
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct chemoselective reduction of plant oils using silane catalysed by Rh(III) complexes at ambient temperature†
Unai
Prieto-Pascual
 a,
Itxaso
Bustos
a,
Zoraida
Freixa
*ab,
Amit
Kumar
*c and
Miguel A.
Huertos
*ab
a,
Itxaso
Bustos
a,
Zoraida
Freixa
*ab,
Amit
Kumar
*c and
Miguel A.
Huertos
*ab
aFacultad de Química, Universidad del País Vasco (UPV/EHU), 20018, San Sebastián, Spain. E-mail: miguelangel.huertos@ehu.es
bIKERBASQUE, Basque Fundation for Science, 48013, Bilbao, Spain
cEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews KY169ST, UK. E-mail: ak336@st-andrews.ac.uk
First published on 21st February 2024
Abstract
We report here a rare example of direct and chemoselective catalytic reduction of various plant oils to unsaturated fatty alcohols and glycerol. The selective reduction process is achieved using 0.5 mol% of a rhodium complex and diphenylsilane at room temperature without using any solvent producing a mixture of unsaturated fatty alcohols in yields >90%. We also observed that the selectivity of the reduction process (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vs. ester) depends on the nature of silane.
C vs. ester) depends on the nature of silane.
Sustainability spotlightAs the world pledges to achieve Net Zero by 2050, the chemical industry faces the significant challenge of rapidly developing new sustainable methods to produce chemicals, fuels and materials from renewable feedstock. Plant oils are important renewable feedstock containing olefin and ester functional groups presenting opportunities for the production of valuable feedstock. We report here a new methodology to directly transform plant oils (olive, coconut, sesame, castor, sunflower oil) to unsaturated fatty alcohols using a rhodium catalyst and silane at ambient temperature without using any solvent. In industry, this process is achieved using two steps and harsh reaction conditions (e.g. 250–350 °C and 100–200 bar). This work addresses UN SDG 12 (Responsible Consumption and Production) and SDG 13 (Climate Action). |
Introduction
Plant oils are important resources produced globally at the scale of 200 million metric tons annually, more than 15% of which is used as industrial feedstock for a range of products such as biodiesel, soap, detergents, lubricants, surfactants and cosmetics.1 With the recent drive in the circular economy, the utilisation of plant oil feedstock to produce petrochemicals has become an attractive approach to divest from fossil oil. The presence of ester and alkene groups in plant oils offers substantial opportunities for chemical manipulation. Accordingly, the transformation of plant oils using several reactions such as reduction, hydrosilylation, epoxidation, metathesis, polymerisation and hydroformylation has been developed in the past.2 Reduction of plant oils and their derivatives, such as fatty acids and their esters, has been a topic of interest for the past few decades. Metal catalysed hydrogenation of double bonds (partially or fully) produces hydrogenated vegetable oils of high interest in the food industry (Pathway A, Fig. 1).3 Further hydrogenation in the presence of a metal catalyst leads to the deoxygenative reduction by the elimination of propane and water to form a long alkane chain also known as green diesel (Pathway B, Fig. 1).4 | ||
| Fig. 1 Different pathways for the reduction of plant oils and the structure of the [Rh(SiSiBu)] complex used in this study. | ||
Another set of products obtained from vegetable oils are fatty alcohols which have a global market size of more than £5 million and have a wide range of applications ranging from industrial/domestic cleaning products to personal care products, food and nutrition additives, plasticisers, detergents and lubricants.5 Unsaturated fatty alcohols can have several advantages over saturated ones as they can have lower melting points, better solubility, and can undergo functional group transformation. They are used as lubricants, additives, and feedstock in agrochemicals.5 The industrial production of fatty alcohols from plant oils is carried out in two steps. First, the transesterification with methanol leads to the formation of fatty acid methyl ester (FAME) which is subsequently hydrogenated to form unsaturated fatty alcohol (Pathway C, Fig. 1). Several heterogeneous and homogeneous catalysts have been reported for the latter step.6 Commercially, heterogeneous catalysts involving Cu/Cr are used under harsh reaction conditions (250–350 °C and 100–200 bar).7
Direct reduction of plant oils or fatty acids to fatty alcohols is challenging and requires corrosion-resistant materials for the reactors, acid-resistant, and glycerol-tolerant catalysts, and harsh reaction conditions producing a large amount of hydrocarbon by-products.5 For example, a patent application claimed the demonstration of this process at 20 to 300 bar H2 and 160 to 250 °C.8 Recently, Zhang and co-workers have reported the direct hydrogenation of Jatropha oil to fatty alcohol using a cobalt-based catalyst.9 However, the reaction conditions were harsh (190 °C, and 40 bar H2) and the major product was the saturated alcohol. Elsevier and de Bruin also reported an example of the hydrogenation of a triglyceride to fatty alcohol using a cobalt-triphos-based homogeneous catalyst at milder conditions (100 °C, 80 bar H2). However, also in this case the saturated alcohol was obtained as the product.10 To the best of our knowledge, the direct reduction of plant oils to unsaturated fatty alcohols is not known in peer-reviewed literature.
