
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuanidine functionalized porous SiO2 as heterogeneous catalysts for microwave depolymerization of PET and PLA†
Éadaoin
Casey
ab,
Rachel
Breen
ab,
Gerard
Pareras
 c,
Albert
Rimola
c,
Justin D.
Holmes
ab and
Gillian
Collins
*ab
c,
Albert
Rimola
c,
Justin D.
Holmes
ab and
Gillian
Collins
*ab
aSchool of Chemistry, University College Cork, Cork, T12 YN60, Ireland. E-mail: g.collins@ucc.ie; Tel: +353 (0)21 4205143
bAMBER Centre, Environmental Research Institute, University College Cork, Cork, T23 XE10, Ireland
cDepartament de Química, Universitat Autònoma de Barcelona, 08193 Bellaterra, Catalonia, Spain
First published on 6th March 2024
Abstract
Chemical recycling is an important strategy to tackle the growing global problem of plastic waste pollution. The development of metal-free catalysts for depolymerization of plastics is attractive as it avoids the use of metal salts, which are potentially damaging to the environment. Here we report a metal-free heterogeneous catalyst for the glycolysis of polyethylene terephthalate (PET) and methanolysis of polylactic acid (PLA). The catalysts are synthesized by covalent surface modification of mesoporous silica (SiO2) with guanidine ligands and evaluated under conventional thermal and microwave-assisted heating. A surface bound cyanoguanidine ligand was found to be the best catalyst leading to 100% PET conversion with 80% BHET yield. The nature of the catalyst support material influenced the catalytic performance of the guanidine ligands with porous SiO2 supports outperforming activated carbon in conventional thermal glycolysis, while the opposite trend was observed with microwave assisted glycolysis. Dedicated density functional theory (DFT) computations were performed to simulate the depolymerization processes, obtain the free energy profiles of the reaction mechanisms, and identify the important role of hydrogen bonding in the reaction mechanism.
Sustainability spotlightChemical recycling a crucial strategy for the transformation of various types of plastic waste into high value monomers and commodity chemicals, but significant challenges such as high energy requirements and efficient catalysts remain. In this context, in this work heterogeneous metal free catalysts are made by covalent attachment of organocatalysts onto a high surface area SiO2 support that can easily be separated and recycled. The use of microwave assisted heating dramatically improves energy efficiency and lowers reaction times for the depolymerization of polyethylene terephthalate (PET). The catalyst is also used for depolymerization of polylactic acid (PLA). PLA is one of the most important bioplastics, and while it is produced from renewable resources, it is highly persistent in the environment and has poor biodegradability outside industrial composting. Chemical recycling has potential to access value-added compounds through open loop recycling of PLA such as the green solvent methyl lactate. This work highlights the importance of UN sustainable development goals sustainable cities and communities (SDG 11), responsible consumption and production (SDG 12) and climate action (SDG 13). |
1. Introduction
Plastics are ubiquitous to our everyday lives owing to their low cost, versatility, and ease of production. They range from lifelong plastic materials for household appliances, building materials and electronics to single use plastics such as shopping bags, straws, food packaging and water bottles. The high demand for single use plastics, which has increased dramatically in the last 50 years from 15 to 311 million tons and is expected to double by 2035, has led to a drastic increase in plastic waste. In recent years, researchers have developed numerous pathways to convert plastic waste into monomers or value-added products, forming a circular economy in the polymer life cycle.1,2 Polyethylene terephthalate (PET) is a thermoplastic polyester, used in wide ranging applications such as textiles and food and beverage packaging and in single use plastics, which are a key contributor to the problem of post-consumer plastic waste. There has been intensive investigation into this polymer with the aim of sustainably converting it into a valuable monomer, moving away from environmentally detrimental practices such as incineration.3,4 Glycolysis is one of the most effective methods for depolymerization of PET as it can be achieved under mild conditions with a relatively low cost. The method involves reacting PET, excess diol, usually ethylene glycol (EG) and a catalyst, with the product bis(2-hydroxyethyl) terephthalate (BHET) being obtained. Many catalysts are effective for this transformation such as metal oxides,5,6 nanoparticles,7–9 metal salts and metal-containing ionic liquids,5,10,11 and organocatalysts.12–18Metal-free catalysts are especially appealing due to their environmentally friendly and sustainable nature, serving as an advantageous option compared to metal-based catalysts. Many metal-free catalysts are homogeneous organocatalysts and Table S1 in the ESI† shows literature reports using different metal-free catalysts. Fukushima and co-workers19 reported the use of a commercially available guanidine catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a highly effective catalyst for the glycolysis of PET in the presence of EG. PET was fully converted and an isolated yield of 78% was obtained at 190 °C, 10 mol% catalyst, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio PET:EG after 325 min. For comparison, when the reaction was completed in the absence of TBD, PET was fully converted after 40 h at 190 °C. Le et al.20 reported the catalytic activity of TBD in a co-solvent assisted glycolysis reaction where 100% conversion of PET and 82% yield of BHET were achieved at 153 °C after 2 h. While organocatalysts show excellent efficiency for PET depolymerization, a key drawback is the lack of easy recovery, which can result in contamination of the monomer product. A combined metal-free and heterogeneous catalyst design is attractive as it provides an easy separation route after the depolymerization reaction. Chipurici et al.21 demonstrated a synthesis of heterogeneous catalysts with guanidine superbases chemically grafted on silica-coated Fe3O4 magnetic nanoparticles for transesterification of waste oil. Fehér et al.13 reported an eco-friendly organocatalyst using TBD supported on silica gel for PET glycolytic depolymerisation, achieving 100% conversion of PET and 88% yield of BHET. The optimal reaction conditions were 190 °C, 15.5 mol% catalyst, an EG:PET molar ratio of 12.6, and 1.7 h reaction time.
:5 ratio PET:EG after 325 min. For comparison, when the reaction was completed in the absence of TBD, PET was fully converted after 40 h at 190 °C. Le et al.20 reported the catalytic activity of TBD in a co-solvent assisted glycolysis reaction where 100% conversion of PET and 82% yield of BHET were achieved at 153 °C after 2 h. While organocatalysts show excellent efficiency for PET depolymerization, a key drawback is the lack of easy recovery, which can result in contamination of the monomer product. A combined metal-free and heterogeneous catalyst design is attractive as it provides an easy separation route after the depolymerization reaction. Chipurici et al.21 demonstrated a synthesis of heterogeneous catalysts with guanidine superbases chemically grafted on silica-coated Fe3O4 magnetic nanoparticles for transesterification of waste oil. Fehér et al.13 reported an eco-friendly organocatalyst using TBD supported on silica gel for PET glycolytic depolymerisation, achieving 100% conversion of PET and 88% yield of BHET. The optimal reaction conditions were 190 °C, 15.5 mol% catalyst, an EG:PET molar ratio of 12.6, and 1.7 h reaction time.
The use of microwave assisted depolymerization of plastics has also received a lot of interest due to the significant reduction in reaction time and energy consumption, improving the energy efficiency for various reactions such as the glycolysis of PET22,23 and polyurethane24 and hydrolysis of polyamides25 and polyolefins.26 Microwave assisted reactions enable rapid volumetric heating and uniform heat transfer resulting in increased reaction rates and product selectivity and increased yields compared to conventional thermal methods.27 The most common catalysts reported for microwave assisted glycolysis of PET are metal salts and metal oxides, and the possibility of combining microwave assisted heating with a heterogeneous metal-free catalyst is a highly attractive depolymerization route for PET.
