related cost-effective and scalable synthesis methods are therefore highly sought after. Herein, DEC was sustainably synthesized from CO2 and ethanol in the presence of acetals as regenerable organic dehydrating agents and dibutyltin(IV) oxide as a catalyst under optimized conditions (ethanol/acetal ratio, temperature, reaction time, presence or absence of Lewis-acidic additive). DEC yield, which was positively correlated with acetal hydrolyzability and negatively correlated with acetal regeneration efficiency (regenerability), increased in the presence of the Lewis-acidic acetal hydrolysis promoter (scandium(III) triflate). Acetal regenerability was strongly impacted by the ethanol/acetone ratio, molecular sieve amount, and the type of acetal regeneration catalyst, e.g., an acidic ion exchange catalyst (Amberlyst) showed high activity and stability for the regeneration of various acetals. The obtained findings pave the way for the use of acetals as sustainable dehydrating agents for direct closed-loop DEC synthesis and thus contribute to the establishment of a greener society.
Sustainability spotlightConversion of CO2 to value-added chemicals coupled with byproduct regeneration helps to minimize the negative environmental impact of modern industry in the context of climate change and sustainability. This work focuses on the development of waste-free synthesis of diethyl carbonate (DEC) from CO2 by which hydrolysis products can be reverted to initial dehydrating agents. The regeneration of the dehydrating agent was simply achieved under ambient conditions using recyclable Amberlyst as a catalyst. Therefore, we believe that this approach could open a new promising direction to valorize CO2 and recycle the waste to produce organic carbonate in a greener fashion, in line with the UN sustainable goals: responsible consumption and production (SDG 12), and climate action (SDG 13). |
Introduction
CO2 valorization, which refers to the conversion of CO2 to value-added products coupled with byproduct regeneration and helps to minimize the negative environmental impact of modern industry in the context of climate change and sustainability, is a key aspect of the circular economy1 and a promising carbon capture and utilization strategy.2,3 Currently, dialkyl carbonates have drawn much attention as valorization products because of their low toxicity, high polarity, and numerous applications in fuel additives, batteries, and polymer synthesis.4Given that diethyl carbonate (DEC) has a higher oxygen content than methyl tert-butyl ether (41% vs. 18%) and is therefore a promising alternative to this fuel additive, cost-effective and scalable DEC synthesis methods are in high demand.5 Compared to traditional procedures such as Bayer phosgenation,6 ENIChem oxidative carbonylation,7,8 and Asahi-Kasei indirect trans-esterification,9 the direct synthesis of DEC from CO2 and ethanol (EtOH) offers the benefit of higher sustainability, with DEC yield strongly depending on the catalyst-dehydrating agent combination due to the severe limitation originated from chemical equilibrium.10 High DEC yields were achieved by combining CeO2 as a catalyst11 with 2-cyanopyridine,12 2-furonitrile,13,14 acetonitrile,15 benzonitrile,16 butylene oxide,17 chloroacetate,18 and orthoesters19 as dehydrating agents, although the efficient regeneration of these agents remains challenging.
The use of chemically recyclable dehydrating agents allows for circular chemical utilization and, hence, more sustainable CO2 valorization. Previously, we reported the sustainable synthesis of DEC from CO2 and tetraethyl orthosilicate (TEOS),20 showing that the obtained byproduct (disiloxane) can be recycled back to TEOS to establish a closed-loop reaction system. Similarly, 2-cyanopyridine and 2-furonitrile can be regenerated from each hydration product, 2-picolinamide and 2-furamide, respectively.21,22 Given that the regeneration step is generally sluggish and requires high temperatures, dehydrating agents that can be simply regenerated under mild conditions are highly sought after.
When selecting an appropriate dehydrating agent, one should consider the trade-off between dehydration performance and regenerability, i.e., strong dehydrating ability allows for high DEC yields at the cost problematic regeneration, whereas high regenerability is typically associated with low DEC yields. In the latter scenario, DEC production can be enhanced through the use of additives. For example, CeO2 was combined with a zeolite cocatalyst (H-FAU) to increase DEC yield (to 72% at maximum) in a process using an acetal (2,2-diethoxypropane) as a dehydrating agent.23,24 The low DEC yield observed in the absence of H-FAU (10%) indicated that the acetal itself did not sufficiently promote the DEC formation. Our group reported that the yield of dimethyl carbonate (DMC) in the system with dibutyltin(IV) oxide (Bu2SnO) as a catalyst and 2,2-dimethoxypropane as a dehydrating agent increased from 17% to 40% after the introduction of [Ph2NH2]OTf as an acidic cocatalyst.25 To date, the regeneration of spent acetals to realize their closed-loop recycling has not yet been reported (Fig. 1).
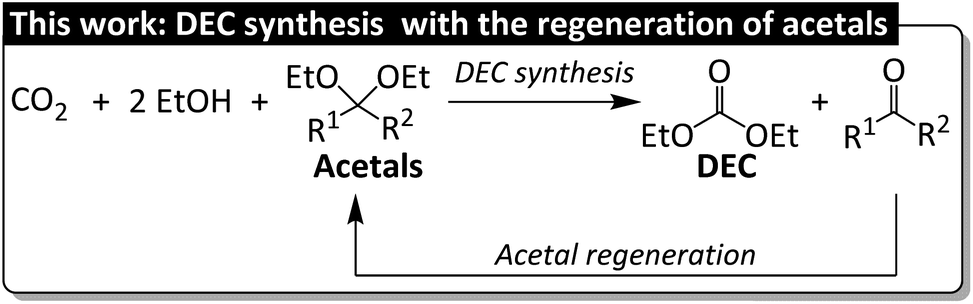 | ||
| Fig. 1 This work related to the DEC synthesis featuring with the regeneration of spent acetal. | ||
The ready availability, ease of regeneration, and benign nature of acetals make them promising dehydrating agents for the sustainable synthesis of DEC despite their inferiority to orthoesters or alkoxysilanes in terms of dehydration ability.26 In particular, acetals can be regenerated by reacting their hydrolysis products (ketones) with (i) orthoesters ((RO)3CH) in the presence of catalysts such as Cu(BF4)2·xH2O27 and 2,3-dichloro-5,6-dicyano-p-benzoquinone28 or (ii) triethoxysilane ((EtO)3SiH) in the presence of antimony(V) catalyst.29 However, these reactions produce unexpected esters and siloxanes as byproducts. In contrast, acetal synthesis from ketones and alcohols, which is promoted by In(OTf)3,30 PPh3-CCl4,31 HCl,32 Eosin Y,33 and Ru-pincer catalysts,34 affords water as the only byproduct and is therefore a more promising route, although the complex preparation of these catalysts and their presence in the homogeneous system present certain complications. Herein, we reported the use of solid acid catalyst for the regeneration of acetal from ethanol.
As a part of our continuous research on the conversion of CO2 into dialkyl carbonates, we herein report the Bu2SnO-catalyzed direct synthesis of DEC from CO2 and EtOH in the presence of acetals (e.g., 2,2-diethoxypropane) as dehydrating agents. Acetal is promising for realizing sustainability because of the simple regeneration, but its dehydrating rate is supported by Lewis acidic cocatalyst Sc(OTf)3 for promoting the corresponding DEC yield. The byproduct of DEC synthesis, namely water, reacts with acetals to produce ketones (Fig. 2a), which subsequently react with EtOH to regenerate acetals (Fig. 2b) and complete the circular synthesis of DEC.