We hypothesise that the reason for the lack of selectivity in the direct reduction of plant oils to unsaturated fatty alcohols is the high temperatures required for the ester reduction. To circumvent this issue, we propose the use of a silane-based reducing agent for this process. Silanes have been used for the reduction of esters to produce ethers,11 arenes (from aryl esters)12 and aldehydes,13 using transition metal catalysts. The reduction of esters to alcohols using silanes has also been reported in the presence of rhodium,14 ruthenium,15 molybdenum,16 iron,17 titanium,18 cobalt19 and zinc.20 Recently, silanes have also been used for the reductive depolymerisation of polyesters using iridium,21 molybdenum,22 and zinc-based23 catalysts to make diols, and for the hydrosilylation of plant oils or fatty acid esters to make silylated esters with potential applications in lubricants.24
We herein report a direct and selective reduction of fatty acid esters and plant oils at ambient temperature using silane as the reducing agent.
Results and discussion
We have recently reported cationic unsaturated hydrido-silyl-rhodium(III) complexes bearing Si,S bidentate ligands as catalysts for the tandem isomerisation–hydrosilylation of alkenes to make linear silanes under mild reaction conditions (room temperature and solvent-free).25 Our studies also demonstrated that ligands with larger steric bulk on the thioether facilitate this process, with the cationic rhodium(III) complex {Rh(H)[SiMe2(o-C6H4SiBu)](PPh3)2}[BArF4] ([Rh(SiSiBu)]) being the most efficient one from the series.25b We therefore decided to use this complex to study its activity and selectivity in the reduction of fatty acid esters and plant oils.We started our investigation by screening the reactivity using simple model substrates (alkenes and esters). Initially, we studied the reactivity of a terminal (1-octene) and an internal (cis-2-octene) alkene towards silanes in the presence of [Rh(SiSiBu)] catalyst (0.5 mol%) at room temperature for 2 hours under neat conditions.25b These have been used as standard reaction conditions in this study. As described in Table 1, using Et3SiH, both substrates transformed into triethyl(octyl)silane with good yields (Table 1, entries 1 and 2).25b However, when Ph3SiH was used, no conversion towards hydrosilylated products was observed (Table 1, entries 3 and 4). Interestingly, when Ph2SiH2 was used, 1-octene was hydrosilylated to form octyldiphenylsilane in >99% yield (Table 1, entry 5) but no hydrosilylation of cis-2-octene was observed (Table 1, entry 6).
