
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CoOOH-catalyzed anodic oxidation of 5-(hydroxymethyl)-furfural under non-alkaline conditions†
Marten Niklas
Gey
 and
Uwe
Schröder
*
and
Uwe
Schröder
*
Institute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17489 Greifswald, Germany. E-mail: uwe.schroeder@uni-greifswald.de
First published on 10th June 2024
Abstract
The oxidation of 5-(hydroxymethyl)-furfural (HMF), a platform chemical of biogenic origin, to 2,5-furandicarboxylic acid (FDCA) is a reaction of high relevance for sustainable production of polymers like polyethylene furanoate. However, a majority of the oxidation processes published to date rely on alkaline conditions, in which the instability of HMF and a resource intensive separation of FDCA are major obstacles for technological realization. In this study, we present the electrochemical oxidation of HMF in non-alkaline acetate and phosphate buffers (pH 5–7) on CoOOH modified electrodes. Current-controlled batch experiments were performed, to obtain optimal conditions with respect to the catalyst loading, current density and reaction temperature. Under optimized conditions, a FDCA yield of 94.7% in the acetate buffer (pH 5) was achieved. Through interval sampling, we were able to observe a consecutive oxidation mechanism during the optimized reaction, which mainly proceeded via the intermediate products 2,5-diformylfuran (DFF) and 5-formyl-2-furancarboxylic acid (FFCA). Furthermore, we observed that humic substances formed during the first reaction steps could also be oxidized to FDCA towards the end of the reaction.
Sustainability spotlightThe exit from the fossil fuel era requires fundamental paradigm shifts not only in the energy sector, but also a complete revolution in the chemical sector. Electroorganic syntheses based on biogenic starting materials simultaneously address the utilisation of regenerative carbon sources for basic and fine chemicals, as well as their energy and chemical-saving conversion. Yet, many electrosyntheses are being carried out under either very acidic or very alkaline conditions – therefore requiring considerable quantities of neutralising bases or acids for downstream processing. Our article is dedicated to the electrochemical oxidation of 5-(hydroxymethyl)-furfural, a biomass-based platform chemical, for the synthesis of 2,5-furandicarboxylic acid, which can, e.g., be considered as a green substitute for terephthalic acid in the production of polymers like PET. The focus of the work was on carrying out HMF oxidation in the neutral pH range, aiming to significantly reduce the amount of chemicals required in downstream processing compared to the usual alkaline reaction media. Our work thereby emphasizes the importance of the following UN sustainable development goals: industry, innovation, and infrastructure (SDG 9), responsible consumption and production (SDG 12) and climate action (SDG 13). |
Introduction
Biomass is an indispensable resource for a sustainable materials and energy economy, as it is the only regenerative carbon source that is available in a sufficiently large quantity to meet the anthropogenic demand for organic substances.1,2 Accordingly, there has been a strongly increasing research effort over the past decades to both identify bulk chemicals obtainable from biomass (i.e. platform chemicals) and to explore their reaction pathways to suitable target molecules for the chemical industry.3 In this context, electrochemistry has proven to be a viable and sustainable alternative to traditional chemical conversion methods, since electrical energy (in the context of green syntheses necessarily from renewable sources) is used to power the reaction, eliminating the need for the addition of reducing or oxidizing reagents on a stoichiometric scale.4An interesting candidate for electrochemical upgrading is 5-(hydroxymethyl)-furfural (HMF), which is produced by dehydration of hexoses like fructose.5,6 HMF is the starting material for a variety of target molecules, e.g. for fuel and polymer applications, which can be obtained by electrochemical reduction or oxidation.7,8 Therefore, both cathodic and anodic reactions of HMF have been studied extensively.9–12 For example, full oxidation of the hydroxy- and the aldehyde-group of HMF leads to 2,5-furandicarboxylic acid (FDCA – see Scheme 1), which has been reported to be an excellent monomer for the production of bio-based polyesters. Due to its structural similarity to terephthalic acid, it can be used to produce polyethylene furanoate (PEF), which exhibits comparable or even enhanced properties as compared to its fossil-based counterpart polyethylene terephthalate (PET).13,14
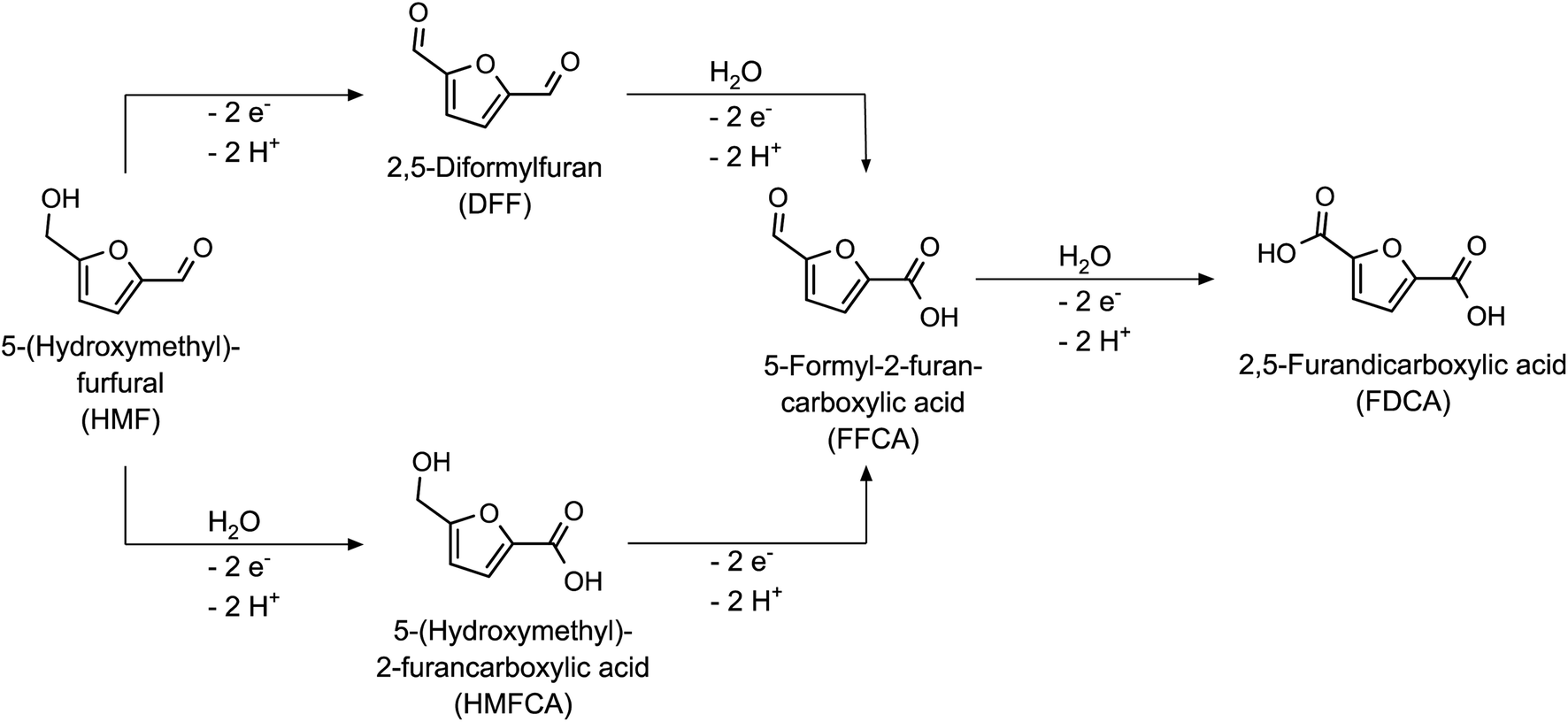 | ||
| Scheme 1 Reaction pathways and intermediates of the HMF oxidation to FDCA. | ||
In the last years, a large number of approaches for the electrochemical oxidation of HMF to FDCA have been proposed. Since noble metal-based electrodes (e.g. Au, Pt, Pd and Ru) exhibit an only insufficient catalytic activity towards a selective HMF oxidation, the search for suitable electrocatalysts has focused on compounds based on earth-abundant (and therefore also less expensive) metals, which are typically coated onto an inert electrode support.15–17 Amongst them, cobalt-based materials like cobalt oxides/hydroxides (e.g., Co3O4 and CoOOH) have been investigated for this purpose based on their known activity to catalyze electrochemical oxidations such as the oxygen evolution reaction (OER).18–20 As a result, a variety of cobalt-based materials with high selectivity towards the anodic HMF oxidation in alkaline media have been found.21–25 Generally, the vast majority of publications on the HMF oxidation rely on strongly alkaline electrolytes (pH 13 and 14), which seems to be logical regarding a better removal of the protons liberated during the HMF oxidation.15,16 However, it has also been shown that HMF undergoes degradation reactions in alkaline solutions leading to the formation of humic substances.26,27 This fact is a major limitation for industrial upscaling, since larger quantities of alkaline HMF solution could not be stored for longer times without risking a loss of educt and a contamination of the electrolyte. Furthermore, the separation of the only sparingly soluble28,29 protonated di-acid via crystallization requires the addition of considerable amounts of mineral acids to the alkaline reaction solution – thus also producing equal amounts of salt waste. To overcome the latter limitations, it is necessary to expand the pH window of the electrocatalytic HMF oxidation from alkaline to the non-alkaline pH values.
