
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the performance of heterogeneous palladium based catalysts in the mild reductive depolymerization of soda lignin through addition of a non-noble metal and tuning of the preparation strategy†
Tibo
De Saegher
a,
Boyana
Atanasova
a,
Pieter
Vermeir
b,
Kevin M.
Van Geem
 c,
Jeriffa
De Clercq
a,
An
Verberckmoes
a and
Jeroen
Lauwaert
*a
c,
Jeriffa
De Clercq
a,
An
Verberckmoes
a and
Jeroen
Lauwaert
*a
aIndustrial Catalysis and Adsorption Technology (INCAT), Department of Materials Textiles and Chemical Engineering (MaTCh), Ghent University, Valentin Vaerwyckweg 1, 9000 Ghent, Belgium. E-mail: Jeroen.Lauwaert@UGent.be
bLaboratory for Chemical Analysis, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Valentin Vaerwyckweg 1, 9000 Ghent, Belgium
cLaboratory for Chemical Technology (LCT), Department of Materials Textiles and Chemical Engineering (MaTCh), Ghent University, Technologiepark 125, 9052 Ghent, Belgium
First published on 8th April 2024
Abstract
Research towards mild reductive depolymerization of lignin is gaining momentum because of its potential for producing sustainable functionalized aromatics, but achieving high yields still relies on expensive noble metal catalysts. This study aims to improve the catalysts' cost effectiveness through addition of a non-noble metal to a Pd nanoparticle catalyst, supported on γ-Al2O3. Six Pd based catalysts (Pd, PdCu, PdNi, PdFe, PdCo, and PdMo) were synthesized and prepared through either calcination or thermal reduction, and their activity and selectivity in lignin depolymerization were evaluated as a function of batch time. Principal component analysis (PCA) of the entire datapool revealed that, albeit to varying degrees, the addition of a secondary metal shifts the behavior of a Pd catalyst more towards that of pure solvolysis and that the preparation strategy has no effect on Pd and PdMo. Regarding activity, it was found that the addition of Cu, Ni, Fe, Co and Mo significantly enhances the catalyst's activity and that the preparation strategy is also important, with calcination being preferred for PdCu and PdFe and thermal reduction for PdNi and PdCo. Using a plethora of analysis techniques to assess the selectivity at increasing depths, it was revealed that the shift in selectivity, as identified in the PCA results, is caused by variations in dehydration of aliphatic OH groups and hydrogenation of aliphatic double bonds. Moreover, due to a size exclusion effect during the reaction, differences in selectivity between the catalysts are most pronounced at lower molecular weights.
Sustainability spotlightAs the effects of climate change become unmistakable, the imperative for transitioning towards a sustainable chemical industry has grown ever more pronounced. In this context, lignin depolymerization under mild conditions will provide a sustainable route for the production of functionalized aromatics, which will serve as essential building blocks for the production of polymers, pharmaceuticals and other high-value chemicals. Within this research, the economic viability of this process, which is currently still hindered by the need for expensive noble based catalysts, is increased through the incorporation of a non-noble metal and tuning of the preparation strategy, drastically reducing catalyst costs, while maintaining the catalytic activity and product selectivities. Consequently, these results contribute to the establishment of responsible consumption and production patterns. |
1. Introduction
Lignocellulosic biomass, comprising cellulose, hemicellulose and lignin, is recognized as the cheapest and most abundant inedible biomass. Moreover, it is anticipated to be the most scalable and economically viable source for bio-fuel and high-value chemical production. Consequently, the depolymerization of lignin has emerged as a subject of growing research interest.1–10 While macropolymer lignin, i.e., extracted from the biomass but not yet depolymerized, is already used for the production of adhesives, emulsifiers, plastics and other products,4,7 selectively depolymerizing said lignin to its functionalized aromatic building blocks would provide sustainable alternatives to fossil based benzene, toluene and xylene (BTX) derivatives, which are crucial platform molecules within the polymer and pharmaceutical industries.5–8A wide range of depolymerization strategies have been developed, which can be categorized as acid catalyzed, base catalyzed, oxidative, reductive, thermal or solvolytic depolymerization. Each of these strategies has its own benefits and drawbacks and the obtained products can range from native-like functionalized aromatic compounds to alkanes, alkenes and aliphatic alcohols and/or mixtures thereof. Moreover, each main category includes different subcategories, for example liquid phase reforming, mild hydroprocessing, harsh hydroprocessing and bifunctional hydroprocessing, all of which fall under the umbrella of reductive depolymerization. Furthermore, for each subcategory, a range of process conditions, such as catalyst type, temperature, solvent, gas atmosphere, and pressure, can be varied, affecting product yield and selectivity.11