|
|
|||
|---|---|---|---|
| Entry | Silane | Alkene | Hydrosilylated product (yield)b |
| a Reaction conditions: alkene (0.25 mmol), R3SiH (0.25 mmol), catalyst 0.5 mol%, solvent-free, 298 K, 2 h. b Yields were calculated based on 1H NMR (CDCl3) analysis using 1,2-dichloroethane (0.0625 mmol) as an internal standard. c Results from previous work.25b | |||
| 1 | Et3SiH | 1-Octene |

|
| (78%) | |||
| 2c | Et3SiH | cis-2-Octene |

|
| (85%) | |||
| 3 | Ph3SiH | 1-Octene | 0% |
| 4 | Ph3SiH | cis-2-Octene | 0% |
| 5 | Ph2SiH2 | 1-Octene |

|
| (>99%) | |||
| 6 | Ph2SiH2 | cis-2-Octene | 0% |

To analyse the ability of this system to catalyse the reduction of an ester, we used ethyl acetate as a model substrate. Aiming for a full reduction of the ester, we used two equivalents of the different hydrosilanes, under standard conditions. When tertiary silanes were used (Et3SiH or Ph3SiH), no conversion was observed. Instead, Ph2SiH2 led to the formation of two equivalents of ethoxydiphenylsilane in 90% yield (Scheme 1a). Additionally, a trace amount of acetaldehyde was observed (Fig. S12, ESI†). We suggest that the reduction occurs in a stepwise manner; after the initial 1,2-hydrosilylation of the ester to form the silyl-protected acetal26 (1-ethoxyethoxy)diphenylsilane, C–O bond cleavage results in a first equivalent of ethoxydiphenylsilane and acetaldehyde. The latter is further hydrosilylated to form a second equivalent of ethoxydiphenylsilane (Scheme 1b).26
 | ||
| Scheme 1 (a) Reaction of ethyl acetate (0.25 mmol) with hydrosilanes (0.5 mmol) using a [Rh(SiSiBu)] complex (0.5 mol%) at 298 K (neat, 16 h). Yields were calculated based on 1H NMR (CDCl3) analysis using 1,2-dichloroethane (0.0625 mmol) as an internal standard. (b) Proposed reaction pathways for the reduction of ethyl acetate using Ph2SiH2. | ||
These preliminary studies on simple substrates suggest that the reactivity of different substrates (internal or terminal alkenes and esters) towards hydrosilylation and/or reduction is strongly dependent on the nature of the silane. Apparently, regardless of the silane used, anti-Markovnikov hydrosilylation of terminal alkenes always occurs. On the other hand for internal alkenes, the formation of the terminal alkyl silane by the tandem isomerisation-hydrosilylation reaction only occurs when using triethylsilane. Finally, ester reduction appears to occur only when diphenylsilane is used. Therefore, a judicious selection of the hydrosilane can be used to control the chemoselectivity in more complex substrates containing both functional groups (i.e. vegetable oils), using [Rh(SiSiBu)] as a catalyst.
To test this hypothesis, we investigated the reactivity of a terminal alkene containing an ester functional group using different loadings of the most reactive silanes (Et3SiH and Ph2SiH2). The reaction of methyl 5-hexenoate with Et3SiH in the presence of 0.5 mol% [Rh(SiSiBu)] led to the complete and selective anti-Markovnikov hydrosilylation of the terminal alkene independently of the amount of hydrosilane used (Scheme 2a and b, Fig. S14 and S15 in the ESI†). Accordingly, the 1H NMR spectrum of the crude reaction mixture showed the formation of compound 1 and unreacted Et3SiH, when an excess (3 equivalents) of triethylsilane was used. This result is consistent with the reactivity pattern described before for the simpler substrates.
 | ||
| Scheme 2 Reaction of methyl 5-hexenoate (0.25 mmol) with Et3SiH (a, 0.25 mmol; b, 0.75 mmol) and Ph2SiH2 (c, 0.25 mmol; d, 0.75 mmol). Yields were calculated based on 1H NMR (CDCl3) analysis using 1,2-dichloroethane (0.0625 mmol) as an internal standard. | ||
Interestingly, when Ph2SiH2 was used as a reducing agent, which was previously shown to be reactive to both terminal alkenes and esters, the main reaction product depended on the equivalents of hydrosilane employed. When 1 equivalent was used, the 1H NMR spectrum of the crude reaction mixture showed the chemo- and regio-selective hydrosilylation of the terminal alkene (forming compound 2). The ester group remained unreacted (Scheme 2c, Fig. S16 in the ESI†). Instead, when 3 equivalents of diphenylsilane were used, compound 3 was obtained (Scheme 2d, Fig. S17 in the ESI†). 3 results from the hydrosilylation of the terminal alkene and the double hydrosilylation of the ester. Accordingly, an equimolecular amount of methoxydiphenylsilane and a small amount of the intermediate aldehyde (<1%) were also present in the 1H NMR spectrum of the crude reaction mixture. These results suggest that, when Ph2SiH2 was used, the hydrosilylation occurred through a stepwise process: first the hydrosilylation of the terminal alkene followed by the reduction of the ester (Fig. S17 in the ESI†).