Since some of the cobalt-based materials show a catalytic activity towards the OER in non-alkaline media (pH ≥ 5) and even exhibit a “catalyst self-healing effect” under pH-neutral conditions,30–33 it is conceivable that this effect may also apply to the conversion of organic molecules like HMF. However, to the best of our knowledge, the use of cobalt-based electrocatalysts for the non-alkaline HMF oxidation has not been reported so far. In this study, we therefore prepared CoOOH modified electrodes based on an electrochemical deposition procedure and investigated these electrodes for the HMF oxidation in acetate- and phosphate-buffered solutions (pH 5 and 7, respectively). In these media, we studied the effects of different CoOOH loadings, current densities and reaction temperatures to optimize the reaction towards a higher HMF conversion rate and FDCA yield. We furthermore examined the possible degradation of the CoOOH catalyst via ICP-OES measurements of the reaction solutions. The gained insights were then used to perform the HMF oxidation under optimized conditions.
Results and discussion
As typical for many electro–organic reactions performed in aqueous media, the narrow potential window of water (thermodynamically 1.23 V) causes strongly competing reactions (namely the hydrogen evolution (HER) on the reductive side and the oxygen evolution (OER) on the oxidative side) when the electro–organic reaction proceeds close to the respective border of the potential window. This also applies for the electrochemical HMF oxidation, which usually lies close to the onset of the oxidative water decomposition – and thus to the OER.34–36 An increasing share of the OER in the overall reaction – e.g., caused by increasing overpotentials – leads to a decreasing Coulomb efficiency (CE), which has to be avoided. This situation requires a compromise to be found between CE and current density: A low current density allows working at low overpotential, but may result in an insufficient substrate turnover. High current densities on the other hand may increase the desired substrate turnover, but may also lead to a decreasing CE due to an increasing overpotential. There are different variables for a given electrocatalytic system to optimize the performance towards a high reaction rate at high CE, which we will analyze in the following sections.Effect of CoOOH loading
The CoOOH electrodes were prepared by the reductive deposition of Co(OH)2 onto titanium sheets from an aqueous Co(NO3)2 solution and subsequent oxidation to CoOOH in an aqueous NaOH solution. To control the CoOOH loading, electrodes were prepared with different amounts of reductive deposition charge (see Table 1).| σ Red | m CoOOH |
|---|---|
| 0.4C cm−2 | 0.17 ± 0.02 mg cm−2 |
| 0.8C cm−2 | 0.25 ± 0.02 mg cm−2 |
| 1.6C cm−2 | 0.48 ± 0.02 mg cm−2 |
| 2.4C cm−2 | 0.76 ± 0.05 mg cm−2 |
A first examination of the electrochemical behavior in acetate (pH 5) and phosphate buffer (pH 7) and of the effect of the CoOOH loading (mCoOOH) was conducted by linear sweep voltammetry (LSV). Starting from the open circuit potential (OCP), oxidative LSVs were measured in 20 mM HMF solutions of both electrolytes (Fig. 1). Reference measurements without HMF are shown for the lowest CoOOH loading. LSVs of uncoated titanium electrodes were also measured, but showed only an insignificant current response in the measured potential range (<0.001 mA cm−2, data not shown).
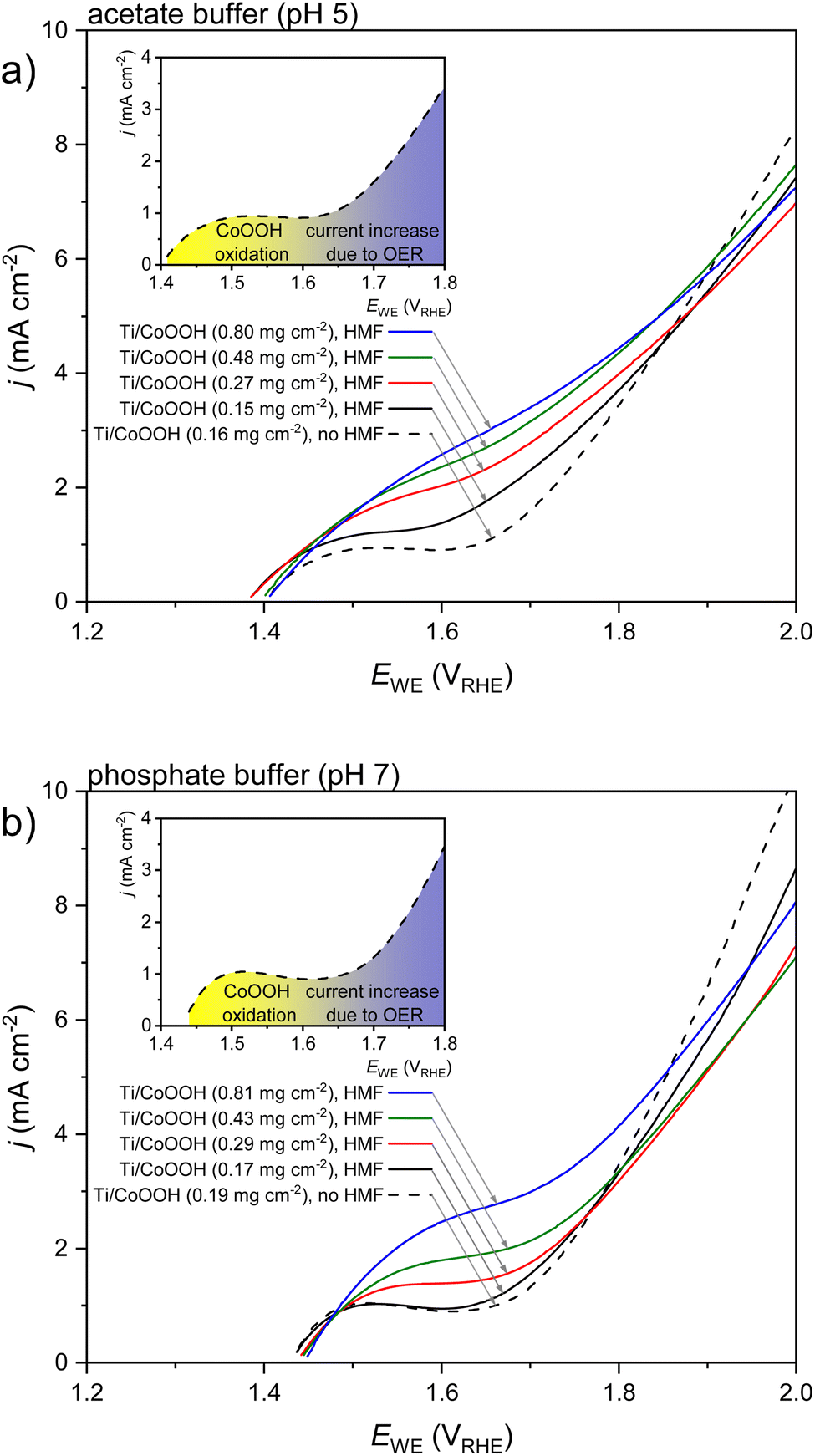 | ||
| Fig. 1 Linear sweep voltammograms of Ti/CoOOH electrodes with different catalyst loadings in the presence of 20 mM HMF (straight lines) (a) in acetate buffer – pH 5 and (b) in phosphate buffer – pH 7. Reference measurements without HMF (black dashed lines) are provided for the lowest catalyst loading. The inset figures schematically illustrate the transition from the CoOOH oxidation to the OER region in the respective electrolyte in the absence of HMF. The scan rate was 10 mV s−1. | ||
In the absence of HMF, an oxidation peak at 1.5 VRHE was recorded for both, acetate and phosphate buffered conditions (see black, dashed curves in Fig. 1a and b). This process can be attributed to the oxidation of CoOOH to CoO2 according to eqn (1).37,38
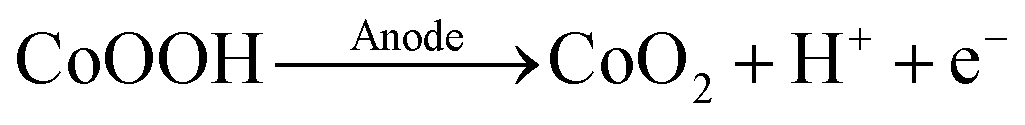 | (1) |
At higher potentials, this process was superimposed by the current increase due to the commencing oxidative water decomposition/the OER. In case of the acetate buffer, an oxidation of the electrolyte (e.g. via a Kolbe reaction under formation of gaseous ethane) may not be excluded.39 The extent of such a side reaction, however, seems low, since in the majority of our results, acetate buffered conditions yielded higher CE values of the HMF oxidation than phosphate buffered experiments. The addition of HMF led to an increasing CoOOH oxidation current, as can be seen by comparing the black solid and dashed curves in Fig. 1a and b. This suggests that the HMF is oxidized by the formed CoO2 at a potential lower than that of the OER. Since this effect is more pronounced in the acetate buffer, it can be deduced that the HMF oxidation is more favored in this electrolyte than in the phosphate buffer. This, however, does not correspond to the expectation concerning an enhanced reactivity at a higher pH value.