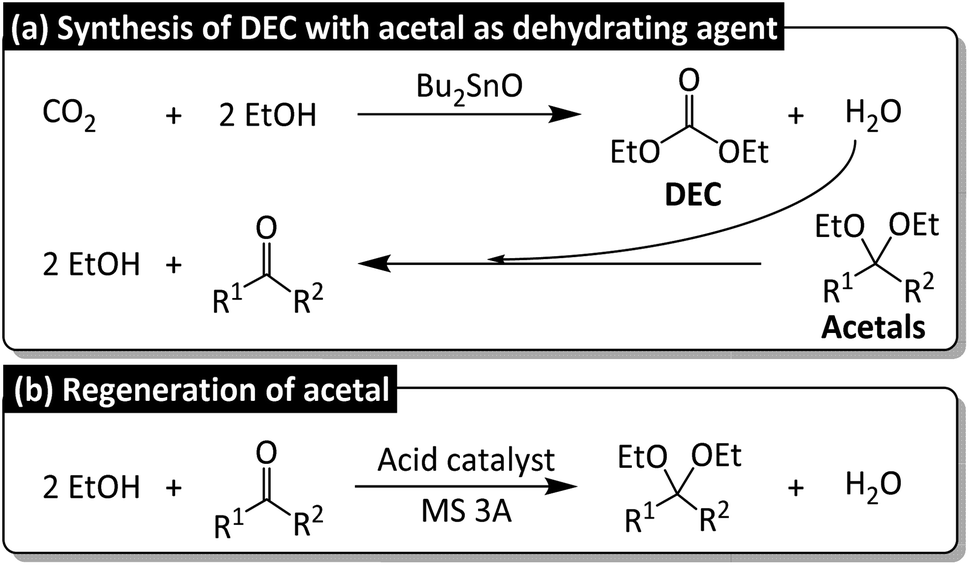 | ||
| Fig. 2 (a) Direct synthesis of diethyl carbonate (DEC) from CO2 and ethanol (EtOH) in the presence of acetals as dehydrating agents, (b) acetal regeneration from corresponding ketones. | ||
Experimental section
Materials
Bu2SnO (98%), scandium(III) triflate (Sc(OTf)3; 99%), and 3 Å molecular sieves (MS3A) were purchased from Sigma Aldrich (USA). Super dry EtOH (99.5%), super dry acetone (99.5%), and Nafion NR-50 were purchased from Wako Pure Chemical Industries (Japan). 2,2-Diethoxypropane (95%), 4-tert-butyltoluene (95%) and Amberlyst 15(H) were purchased from Tokyo Chemical Industries Co., Ltd. (Japan), and Alfa Aesar (USA) respectively. H-FAU (SiO2/Al2O3 molar ratio = 5.5) was received from Tosoh Corporation (Japan). All compounds were used without further purification or pretreatment.DEC synthesis
All reactions and operations involving moisture-sensitive compounds (all acetals and super dry EtOH) were performed in a nitrogen-filled glove box. General DEC syntheses from CO2 and EtOH in the presence of Bu2SnO and acetals were carried out in an autoclave with an inner volume of 10 mL. The following example can be viewed as representative. The autoclave was charged with a magnetic stirrer bar, 2,2-diethoxypropane (1.7 mL, 10.7 mmol), Bu2SnO (0.54 g; 20 mol% with respect to 2,2-diethoxypropane), Sc(OTf)3 (2.1 mg, 4.3 μmol; 0.04 mol% with respect to 2,2-diethoxypropane with ratio of Sn/Sc = 500), and EtOH (2.4 mL, 42.5 mmol; EtOH/acetal = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the stated order, sealed, purged five times with 5 MPa CO2 (∼3 g of CO2), maintained under pressure for 10 min, heated to 160 °C for the required amount of time, and cooled to room temperature using an ice bath.
:1) in the stated order, sealed, purged five times with 5 MPa CO2 (∼3 g of CO2), maintained under pressure for 10 min, heated to 160 °C for the required amount of time, and cooled to room temperature using an ice bath.
Reaction mixtures were analyzed by gas chromatography (GC; internal standard = 4-tert-butyltoluene, injection temperature = 210 °C, detection temperature = 220 °C, initial temperature = 40 °C, and final temperature = 210 °C) using a Shimadzu GC-2014 instrument equipped with a flame ionization detector and an Rtx column. The column temperature was increased to 50 °C at a heating rate of 10 °C min−1, held for 9 min, increased to 110 °C at the same heating rate, held for 12 min, and finally increased to 210 °C at heating rate of 50 °C min−1. The carrier gas (nitrogen) was supplied at a pressure of 114 kPa and a flow rate of 3 mL min−1. DEC yield and material balance (MB) were calculated as
| DEC yield based on acetal (%) = 100% × (mmol DEC/initial mmol acetal), |
| MB (%) = 100% × (mol ketone + mol remaining acetal)/initial mol acetal. |
Acetal synthesis
Similar to DEC, acetals were synthesized in a nitrogen-filled glove box. The following example can be viewed as representative. A 50 mL round-bottom flask was charged with a magnetic stirrer bar, EtOH (18.4 g; 0.4 mol), acetone (5.8 g, 0.1 mol), H-FAU (1.275 g; 0.9 mmol H+), and MS3A (10 g), and the mixture was stirred under ambient conditions for the required amount of time. The content of water adsorbed by the molecular sieve (MS) was determined by Karl-Fischer titration (KEM ADP-611) to control the reaction progress. The reaction mixture was analyzed by GC as above. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Bruker ADVANCE III HD instrument (1H NMR at 400 MHz, 13C{1H} NMR at 100.7 MHz). Acetal yield MB were calculated as| Acetal yield (%) = 100% × (mol acetal/initial mol ketone), |
| MB (%) = 100% × (mol acetal + mol remaining ketone)/initial mol ketone. |
Catalyst reusability testing in normal reactor