Increasing the complexity of the substrate, we studied the reactivity of the [Rh(SiSiBu)] towards the reduction of a fatty acid ester (ethyl oleate) containing an internal alkene. As expected, using Ph2SiH2 (2 equivalents), the chemoselective reduction of ester towards the silyl ether 4 was observed in 70% yield (Scheme 3). The 1H NMR spectrum of the crude reaction mixture (Fig. S19 in the ESI†) also showed the presence of a small amount of aldehyde (4%) and silyl-protected acetal (8%). These results suggest a mechanism analogous to that proposed for the reduction of ethyl acetate (Scheme 1b).
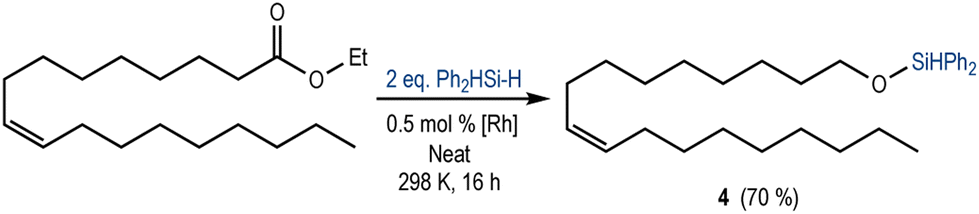 | ||
| Scheme 3 Catalytic hydrosilylation of ethyl oleate (0.25 mmol) using 0.50 mmol of Ph2SiH2. Yields were calculated based on 1H NMR (CDCl3) analysis using 1,2-dichloroethane (0.065 mmol) as an internal standard. | ||
We decided to apply this catalytic protocol towards the chemoselective reduction of plant oils, as most of them contain internal CC bonds and ester functional groups. We initially explored the use of olive oil as a substrate. Olive oil is mainly formed by triglyceride esters of linoleic, palmitic and oleic acid, the latter being the most abundant one. Olive oil was reacted with 6 equivalents of Ph2SiH2 under standard conditions. Interestingly, the 1H NMR spectra of the reaction crude after 16 h showed ∼78% conversion of olive oil to a mixture of aldehydes (∼13%), and silyl ethers (∼65%) originating from the three triglyceride chains (Table 2, entry 1; Fig. S21 in the ESI†). As expected, the CC bond was found to be unreacted and the selectivity towards silyl ether was found to be 100%. When the reaction time was extended to 72 h, the 1H NMR spectrum of the crude reaction mixture in CDCl3 showed an almost complete consumption of the olive oil, and the formation of the corresponding silyl ethers in quantitative yields (Fig. 2 and S22 in the ESI†). Hydrolysis of these silyl ethers under basic conditions resulted in the formation of the desired fatty alcohols as well as glycerol in quantitative yields (Fig. 2, S23 and S24 in the ESI†).