Recently, there has been great interest in biodegradable plastics such as polylactic acid (PLA) as an alternative to fossil-fuel derived plastics. The PLA market has increased exponentially in recent years as its physical properties are similar to those of non-biodegradable plastics such as PET. While often branded as an eco-friendly biodegradable plastic, PLA only degrades at an appreciable rate under specific conditions such as those generated in industrial composing facilities. In the natural environment, PLA persists for hundreds of years.28 Therefore, instead of disposing of PLA to composting sites, upcycling strategies are needed to enable more sustainable use of PLA. Methanolysis can be used to transform PLA into methyl lactate (MeLa), which is attractive as a green solvent. Several literature reports have shown inorganic metal salts such as FeCl3 and ZnAc to be active catalysts. Román-Ramírez et al.28 produced MeLa from the chemical degradation of PLA using an ethylenediamine Zn(II) complex. Petrus and co-workers,29 synthesised multiple alkyl lactates using metal containing catalysts at temperatures ranging from 80 to 260 °C and reported that a combination of high temperatures and pressure with excess alcohol leads to an increase in the lactate production and yield.
This article describes the process of creating guanidine-based heterogeneous catalysts, achieved through surface functionalization of porous SiO2 supports and highlights the adaptability of these catalysts in various applications, including PET glycolysis for the production of BHET, butanolysis of PET to form bishydroxybutyl terephthalate (BHBT), and PLA methanolysis to yield MeLA. The catalysts can be used in both conventional thermal and microwave assisted depolymerisation. Density functional theory (DFT) calculations were used to model the surface bound guanidine ligands in the PET and PLA depolymerization reactions to obtain unique atomic scale information on the processes and rationalize the experimental findings.
2. Results and discussion
2.1 Synthesis and characterization of guanidine functionalized SiO2
The use of porous SiO2 as a support enables surface modification via functionalization with organic ligands. Mesoporous cellular foam SiO2 was functionalized with three different guanidine precursors using a one pot method (see the ESI† for experimental details) to produce SiO2-DCG (1), SiO2-EDG (2) and SiO2-DCD (3) catalysts, as illustrated in Fig. 1.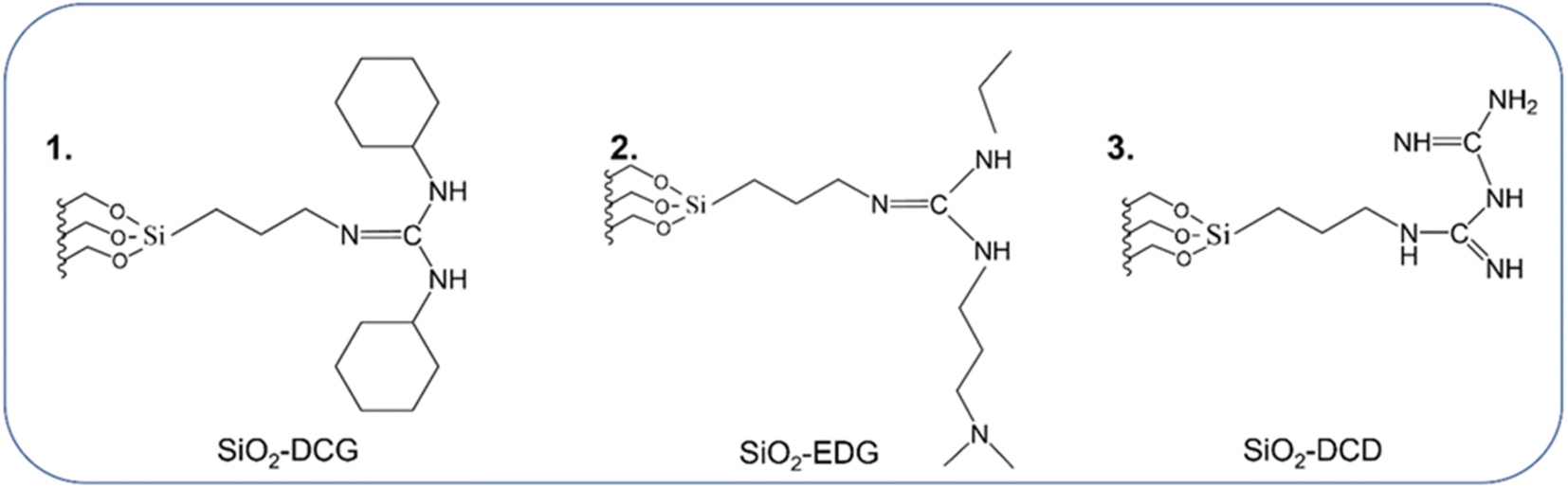 | ||
| Fig. 1 Schematic illustrating the three guanidine-type ligands used to functionalize mesoporous cellular foam SiO2via alkoxysilane attachment. | ||
Surface area and porosity measurements were carried out using N2-physisorption at −196 °C (see the ESI† for details). Fig. 2 shows the N2-physisorption isotherms of bare and modified SiO2. The catalysts showed a typical type-IV isotherm with an H1 hysteresis loop which is a typical feature for this type of large pore mesoporous material. The surface area of the catalyst decreased after functionalization from 412 m2 g−1 for bare SiO2 to 182 m2 g−1, 101 m2 g−1 and 38 m2 g−1, for DGC, ECG and DCD modified SiO2, respectively (see Table S2†). Similar reductions in surface area have been reported in the literature for amino functionalized SBA-15 and MCM-41, indicating that a degree of pore blockage occurred on functionalization.30 The SiO2-DCD catalyst has a substantially reduced surface area, when compared to the -EDG and -DCG ligands, which may be attributed to the smaller size of the DCD ligands. Surface functionalization of SiO2 using alkoxysilane precursors can also produce multi-layered silyated surfaces due to multiple surface attachments and/or silane cross-linking.31Fig. 2(b) also shows the decrease in pore diameter after modification, which is associated with functionalization of the internal pores.30 The pore size of bare SiO2 is 102 Å, which decreases for the SiO2-EDG (82 Å) and SiO2-DCD (79 Å) catalysts but remains similar after DCG modification (100 Å), which may be attributed to the bulky cyclohexyl side groups of the guanidine ligand. Fig. 3(a) shows the scanning electron microscopy (SEM) image of bare SiO2, and Fig. 3(b) shows the transmission electron microscopy (TEM) image of mesoporous cellular foam SiO2. Higher resolution SEM images of MCF, -DCG, -EDG and -DCD modified SiO2 are shown in Fig. 3(c–f), respectively, and ESI Fig. S1.†
 | ||
| Fig. 2 N2-physisorption isotherms of bare and modified SiO2 showing (a) the pore volume and (b) the pore size distribution. | ||
 | ||
| Fig. 3 (a–c) SEM and TEM images of bare mesoporous cellular foam SiO2 and after functionalization with the (d) DCG, (e) EDG and (f) DCD ligands. | ||
XPS was employed to verify successful functionalization of the guanidine ligands on the SiO2 support. The survey spectra show the appearance of N 1s and C 1s peaks after functionalization (ESI Fig. S2†). The N 1s core level shown in Fig. 4(a), of the three catalysts revealed the presence of a dominant peak at a binding energy (BE) of 400.2, 400.2 and 399.8 eV, for SiO2-DCG, EDG and DCD catalysts, respectively, which is characteristic of amine species.32 A small shoulder peak was observed at a higher BE assigned to protonated N species. Covalent attachment of the alkoxysilane ligands is further evidenced by the O 1s and Si 2p core levels, as shown in Fig. 4(b and c). The O 1s of bare SiO2 is centred at 533.8 eV and is downshifted to 532 eV after functionalization, associated with the loss of Si–OH groups due to covalent modification of the surface.33 This downshift in the BE is also observed in the Si 2p core level scans of functionalized SiO2.