Hence, it is imperative that the depolymerization strategy and conditions are tailored towards the intended application of the resulting product pool to maximize the valorization potential of lignin. The aforementioned production of sustainable polymers and pharmaceuticals from lignin-derived compounds would primarily require functionalized aromatic compounds.5–8 Within this context, mild hydroprocessing, also known as mild reductive depolymerization, is a promising strategy, as it has demonstrated high selectivities towards such functionalized aromatic products.11 Moreover, operating at temperatures below 320 °C and pressures between 10 and 100 bar, this approach allows the use of green solvents such as isopropyl alcohol or ethanol, either pure or in mixtures with water. Furthermore, reaction temperatures and pressures far below 320 °C and 100 bar have been implemented successfully.12–18 Additionally, this strategy can also be integrated with the extraction of lignin from the biomass, which is typically performed beforehand within a dedicated pulping process. This integrated process is often referred to as reductive catalytic fractionation (RCF).2,10,11
Despite its promise, both mild reductive depolymerization of lignin and RCF currently face challenges, including low overall yields of desired products. Typically, the use of expensive noble metal based, i.e., platinum (Pt), palladium (Pd) or ruthenium (Ru), catalysts, is required to achieve sufficient depolymerization under mild conditions, with satisfactory selectivities towards low molecular weight aromatics, rendering the process economically unviable.11,12 Within the literature, it has been noted than Pd exhibits significantly less hydrodeoxygenation activity than Ru and Pt, making it a preferred choice for the production of lignin-derived functionalized aromatic compounds, despite its higher cost.19,20 As a result, Pd based nanoparticle catalysts, supported on various supporting materials such as carbon,21,22 alumina23 or zeolites24,25 have been implemented successfully to depolymerize lignin under mild reductive conditions.11 Nonetheless, the high cost of Pd inhibits its application as a catalyst within actual industry. Hence, a primary research goal in the mild reductive depolymerization of lignin is to reduce Pd catalyst synthesis costs without sacrificing catalyst activity or selectivity towards the desired products. One strategy to achieve this is the incorporation of non-noble metals. This has been proven to create synergetic effects and, hence, alter the activity of the noble metal. Furthermore, as non-noble metals typically have a higher oxyphilicity than Pd, they interact with functional groups in a different way. As a result, they not only impact the catalyst activity but also the selectivity.26 It has been noted in the literature that the addition of non-noble metals to Pd catalysts can, on the one hand suppress unwanted hydrogenation of aromatics but, on the other hand, also cause hydrodeoxygenation reactions, which are also undesirable.12,14–16,27–29 Therefore, careful selection of secondary metals is key to ensuring a boost in catalyst activity while still producing desirable products, suitable for the production of both polymers and pharmaceuticals.5–8 The use of nickel (Ni), copper (Cu) or cobalt (Co) instead of Pd catalysts for the mild reductive depolymerization of lignin or RCF of biomass has been successful, albeit at elevated temperatures, i.e., above 240 °C.12,14–18,29 Moreover, monometallic Ni demonstrates acceptable yields at lower temperatures, though still significantly lower than Pt, Pd or Ru and often with the additional aid of acid co-catalysts. Despite this, Ni remains a strong candidate for a secondary metal to be added to a Pd catalyst.30,31 Concerning selectivity, addition of Ni or Fe (iron) to Pd catalysts has also been observed to inhibit the hydrogenation of aromatics.28,29 Additionally, monometallic Co based catalysts have been shown to facilitate the oxidation of the α-OH group within the β-O-4 linkage, significantly lowering the bond dissociation energy of the ether linkage, making Co another compelling secondary metal.32–34 Finally, molybdenum (Mo) is also selected as a candidate as monometallic Mo has also been found to catalyze the reductive depolymerization of lignin, however, at temperatures above 250 °C.35,36 To the best of our knowledge, a study systematically comparing supported PdX catalysts (X = Cu, Ni, Fe, Co or Mo) for the mild reductive depolymerization of lignin has not yet been performed. Therefore, the primary objective of this study is to address and fill this research gap. Additionally, the assessment of both calcination and a sequential process involving calcination, thermal reduction and passivation as preparation strategies for all catalysts introduces an additional dimension to the current state of the art in this field. The final objective of this study is to introduce the use of principal component analysis (PCA) into the field of lignin depolymerization.