While comparing the oxidation behavior of MOOH (M = Ni, Co, Fe) in 0.1 M KOH solution, Choi et al. observed a similar oxidation process for CoOOH and NiOOH in the same (pH-corrected) potential region when HMF or one of the intermediate oxidation products was present in the solution.40 However, in their system no MOOH oxidation process was observed in the absence of educt. The same workgroup recently established a mechanism for the oxidation of alcohols and aldehydes based on the related NiOOH/NiO2 redox system. According to this model, the deprotonated alcohol is oxidized after adsorption to the NiO2 surface by a hydride transfer from the α-C atom, thus reducing the NiO2 to Ni(OH)O−. This species is afterwards regenerated by the anode.41–43 It is possible that an analogous mechanism is present for the CoOOH/CoO2 system.
The LSVs also showed a decreased slope of the water electrolysis current in the presence of HMF, indicating an inhibition of the OER (Fig. 1). This is a typical effect during electroorganic oxidation (as well as reduction) reactions and has been observed in other systems before.9,34,44 Higher catalyst loadings led to a further increase of the CoOOH/CoO2 oxidation current, showing that an increasing number of reaction sites is active on the surface. This effect was also observed in the absence of HMF (Fig. S3 in the ESI†).
To study the impact of the catalyst loading on the HMF oxidation in more detail, electrolysis experiments of 20 mM HMF solutions were carried out to determine the HMF conversion rates and the product yields. HPLC analysis of the reaction solution before and after the reaction allowed deriving the performance parameters of the respective reaction: The HMF conversion (X), as well as the yields (Y) and Coulomb efficiencies (CE) of all oxidation products were calculated according to their standard definitions (eqn (S1)–(S3) in the ESI†). The CE of the desired reaction products (final product FDCA and intermediate products DFF, HMFCA, FFCA – see Scheme 1) was summed up to obtain a total Coulomb efficiency (CEtot) for the whole HMF-to-FDCA oxidation process (eqn (S4) in the ESI†). In order to assess the loss of substance due to side reactions to unknown products, the mole balance (MB) was also calculated (eqn (2)).
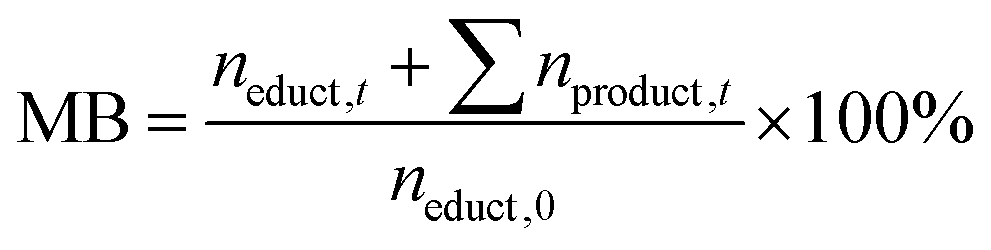 | (2) |
Furthermore, the stability of the CoOOH coatings was evaluated indirectly by determining the amount of cobalt ions in the reaction solutions after the respective electrolysis experiments via ICP-OES. The percentage of remaining CoOOH on the electrode (aCoOOH) was then calculated based on the ratio of cobalt ions in the solution after the reaction and the initially deposited amount of CoOOH on the electrode (eqn (3)).
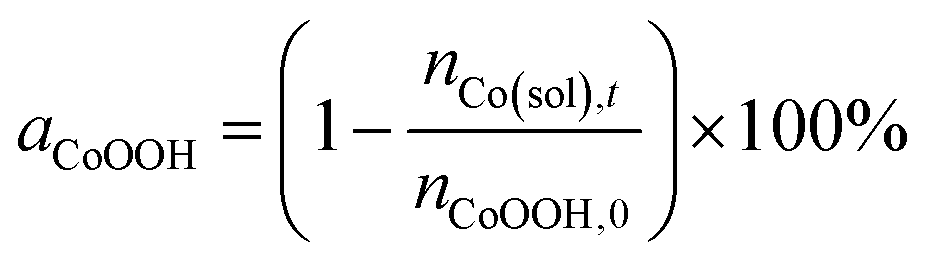 | (3) |
The electrolysis experiments were carried out galvanostatically, by applying a current density of 1.0 mA cm−2 (relative to the geometric anode surface area) until 6 Faraday equivalents (F) of charge – representing the amount of charge needed to fully convert the present HMF to FDCA at CEtot = 100% – was transported. The current-controlled instead of potential-controlled approach was chosen in order to have a more predictable timing of the experiments. Here, a possible inhibition of the reaction process would be observed by an increase of the working electrode (WE) potential instead of a decrease in current, which would extend the reaction time significantly. Also, data of current-controlled measurements have a higher relevance for industrial upscaling, since potential-controlled experiments are difficult to be performed in large reactors.4 The resulting performance parameters of electrodes with CoOOH loadings between 0.22 and 0.74 mg cm−2 are shown in Fig. 2. The proportion of the individual products to the total CE is shown in Fig. S1a in the ESI.†
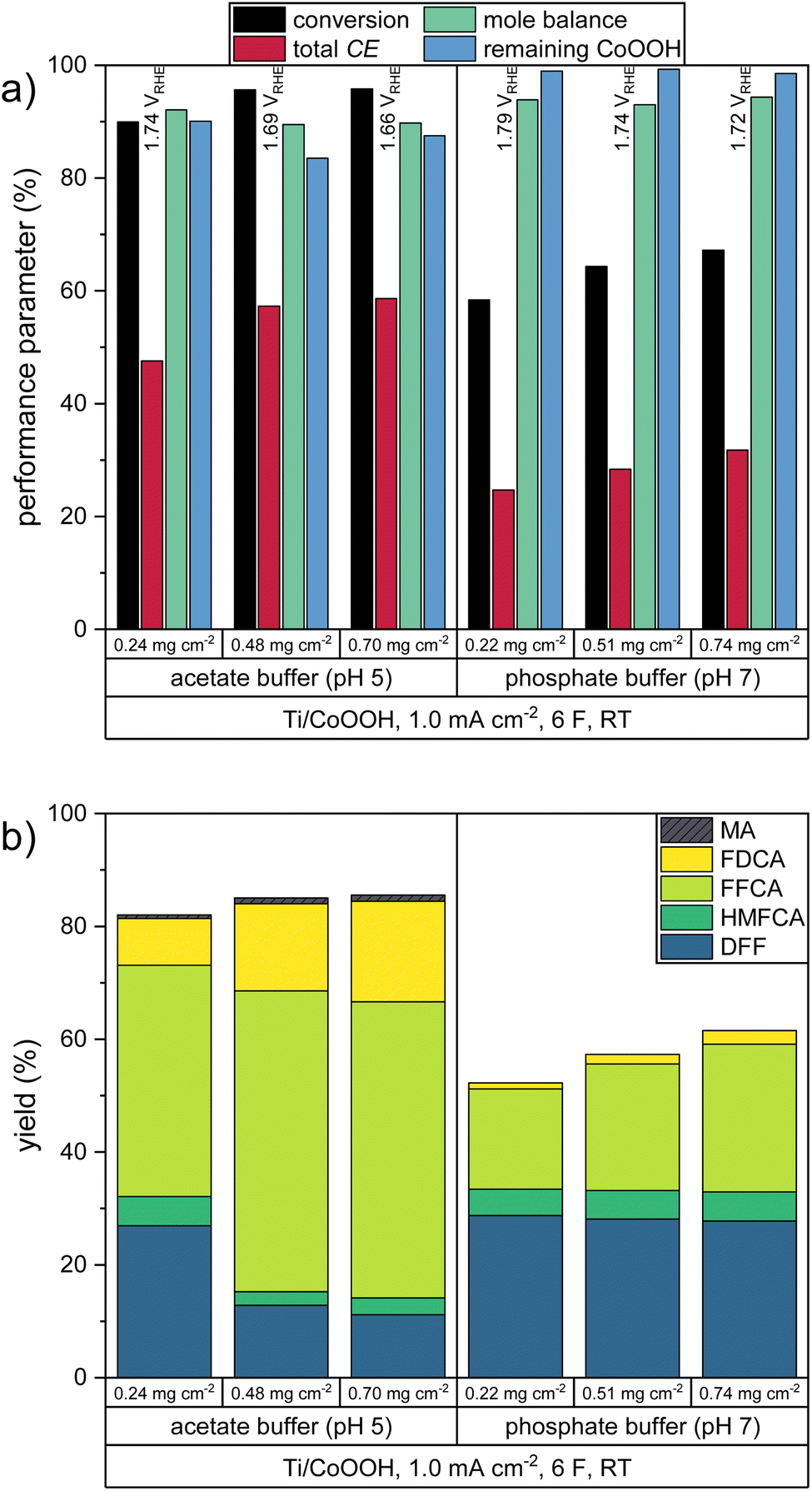 | ||
| Fig. 2 Performance parameters of the HMF oxidation with different CoOOH loadings: (a) HMF conversion, total Coulomb efficiency, mole balance, remaining percentage of CoOOH and (b) product yields. Average WE potentials of the respective experiment are indicated. | ||
All reactions yielded a mixture of the intermediate products and FDCA. The acetate buffer reactions showed a higher selectivity for the HMF-to-FDCA process with total CE values of 47.5–58.6% – thereby increasing with increasing CoOOH loadings. The same trend was observed for the phosphate buffered reaction. Here however, only total CE values of 24.6–31.8% were obtained, resulting in a lower HMF conversion and lower overall yields of oxidation products. By examining the product mixture in both electrolytes, it is clear that the HMF oxidation follows a consecutive reaction mechanism, with the consecutive oxidation steps becoming progressively slower. Thus, the final step – the oxidation of FFCA to FDCA – proceeds at the lowest rate and is only accelerated when a sufficiently high concentration of FFCA is provided. Furthermore, it is evident that the oxidation via the DFF pathway is favored over the HMFCA pathway (Scheme 1), as HMFCA is only obtained in small amounts (Fig. 2b). This is in agreement with respective literature on HMF oxidation.26,34,45 Therefore, DFF was obtained as the major product in the phosphate buffer reactions with a similar yield of 27.8–28.7%, with a higher catalyst loading leading to an acceleration of the further oxidation steps and thus to higher yields of FFCA and FDCA. The higher total CE in the acetate buffer reactions enabled the oxidation to proceed further, so that FFCA was obtained as the major product. Here, the higher catalyst loading resulted in higher FDCA yields and lower DFF yields.