 | ||
| Fig. 4 XPS analysis of core level scans (a) N 1s, (b) O 1s and (c) Si 2p for SiO2, SiO2-DCG, SiO2-EDG and SiO2-DCD catalysts. (d) ATR-FTIR spectra of bare and guanidine functionalized SiO2 catalysts. | ||
FTIR was employed to investigate the surface of the modified SiO2 catalysts as shown in Fig. 4(d). Peak assignment in this region can be challenging because of overlapping contributions from several species such as Si–O–Si and Si–O–C, but changes in the spectral region on functionalization are clearly identified.34 Bare SiO2 particles exhibit peaks at 1060 cm−1 and 950 cm−1 which can be indexed to Si–O–Si stretching and the Si–OH bond stretch, respectively.35 For the EDG and DCD catalysts there is a significant decrease in the Si–OH peak and a strong peak emerges at 1038 cm−1, which is in excellent agreement with the Si–O–C stretching vibration. For the DCG catalyst, this peak is less defined and a small decrease in the Si–OH peak is observed, which may indicate a weaker covalent attachment, likely to originate from the bulky side groups. The modified SiO2 catalyst shows a peak at 1636 cm−1 associated with N–H bending,34 correlating well with the N 1s XPS, although peaks in this region have also been attributed to adsorbed water, formed from the condensation reaction of surface Si–OH and the alkoxysilane precursor. The SiO2-DCD ligand has an additional peak at 1560 cm−1 assigned to primary amine N–H stretching vibrations. Both FTIR and XPS indicate covalent attachment of the guanidine ligands on the support.
TGA was used to assess the thermal stability of each of the SiO2 guanidine catalysts and to estimate the amount of organic species present after functionalization, as shown in ESI Fig. S3.† The initial weight loss observed in all the guanidine grafted SiO2 catalysts is attributed to water and residual solvent molecules on the surface. The second weight-loss step can be assigned to the release of organosilanes and the guanidine derivatives from the silica surface. Based on mass loss observed between 120 °C and 350 °C, the organic content was found to be 23%, 20% and 25% for DCG, EGC and DCD functionalized SiO2, respectively. The EDG and DCD catalysts have a slightly higher thermal stability, with mass loss not occurring until ∼250 °C, compared to the DCG ligand where mass loss starts to occur at ∼220 °C.
2.2 Catalytic evaluation of heterogeneous guanidine catalysts
The catalytic performance of the three SiO2 supported guanidine catalysts was investigated for the depolymerization of PET to BHET during conventional thermal and microwave assisted glycolysis. Fig. 5(a–c) compare the reaction time series (0–4 h) for each catalyst, evaluating PET conversion and isolated BHET yields obtained under conventional thermal conditions at 190 °C for 3 h with 100 mg of the catalyst. Bare SiO2 did not catalyse the reaction to any extent. The SiO2-DCD catalyst displayed the best catalytic activity, which during thermal depolymerization led to full PET conversion, and the isolated yield of BHET was 70%. 1H NMR of isolated BHET can be found in ESI Fig. S4.† Analysis of the filtrate after recrystallization revealed residual BHET in the solution, but no dimers were detected by NMR. The superior performance of the SiO2-DCD catalyst may be due to the structure having a more nitrogen-rich environment, which has been reported to be beneficial in increasing the catalytic activity.7,36 The DCD ligands also have less steric constraints compared to the EDG and DCG catalysts, associated with the bulky side groups, and steric effects have been shown to lower the performance of metal containing ionic liquids.22 At longer reaction times, the BHET yield decreased, which is indicative of dimerization of the BHET monomer.37 Increasing the PET:EG ratio decreased conversions under thermal conditions, and the optimal ratio was found to be 1:5.
 | ||
| Fig. 5 Reaction time series for the depolymerization of PET to BHET under conventional thermal heating using optimised conditions (190 °C, PET:EG 1:5) for the (a) DCG, (b) EDG and (c) DCD modified SiO2 catalysts. Reaction time series for microwave assisted heating under optimized conditions (210 °C, PET:EG 1:10) for (d) DCG, (e) EDG and (f) DCD modified SiO2 catalysts. | ||
The catalysts were then evaluated using microwave assisted heating instead of conventional heating. Reactions run under the same conditions and 190 °C, led to full conversion of PET but resulted in an inferior yield of 58%. An optimization study of the reaction parameters for microwave assisted glycolysis was initially performed, as shown in the ESI in Table S3 and Fig. S5.† Different optimal parameters are observed for reactions under thermal and MW assisted heating. It was found that increasing the temperature greatly increased PET conversion while lowering reaction times (30 min). Each of the catalysts performed best at 210 °C and a higher PET:EG ratio (1:10), and sufficient agitation of the reaction mixture by using a high stirring speed (960 rpm) was also important. Fig. 5(b–d) show reaction time series (0–40 min) for each catalyst for evaluating PET conversion and isolated BHET yields obtained under microwave-assisted heating. Optimum reaction conditions were 210 °C, 30 min, 960 rpm, 100 mg catalyst and 1:10 PET:EG ratio, giving an 80% BHET yield. The significant reduction in reaction time associated with efficient microwave heating38 is a clear advantage of using a microwave assisted route.
The cyanoguanidine ligand i.e. SiO2-DCD, emerged as the most effective catalyst in both conventional heating and microwave assisted routes but the catalyst reactivity trend observed during microwave assisted glycolysis was different to that observed during conventional thermal glycolysis. The SiO2-EDG catalyst was sluggish under conventional heating but demonstrated a favourable performance under microwave heating. The SiO2-DCG catalyst performed well under thermal conditions (93% PET conversion) but was the least effective catalyst under microwave conditions, despite the higher temperature, resulting in only 50% PET conversion. TGA showed that the SiO2-DCG ligand had the lowest thermal stability, and this may have contributed to the poor performance of this ligand under microwave assisted heating compared to conventional thermal heating. Microwave heating is fundamentally different from conventional heating because microwaves can penetrate deep into the sample, heating it volumetrically, as opposed to conventional thermal heating, which heats materials from the outside via standard heat-transfer mechanisms, i.e., through convection, conduction and radiation. The acceleration effect of microwaves on reaction kinetics associated with volumetric heating is a well-known and reported phenomenon, and the specific mechanisms occurring within particular reactions often remain unclear.39
The best microwave-responsive catalysts are those that can absorb microwave irradiation, and while SiO2 is a microwave absorber, carbon-based catalysts have also been widely used in microwave assisted reactions such as biomass conversion, phenol oxidation and NOx reduction.40 To evaluate the role of the support material, the poorest performing guanidine ligand during microwave glycolysis, DCG was grafted onto acid-treated activated carbon and evaluated under the same reaction conditions. The PET conversion obtained using the DCG ligand increased from 50% using a SiO2 support to 100% using activated carbon. Interestingly, the same trend was not observed when the reactions were conducted under conventional heating, with PET conversion using carbon supported catalysts decreasing to 63% compared to that using a SiO2 support (93%) (see ESI Fig. S6†). This result demonstrates the important role of the support material in the catalytic behaviour of heterogeneous catalysts for microwave assisted glycolysis compared to conventional heating. While the exact origin of these support effects requires further investigation, they are likely related to differences in the heating characteristics and thermal stability of the two materials under microwave and conventional thermal heating. The recyclability performance of the catalyst was also tested over five reaction cycles for the DCD catalyst under both thermal and MW assisted heating (see ESI Fig. S7†). The MW assisted recyclability test displayed slightly better conversion (100, 100, 94, 90, and 84%) than the thermal heating reactions (100, 100, 90, 85, and 81%). Interestingly, the recyclability performance of DCG was inferior, decreasing over successive cycles. This difference in recyclability performance may be due to the lower thermal stability of the DCG catalyst as indicated by TGA.