The first reaction was performed according to the procedure described above. At the end of the reaction, the mixture was analyzed by GC, and the solid and liquid phases were separated by centrifugation at 4000 rpm for 6 min. The thus isolated H-FAU was calcined at 500 °C for 3 h and vacuum-dried for 20 h prior to reuse. Spent Nafion NR-50 and Amberlyst 15(H) were separated from MS3A powder using a sieve and dried in a vacuum oven at 120 °C for 20 h prior to reuse. Activity test was carried out for a second time with the substrate/catalyst ratio adjusted accordingly.Catalyst reusability testing in SpinChem reactor
Unlike the reaction in the normal reactor, that in the four-compartment SpinChem reactor was performed with the catalyst and MS loaded in separate compartments. The MS and ion-exchange catalysts (Nafion and Amberlyst) were present as pellets, which facilitated the separation and collection of the spent catalyst. The spent catalyst was dried in a vacuum oven at 120 °C for 20 h prior to reuse.Results and discussion
Synthesis of DEC from CO2 and EtOH
To optimize the conditions of DEC synthesis, we explored the effects of the EtOH/acetal molar ratio, temperature (160–220 °C), and reaction time using Bu2SnO as a catalyst and 2,2-diethoxypropane (1) as a dehydrating agent (Table 1). The amount of DEC initially increased with increasing EtOH/acetal molar ratio (from 0.9 mmol at 2/1 (entry 2) to 1.6 mmol at 4/1 (entry 4)) but decreased when this ratio became overly large (entries 5 and 6). The optimum temperature was identified as 180 °C (1.6 mmol of DEC, entry 4). It was found that side reactions such as the thermal decomposition of 1 and aldol condensation of acetone to form 2-ethoxypropene and mesityl oxide was observed from 0.09 to 1.54 and 0 to 0.05 mmol (Fig. S1†), respectively, thus resulting in decreased MB (Table S1†). These side products were also detected in previous studies when 1 was used as a dehydrating agent for DEC synthesis.23 Prolonging the reaction time increased the DEC amount to 1.3 mmol but decreased MB (entry 8). Thus, the conditions of entry 4 were selected as the optimal for further experiments.|
|
|||||
|---|---|---|---|---|---|
| Entry | EtOH/acetal (mol mol−1) | T (°C) | t (h) | DECb (mmol) | MBc (%) |
| a Reaction conditions: 5 MPa CO2 at room temperature, 20 mol% Bu2SnO relative to acetal. b DEC determined by GC using 4-tert-butyltoluene as internal standard. c MB = material balance (%). | |||||
| 1 | 2/1 | 160 | 20 | 0.5 | 92 |
| 2 | 2/1 | 180 | 20 | 0.9 | 85 |
| 3 | 4/1 | 160 | 20 | 0.8 | 90 |
| 4 | 4/1 | 180 | 20 | 1.6 | 85 |
| 5 | 10/1 | 180 | 20 | 1.3 | 85 |
| 6 | 20/1 | 180 | 20 | 0.8 | 85 |
| 7 | 20/1 | 200 | 20 | 1.0 | 77 |
| 8 | 20/1 | 200 | 48 | 1.3 | 65 |
| 9 | 20/1 | 220 | 20 | 0.8 | 71 |
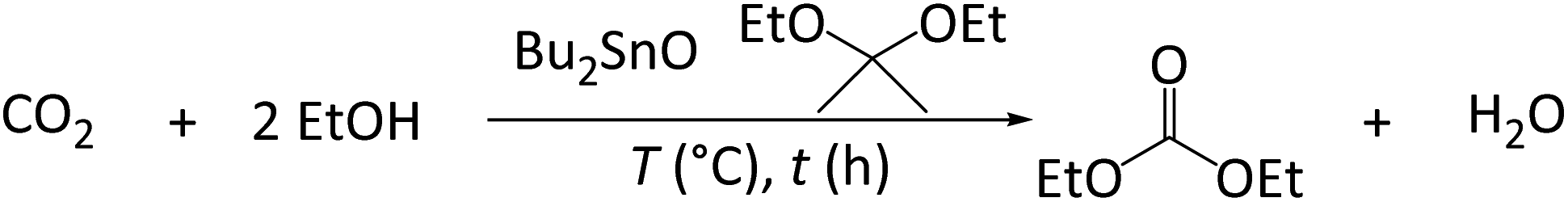
Acid additives can promote the hydrolysis of acetals and thus accelerate the direct synthesis of organic carbonates, e.g., an acidic triflate was shown to enhance the catalytic performance of Bu2SnO in DMC synthesis,25 and an CeO2 + H-FAU physically mixed catalysts was shown to effectively promote DEC formation.23 Therefore, we examined the effect of Sc(OTf)3 as an acid additive at different EtOH/acetal ratios and temperatures of 160 and 180 °C (Fig. 3).
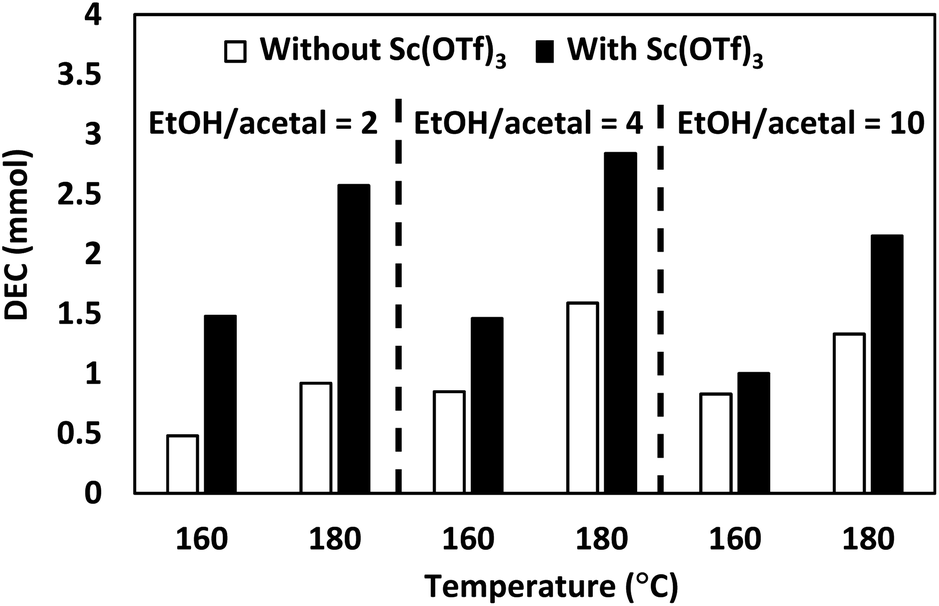 | ||
| Fig. 3 Effect of Sc(OTf)3 on DEC synthesis. Reaction conditions: 5 MPa CO2 at room temperature, 20 mol% Bu2SnO relative to acetal, 0.04 mol% Sc(OTf)3 relative to acetal (Sn/Sc = 500), 20 h. DEC amount was determined by GC using 4-tert-butyltoluene as internal standard. | ||
The addition of Sc(OTf)3 increased DEC production under all conditions. In particular, the introduction of this additive increased the amount of DEC three-fold (from 0.5 to 1.5 mmol) at 160 °C and EtOH/acetal ratio = 2 and almost two-fold (from 1.6 to 2.8 mmol of DEC) at 180 °C and EtOH/acetal ratio = 4. These results suggested that Sc(OTf)3 effectively promoted acetal hydrolysis at various temperatures and EtOH/acetal ratios. The effect of acetal screening for the DEC synthesis was subsequently examined in the absence and presence of Sc(OTf)3 (Fig. 4).