Additionally, a small amount of maleic acid (MA) was formed in the acetate buffer reactions. This was probably the product of a ring-opening reaction (Scheme 2), which was already reported for the acidic oxidation of furfural and HMF.34,46
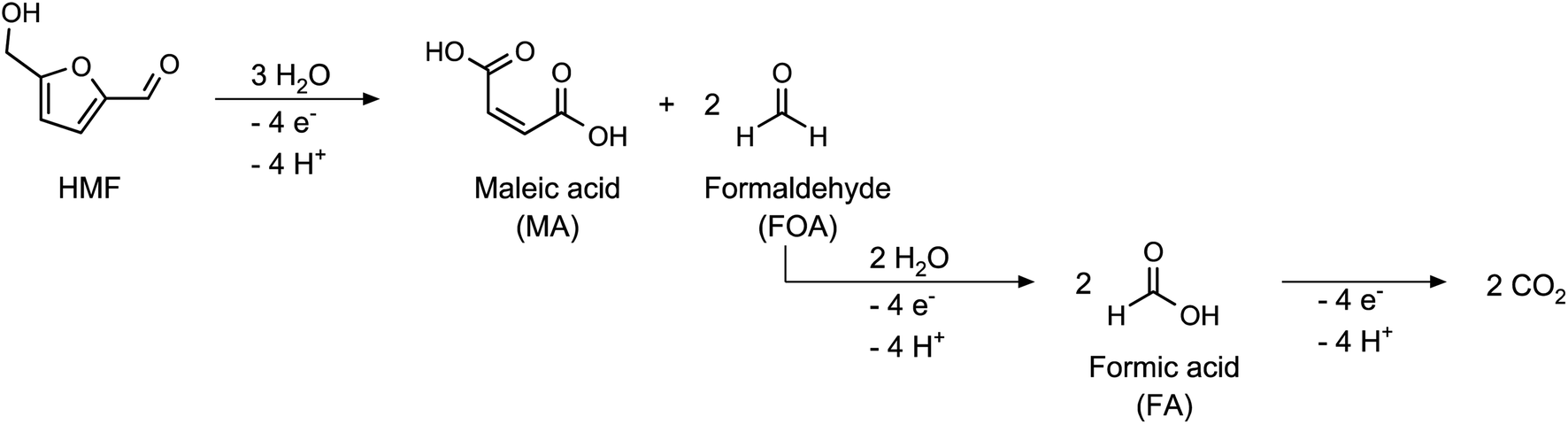 | ||
| Scheme 2 Oxidation of HMF to maleic acid (MA). | ||
Since the formation of MA involves the cleavage of two C1 species from the HMF molecule, it is an unwanted side reaction of the FDCA synthesis. Therefore, the CE of the MA formation process was not included in the calculation of the total CE (see eqn (S4) in the ESI†). However, the obtained mole balances of only 89.5–94.3% (after consideration of MA) indicate the presence of other side products which were not identified. We assume that these could be the products of further C–C-bond cleavage reactions, either starting from HMF or MA. Since the C1 species formaldehyde and formic acid was not found in the product solutions, it is assumed that a full oxidation to CO2 occurred. Since CO2 is only sparingly soluble at pH 5, its presence could only be confirmed by a gas phase analysis, which was not conducted in this study.
In all experiments, the WE potential showed only a slight increase of less than 0.10 V after an initial increase from the equilibrium state (Fig. S4 in the ESI†). For both electrolytes the use of a higher catalyst loading resulted in a lower average WE potential, with slightly higher values obtained in the phosphate buffer reactions (Fig. 2a). By following the respective explanation for an analogous mechanism at NiOOH/NiO2 electrodes by Choi et al.,41 it can be assumed that the anodic reoxidation of CoOOH to CoO2 in the catalytic cycle is the rate-determining step, thus making the overall process dependent on the potential. This can be supported by cyclic voltammograms of the HMF-free electrolytes (Fig. S5†), which show a large peak separation of the considered cobalt oxide system and thus indicate a relatively low electron transfer rate. Therefore, in this galvanostatic experiment, the measured WE potential can be used as an indicator for the driving force necessary to achieve the CoOOH reoxidation at sufficient rate to sustain the required current flow. Through a higher loading of the electroactive catalyst, the charge transfer can be maintained at a lower driving force, resulting in less positive oxidation potentials. Due the lower potentials, the participation of the OER to the catalytic cycle is also decreased, leading to higher CE values of the HMF oxidation. This is in agreement with the results obtained in the LSV measurements (Fig. 1). In other words, a low WE potential can be seen as an indication of a good HMF convertibility in the corresponding system.
The catalytic coating showed a comparatively high stability in the phosphate buffer reactions with 98.5–99.3% of the CoOOH remaining at the electrode after the transfer of 6 F. In the acetate buffer reactions only 83.5–90.1% of the catalyst remained at the electrode, which could be a result of the higher HMF turnover. However, it is assumed that the different film stability may also be influenced the different pH values of the buffer systems as well as by variances in the CoOOH morphologies due variations in the electrode preparation. Thus, comparing the acetate buffer reactions, the lowest remaining fraction of the catalyst was found after the reactions with a CoOOH loading of 0.48 ± 0.02 mg cm−2. To study the electrochemical stability of the CoOOH catalyst, cyclic voltammograms were recorded before, during and after a galvanostatic HMF oxidation in both electrolytes (Fig. S6 in the ESI†). Here, a decrease of the peak currents of the CoOOH/CoO2 redox couple was measured over the course the reaction, indicating an electrochemical inactivation of the film. This effect was more pronounced in the acetate buffer, which could again be an effect of a higher HMF turnover.