|
|
|||
|---|---|---|---|
| Entry | Plant oil | Aldehydes (% yield) | Silyl ethers (% yield) |
| a Reaction conditions: oil (0.1 mmol), Ph2SiH2 (0.2 mmol), catalyst 0.5 mol%, solvent-free, at room temperature. The amount of products (16 hours) is estimated based on 1H NMR (CDCl3) analysis using 0.05 mmol of 1,1,2,2-tetrachloroethane as an internal standard. | |||
| 1 | Olive | 13 | 65 |
| 2 | Coconut | 28 | 51 |
| 3 | Castor | 22 | 65 |
| 4 | Sesame | 24 | 71 |
| 5 | Sunflower | 25 | 64 |
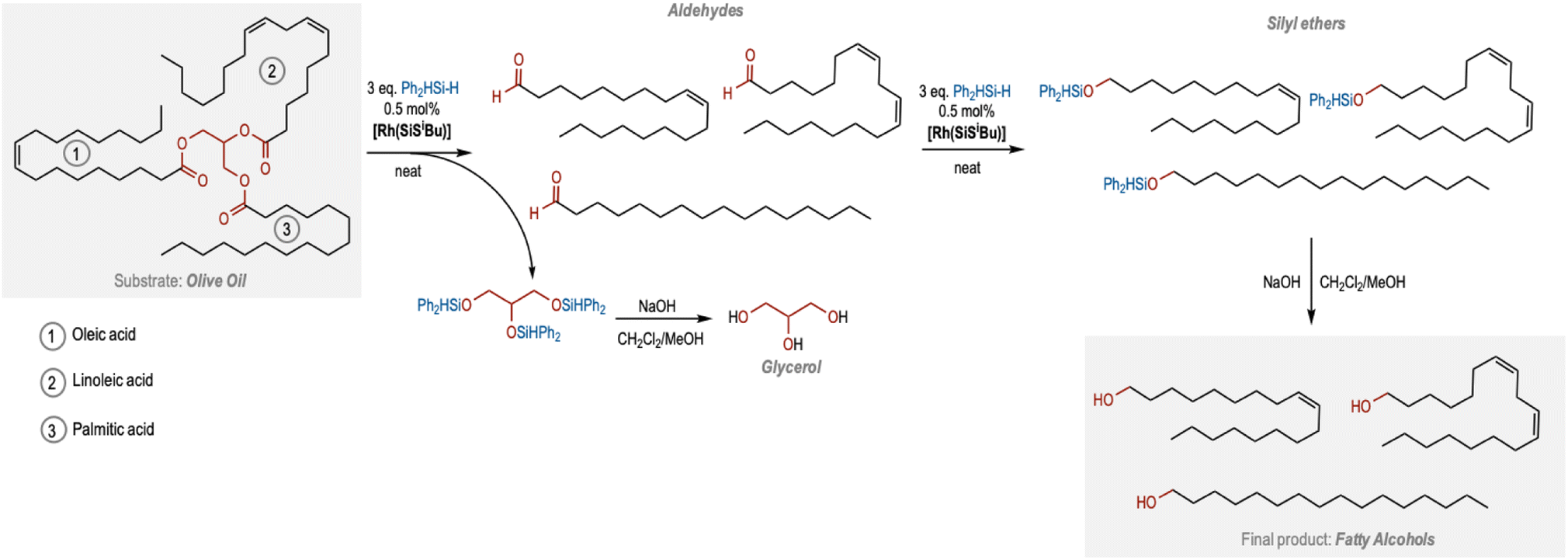 | ||
| Fig. 2 Proposed pathway to produce fatty alcohols from plant oils. | ||
This methodology was extended to the reduction of other plant oils (castor, sesame, and sunflower oil) including an unsaturated plant oil (coconut oil). The results obtained are presented in Table 2. Interestingly, in all the cases good conversions were achieved in 16 h, producing the corresponding silyl ethers as major products (Fig. S26, S28, S30 and S32 in the ESI†). An attempt to reuse the catalyst after reduction of olive oil for 24 h as per the conditions described in Table 2, entry 1 was not successful. The reaction using the recovered catalyst did not lead to the production of any fatty alcohol (see ESI, Section 7†).
Based on these studies, we have outlined a pathway to produce unsaturated fatty alcohols from plant oils (Fig. 2). We suggest that the three equivalents of silanes first cleave the triglyceride esters to produce three aldehyde molecules derived from the corresponding fatty acid esters along with the silyl ether of glycerol that upon hydrolysis leads to the formation of glycerol. The three saturated/unsaturated aldehydes undergo the second CO reduction to form the three silyl ether chains that upon hydrolysis lead to the formation of fatty alcohols (Fig. 2). Remarkably, the internal fatty acid unsaturations remain intact throughout the process.