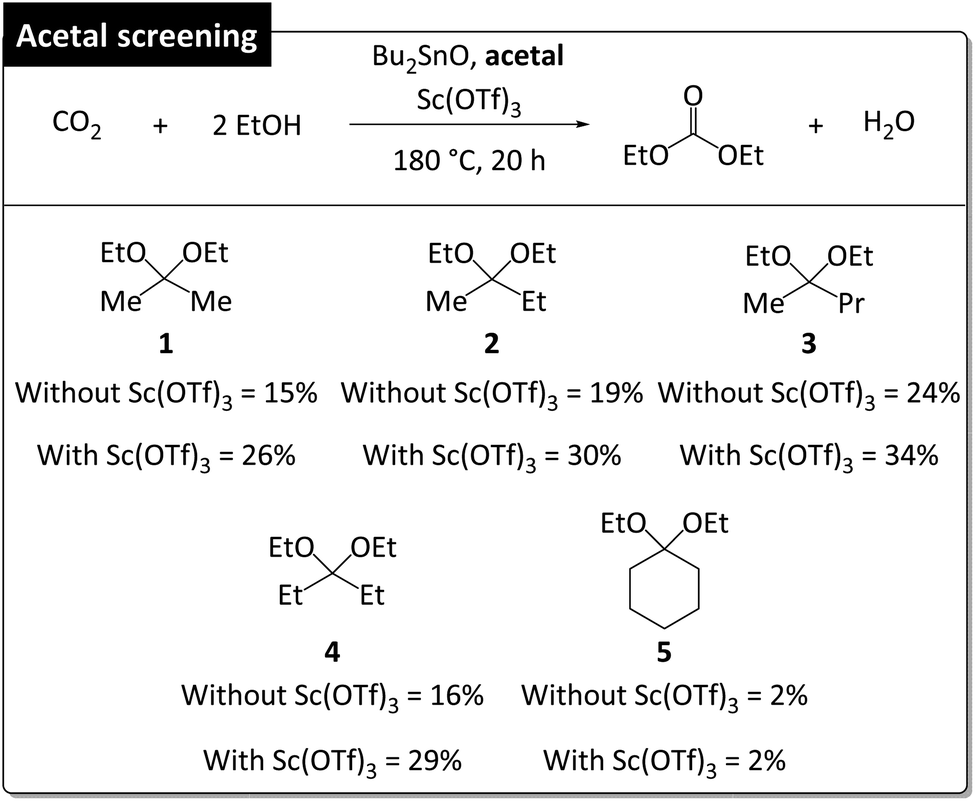 | ||
| Fig. 4 Screening of acetals as dehydrating agents for Bu2SnO-catalyzed DEC synthesis with and without Sc(OTf)3. Reaction conditions: 5 MPa CO2 at room temperature, 40 mmol EtOH, 10 mmol acetal (EtOH/acetal = 4), 20 mol% Bu2SnO relative to acetal, 0.04 mol% Sc(OTf)3 relative to acetal (Sn/Sc = 500), 180 °C, 20 h. DEC yield (%) was determined by GC using 4-tert-butyltoluene as internal standard compound. | ||
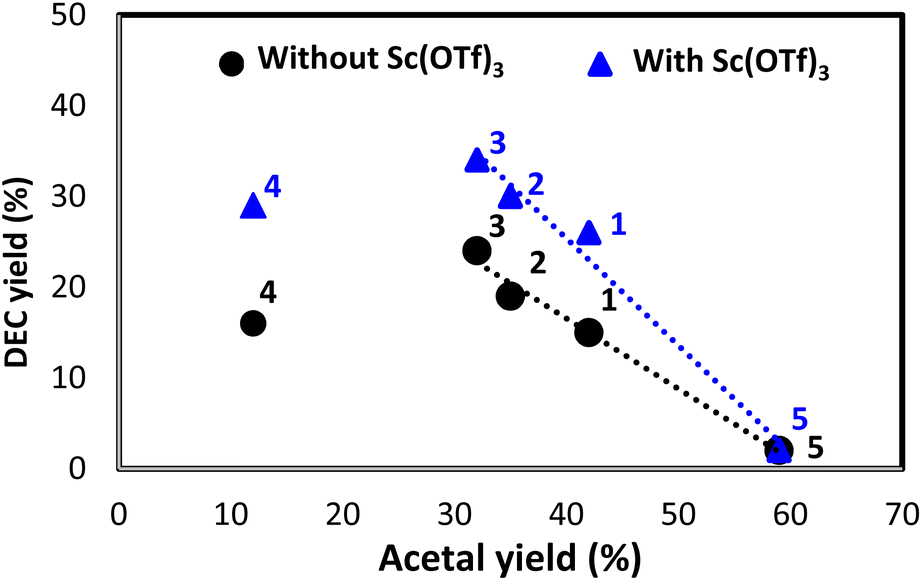
With these optimized conditions in hand, we screened different acetals in the absence and presence of Sc(OTf)3 at 180 °C, 20 h, and EtOH/acetal = 4/1 (Fig. 4). The length of the alkyl substituent of acetals (1–3) was positively correlated with DEC amount irrespective of the presence of Sc(OTf)3, which was ascribed to the fact that ketones with longer chains are less readily converted into acetals, i.e., longer-chain acetals are hydrolyzed more readily (Table S3†).35,36 2,2-Ethoxypentane (3) showed the highest DEC yield (34% with Sc(OTf)3 and 24% without Sc(OTf)3). Acetal 4 (DEC yield = 29% with Sc(OTf)3 and 16% without Sc(OTf)3) performed similarly to 1 (DEC yield = 26% with Sc(OTf)3 and 15% without Sc(OTf)3) despite being more readily hydrolyzable. 3-Ethoxy-2-pentene (m/z = 114) was detected as a major byproduct (Fig. S2†), accounting for the low DEC yield. In the case of 5, Sc(OTf)3 had no positive effect on DEC yield (2%), i.e., 5 was a poor dehydrating agent for DEC synthesis. Thus, DEC yield was strongly influenced by acetal hydrolyzability.
The increased DEC yields observed in the presence of Sc(OTf)3 for all acetals except 5 (26–34% vs. 15–24% without Sc(OTf)3) indicated that this additive promoted acetal hydrolysis and was well tolerated for catalyzing the hydrolysis of various acetals. Notably, Sc(OTf)3 did not adversely affect MB when acetals 2–4 were used (Table S4†). Apart from having a low dehydrating ability, 5, engaged in side reactions, which resulted in a substantial MB decrease and low DEC yield. High MBs can be achieved when acetal hydrolysis affords ketones as the sole products (Fig. 5). These are general illustrative reactions from DEC synthesis using acetal as the dehydrating agent.
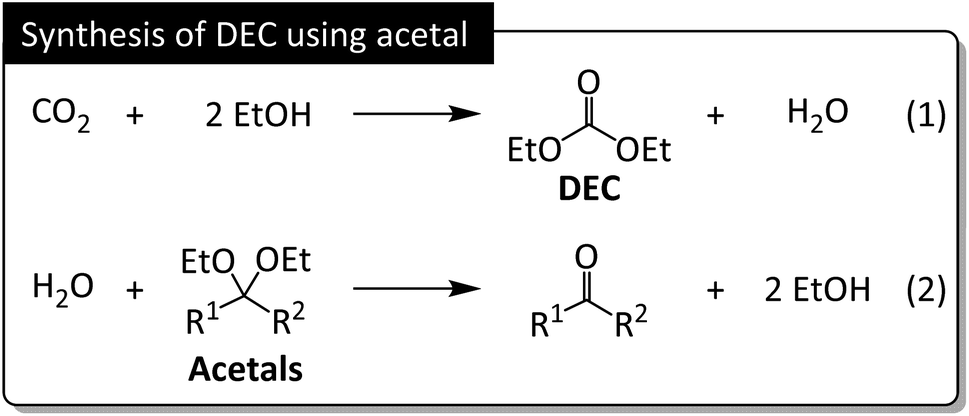 | ||
| Fig. 5 DEC synthesis from CO2 and EtOH. Acetals exert their dehydrating effect by reacting with water to generate ketones and EtOH. | ||
One of the main issues regarding the use of organic dehydrating agents in the synthesis of organic carbonates is finding an effective regeneration process. The regeneration of orthoesters remains unknown,19 and that of 2-cyanopyridine,22,37 furanonitrile21 and alkoxysilanes20 requires high temperatures. Hence, easily regenerable dehydrating agents are required to realize high sustainability. To clarify the potential sustainability of the current reaction, we examined the regeneration of acetals from the corresponding ketones under ambient conditions.