Currently, there are many contradictory insights to be found in the literature. For example, several studies suggest that the synthesis of highly dispersed Pd nanoparticles can achieve high degrees of depolymerization at low Pd loadings.13 However, in contrast, other studies seem to demonstrate that bulk Pd sites exhibit substantially increased depolymerization activity and attribute this to the facilitation of multidentate adsorption of lignin molecules.24 Thus, although specific studies have reported some trends between catalyst properties and their performance in lignin depolymerization, to the best of our knowledge, the underlying mechanisms still largely remain unknown. This is, in our opinion, largely due to the chemical complexity and heterogeneity of lignin, which not only causes a very complex behavior, but also negates the ability to define clear parameters to unambiguously assess and fairly compare catalyst performances, such as conversion, turnover number (TON), turnover frequency (TOF) or site time yield (STY).29 The objective of this study, to investigate the impact of 5 different secondary metals and the preparation strategy on the performance of a Pd catalyst only complicates things further. Therefore, to mitigate this, a strategy of in-depth product pool characterization as a function of reaction time, rather than the characterization of the catalysts themselves, combined with PCA is proposed. A plethora of analysis techniques and data analysis protocols are implemented to study catalyst activity and selectivity in an in-depth manner. The reduction in mass average molar mass (Mw), derived from the analysis of the lignin stock solution and product pools with gel permeation chromatography with a refractive index detector (GPC-RID), is used as a parameter for catalyst activity. Selectivity has been assessed with a plethora of analysis techniques and with an increasing depth, i.e., starting with the differences in average functionalities within the entire product pool through phosphorus nuclear magnetic resonance spectroscopy (31P-NMR), followed by differences for targeted (groups of) compounds through deconvolution of GPC chromatograms and, finally, the molar concentrations of the main monomeric products through an in-house two-dimensional GPC-HPLC-UV/VIS analysis technique.37,38 PCA is employed to identify overarching trends present within the entire dataset, which are, subsequently, evaluated in more detail.
In summary, in this work, a novel and systematic methodology is employed for the assessment of the catalyst activity and selectivity in the mild reductive depolymerization of lignin. The main objective is to identify calcined or reduced and passivated PdX catalysts that offer lower synthesis costs, without compromising catalyst activity or steering the selectivity towards unwanted products, i.e., avoiding hydrogenation of aromatic rings and hydrodeoxygenation of oxygen containing functionalities. Consequently, this study significantly expands on the state of the art as such catalysts are essential in increasing the economic viability and sustainability of mild reductive depolymerization of lignin and RCF of biomass. Furthermore, the proposed research methodology is anticipated to have a profound impact on the future conduct of lignin research.