Effect of current density
The current density is an important reaction parameter defining the number of turnovers per time unit and electrode area. Thus, it is desirable to perform the reaction at the highest possible current density to shorten the reaction time or to reduce the electrode surface needed to convert a specific amount of substance. To study this influence of the current density on the HMF oxidation, we increased the current density from 1.0 mA cm−2 to 4.0 and 7.5 mA cm−2 for CoOOH electrodes with mCoOOH = 0.25 ± 0.02 mg cm−2. The resulting performance parameters in the acetate and phosphate buffered solutions are shown in Fig. 3. The share of the individual products in the total CE is shown in Fig. S1b in the ESI.†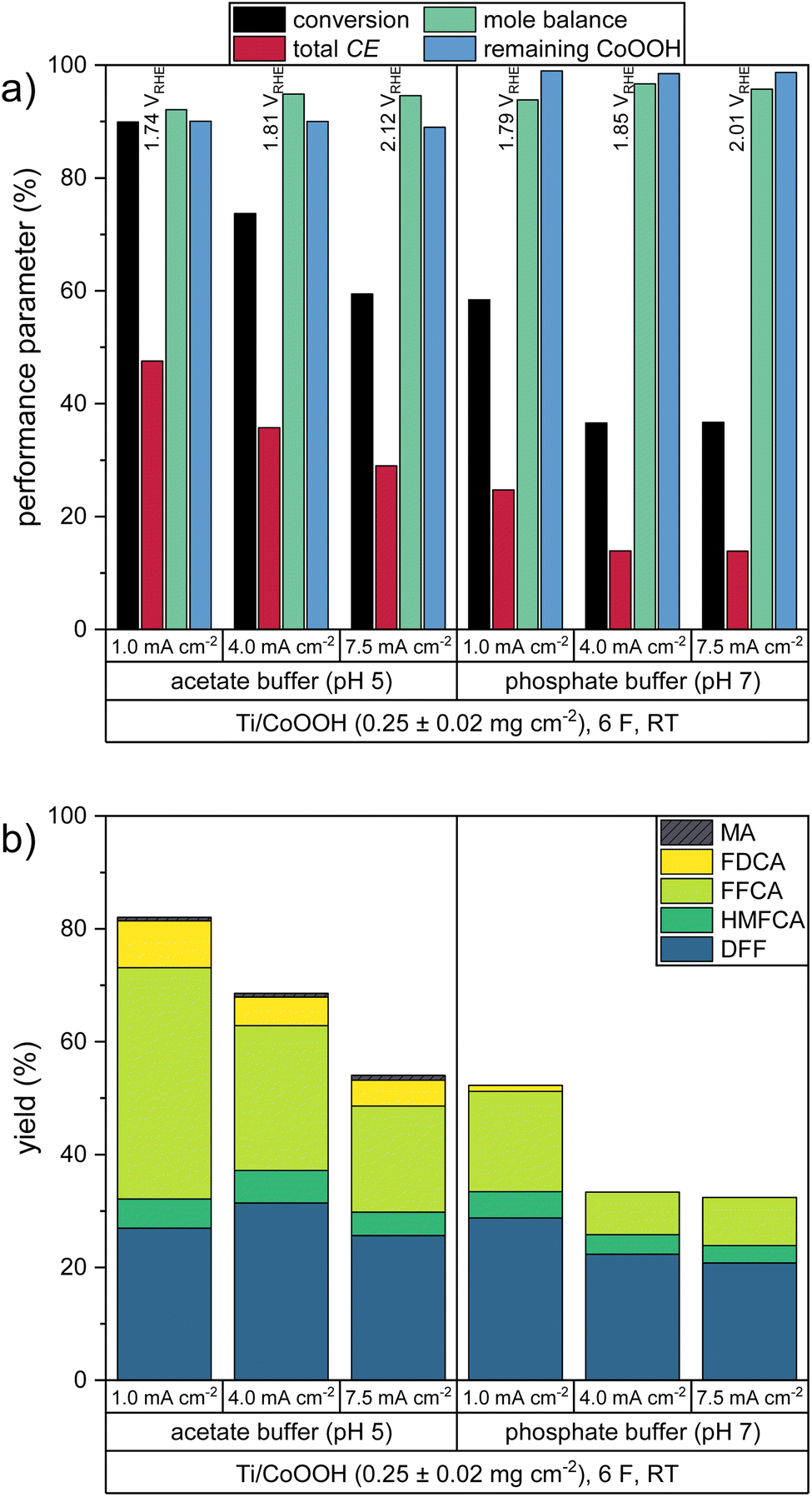 | ||
| Fig. 3 Performance parameters of the HMF oxidation at different current densities: (a) HMF conversion, total Coulomb efficiency, mole balance, remaining percentage of CoOOH and (b) product yields. Average WE potentials of the respective experiment are indicated. | ||
In both electrolytes, the higher current densities led to a significant increase of the measured WE potentials, indicating that a higher driving force was necessary to sustain the CoOOH reoxidation at increased rates. Accordingly, these higher WE potentials resulted in a more pronounced participation of the OER (Fig. 1) and thus to a decrease in the total CE and to smaller HMF conversions.
Since similar product mixtures as in the previous experiments were obtained, it can be deduced, that the same consecutive HMF oxidation mechanism took place and individual oxidation steps were not accelerated by the higher current densities. Compared to experiments conducted at 1.0 mA cm−2 the higher current densities led to slightly higher mole balances. This effect, however, may also be attributed to the reduced duration of the experiment at increasing current density, leading to a smaller time window for non-electrochemical side reactions such as the formation of humic substances. Overall, a low current density is shown to be necessary for a good reaction performance.
The stability of the CoOOH coating was found to behave independently of the current density in both electrolytes, which also makes clear that higher OER rates do not increase catalyst degradation. It can also be deduced that the coating is also not physically removed due to O2 gas evolution.
Effect of temperature
In order to enhance the reaction kinetics of the consecutive oxidation, we studied the influence of elevated reaction temperatures. Since the previous experiments showed that especially the last oxidation step – the oxidation of FFCA to FDCA – is rather slow, a higher temperature could increase the yield of the end product substantially. Thus, we performed oxidation reactions at 1.0 mA cm−2 on CoOOH electrodes with mCoOOH = 0.25 ± 0.02 mg cm−2 at 40, 60 and 80 °C in both electrolytes (Fig. 4). The share of the individual products in the total CE is shown in Fig. S1c in the ESI.†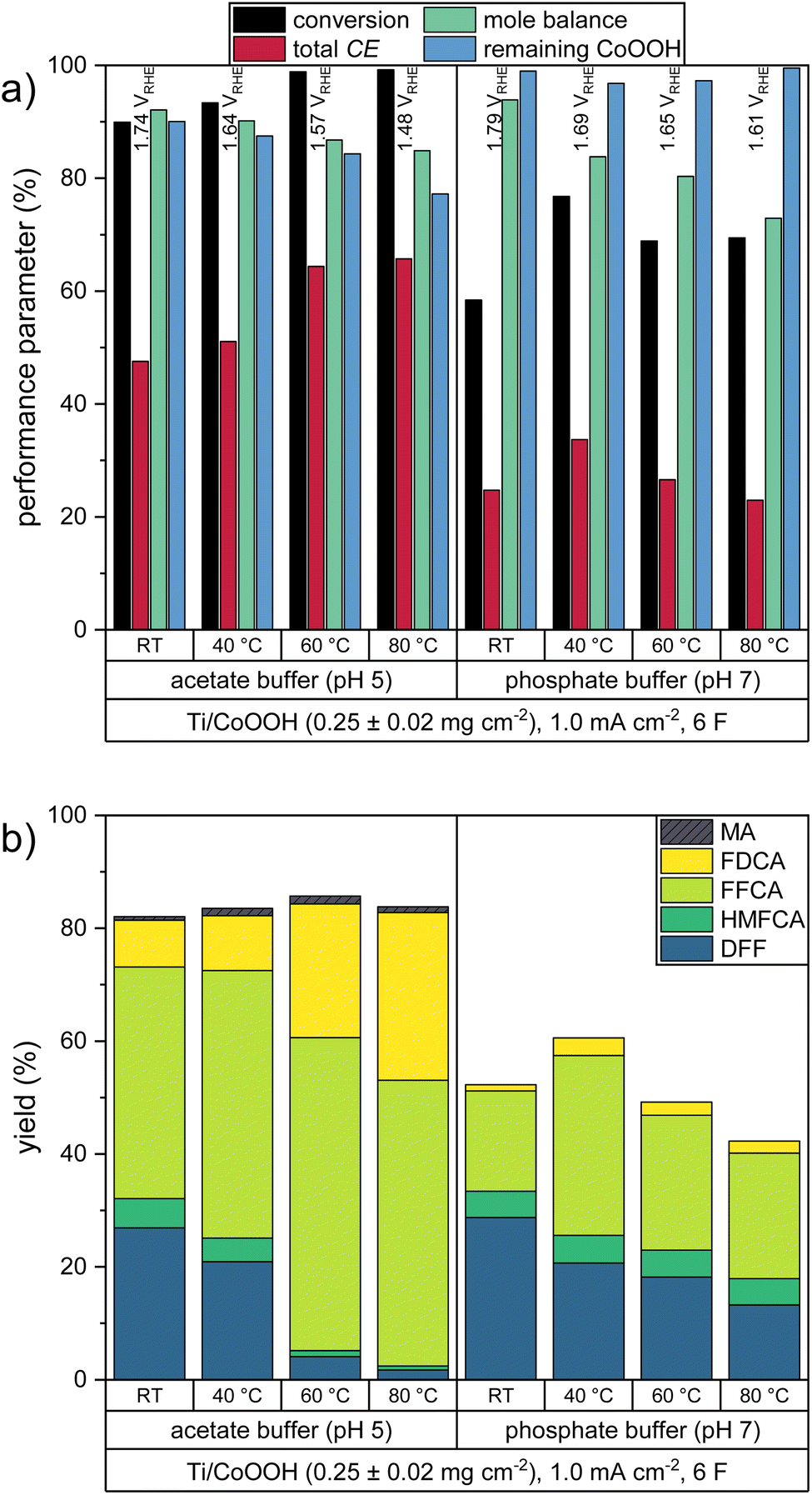 | ||
| Fig. 4 Performance parameters of the HMF oxidation at different temperatures: (a) HMF conversion, total Coulomb efficiency, mole balances, remaining percentage of CoOOH and (b) product yields. Average WE potentials of the respective experiment are indicated. | ||
For the two electrolyte systems, the effect of higher temperatures differs significantly. In the acetate buffer, the total CE increased significantly up to a value of 65.8% (80 °C), while the measured WE potential decreased. Accordingly, an almost complete HMF conversion was observed at 60 and 80 °C, while FDCA yields of 23.6% and 29.7% were obtained. The yield of the side product MA was not significantly increased. In the phosphate buffered solutions, the highest total CE value of 33.7% was obtained at 40 °C. Further temperature increase favored the OER, leading to smaller CE values and HMF conversion rates.