Glycolysis of PET using EG to form the valuable BHET monomer has been intensively studied as a key reaction for chemical recycling of PET, but glycolysis of PET can also be carried out using other diols such as propyl glycol, 1,4-butanediol (BD) and triethylene glycol.41 These reactions, while much less explored in comparison to glycolysis with EG, have numerous applications such as the formation of oligomeric precursors that can be used as building blocks to synthesize other polymers with higher economic value such as polyurethane foams and unsaturated polyesters applied as polymer concrete and mortar materials.42,43 One example is the production of the polyester polybutylene terephthalate (PBT) from recycled PET by using BD in place of EG. The use of traditional metal-based catalysts such as metal salts and oxides is problematic, as they efficiently catalyse the cyclodehydration reaction of BD to form THF.44 Therefore, the use of a metal free catalyst offers a distinct advantage to limit this undesired side reaction for the depolymerization pathway, as illustrated in Fig. 6(a). Glycolysis of PET using BD was carried out using the SiO2-DCD catalyst at 210 °C under microwave heating. Optimum mass loss of PET and formation of the desired bis(4-hydroxybutyl) terephthalate (BHBT) product was achieved after 6 h, as shown in the reaction time series in ESI Fig. S8.† The NMR analysis of the crude product shown in Fig. 6(b) confirms the formation of the BHBT product with a singlet at δ = 8.09 ppm corresponding to the aromatic protons. A distinguishable feature of the BHBT NMR spectrum is the presence of 2 quintets at δ = 1.76 and δ = 1.55 ppm arising from CH2 groups, labelled c and d, respectively. A peak arising from the OH protons appears at δ = 4.45 ppm, in good agreement with literature reports for 1H NMR of mono(4-hydroxybutyl) terephthalate.45 The 1H NMR analysis results of THF, 1,4-BD and EG are also shown in Fig. 6(b) and indicate that the undesired cyclodehydration reaction of BD to THF did not occur using the metal-free catalysts.
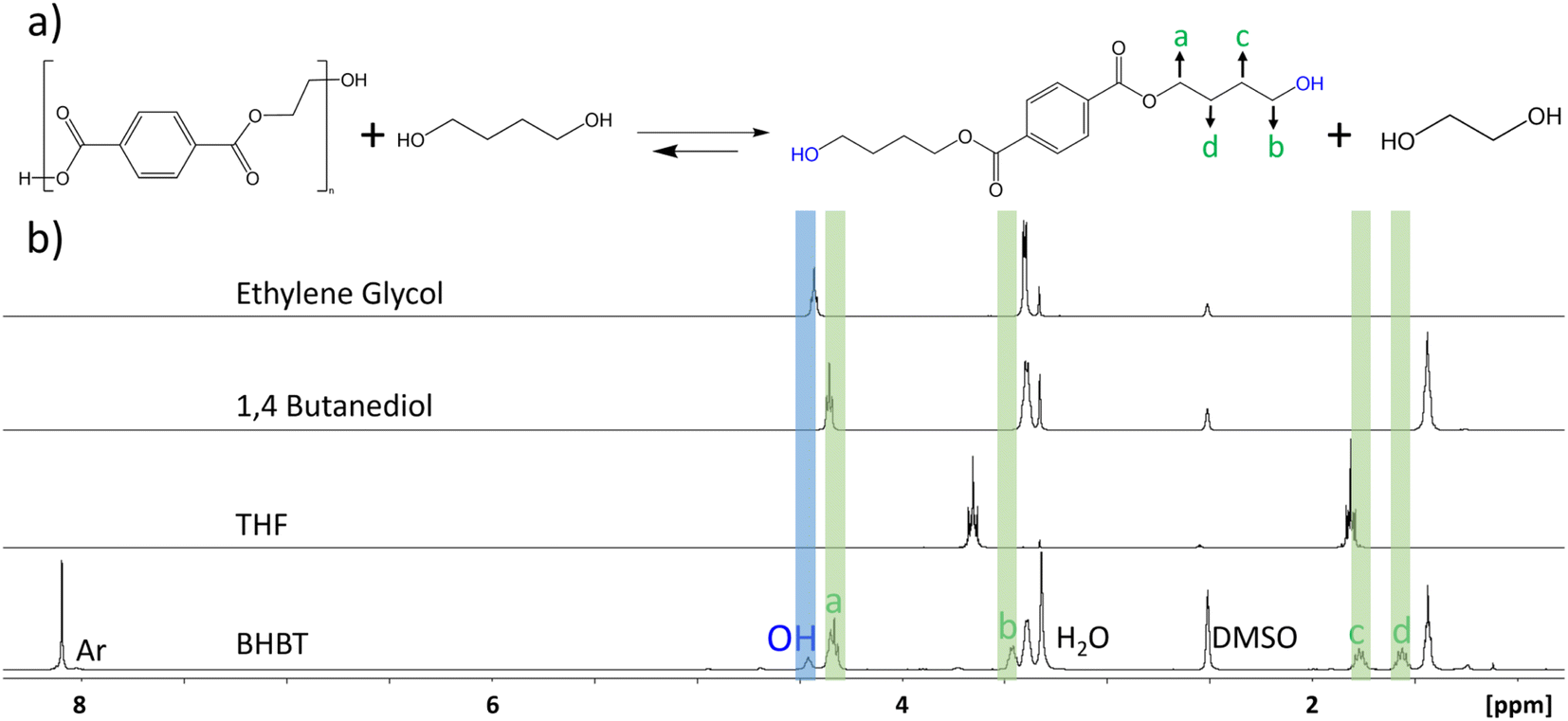 | ||
| Fig. 6 (a) Reaction scheme showing glycolysis of PET using 1,4-butanediol. (b) NMR analysis of the reaction product showing the formation of bis(4-hydroxybutyl) terephthalate (BHBT) and no undesired THF product. | ||
We further investigated the versatility of the metal-free SiO2-guanidine catalysts by evaluating their applicability in the methanolysis of PLA to methyl lactate (MeLa) under both thermal and microwave assisted heating. Methanolysis of PLA is typically carried out using a metal catalyst in a pressurized vessel at 120 °C with reaction times reported up to 3 days.28,46,47 The reaction scheme for depolymerization of PLA to MeLa can be seen in Fig. 7, where methanol (MeOH) and THF are used as the protic source and solvent, respectively. Initially, an optimization study was performed thermally to find the best reaction conditions. The role of THF is to increase the solubility of PLA, and reaction did not proceed in the absence of THF. Depolymerization was carried out under standard reflux conditions but resulted in low PLA conversions due to the low boiling points of MeOH and THF. Therefore, sealed glass vessels were used to achieve higher temperatures for the reaction. Lamberti et al.48 reported that the mechanism is a two-step reversible reaction in which the PLA chains are quickly converted to chain end oligomers, which then slowly form the product MeLa. As the reaction proceeds, the amount of internal oligomers decreased as the polymer chains were degraded, producing more chain end oligomers and subsequently the MeLa monomer. Reactions with a higher yield of MeLa had a lower percentage of internal oligomers and a higher percentage of chain end oligomers as the