2. Materials and methods
2.1. Catalyst preparation
A series of different gamma alumina (γ-Al2O3) supported nanoparticle catalysts were synthesized using incipient wetness impregnation of 0.75 mL of precursor solution per 0.95 g of γ-Al2O3 (Sasol Puralox sCCa 150/200). For this study, γ-Al2O3 is selected as the support for the metals, because of its low cost, high mechanical strength and temperature resistance, ease of impregnation with metal precursor solutions, and acidic character. According to the literature, the acid sites on the surface of γ-Al2O3 can act as cocatalysts for the reductive depolymerization of lignin.24 The precursors used for Pd, Cu, Ni, Fe, Co and Mo were Pd(NO3)2·2H2O (Sigma-Aldrich, 98%+), Cu(NO3)2·5H2O (Chem-Lab, 98%+), Ni(NO3)2·6H2O (Sigma-Aldrich, 97%+), Fe(NO3)3·9H2O (Sigma-Aldrich, 98%+), Co(NO3)2·6H2O (Chem-Lab, 98%+) and (NH4)6Mo7O24·4H2O (Sigma-Aldrich, 99%+), respectively, which were dissolved in H2O (Chemlab, HPLC grade). The catalyst series consists of 6 monometallic catalysts containing 5 wt% of either Pd, Cu, Ni, Fe, Co or Mo and 5 bimetallic catalysts combining Pd with one of the non-noble metals with a total metal content of 5 wt% in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The bimetallic catalysts were all synthesized through impregnation of a precursor solution containing both metals, except for PdMo. For the latter, two separate 0.375 mL solutions containing one of both precursors were impregnated simultaneously, due to the limited mutual solubility of Pd(NO3)2·2H2O and (NH4)6Mo7O24·4H2O. All catalysts were subsequently heated to 60 °C at a rate of 2 °C min−1, dried at this temperature for 16 h, further heated to 450 °C at 20 °C min−1 and finally calcined at this temperature for 4 h. The resulting catalysts are labeled Pd-calc or (Pd)X-calc with ‘X’ being Cu, Ni, Fe, Co or Mo. Half of the amount of each calcined catalyst was, subsequently, thermally reduced and passivated using an Autochem II series from Micromeritics. During thermal reduction, the catalysts were purged at 120 °C for 1 h and 200 °C for 5 min under an argon (Ar) atmosphere, reduced at 500 °C for 1 h with 5% H2 in Ar at 60 cm3 min−1 and passivated at 50 °C for 1 h with 1% nitrogen oxide (N2O) in helium (He) at 10 cm3 min−1. These catalysts are labeled Pd-red or (Pd)X-red. Hence, in total 20 catalysts were prepared, 5 monometallic and 5 bimetallic ones, both in calcined and reduced states.
:1. The bimetallic catalysts were all synthesized through impregnation of a precursor solution containing both metals, except for PdMo. For the latter, two separate 0.375 mL solutions containing one of both precursors were impregnated simultaneously, due to the limited mutual solubility of Pd(NO3)2·2H2O and (NH4)6Mo7O24·4H2O. All catalysts were subsequently heated to 60 °C at a rate of 2 °C min−1, dried at this temperature for 16 h, further heated to 450 °C at 20 °C min−1 and finally calcined at this temperature for 4 h. The resulting catalysts are labeled Pd-calc or (Pd)X-calc with ‘X’ being Cu, Ni, Fe, Co or Mo. Half of the amount of each calcined catalyst was, subsequently, thermally reduced and passivated using an Autochem II series from Micromeritics. During thermal reduction, the catalysts were purged at 120 °C for 1 h and 200 °C for 5 min under an argon (Ar) atmosphere, reduced at 500 °C for 1 h with 5% H2 in Ar at 60 cm3 min−1 and passivated at 50 °C for 1 h with 1% nitrogen oxide (N2O) in helium (He) at 10 cm3 min−1. These catalysts are labeled Pd-red or (Pd)X-red. Hence, in total 20 catalysts were prepared, 5 monometallic and 5 bimetallic ones, both in calcined and reduced states.