Acetal regeneration
Acetal regeneration was first examined by reacting acetone as a model substrate with EtOH in the presence of H-FAU as a catalyst. Given the reversibility of acetal formation from alcohols and carbonyl compounds, water removal is required to overcome the equilibrium limitation.32 Herein, we used MS3A to bind water and monitored the amount of MS3A-bound water using Karl-Fischer titration to control the reaction progress. The effects of the EtOH/acetone ratio and MS loading on acetal formation are given in Fig. 6 associated with monitoring H2O amount adsorbed by MS3A.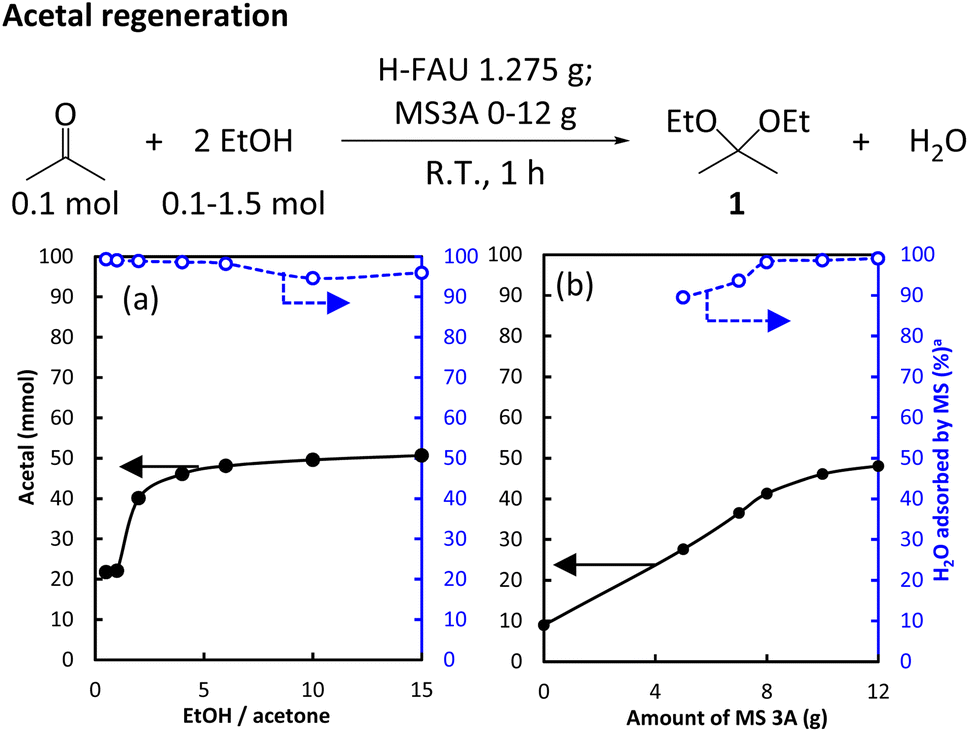 | ||
| Fig. 6 (a) Effect of EtOH/acetone ratio on the formation of 1. Reaction conditions: 0.1 mol acetone, 1.275 g H-FAU, 10 g MS3A, 1 h, ambient conditions. (b) Effect of MS3A loading. Reaction conditions: 0.1 mol acetone, 0.4 mol ethanol, 1.275 g H-FAU, 1 h, ambient conditions. awt% of total H2O adsorbed by MS after the reaction based on the acetal formation. | ||
A 1 h reaction under ambient conditions afforded 22 mmol of 1 at an EtOH/acetone ratio of 1 and twice this amount (51 mmol of 1) at a higher EtOH/acetone ratio of 15 (Fig. 6a). The yield of 1 hardly changed at EtOH/acetone ratios of >4. Given that MS adsorbed more than 95% of water formed (Table S5†), this behavior could not be caused by MS3A saturation with water. The zeolite catalyst turned red after exposure to the acetal but remained white when exposed to acetone (Fig. S3†). The use of acetal-exposed catalyst drastically decreased the acetal yield to 3%, which suggested that the acetal deactivated the catalysts. The addition of 1 to the mixture of acetone and EtOH decreased the 1 yield to 10% (Table S6†). In the presence of 1, the use of an acetal-pretreated catalyst hindered acetal production (Table S7†). These results indicated that the catalyst was deactivated by the acetal itself, which explains the saturation behavior observed at 50% acetal yield.
The regeneration of acetals from the corresponding ketones is a moisture-sensitive process, and the presence of dehydrant is necessary to avoid shifting the equilibrium back to the reactant. In the absence of MS3A, the yield of 1 was as low as 9%, increasing 28% and 48% at MS loadings of 5 g to 12 g MS3A, respectively (Fig. 6b). In line with the increased yield of 1, the amount of water adsorbed by MS also increased (because of the increased MS loading). Notably, side reactions comprising aldol condensation were hardly observed when the effect of MS loading was evaluated, which resulted in high MB (Table S8†).
Acid catalysis is a common feature of successful acetal formation, which is, however, mainly focused on the development of homogeneous catalysts.38 Encouraged by the catalytic performance of H-FAU, we evaluated the performances of solid acid catalysts at the same amount of H+ (0.09 mmol) under optimal conditions (Fig. 7a). Unlike in a previous report on isomerization and alkylation reactions, sulphated zirconia (SO42−/ZrO2) with superacidic sites was less active than H-FAU (27 vs. 46%),39i.e., strongly acidic sites were less favorable for acetal formation. The two main types of ion-exchange resins based on styrene-based sulfonic acids (Amberlyst and Dowex resins) and the perfluorosulfonic acid–based resin (Nafion, Table S11†) afforded yields of 45, 27, and 41%, respectively. The three highly active catalysts (H-FAU, Nafion, Amberlyst) were then subjected to a reusability test (Fig. 7b). H-FAU exhibited poor stability, as revealed by its marked activity decrease in the second and third cycles, whereas ion-exchange catalysts remained stable. The red color of spent catalysts indicated the high affinity of organic compounds (mostly acetal product) to the catalyst surface. Annealing at 500 °C restored the white color of the catalyst but resulted in an activity drop, which was ascribed to zeolite deactivation due to deprotonation of H-FAU which probably attached in MS. Given that H-FAU and MS powder were used in the form of a physical mixture, the active protons of the catalyst were prone to become attached to MS. This result inspired us to use the SpinChem reactor allowing for reactions with the catalyst and MS located in separate compartment (Fig. 8).