depolymerization process proceeds, which can be observed with the NMR analysis to evaluate the product distribution, as shown in Fig. 7. The 1H NMR of the products matched previously reported values which are internal (int) methine (5.09–5.21 ppm), chain end methine (CE) (4.30–4.39 ppm/5.09–5.21 ppm), and MeLa (4.23–4.29 ppm).28 Using the sealed glass vessels, under reaction conditions of 130 °C, 3 h, 250 rpm and a 2:3 ratio of MeOH:THF, full conversion of PLA was achieved with the yield of MeLa and both chain end and internal methine oligomers being 17, 54, and 29%, respectively. Román-Ramírez and co-workers,28 achieved a similar MeLa yield using a Zn catalyst under thermal conditions. Due to the equilibrium reaction between the oligomers and MeLa, removing the MeLa as it is formed can shift the reaction equilibrium and increase the yield. There have also been reports of using an autoclave to perform the degradation of PLA to MeLa, where the higher pressure can also increase MeLa yield.48 On carrying out the reaction in an autoclave, it was possible to achieve full conversion of PLA and further increase the yield of MeLa to 42% after 72 h (Fig. 7(c)). The higher MeLa yields and minimal internal methine oligomers demonstrate the beneficial impact of the high pressure on an autoclave reactor; however the use of long reaction times is not desirable. Therefore, the use of microwave assisted methanolysis was next explored, and Table S4 in the ESI,† summarizes the optimization of the reaction conditions. While microwave heating did not lead to further enhancements in MeLa yield, it did significantly reduce the reaction time. After 30 min PLA was fully converted yielding 30, 62, and 8% MeLa and chain end and internal methine oligomers, respectively, which substantially reduces the reaction time.
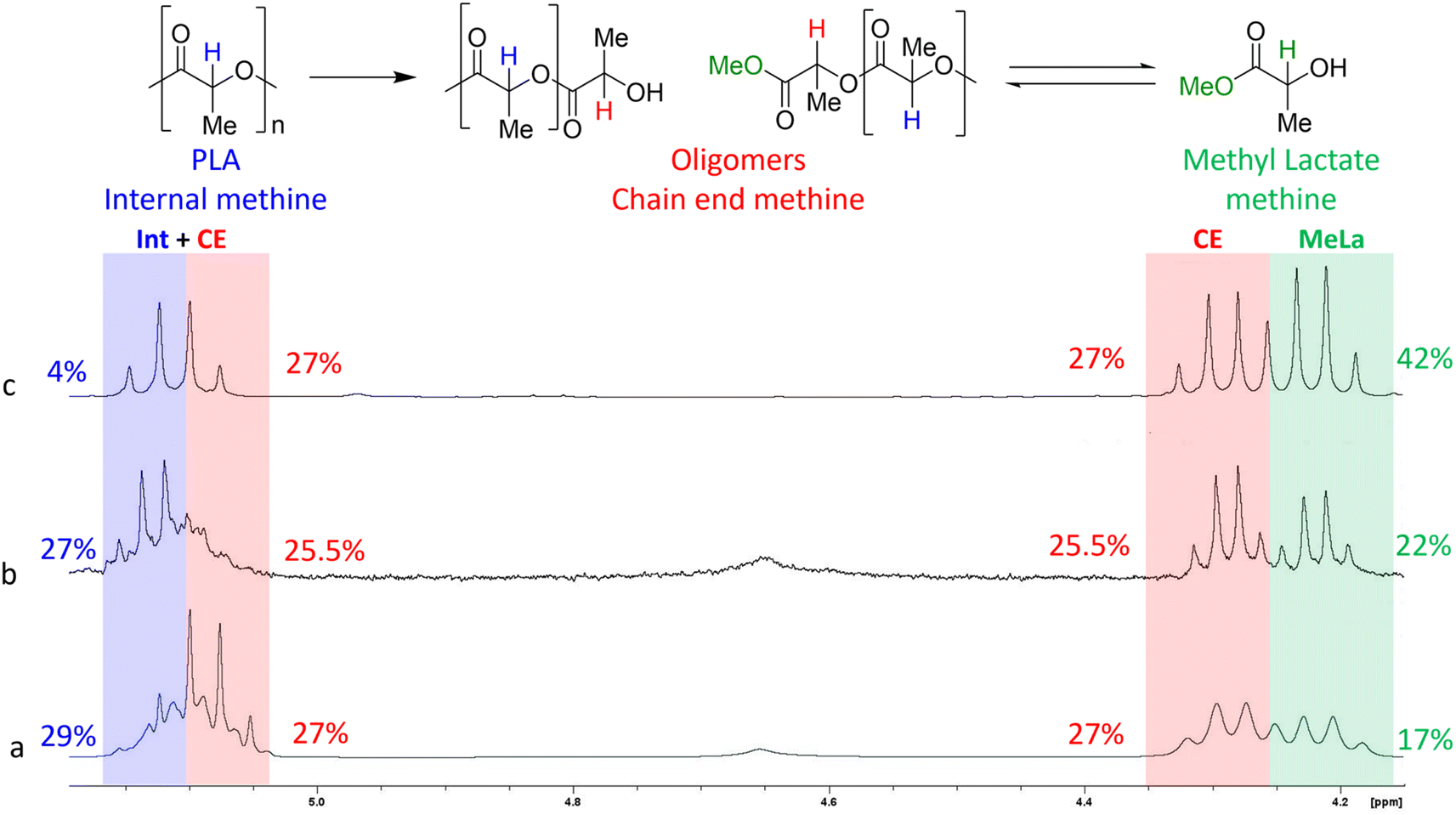 | ||
| Fig. 7 1H NMR stacked spectra of the products obtained from the methanolysis of PLA to MeLa for the (a) sealed vessel reaction at 4 h, (b) 20 h reaction in an autoclave and (c) 72 h autoclave reaction. Each reaction was performed at 130 °C with 0.05 g catalyst loading. | ||
2.3 Mechanistic insights into cyanoguanidine functionalized SiO2 using density functional theory
Surface bound guanidines supported on mesoporous SiO2 and, in particular, dicyandiamide ligands are effective catalyst for PET glycolysis, butanolysis and PLA methanolysis. DFT calculations were carried out to gain further insights into the catalytic role of the most reactive guanidine-derivative. Geometry optimizations were carried out using the semi-local Perdew–Burke–Ernzerhof (PBEsol) function,49 combined with a double-ζ basis set (DZVP-MOLOPT-SR-GTH Gaussian basis set) for all the atom types, together with Grimme's D3(BJ) dispersion correction of the potential energy,50 and a cutoff set at 500 Ry for the plane wave auxiliary basis set. Core electrons were described with the Goedecker–Teter–Hutter pseudopotentials51 and valence ones with a mixed Gaussian and plane-wave (GPW) approach.52To prove the accuracy of the DFT PBE-D3(BJ) method used in this work, a preliminary benchmarking study with a reaction model (see the Experimental section for details and Fig. S9†) was performed. In this benchmark, we optimized the (non-catalysed) reaction model at the PBE-D3(BJ)/DZVP level, and then we performed single point energy calculations at the DLPNO-CCSD(T)53 level of theory. Calculations were performed with the ORCA 5.0.3 programme suite.54 The results illustrated in Table S5† show that the relative error for the energy barrier is only 4.6% (energy barriers of 36.05 and 37.79 kcal mol−1 at PBE-D3(BJ) and DLPNO-CCSD(T), respectively), indicating that the method used is accurate and reliable for predicting the energy barriers of the reactions investigated in this work.