Moreover, we speculate that both the steric and electronic factors of silanes are important in the selectivity of the reaction. A detailed study to elucidate a catalytic cycle involving organometallic intermediates is currently ongoing.
Conclusion
In conclusion, we report here the effect of silanes (nature and amount) on the selectivity of reduction of esters and CC bonds. For example, (a) triethylsilane performs hydrosilylation of both terminal and internal alkenes but doesn't reduce an ester group, (b) triphenylsilane is unreactive towards both alkenes (terminal or internal) and esters whereas (c) diphenylsilane reduces an ester to alcohol and performs hydrosilylation of a terminal alkene but not of an internal alkene. We have utilised this reactivity to demonstrate a rare example of the direct and chemoselective reduction of plant oils to unsaturated fatty alcohols under mild reaction conditions (298 K under neat conditions). This was achieved using 0.5 mol% rhodium catalyst and Ph2SiH2 (6 equivalents relative to plant oil, two for the reduction of each carbonyl bond).
Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/17b8990a-2901-4d90-aa94-6786412a3c21.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is part of the projects PID2019-111281GB-I00 funded by MCIN/AEI/10.13039/501100011033 and IT1741-22 and IT1553-22 funded by Gobierno Vasco. The authors thank SGiker for support. Universidad del País Vasco (UPV/EHU) (U. P.) and IKERBASQUE (M. H. and Z. F.) are acknowledged for personnel funding. A. K. acknowledges a UKRI Future Leaders Fellowship for funding (MR/W007460/1).References
- A. S. Carlson, Biochimie, 2009, 91, 665–670 CrossRef PubMed.
- G. Karmakar, P. Ghosh, K. Kohli, B. K. Sharma and S. Z. Erhan, in Innovate Uses of Agricultural Products and Byproducts, ACS, 2020, pp. 1–31 Search PubMed.
- J. W. Veldsink, M. J. Bouma, N.-H. Schöön and A. A. C. M. Beenackers, Catal. Rev.: Sci. Eng., 1997, 39, 253–318 CrossRef CAS.
- G. D. V. Nolfi, K. Gallucci and L. Rossi, Int. J. Environ. Res. Public Health, 2021, 18, 13041 CrossRef PubMed.
- M. A. Sánchez, G. C. Torres, V. A. Mazzieri and C. L. Pieck, J. Chem. Technol. Biotechnol., 2017, 92, 27–42 CrossRef.
- C. Hu, D. Creaser, S. Siahrostami, H. Grönbeck, H. Ojagh and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 2427–2444 RSC.
- (a) D. S. Brands, G. U.-A. Sai, E. K. Poels and A. Bliek, J. Catal., 1999, 186, 169–180 CrossRef CAS; (b) R. D. Rieke, D. S. Thakur, B. D. Roberts and G. T. White, J. Am. Oil Chem. Soc., 1997, 74, 341–345 CrossRef CAS.
- F.-J. Carduck, J. Falbe, T. Fleckenstein and J. Pohl, US Pat., 4982020, 1991 Search PubMed.
- W. Jia, G. Xu, X. Liu, F. Zhou, H. Ma, Y. Zhang and Y. Fu, Energy Fuels, 2018, 32, 8438–8446 CrossRef CAS.
- T. J. Korstange, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef PubMed.
- (a) S. Hosokawa, M. Toya, A. Noda, M. Morita, T. Ogawa and Y. Motoyama, ChemistrySelect, 2018, 3, 2958–2961 CrossRef CAS; (b) S. Xu, J. S. Boschen, A. Biswas, T. Kobayasi, M. Pruski, T. L. Windus and A. D. Sadow, Dalton Trans., 2015, 44, 15897–15904 RSC; (c) M. Yato, K. Homma and A. Ishida, Tetrahedron, 2001, 57, 5353–5359 CrossRef CAS.