The higher temperatures led to a decrease of the mole balance in both electrolytes. It can be presumed that this effect is caused by a facilitated self-polymerization reaction of HMF leading to the formation of humic substances. This effect was also visible by a color change of the light yellow HMF solutions to yellow at 40 °C and orange at 60–80 °C.
As in the CoOOH loading experiments, a dependence between the catalyst stability and the reaction progress (i.e. the number of turnovers) could be observed. Thus, in the acetate buffer reactions the stability decreased at higher temperatures, with only 77.2% of the CoOOH remaining on the electrode at 80 °C. Interestingly, in the phosphate buffer reactions, the minimal stability was obtained at 40 °C with 96.8% of the CoOOH remaining.
Optimized reaction conditions
The gained insights of the previous experiments were used to carry out an electrolysis experiment under optimized reaction conditions. As the highest total CE was obtained in the acetate buffer at 80 °C, we repeated this experiment with a higher catalyst loading of 0.81 ± 0.01 mg cm−2. Since higher current densities resulted in lower CE values, the current density of 1.0 mA cm−2 was maintained as a compromise between high CE and low measuring time. In order to test whether this optimized system is able to fully convert the present HMF to FDCA, the experiment was extended to a charge transport of 24 F (four times the charge theoretically required at CEtot = 100%). To study the progression of the consecutive oxidation mechanism, HPLC samples were taken in intervals during the reaction. The resulting performance parameters are depicted in Fig. 5. The numerical values are listed in Table S4 in the ESI.†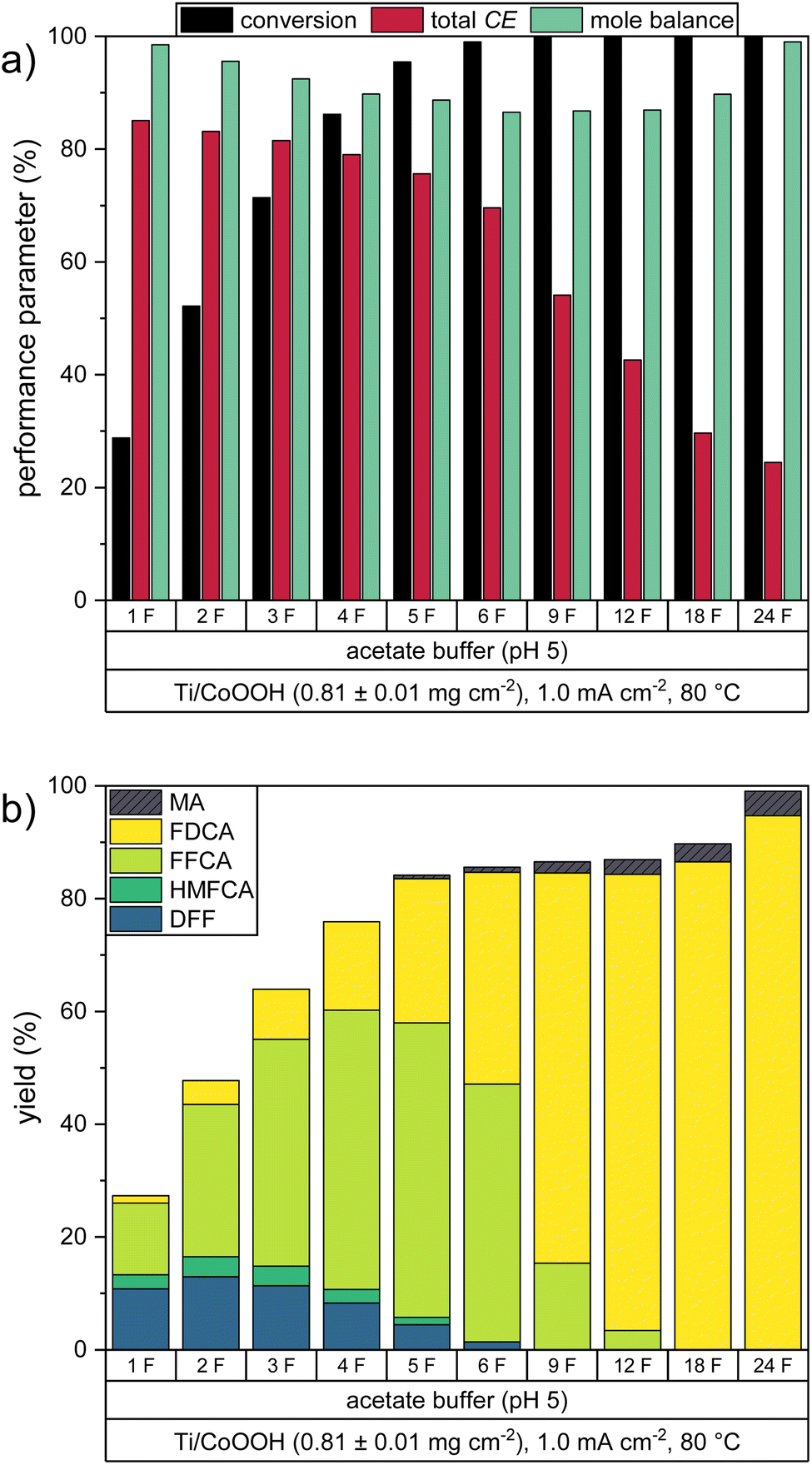 | ||
| Fig. 5 Performance parameters of a HMF oxidation under optimized conditions (20 mM HMF in acetate buffer (pH 5), 80 °C, mCoOOH = 0.81 ± 0.01 mg cm−2): (a) HMF conversion, total Coulomb efficiency, mole balance, remaining percentage of CoOOH and (b) product yields. | ||
As in the previous acetate buffer reaction at 80 °C, the first two oxidation steps from HMF to FFCA proceeded almost completely within the charge transport of 6 F. Both DFF and HMFCA yield reached their maximum of 13.0% and 3.5% after 2 F, while a maximal FFCA yield of 52.2% was reached after 5 F. The higher CoOOH loading caused an increase in the total CE, which was 7.9% higher after 6 F, than in the previous experiments. During this process, the formation of humic substances could be observed by a yellow coloration of the HMF solution, also causing the mole balance to decrease.
The final oxidation step from FFCA to FDCA caused a rapid decrease of the total CE, so that the FFCA yield was only decreased to 3.4% after 12 F, giving a FDCA yield of 80.9%. At this point, the mole balance reached a minimum of 86.9%. Interestingly, further oxidation led to an increase in the mole balance, thus resulting in a final FDCA yield of 94.7% after 24 F. During this phase, a decolorization of the solution to the original light yellow color was observed. Based on these observations, we assume that this was caused by oxidation of the humic substances. During the entire reaction, only a negligible amount of MA was formed, with a yield of 4.4% obtained after 24 F.
ICP-OES measurements of the product solution after the oxidation revealed that 83.9% of the CoOOH remained on the electrode. Thus, a higher percentage of CoOOH remained on the electrode as in the respective experiment with 0.25 ± 0.02 mg cm−2 CoOOH, where only 6 F of charge were transported (Fig. 4a). This demonstrates again that the catalyst stability is also influenced by other factors than the turnover number (e.g., morphology). A catalyst loss in this order of magnitude would be problematic for an industrial upscaling. Also, it is possible that the observed decrease in the reaction rate was partially caused by this catalyst loss or by the electrochemical inactivation observed before. Thus, it is important to study the exact mechanisms of catalyst inactivation and detachment during the oxidation to gain insight on how the stability can be increased. This however was not part of this study.