The SiO2-DCD catalyst surface model was constructed by attaching the DCD ligand on the outermost silicon atom of the SiO2 surface. However, guanidine when supported on SiO2 can present two tautomeric forms: one composed of a primary amine, two secondary amines and two imines and the other comprising two primary amines, one secondary amine and two imines, as illustrated in ESI Fig. S10.† Therefore, initial calculations were carried out to identify the more stable configuration, and the results indicate that the system with two primary imines (Fig. S8(b)†) is 5 kcal mol−1 more stable. Therefore, when we refer to the SiO2-DCD catalyst, we are only considering this structure.
The glycolysis of PET is the molecular degradation of the polymer by EG through a trans-esterification reaction, where the C–O ester bond breaks and is transformed into hydroxyl groups forming BHET. In order to simulate the reaction processes, we modelled the polymer considering the oligomer with only two repetitive units of PET (see Fig. 8(A)) and using EG as the solvent, adopting a continuum model (see the Experimental section in the ESI†). Briefly, the reaction proceeds through a transesterification of the ester by EG, where the ester carbon atom is attacked by the lone pair electrons of the hydroxyl group of EG forming a C–O bond, followed by a proton transfer from the hydroxyl group of EG to the ester O atom of PET. This results in the breaking of oligomer 1, forming the BHET product 2, through the transition state TS1–2, as illustrated in Fig. 8(A). The same mechanism is proposed for the SiO2-DCD system, in which the catalyst can activate the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of PET by stabilizing [CO⋯H–N] hydrogen bonds, making the bonds more prone to nucleophilic attack of EG (Fig. 8(B)). The formation of H-bonds has been shown to be beneficial for PET degradation using ionic liquid catalysts.55
O bond of PET by stabilizing [CO⋯H–N] hydrogen bonds, making the bonds more prone to nucleophilic attack of EG (Fig. 8(B)). The formation of H-bonds has been shown to be beneficial for PET degradation using ionic liquid catalysts.55
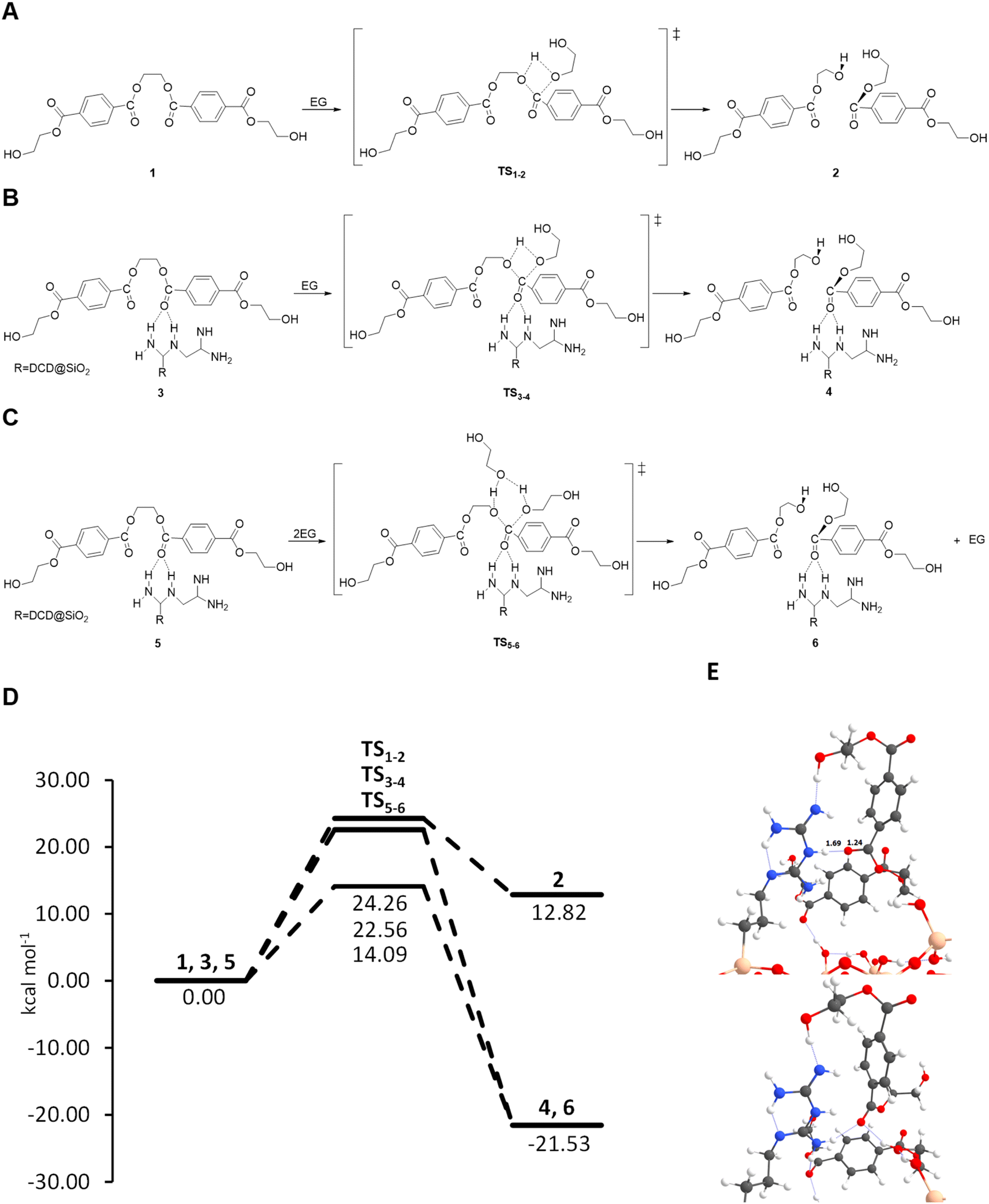 | ||
| Fig. 8 (A–C) Reaction mechanisms proposed for PET depolymerization, uncatalyzed and catalysed by SiO2-DCD, considering one and two units of EG, respectively. (D) Energy profiles presenting the relative Gibbs free energies at 190 °C (in kcal mol−1) considering EG as the solvent for the proposed uncatalyzed and catalysed reaction mechanisms. (E) Optimized structures for reactant 3 and product 4; relevant distances are in Angstroms (Å) and the hydrogen bonds are represented with blue dotted lines. Colour scheme: grey for C, red for O, white for H, blue for N and beige for Si. | ||
Energetic data collected in the energy profiles displayed in Fig. 8(D) show a subtle reduction of the free energy barrier of around 2 kcal mol−1 for the SiO2-DCD system compared to that of the uncatalyzed process. As mentioned above, the catalytic activity of the DCD ligand is associated with activation of the CO bond of the oligomer via hydrogen bonds with the amino groups. This activation is relatively low, as the H-bonds do not significantly perturb the CO bond length (1.24 and 1.22 Å in SiO2-DCD and isolated DCD, respectively), see Fig. 8(E). A more significant effect relates to the change in the thermodynamics. While the uncatalyzed reaction shows an endergonic character, the catalyzed reaction is exergonic, with a favourable reaction free energy of −21.53 kcal mol−1 at 190 °C. This change in the thermodynamics is due to the different anchoring positions offered by the amino groups on the SiO2 surface that generate a net gain in hydrogen bonds with the newly formed BHET product, thereby inducing an extra stabilization effect, which is not possible in the uncatalyzed reaction.