- (a) H. Yue, L. Guo, S.-C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef CAS PubMed; (b) A. Cook, S. Prakash, Y.-L. Zheng and S. G. Newman, J. Am. Chem. Soc., 2020, 142, 8109–8115 CrossRef CAS PubMed.
- (a) J. Nakanishi, H. Tatamidani, Y. Fukumoto and N. Chatani, Synlett, 2006, 6, 869–872 Search PubMed; (b) H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Angew. Chem., Int. Ed., 2013, 52, 8045–8049 CrossRef CAS PubMed; (c) C. Chen and M. Brookhart, Angew. Chem., Int. Ed., 2012, 51, 9422–9424 CrossRef PubMed.
- T. Ohta, M. Kamiya, K. Kusui, T. Michibata, M. Nobutomo and I. Furukawa, Tetrahedron Lett., 1999, 40, 6963–6966 CrossRef CAS.
- K. Matsubara, T. Iura, T. Makai and H. Nagashima, J. Org. Chem., 2002, 67, 4985–4988 CrossRef PubMed.
- A. C. Fernandes and C. C. Romão, J. Mol. Catal. A: Chem., 2006, 253, 96–98 CrossRef CAS.
- (a) D. Bézier, G. T. Venkanna, L. C. Misal Castro, J. Zheng, T. Roisnel, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2012, 354, 1879–1884 CrossRef; (b) S. R. Tamang, A. F. Cozzolino and M. Findlater, Org. Biomol. Chem., 2019, 17, 1834–1838 RSC.
- (a) T. K. Mukhopadhyay, C. L. Rock, M. Hong, D. C. Ashley, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2017, 139, 4901–4915 CrossRef CAS PubMed; (b) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed; (c) C. M. Kelly, R. McDonald, O. L. Sydora, M. Stradiotto and L. Truculet, Angew. Chem., Int. Ed., 2017, 56, 15901–15904 CrossRef CAS PubMed; (d) S. A. C. Sousa, S. Realista and B. Royo, Adv. Synth. Catal., 2020, 362, 2437–2443 CrossRef CAS.
- V. Rysak, A. Descamps-Mandine, P. Simon, F. Blanchard, L. Burylo, M. Trentesaux, M. Vandewalle, V. Colliére, F. Agbosou-Niedercorn and C. Michon, Catal. Sci. Technol., 2018, 8, 3504–3512 RSC.
- O. O. Kovalenko and H. Adolfsson, Chem.–Eur. J., 2015, 21, 2785–2788 CrossRef CAS PubMed.
- L. Monsigny, J.-C. Berhet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- B. F. S. Nunes, M. C. Oliveira and A. C. Fernades, Green Chem., 2020, 2, 2419–2425 RSC.
- A. C. Fernades, ChemSusChem, 2021, 14, 4228–4233 CrossRef PubMed.
- (a) A. Behr, F. Naendrup and D. Obst, Eur. J. Lipid Sci. Technol., 2002, 104, 161–166 CrossRef CAS; (b) T. Huber, D. Firlbeck and H. M. Riepl, J. Organomet. Chem., 2013, 744, 144–148 CrossRef CAS; (c) X. Zhu, Z. Yun and X. Gui, Can. J. Chem. Eng., 2022, 100, 254–260 CrossRef CAS.
- (a) S. Azpeitia, M. A. Garralda and M. A. Huertos, ChemCatChem, 2017, 9, 1901–1905 CrossRef CAS; (b) U. Prieto, S. Azpeitia, E. San-Sebatian, Z. Freixa, M. A. Garralda and M. A. Huertos, ChemCatChem, 2021, 13, 1403–1409 CrossRef CAS; (c) U. Prieto-Pascual, A. Matínez de Morentin, D. Choquesquillo-Lazarte, A. Rodríguez-Diéguez, Z. Freixa and M. A. Huertos, Dalton Trans., 2023, 52, 9090–9096 RSC.
- S. Xu, J. S. Boschen, A. Biswas, T. Kobayashi, M. Pruski, T. L. Windus and A. D. Sadow, Dalton Trans., 2015, 44, 15897–15904 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00481c |
| This journal is © The Royal Society of Chemistry 2024 |