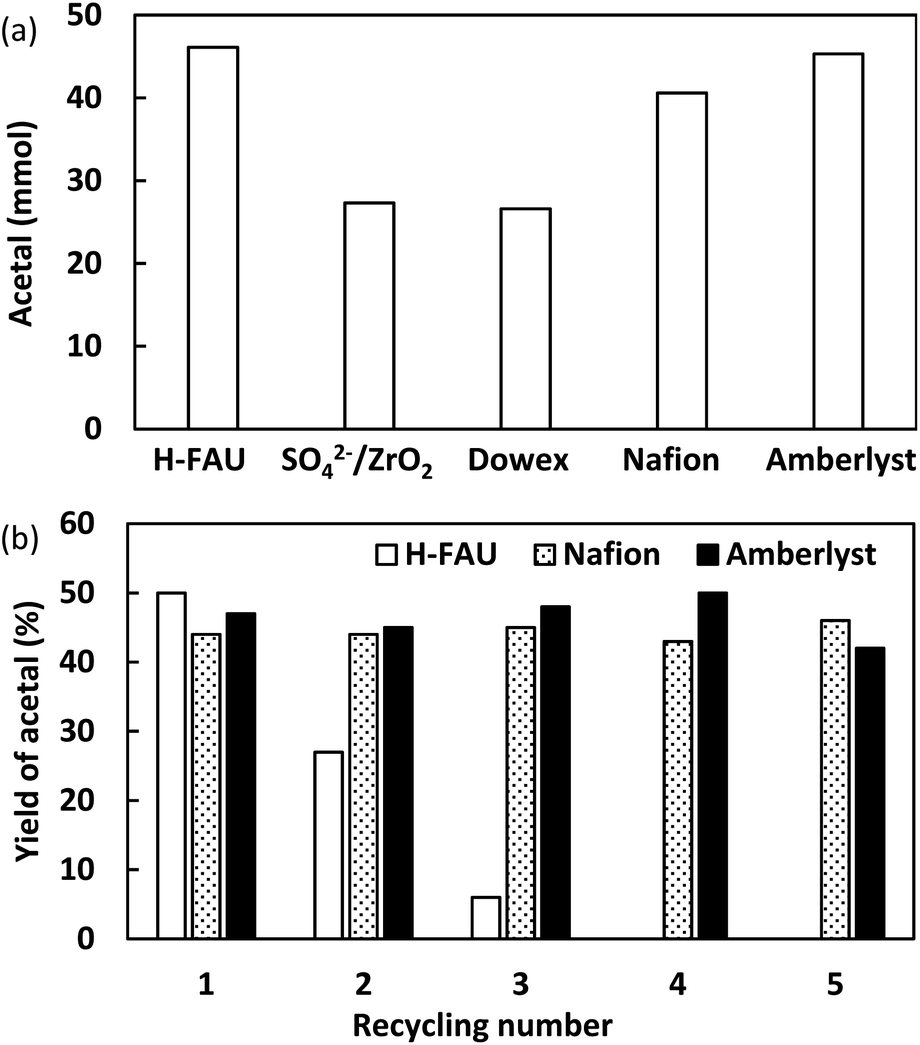 | ||
| Fig. 7 (a) Catalyst screening. Reaction conditions: 0.1 mol acetone, 0.4 mol ethanol, 10 g MS3A, 0.09 mmol H+, H-FAU → 1 h, SO42−/ZrO2, Dowex → 2 h, Nafion → 3 h, and Amberlyst → 2 h, ambient conditions. (b) Reusability test of various catalysts. Acetal quantitation was achieved by GC using 4-tert-butyltoluene as an internal standard. | ||
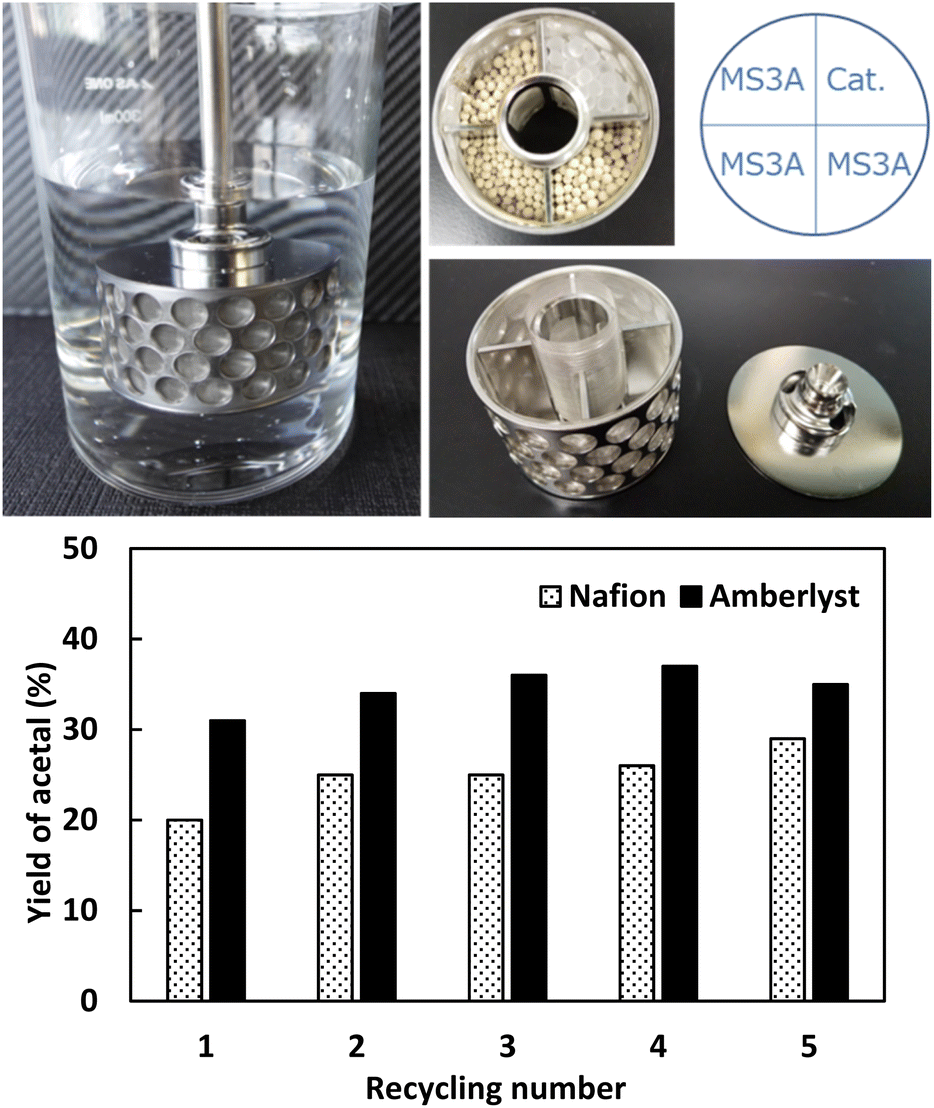 | ||
| Fig. 8 (top) Photograph of SpinChem reactor and (bottom) results of Nafion and Amberlyst reusability testing in the same reactor. | ||
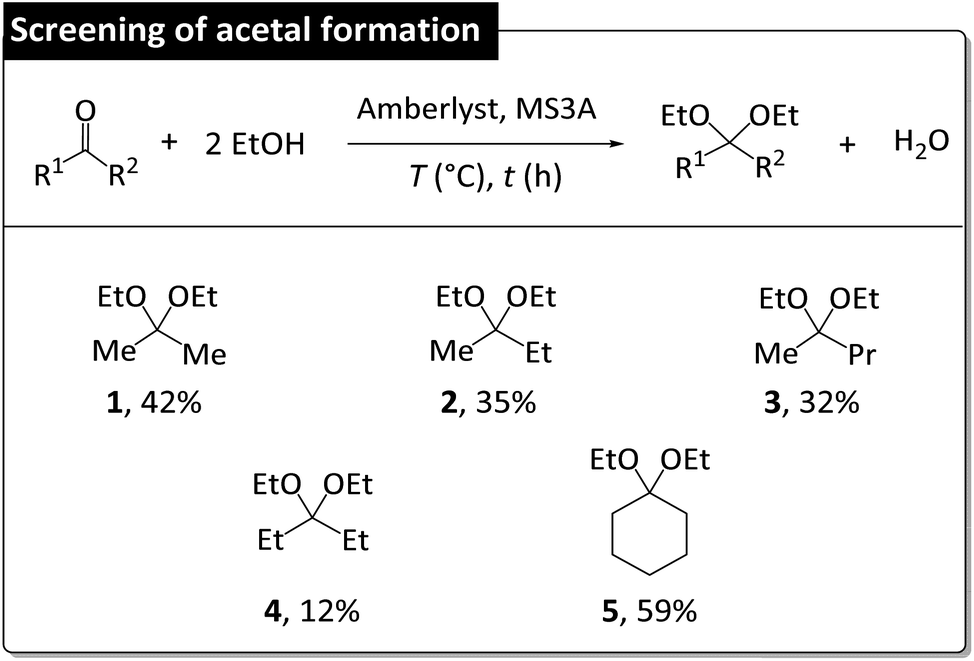 | ||
| Fig. 9 Scope of acetal regeneration from corresponding ketones. Reaction conditions: 0.1 mol ketone, 0.4 mol EtOH, 10 g MS3A, 0.2 g Amberlyst, room temperature, 1 h. Acetal yield (%) determined by GC using 4-tert-butyltoluene as internal standard. | ||
The SpinChem reactor contains four compartments attached in a stirrer shaft and therefore allowed for higher recycling effectivity.40,41 The catalyst was set in one compartment of this reactor, and the other compartment was charged with MS. Compared with normal reactions, those carried out in SpinChem reactor featured lower yields. For example, Amberlyst showed a steady activity at a level of 45% yield for five consecutive runs in the normal reactor, whereas its activity in the SpinChem reactor was as low as 33% yield. The activity of H-FAU stabilized at an acetal yield of 27% after four runs, which suggested that the SpinChem reactor setup increased catalyst durability (Fig. 8). Compared to the zeolite catalyst, ion-exchange catalysts exhibited higher resilience in both reactors, with Amberlyst showing higher activity than Nafion. Therefore, we used Amberlyst as a catalyst for the regeneration of acetals previously used for DEC synthesis (Fig. 8).