The results show that SiO2-DCD alone, although favouring the thermodynamics with respect to the uncatalyzed systems, does not exert strong catalytic effects since it does not induce a significant energy barrier decrease, with a relatively high free energy barrier (22.56 kcal mol−1). This does align with the experimental observations, which demonstrate fast PET degradation in the presence of the catalyst. To rationalise this, the role of the EG in the reaction was next considered. EG behaves as the reactant and solvent and can directly participate in the proton transfer elementary step. Without accounting for the participation of the solvent, the transition state presents a 4-membered ring, which is geometrically highly strained, as illustrated in Fig. 8(B), and accordingly inidcates a high energy barrier. In contrast, if EG can assist the proton transfer by receiving one proton and donating another one (establishing a proton relay mechanism) the TS structure presents a 6th-membered ring, which is geometrically less strained, thereby presenting a lower energy barrier (Fig. 8(C)). To evaluate the role of EG, a benchmarking study with n = 2, 3 and 4 EG molecules has been carried out with the uncatalyzed system. The results indicate that with two units of EG there is a reduction of the barrier by 5 kcal mol−1. Interestingly, an increase of the number of EG molecules does not further decrease the free energy barrier, which remains at ca. 17 kcal mol−1, as the barriers show a reduction of 7.20 and 7.83 kcal mol−1 (three and four EG units, respectively) compared with that of the uncatalyzed reaction (see Table S6 in the ESI†). After assessing the crucial role of EG, we next computed the reaction on the SiO2-DCD catalysts considering two EG molecules, and the results indicate a decrease in the free energy barrier to 14.09 kcal mol−1, correlating with the experimental findings.
2.4 PLA degradation mechanism
Similar to PET degradation, PLA depolymerization by the addition of protic low molecular weight alcohols, in this case MeOH, proceeds by the transesterification of PLA. As a first step, PLA breaks forming the oligomer chain end methine, followed by a second transesterification of the oligomer, finally leading to the MeLa product. The reaction process modelled here to assess the catalytic role of SiO2-DCD is focused only on the last step of depolymerization, i.e. the oligomer cleavage in two units of MeLa, using THF as the solvent, and adopting a continuum model (see the Experimental section for computational details). The reaction proceeds through nucleophilic attack of the carbonyl group by the lone pair electrons on the hydroxyl group of MeOH, which is followed by a proton transfer from MeOH to the ester O, resulting in breaking of oligomer 1 forming the MeLa product 2 (see Fig. 9(A)). | ||
| Fig. 9 (A–C) Reaction mechanisms proposed for the PLA depolymerization uncatalyzed and catalysed by SiO2-DCD considering one and two units of EG, respectively. (D) Energy profiles presenting the relative Gibbs free energies at 130 °C (in kcal mol−1) considering THF as the solvent, for the proposed uncatalyzed and catalysed reaction mechanisms. (E) Optimized structures for reactant 3 and product 4; relevant distances are in Angstroms (Å) and the hydrogen bonds are represented with blue dotted lines. Colour scheme: grey for C, red for O, white for H, blue for N and beige for Si. | ||
The energy profile shown in Fig. 9(D) reveals that the free energy barrier for the transesterification process is reduced by ∼2 kcal mol−1 when using the SiO2-DCD system with respect to that of the uncatalyzed process. Similar to the PET mechanism, the capability of the SiO2-DCD catalyst to activate the CO bond in PLA is rather limited; in this case the CO bond is enlarged from 1.23 Å in isolated PLA to 1.24 Å in SiO2-DCD, which is translated to a slight energy barrier decrease (see Fig. 9(E)). Despite the small reaction enhancement, the presence of the catalyst also has an effect on the thermodynamics of the process. As mentioned above, the breaking of the oligomer to finally form the MeLa product is an equilibrium reaction, which is confirmed by the computed energy values for the uncatalyzed process (i.e., slightly endergonic, with a reaction free energy at 190 °C of +2.06 kcal mol−1). However, in the presence of the SiO2-DCD catalyst, the MeLa product is stabilized, now presenting an exergonic character with a reaction free energy of −2.02 kcal mol−1. Similar to PET degradation, the SiO2-DCD surface provides different amino binding positions, creating additional hydrogen bonds with MeLa (Fig. 9(E)), therefore providing extra stabilization of the product. However, for PLA, the product stabilization is less significant compared to that for PET degradation, and the equilibrium between the oligomer chains and the MeLa product is still present. Despite the slight decrease in the reaction energy barrier, this reaction can also be catalysed by the MeOH solvent which can participate in a proton relay mechanism due to being a protic solvent with the capability to exchange protons. We simulated this process considering the participation of two units of MeOH (one being the reactant and the other the solvent proton transfer assistant) and found that the new barrier is located at 6.36 kcal mol−1 (see Fig. 9(D)). Similar to that observed for PET degradation, the participation of the reactant in the proton transfer step plays an important role in the mechanism.
3. Conclusions
This work demonstrates that SiO2 and carbon supports covalently functionalized with different guanidine ligands are effective heterogeneous metal free catalysts for the depolymerization of PET and PLA. The three catalysts displayed good catalytic activity with high PET conversions by both thermal and microwave assisted methods. Different optimal conditions were found for the catalysts under conventional heating and microwave assisted heating. The SiO2-DCD catalyst, which is a cyanoguanidine ligand, performed the best, and this is likely due to its nitrogen rich environment. The nature of the support material influenced the catalytic performance, and the use of activated carbon as a support material was beneficial for microwave-assisted glycolysis. To assess the versatility of the surface immobilized guanidine catalysts, the methanolysis of PLA to MeLa, was demonstrated with microwave assisted heating greatly improving the reaction time. DFT simulations provide a deep understanding of the reaction mechanisms for both PET and PLA degradation processes, concretely unveiling the role of the SiO2-DCD catalyst, which through its ability to create a net of hydrogen bonds is not only able to enhance the reaction processes kinetically but also thermodynamically by stabilizing both BHET and MeLA products. The catalytic role of the corresponding additional reactive molecules (EG and MeOH) is also highlighted when they participate in the reaction by assisting proton transfer processes, which inidcates an additional reduction of the free energy barriers. This work demonstrates that guanidine functionalized SiO2 based catalysts are versatile heterogeneous catalysts for chemical recycling of both PET and PLA to high quality monomers and commodity chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Science Foundation Ireland (AMBER Grant No. 12/RC2278_P2). G. P. and A. R. gratefully acknowledge the computer resources and technical support provided by the Barcelona Supercomputing Centre (CNS-BSC) and the Consorci de Serveis Universitaris de Catalunya (CSUC). We thank the Advanced Microscopy Laboratory at Trinity College Dublin for TEM analysis, Bernal Institute University of Limerick for access to XPS facilities and Tyndall National Institute for access to SEM facilities.References
- R. Banu and G. Sharmila, Catal. Sci. Technol., 2023, 13, 2291–2302 RSC.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- Y. Luo, E. Selvam, D. G. Vlachos and M. Ierapetritou, ACS Sustainable Chem. Eng., 2023, 11, 4209–4218 CrossRef CAS.