2.2. Determining the metal content
All Pd containing catalysts, i.e., all Pd(X)-calc and Pd(X)-red catalysts, were characterized with inductively coupled plasma-optical emission spectroscopy (ICP-OES) to determine the Pd, Cu, Ni, Fe, Co and Mo content. The ICP-OES analyses were performed with an IRIS Intrepid II XSP instrument. Before analysis, destruction of the supporting γ-Al2O3 is performed by mixing 0.025 g of catalyst with 2 mL of HNO3 (65%), 2 mL of HCl (37%) and 2 mL of HF (40%) solutions inside of a destruction tube. The latter is then heated to 250 °C at 8.33 °C min−1 and held at this temperature for 20 min by means of a microwave. The resulting mixture, wherein no solid catalyst is present anymore, is diluted quantitatively and analyzed through ICP-OES.2.3. Catalyst performance testing in the mild reductive depolymerization of lignin
A depolymerization reaction is performed by loading a reactor vessel with 0.05 g of catalyst, 5.25 mL of ethanol (100% Abs, Chemlab), 2.25 mL of H2O (HPLC grade, Chemlab) and 2.5 mL of the miscanthus lignin stock solution. Afterwards, the reactor vessel is closed off, flushed with hydrogen gas (H2) for 10 seconds and finally pressurized to 10 bar with H2. The reaction is started when the reactor vessel is placed inside of the heating block, which has been preheated to 200 °C. As a result, the temperature inside of the reactors is 180 °C. All catalysts as well as the experiments without the catalyst (0Cata) and in the presence of pristine γ-Al2O3 (Al2O3) were studied at reaction times of 3 h (with the exception of Pd-calc and Pd-red, which were tested at 2 h), 6 h and 20 h. However, note that, in order to account for slight deviations in metal contents between the various catalysts, the catalyst performance was assessed as a function of batch time (τ (mmol Pd s)), which is defined as the molar amount of Pd added to the reactor (nPd, mmol Pd) multiplied by the reaction time (Δt, s), and can be calculated from eqn (1), in which mcatalyst (g) represents the catalyst mass added to the reactor, wt%Pd is the weight percentage of Pd on the catalyst support as determined through ICP-OES analysis and MMPd, the molar mass of Pd (g mol−1).
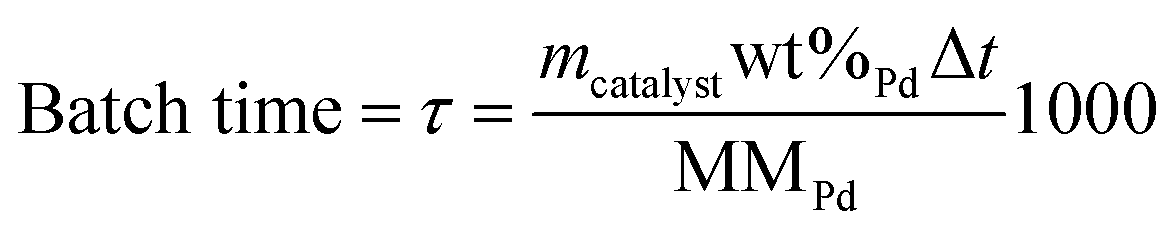 | (1) |
After the desired reaction time is reached, the reactor vessel is removed from the heating block, cooled down to ambient temperature in water and depressurized. The reactor effluent is extracted from the reactor using a syringe equipped with a 0.25 μm PTFE filter. Three 2 mL samples of each reactor effluent were evaporated at 80 °C for >16 h and stored at −4 °C in closed glass vials for later analysis.