The screening of acetal formation was performed using the normal reaction system, as it resulted in higher catalyst activity. The formation of 1 from acetone proceeded in 42% yield, whereas lower yields (35% of 2 and 32% of 3) were obtained for substrates with longer alkyl chains. The regeneration of 4 proceeded in 12% yield. The regeneration of 5 proceeded in 59% yield, which reflected the elevated reactivity of the parent cyclic ketone and agreed with a previous study according to which the feasibility of acetal formation is determined by acetal hydrolyzability.32 That is, water-sensitive acetals were effective for DEC synthesis but were more difficult to regenerate.
The correlation between acetal and DEC yields observed with/without Sc(OTf)3 (Fig. 11) was negative, with the trendline slope becoming steeper upon the introduction of Sc(OTf)3. The more water-tolerant acetals (e.g., 5) affording low DEC yields showed higher regeneration reactivities, and vice versa, which is in line with a previous report (Table S3†).36 Besides, a hemiacetal of 5 was obtained, accounting for about one third of the acetal formed at the beginning of the reaction (Table S16†). The amount of this hemiacetal decreased with time, as expected for an intermediate. The datapoints of 4 was outliers because of the occurrence of a prominent side reaction during the synthesis of DEC in this case (Fig. S2†). The presence of catalyst and dehydrating agent is significant for the DEC synthesis because the reaction was not observed without their existence (Table S19,† entries 1–3). The addition of only Sc(OTf)3 slightly improve the yield of DEC, but Bu2SnO catalyzing the reaction boosted the DEC yield significantly (entries 4–6). This result is lower than previous reports applying TEOS, orthoester and 2-cyanopyridine as dehydrating agent or CeO2 as a catalyst (entries 7–13). However, this work reports not only focusing on the DEC synthesis but the regeneration of spent acetal is also discussed. The fact that the regeneration of acetals proceeded under ambient conditions demonstrated the feasibility of the regeneration process and the closed-loop green synthesis of DEC (Fig. 10).