Conclusions
This study showed that CoOOH electrodes, which have been prepared in alkaline media, can be used to catalyze the anodic oxidation of HMF to FDCA outside of the alkaline pH range via the CoOOH/CoO2 redox couple. Comparative experiments were carried out in an acetate buffer (pH 5) and a phosphate buffer (pH 7), which showed different properties in terms of reaction selectivity and catalyst stability. Generally, the reactions in the acetate buffer resulted in higher Coulomb efficiencies and lower catalyst stabilities, while the opposite was the case for the phosphate buffer reactions. By variation of CoOOH loading, current density and reaction temperature, optimal conditions were determined for both electrolytes. Here, the best results were obtained in the acetate buffer with a high CoOOH loading (0.76 ± 0.05 mg cm−2), a low current density (1.0 mA cm−2) as well as a high temperature (80 °C). Under these conditions, a long-term experiment was performed to investigate the performance parameters during the consecutive oxidation. Here, a FDCA yield of 37.6% was obtained after 6 F. After 24 F an almost complete conversion of HMF to FDCA with a yield of 94.7% could be achieved. The initial total Coulomb efficiency was 85.1%, which however dropped significantly during the last oxidation step from FFCA to FDCA. The obtained values are in a comparable order of magnitude to other published cobalt oxide-based catalysts in the alkaline pH region (Table 2).| Electrode | Electrolyte | c HMF | Conditions | Y FDCA | CEFDCA | Ref. |
|---|---|---|---|---|---|---|
| Unmodified cobalt oxide catalysts | ||||||
| Ti/CoOOH | Acetate buffer (pH 5) | 20 mM | 1.0 mA cm−2 (6 F) | 37.6% | 38.6% | This work |
| Ti/CoOOH | Acetate buffer (pH 5) | 20 mM | 1.0 mA cm−2 (24 F) | 94.7% | 24.5% | This work |
| FTO/CoOOH | 0.1 m KOH (pH 13) | 5 mM | 1.56 VRHE (6 F) | 35.1% | 35.1% | 40 |
| Cpaper/CoO | 1.0 m KOH (pH 14) | 10 mM | 1.43 VRHE (6.1 F) | 42% | 57.9% | 22 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Modified cobalt oxide catalysts | ||||||
| Cpaper/CoO–CoSe | 1.0 m KOH (pH 14) | 10 mM | 1.43 VRHE (6.1 F) | 99% | 97.9% | 22 |
| Nifoam/NiCo2O4 | 0.1 m KOH (pH 13) | 10 mM | 1.55 VRHE (6 F) | 90% | 92–99% | 24 |
| Cufoam/CoP | 1.0 m KOH (pH 14) | 50 mM | 1.423 VRHE (6 F) | 90% | 100% | 25 |
It was also found that higher temperatures also increase the formation rate of humic substances, even under these non-alkaline conditions. Surprisingly, it was shown that these humic compounds can also be converted to FDCA at the end of the reaction. Furthermore, only a low yield of the previously reported side product MA was produced in this system.
Even though an almost full conversion to FDCA could be realized at pH 5, the decrease of the Coulomb efficiency towards the end of the reaction and the limited stability of the CoOOH catalyst at simultaneously low current densities remain as challenges. Thus, it will be the goal of future studies to modify the CoOOH catalyst and explore new electrolytes in order to increase the selectivity and stability. For instance, previous results in the alkaline pH range show that chemically modified cobalt oxide catalysts exhibit considerably higher Coulomb efficiencies with simultaneously higher current densities than the unmodified oxide (Table 2). Higher HMF concentrations could also inhibit the OER further and thus increase the selectivity of the process. Thus, although the influence of this parameter on the Coulomb efficiency was not evaluated here, LSV measurements showed an increased current at higher HMF concentrations (Fig. S7 in the ESI†). Furthermore, the transition to a continuous operation (i.e., electrochemical flow cell) would increase conversion per area through a higher electrode surface to solution volume ratio, allowing the conversion of higher HMF concentrations – including a subsequent product isolation. By online monitoring of individual parameters (e.g., product concentration or pH), settings could be optimized for the individual consecutive oxidation steps to improve the total Coulomb efficiency. Thus, it was recently shown for the Kolbe electrolysis of valeric acid in a flow cell reactor, that online monitoring and adjustment the pH value allowed a continuous operation with product isolation, maintaining a high Coulomb efficiency.47
Experimental section
Chemicals
The following chemicals were purchased commercially and were used without further purification: Co(NO3)2·6H2O (≥99%, Carl Roth), NaOH (≥99%, Carl Roth), HMF (98%, abcr), DFF (98%, TCI), HMFCA (98%, TCI), FFCA (98%, TCI), FDCA (98%, Alfa Aesar), MA (>99.0%, Fisher Scientific), CH3COOH (≥99%, Carl Roth), CH3COONa (≥99%, Carl Roth), NaH2PO4·2H2O (≥99%, Carl Roth) and Na2HPO4·2H2O (≥99%, Merck). Deionized water was used as solvent in all experiments.Acetate buffer (CH3COOH/CH3COO−, pH 5) and phosphate buffer (H2PO4−/HPO42−, pH 7) solutions with a total anion concentration of 1.0 mol L−1 were prepared from the respective acid and sodium salts.
Electrochemical setup
All electrochemical experiments were conducted using an Autolab PGSTAT20 or a PGSTAT302 potentiostat/galvanostat (METROHM GmbH, Germany) with a three-electrode arrangement. Here, titanium sheets (2 × 5 cm, chemPUR, Germany) served as working electrode and platinum sheets (2 × 5 cm, chemPUR, Germany) were used as counter electrodes. Potentials were measured against Ag/AgCl sat. KCl electrodes (Sensortechnik Meinsberg, Germany). The potentials measured during the HMF oxidation experiments were afterwards converted to the reversible hydrogen electrode (RHE) using eqn (4).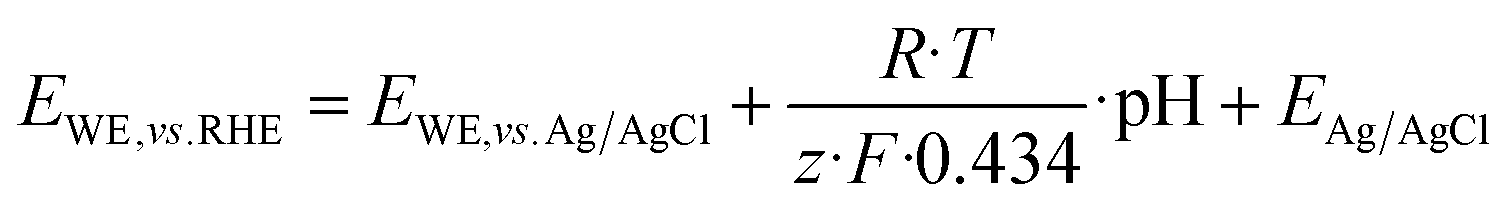 | (4) |
Preparation of the CoOOH working electrodes
The titanium working electrodes were freshly coated with CoOOH before each experiment. Therefore, the titanium sheets were stored in 20% HCl solution for 30 min and were then polished with fine-grained sandpaper to remove the passivating TiO2 layer. Afterwards, the electrode surface was roughened with coarse-grained sandpaper to enable better adhesion of the coating.The CoOOH coatings were prepared on the titanium electrodes by reductive deposition of Co(OH)2 from an aqueous Co(NO3)2 solution and subsequent oxidation to CoOOH in an aqueous NaOH solution. This technique was adapted from typical coating methods, which have been described in previous works.40,48 The deposition of Co(OH)2 is based on the reduction of NO3−, leading to the formation of OH− at the working electrode surface according to eqn (5). This causes the water-insoluble Co(OH)2 to precipitate on the electrode (eqn (6)).
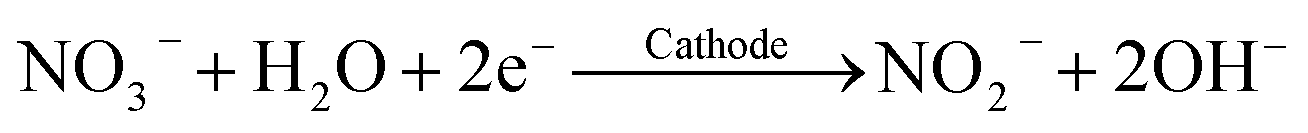 | (5) |
| Co2+ + 2OH− → Co(OH)2 | (6) |
For Co(OH)2 deposition, a single-compartment electrochemical cell with a filling volume of 40 mL was used. The titanium working electrode as well as the platinum counter electrode were immersed into a 50 mM Co(NO3)2 solution to give a projected surface area of 7 cm2 each. The distance between the working and counter electrode was approximately 1 cm. A linear sweep voltammogram (LSV) of this system revealed the onset potential of NO3− reduction at −0.81 VAg/AgCl (Fig. S8a in the ESI†). Thus, the deposition was performed by applying a constant potential of −1.0 VAg/AgCl to the working electrode. It was automatically stopped after a defined charge density σRed was transferred (0.4, 0.8, 1.6 or 2.4C cm−2). The adjustment of this value allowed the regulation of the CoOOH loading, which however could not be determined from this measurement, since the charge transport does not directly correlate with the amount of deposited Co(OH)2. During the deposition, the solution was stirred at 500 rpm. The electrodes were coated on both sides.
Afterwards, the Ti/Co(OH)2 electrodes were rinsed with water, dried on air and were then transferred to a 1.0 M NaOH solution for oxidation. To characterize the oxidation behavior of the Co(OH)2 film, a cyclic voltammogram was recorded (Fig. S8b in the ESI†). Here, within the potential window, a single oxidation process was measured at 0.28 VAg/AgCl, which can be assigned to the formation of CoOOH according to eqn (7).19,38 The onset potential of the OER was measured at 0.54 VAg/AgCl.
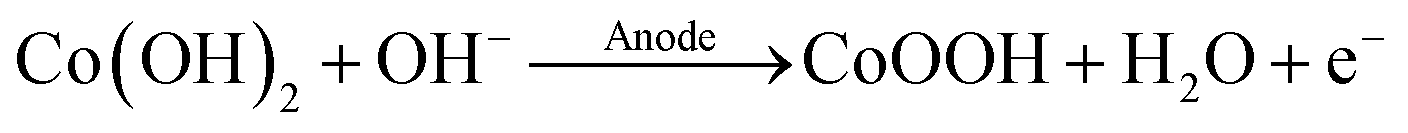 | (7) |
The corresponding reduction process peaked at 0.00 VAg/AgCl with the peak height reaching only 24% of the respective oxidation peak. This effect was observed before and has been attributed to a quasi-reversible behavior of the Co(OH)2/CoOOH redox couple.19,20
According to this measurement, a constant potential of 0.4 VAg/AgCl was applied for the oxidation of the deposited Co(OH)2 electrodes while the solution was stirred at 500 rpm. Here, the reaction was automatically stopped after the measured current density was below 0.35 mA cm−2. The electrodes were then again rinsed with water and dried on air before being used for further measurements.