2.4. Product pool analysis
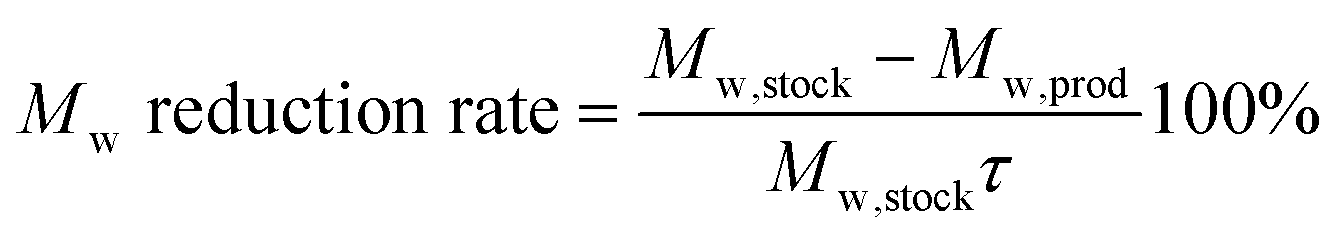 | (2) |
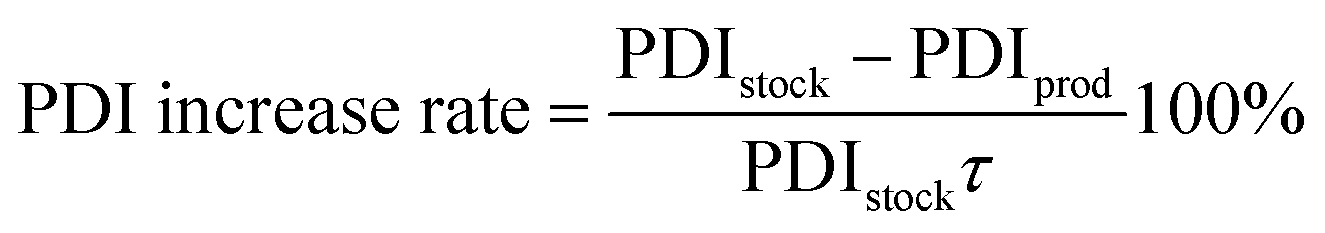 | (3) |
The GPC-UV/VIS data are deconvoluted using 6log normal curves, which were selected to account for natural peak tailing within GPC. In such a deconvolution, the objective function L (eqn (4)) was minimized. The first term in this function represents the absolute difference between the experimental GPC-UV/VIS chromatogram (Yi,exp) and the calculated composite of the deconvoluted curves (Yi,calc) at any given retention time. The second term represents the difference between the derivatives of the experimental 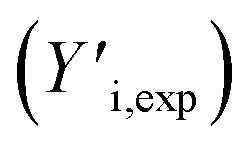 and calculated
and calculated 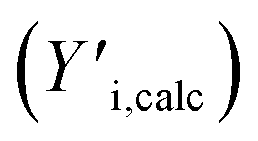 curves and was added to better match the experimentally observed shape of the molecular weight distributions. Based on preliminary tests, the weight factors, w1 and w2, were set at 30 and 1, respectively. The resulting deconvoluted peak areas are normalized through division by the total area of the respective GPC-UV/VIS chromatogram and can be used to qualitatively assess differences in product selectivity between catalysts. In particular, the three discrete peaks between 19.5 min and 23.5 min (≈40–400 g mol−1), which contain most low molecular weight compounds, i.e., monomers to trimers/tetramers, are of interest to study the catalyst's effect on the product pool. Previous research has concluded that group 1 mainly contains monomers with propyl and propenyl chains as para-substitutions, group 2 contains monomers with propyl or propenyl chains with oxygen functionalities and group 3 mainly contains dimeric products.37,38
curves and was added to better match the experimentally observed shape of the molecular weight distributions. Based on preliminary tests, the weight factors, w1 and w2, were set at 30 and 1, respectively. The resulting deconvoluted peak areas are normalized through division by the total area of the respective GPC-UV/VIS chromatogram and can be used to qualitatively assess differences in product selectivity between catalysts. In particular, the three discrete peaks between 19.5 min and 23.5 min (≈40–400 g mol−1), which contain most low molecular weight compounds, i.e., monomers to trimers/tetramers, are of interest to study the catalyst's effect on the product pool. Previous research has concluded that group 1 mainly contains monomers with propyl and propenyl chains as para-substitutions, group 2 contains monomers with propyl or propenyl chains with oxygen functionalities and group 3 mainly contains dimeric products.37,38
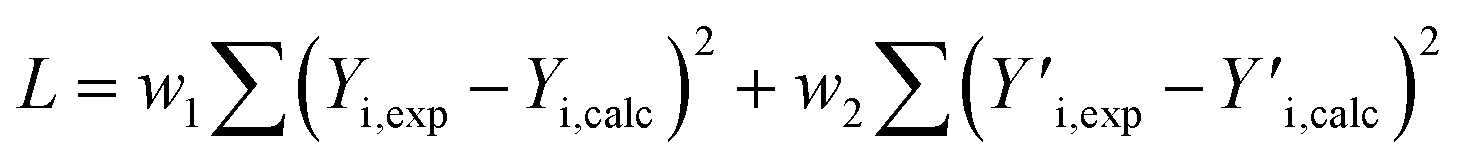 | (4) |
The 95% confidence intervals for the Mw reduction (rate), PDI increase (rate), total area under the GPC-UV/VIS chromatogram and the normalized group areas were determined from 6 repeated reactions (reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg of lignin and 50 mg of Pd-calc, 6 h reaction time). Hence, these intervals comprise experimental errors arising from the catalyst performance testing, sample analysis and the deconvolution procedure.