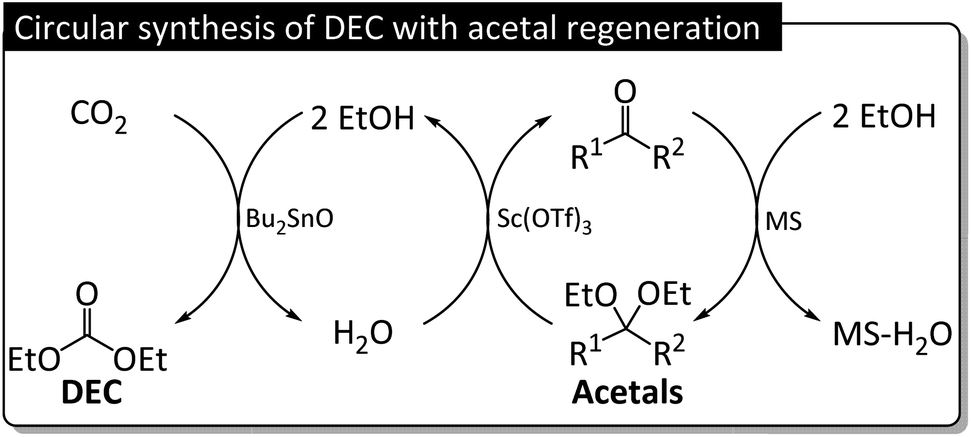 | ||
| Fig. 10 Illustration of DEC synthesis using acetals as regenerable dehydrating agents. | ||
Conclusion
Sustainable DEC synthesis was realized using acetals as regenerable dehydrating agents, with the optimal conditions identified as EtOH/acetal ratio = 4, temperature = 180 °C, and duration = 20 h. The addition of Sc(OTf)3 as an acetal hydrolysis catalyst increased DEC yields under all conditions. Acetal hydrolyzability was positively correlated with DEC yield, and acetal regenerability was strongly affected by the. EtOH/acetal ratio and MS loading. Acetal formation poisoned the zeolite catalyst and terminated the catalytic process at an acetal yield of 50%. Ion-exchange acid catalysts, namely Nafion and Amberlyst, exhibited high activity and stability for acetalization in normal and SpinChem reactors. Amberlyst could also be used to regenerate acetals previously utilized for DEC synthesis. DEC yield was negatively correlated with acetal regenerability, and the slope of the corresponding trendline increased upon the introduction of Sc(OTf)3. This result indicates that the trade-off between acetal dehydration power and regeneration ability should be considered to improve the ecofriendliness and industrial feasibility of organic carbonate synthesis. This report opens a promising prospective in industrial application for the development of sustainable synthesis of DEC.Author contributions
Wahyu S. Putro: conceptualization, investigation, writing, reviewing, and editing the original paper. Seiichiro Ijima: investigation, data validation, and formal analysis. Seiji Matsumoto: writing, reviewing, and editing. Satoshi Hamura: writing, reviewing, and editing. Mizuho Yabushita: writing, reviewing, and editing. Keiichi Tomishige: writing, reviewing, and editing. Norihisa Fukaya: conceptualization, supervising, reviewing, and editing the original paper. Jun-Chul Choi: conceptualization, supervising, reviewing, and editing the original paper.Conflicts of interest
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank Dr Tadahiro Nitta for providing the experimental assistance.References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- K. Tomishige, Y. Gu, Y. Nakagawa and M. Tamura, Front. Energy Res., 2020, 8, 117 CrossRef.
- S. Huang, B. Yan, S. Wang and X. Ma, Chem. Soc. Rev., 2015, 44, 3079–3116 RSC.
- K. Shukla and V. C. Srivastava, RSC Adv., 2016, 6, 32624–32645 RSC.
- Z. Kricsfalussy, H. Steude, H. Waldmann, K. Hallenberger, W. Wagner and H.-J. Traenckner, US Pat., 5523452, 1996 Search PubMed.
- U. Romano, F. Rivetti and N. Di Muzio, US Pat., 4318862, 1982 Search PubMed.
- K. Tomishige, T. Sakaihori, S. I. Sakai and K. Fujimoto, Appl. Catal., A, 1999, 181, 95–102 CrossRef CAS.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
- Y. Gu, K. Matsuda, A. Nakayama, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustain. Chem. Eng., 2019, 7, 6304–6315 CrossRef CAS.
- W. Sun, P. Li, M. Yabushita, Y. Nakagawa, Y. Wang, A. Nakayama and K. Tomishige, ChemSusChem, 2016, 16, e202300768 CrossRef PubMed.
- M. Honda, S. Kuno, N. Begum, K. I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165–170 CrossRef CAS.
- M. Honda, S. Kuno, S. Sonehara, K. I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, ChemCatChem, 2011, 3, 365–370 CrossRef CAS.
- E. Leino, P. Mäki-Arvela, K. Eränen, M. Tenho, D. Y. Murzin, T. Salmi and J. P. Mikkola, Chem. Eng. J., 2011, 176–177, 124–133 CrossRef CAS.
- A. A. Marciniak, O. C. Alves, L. G. Appel and C. J. A. Mota, J. Catal., 2019, 371, 88–95 CrossRef CAS.
- W. S. Putro, Y. Munakata, S. Ijima, S. Shigeyasu, S. Hamura, S. Matsumoto, T. Mishima, K. Tomishige, J.-C. Choi and N. Fukaya, J. CO2 Util., 2022, 55, 101818 CrossRef CAS.
- W. S. Putro, A. Ikeda, S. Shigeyasu, S. Hamura, S. Matsumoto, V. Y. Lee, J.-C. Choi and N. Fukaya, ChemSusChem, 2020, 14, 842–846 CrossRef PubMed.
- Y. Nagasaki, M. Tamura, M. Yabushita, Y. Nakagawa and K. Tomishige, ChemCatChem, 2022, 14, 1–8 CrossRef.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. I. Fujimoto and K. Tomishige, ChemSusChem, 2013, 6, 1341–1344 CrossRef CAS PubMed.
- T. Chang, M. Tamura, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Green Chem., 2020, 22, 7321–7327 RSC.
- T. Chang, M. Yabushita, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Catal. Sci. Technol., 2023, 13, 5084–5093 RSC.
- J.-C. Choi, K. Kohno, Y. Ohshima, H. Yasuda and T. Sakakura, Catal. Commun., 2008, 9, 1630–1633 CrossRef CAS.
- T. Sakakura, J.-C. Choi, Y. Saito and T. Sako, Polyhedron, 2000, 19, 573–576 CrossRef CAS.
- R. Kumar and A. K. Chakraborti, Tetrahedron Lett., 2005, 46, 8319–8323 CrossRef CAS.
- B. Kaimi and A. M. Ashtiani, Chem. Lett., 1999, 28, 1199–1200 CrossRef.
- R. A. Ugarte and T. W. Hudnall, Green Chem., 2017, 19, 1990–1998 RSC.
- B. T. Gregg, K. C. Golden and J. F. Quinn, Tetrahedron, 2008, 64, 3287–3295 CrossRef CAS.
- N. S. Radulović and M. S. Nešić, RSC Adv., 2016, 6, 93068–93080 RSC.
- J. L. Dong, L. S. H. Yu and J. W. Xie, ACS Omega, 2018, 3, 4974–4985 CrossRef CAS PubMed.
- H. Yi, L. Niu, S. Wang, T. Liu, A. K. Singh and A. Lei, Org. Lett., 2017, 19, 122–125 CrossRef CAS PubMed.
- C. Gunanathan, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2009, 131, 3146–3147 CrossRef CAS PubMed.
- J. M. Bell, D. G. Kubler, P. Sartwell and R. G. Zepp, J. Org. Chem., 1965, 30, 4284–4292 CrossRef CAS.
- M. J. Huggins and D. G. Kubler, J. Org. Chem., 1972, 40, 2813–2815 CrossRef.
- H. Ohno, M. Ikhlayel, M. Tamura, K. Nakao, K. Suzuki, K. Morita, Y. Kato, K. Tomishige and Y. Fukushima, Green Chem., 2021, 23, 457–469 RSC.
- A. Clerici, N. Pastori and O. Porta, Tetrahedron, 1998, 54, 15679–15690 CrossRef CAS.
- A. Corma, M. I. Juan-Rajadell, J. M. López-Nieto, A. Martinez and C. Martínez, Appl. Catal., A, 1994, 111, 175–189 CrossRef CAS.
- H. Mallin, J. Muschiol, E. Byström and U. T. Bornscheuer, ChemCatChem, 2013, 5, 3529–3532 CrossRef CAS.
- S. Pithani, S. Karlsson, H. Emtenäs and C. T. Öberg, Org. Process Res. Dev., 2019, 23, 1926–1931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00367a |
| This journal is © The Royal Society of Chemistry 2024 |