Since the amount of transported charge QOx corresponds to the amount of oxidized Co sites, this method allowed the determination of the CoOOH loading mCoOOH on the electrode according to Faraday's law (eqn (8)). Average CoOOH loadings mCoOOH of the used deposition charges σRed are stated in Table 1.
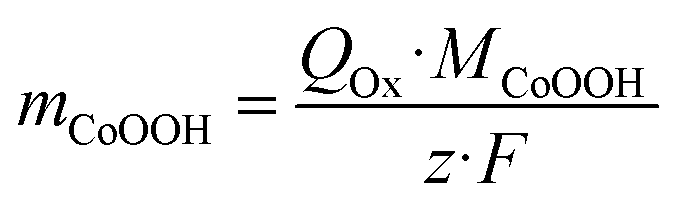 | (8) |
Exemplary current and charge curves of the potentiostatic Co(OH)2 oxidation are provided in Fig. S9 in the ESI.† The quantification of the catalyst loading via the oxidation charge was validated with exemplary gravimetric experiments measuring the weight of the electrode before deposition and after deposition and oxidation (data not shown). The gravimetric data also proved a one-electron oxidation of Co(OH)2 to CoOOH.
After each measurement, the electrodes were stored in 20% HCl solution to remove the CoOOH coating.
HMF oxidation
For all HMF oxidation experiments, separated H-type glass reactors were used, with the anodic (working electrode) half-cell containing 50 mL of a HMF solution in the respective electrolyte (“reaction solution”) and the cathodic (counter electrode) half-cell containing 50 mL of the electrolyte. The half-cells were separated with a cation exchange membrane (Fumasep FKE-50, Fumatech Germany). The working and counter electrodes were immersed to give a surface area of 10 cm2 each. The distance between the electrodes was approximately 9 cm.For experiments at room temperature, single-wall H-type cells were used in combination with a water bath to ensure constant temperature during the measurement and to balance any possible temperature increase caused by ohmic heating. Experiments at higher temperatures were conducted in double-wall H-type cells, which were heated with by a thermostat (Ecoline E103, Lauda, Germany) using a water/ethylene glycol mixture (50/50%vol) as heat transfer medium.
In the galvanostatic experiments, the respective current was applied until the required charge (i.e. 6 or 24 F, corresponding to reaction times of 15.8 and 63.2 h) was transported. During the reaction, the HMF solution was stirred at 500 rpm. In the 6 F experiments, 1 mL of the reaction solution was withdrawn before and after the experiment, filtered and analyzed via HPLC. In the 24 F experiment, samples of 200 μL were withdrawn and analyzed via HPLC without filtration. In addition, 5 mL of each reaction solution was taken after the reaction and used for ICP-OES analysis without filtration.
All experiments were conducted in duplicates. The mean values of both measurements were used for Fig. 2–5. Error bars were not depicted in the figures to maintain the clarity. Instead, numeric mean values and standard deviations of all reactions can be found in Tables S1–S4 in the ESI.† The stated potentials represent average potentials measured over the entire course of the experiments. Exemplary potential curves of all optimization experiments are shown in Fig. S4 in the ESI.†
HPLC analysis
Samples were analyzed using a 1260 Infinity II HPLC system (Agilent, USA) with a REZEX ROA Organic Acid column (300 × 7.8 mm, Phenomenex, USA). The substances were separated at a column temperature of 70 °C with an isocratic method. Here, an aqueous H2SO4 solution (2.5 mM) at a flow rate of 0.6 mL min−1 was used as mobile phase. Quantitative analysis of HMF, DFF, HMFCA, FFCA, FDCA and MA was accomplished using the signals of a refractive index detector and a diode array detector. These substances were calibrated using standard solutions made from the pure substances specified above. The presence of formaldehyde and formic acid was checked qualitatively.ICP-OES analysis
To analyze the reaction samples with ICP-OES, the organic components had to be removed from the solution. Therefore, 1 mL of every sample was mixed with 5 mL HNO3 (conc.). The mixtures were transferred into PTFE tubes and digested with a Mars 5 microwave digestion system (CEM Corp., USA). Here, the temperature was increased to 90 °C within 10 min and afterwards held constant at 90 °C for 20 min while the microwave was set to 448 W (56% of 800 W). After cooling down, the digested samples were diluted with DI water to give 25 mL of solution.The diluted samples were analyzed with an Optima 2100 DV ICP-OES system (PerkinElmer, USA) with a nebulizer flow rate of 0.6 L min−1. The concentration of the dissolved cobalt was determined via the intensity of the 228.616 nm emission line. For each sample the mean value of three consecutive measurements was used. For calibration a cobalt standard solution (1000 ± 2 mg L−1, Merck) was used.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the German Federal Ministry for Economic Affairs and Climate change under the ElektroSyn project (Project ID 03EN2071D). The authors would like to thank Anja Albrecht and Gabriele Meyer for the analysis of the ICP-OES samples.References
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., 2011, 123, 10686–10694 CrossRef.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- T. Lenk, V. Rueß, J. Gresch and U. Schröder, Green Chem., 2023, 25, 3077–3085 RSC.
- O. Simoska, Z. Rhodes, S. Weliwatte, J. R. Cabrera-Pardo, E. M. Gaffney, K. Lim and S. D. Minteer, ChemSusChem, 2021, 14, 1674–1686 CrossRef CAS PubMed.
- L. Guo, X. Zhang, L. Gan, L. Pan, C. Shi, Z. Huang, X. Zhang and J. Zou, Adv. Sci., 2023, 10, 2205540 CrossRef CAS PubMed.
- A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407–6422 RSC.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 1–18 CrossRef PubMed.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- D. A. Giannakoudakis, J. C. Colmenares, K. S. Triantafyllidis and D. Tsiplakides, ACS Sustain. Chem. Eng., 2021, 9, 1970–1993 CrossRef CAS.
- R. Latsuzbaia, R. J. M. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- S. Zhang, T. Yu, H. Wen, Z. Ni, Y. He, R. Guo, J. You and X. Liu, Chem. Commun., 2020, 56, 15387–15405 RSC.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- Z. Zhou, C. Chen, M. Gao, B. Xia and J. Zhang, Green Chem., 2019, 21, 6699–6706 RSC.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- R. Zhang, S. Jiang, Y. Rao, S. Chen, Q. Yue and Y. Kang, Green Chem., 2021, 23, 2525–2530 RSC.
- L. Gao, Y. Bao, S. Gan, Z. Sun, Z. Song, D. Han, F. Li and L. Niu, ChemSusChem, 2018, 11, 2547–2553 CrossRef CAS PubMed.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 CrossRef CAS.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- H. A. Rass, N. Essayem and M. Besson, ChemSusChem, 2015, 8, 1206–1217 CrossRef PubMed.
- Y. Zhang, X. Guo, P. Tang and J. Xu, J. Chem. Eng. Data, 2018, 63, 1316–1324 CrossRef CAS.
- H. B. Rose, T. Greinert, C. Held, G. Sadowski and A. S. Bommarius, J. Chem. Eng. Data, 2018, 63, 1460–1470 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14431–14442 CrossRef CAS PubMed.
- S. Dey, B. Mondal and A. Dey, Phys. Chem. Chem. Phys., 2014, 16, 12221–12227 RSC.
- S. R. Kubota and K.-S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- J. Weidner, S. Barwe, K. Sliozberg, S. Piontek, J. Masa, U. P. Apfel and W. Schuhmann, Beilstein J. Org. Chem., 2018, 14, 1436–1445 CrossRef PubMed.
- D. H. Nam, B. J. Taitt and K.-S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- J. G. McAlpin, Y. Surendranath, M. Dincã, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882–6883 CrossRef CAS PubMed.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. Glasstone and A. Hickling, J. Chem. Soc., 1934, 1878–1888 RSC.
- B. J. Taitt, D. H. Nam and K.-S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- M. T. Bender and K.-S. Choi, ChemSusChem, 2022, 15, e202200675 CrossRef CAS PubMed.
- M. K. Goetz, M. T. Bender and K.-S. Choi, Nat. Commun., 2022, 13, 5848 CrossRef CAS PubMed.
- P. Nilges, T. R. Dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- S. R. Kubota and K.-S. Choi, ACS Sustain. Chem. Eng., 2018, 6, 9596–9600 CrossRef CAS.
- P. Drögemüller, T. Stobbe and U. Schröder, ChemSusChem, 2024, 17, e2023009 CrossRef PubMed.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, Appl. Catal., B, 2021, 297, 120396 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00050a |
| This journal is © The Royal Society of Chemistry 2024 |