- F. Cao, L. Wang, R. Zheng, L. Guo, Y. Chen and X. Qian, RSC Adv., 2022, 12, 31564–31576 RSC.
- A. Bohre, P. R. Jadhao, K. Tripathi, K. K. Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142 CrossRef CAS PubMed.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, ACS Eng. Au, 2023, 3, 37–44 CrossRef CAS PubMed.
- É. Casey, R. Breen, J. S. Gómez, A. P. M. Kentgens, G. Pareras, A. Rimola, J. D. Holmes and G. Collins, ACS Sustainable Chem. Eng., 2023, 11, 15544–15555 CrossRef.
- Y. Jo, E. J. Kim, J. Kim and K. An, Green Chem., 2023, 25, 8160–8171 RSC.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- M. Khoonkari, A. H. Haghighi, Y. Sefidbakht, K. Shekoohi and A. Ghaderian, Int. J. Polym. Sci., 2015, 2015, 124524 Search PubMed.
- N. G. Bush, C. H. Dinh, C. L. Catterton and M. E. Fieser, RSC Sustainability, 2023, 1, 938–947 RSC.
- L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustainable Chem. Eng., 2020, 8, 13362–13368 CrossRef CAS.
- Z. Fehér, J. Kiss, P. Kisszékelyi, J. Molnár, P. Huszthy, L. Kárpáti and J. Kupai, Green Chem., 2022, 24, 8447–8459 RSC.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- N. E. Kamber, Y. Tsujii, K. Keets, R. M. Waymouth, R. C. Pratt, G. W. Nyce and J. L. Hedrick, J. Chem. Educ., 2010, 87, 519–521 CrossRef CAS.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122–3131 RSC.
- S. Kaiho, A. A. R. Hmayed, K. R. Delle Chiaie, J. C. Worch and A. P. Dove, Macromolecules, 2022, 55, 10628–10639 CrossRef CAS.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- N. H. Le, T. T. Ngoc Van, B. Shong and J. Cho, ACS Sustainable Chem. Eng., 2022, 10(51), 17261–17273 CrossRef CAS.
- P. Chipurici, A. Vlaicu, I. Călinescu, M. Vînătoru, C. Busuioc, A. Dinescu, A. Ghebaur, E. Rusen, G. Voicu, M. Ignat and A. Diacon, Sci. Rep., 2021, 11, 17518 CrossRef CAS PubMed.
- F. Scé, I. Cano, C. Martin, G. Beobide, Ó. Castillo and I. de Pedro, New J. Chem., 2019, 43, 3476–3485 RSC.
- E. Selvam, Y. Luo, M. Ierapetritou, R. F. Lobo and D. G. Vlachos, Catal. Today, 2023, 418, 114124 CrossRef CAS.
- R. Donadini, C. Boaretti, A. Lorenzetti, M. Roso, D. Penzo, E. Dal Lago and M. Modesti, ACS Omega, 2023, 8, 4655–4666 CrossRef CAS PubMed.
- U. Češarek, D. Pahovnik and E. Žagar, ACS Sustainable Chem. Eng., 2020, 8, 16274–16282 CrossRef PubMed.
- E. Selvam, P. A. Kots, B. Hernandez, A. Malhotra, W. Chen, J. M. Catala-Civera, J. Santamaria, M. Ierapetritou and D. G. Vlachos, Chem. Eng. J., 2023, 454, 140332 CrossRef CAS.
- P. D. Muley, Y. Wang, J. Hu and D. Shekhawat, in Catalysis: Volume 33, The Royal Society of Chemistry, 2021, vol. 33, pp. 1–37 Search PubMed.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- A. K. Meka, A. Gopalakrishna, C. Iriarte-Mesa, P. Rewatkar, Z. Qu, X. Wu, Y. Cao, I. Prasadam, T. I. Janjua, F. Kleitz, T. Kumeria and A. Popat, Mol. Pharm., 2023, 20, 2966–2977 CrossRef CAS PubMed.
- W. Yoshida, R. P. Castro, J.-D. Jou and Y. Cohen, Langmuir, 2001, 17, 5882–5888 CrossRef CAS.
- F. Truica-Marasescu and M. R. Wertheimer, Plasma Processes Polym., 2008, 5, 44–57 CrossRef CAS.
- N. Paengjun, K. Vibulyaseak and M. Ogawa, Sci. Rep., 2021, 11, 3210 CrossRef CAS PubMed.
- R. Tian, O. Seitz, M. Li, W. Hu, Y. J. Chabal and J. Gao, Langmuir, 2010, 26, 4563–4566 CrossRef CAS PubMed.
- R. M. Pasternack, S. Rivillon Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963–12971 CrossRef CAS PubMed.
- J. Zhang, Z. Huang, Y. Xie and X. Jiang, Chem. Sci., 2022, 13, 1080–1087 RSC.
- S. Lalhmangaihzuala, Z. Laldinpuii, C. Lalmuanpuia and K. Vanlaldinpuia, Polymers, 2021, 13(1), 37 CrossRef CAS PubMed.
- A. de la Hoz, Á. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- V. Palma, D. Barba, M. Cortese, M. Martino, S. Renda and E. Meloni, Catalysts, 2020, 10, 246 CrossRef CAS.
- T. Ji and J. Zhu, in Responsive Nanomaterials for Sustainable Applications, ed. Z. Sun and T. Liao, Springer International Publishing, Cham, 2020, pp. 65–91 Search PubMed.
- S. H. Mansour and N. E. Ikladious, Polym. Test., 2002, 21, 497–505 CrossRef CAS.
- M. Y. Abdelaal, T. R. Sobahi and M. S. I. Makki, Constr. Build. Mater., 2011, 25, 3267–3271 CrossRef.
- Y. Öztürk and G. Güçlü, Polym.-Plast. Technol. Eng., 2005, 43, 1539–1552 CrossRef.
- M. Cho, J. Yang, S. Noh, H. Joe and M. Han, Korean Chem. Eng. Res., 2016, 54, 437–442 CrossRef CAS.
- V. Perz, K. Bleymaier, C. Sinkel, U. Kueper, M. Bonnekessel, D. Ribitsch and G. M. Guebitz, Data Brief, 2016, 7, 291–298 CrossRef PubMed.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- C. Alberti and S. Enthaler, ChemistrySelect, 2020, 5, 14759–14763 CrossRef CAS.
- F. M. Lamberti, A. Ingram and J. Wood, Processes, 2021, 9(6), 921 CrossRef CAS.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- H. Yao, X. Lu, L. Ji, X. Tan and S. Zhang, Ind. Eng. Chem. Res., 2021, 60, 4180–4188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00425b |
| This journal is © The Royal Society of Chemistry 2024 |