Several low monomeric compounds were quantified directly within the GPC-HPLC-UV/VIS heatmaps according to a protocol published elsewhere.2 The mass yield of each compound is calculated as the mass formed of the product divided by the amount of lignin added to the system (265 mg). The compounds that were identified through calibration with standards are eugenol (EUG, 99+%, Sigma-Aldrich), isoeugenol (I-EUG, 99+%, Sigma-Aldrich), vanillin (V, 99%, Sigma-Aldrich), 4-hydroxy-3-methoxycinnamaldehyde (HMCA, 96%, Sigma-Aldrich), 4-hydroxy-3-methoxyphenylacetone (HMPA, 96%, Sigma-Aldrich), 2-methoxy-4-propylphenol (MPP, 99+%, Sigma-Aldrich), ethyl hydroferulate (EHF, 90%, Biosynth), dihydroconiferylalcohol (DHCA, 97%, Ambeed), coniferyl alcohol (CA, Alfa Aesar, 98%), sinapyl alcohol (SA, Sigma-Aldrich, 80%) and 4-propylsyringol (4PS, 90%, Chemspace) and are depicted in Fig. 1. Besides quantification of isolated monomeric products, the center point, i.e., the 1D and 2D coordinates of the weighted average of the entire heat map, were also determined utilizing a Python algorithm.
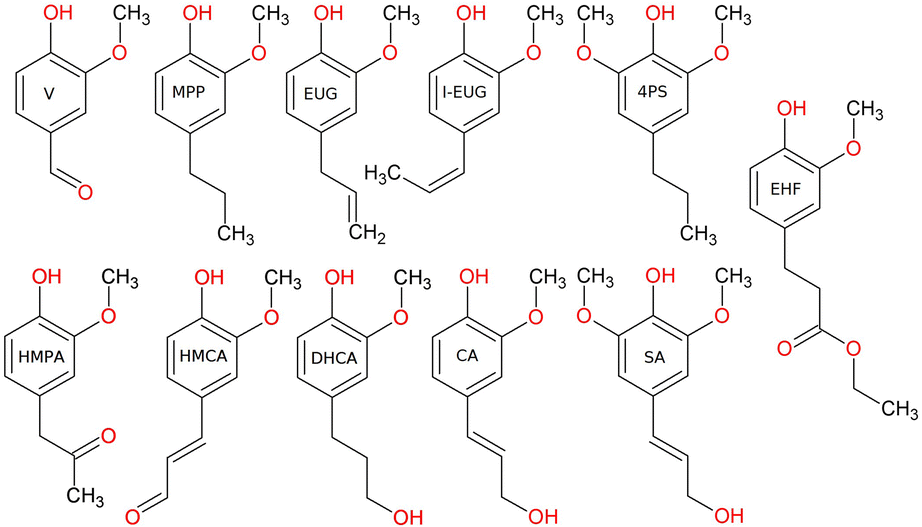 | ||
| Fig. 1 Chemical structure and corresponding abbreviations for all quantified monomeric products. | ||
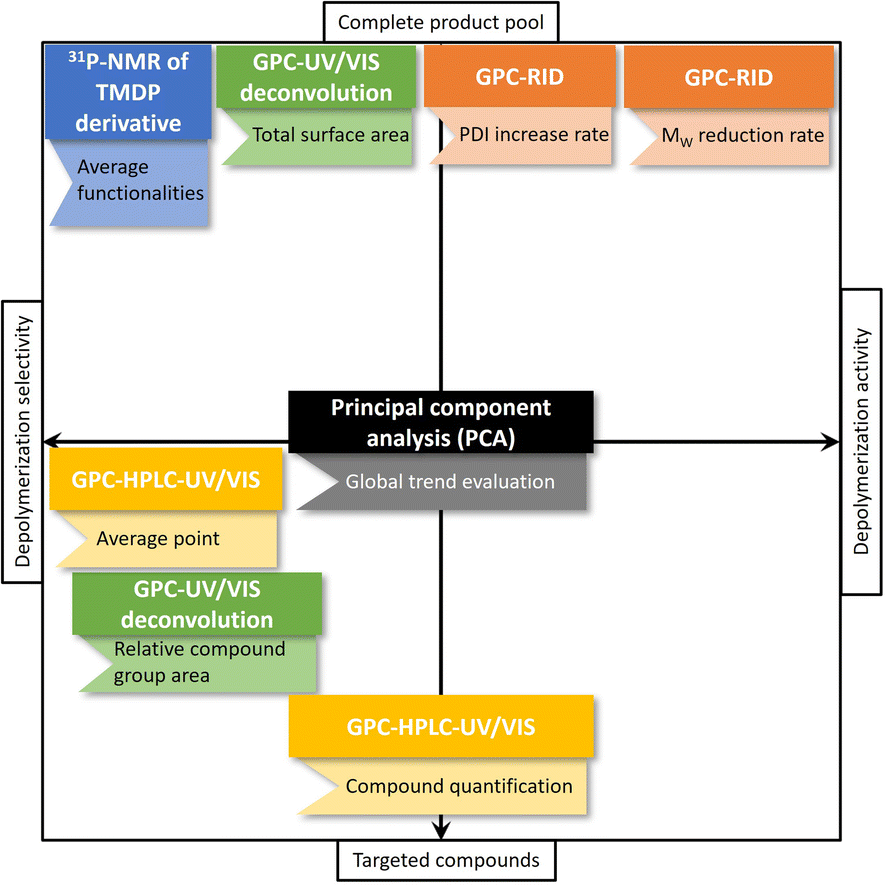 | ||
| Fig. 2 All analysis techniques performed on the lignin feedstock and depolymerization product pools, divided based on whether they analyze the entire product pool or targeted compounds and on whether they assess depolymerization activity or selectivity. | ||
3. Results and discussion
3.1. Global trend evaluation through PCA
Twelve Pd containing catalysts, i.e., Pd(X)-calc and Pd(X)-red catalysts, have been prepared and assessed for their performance in the mild reductive depolymerization. Additionally, the depolymerization was also evaluated without a catalyst (0Cata), which can be considered solvolysis, albeit under a hydrogen atmosphere. Furthermore, the same reaction was performed with pristine γ-Al2O3 (Al2O3). All resulting product pools were thoroughly analyzed by means of GPC-RID (Mw and PDI), GPC-UV/VIS (total area and relative peak areas for groups 1, 2 and 3), 31P-NMR (aliphatic OH content, phenolic OH content, carboxylic acid content, condensed phenolic content and uncondensed phenolic content) and GPC-HPLC-UV/VIS (average first and second dimension times (1tR and 2tR) and molar concentrations for V, DHCA, HMPA, HMCA, EHF, EUG, I-EUG, 4PS, MPP, CA and SA).All of the data derived from these techniques were used as inputs to the PCA. According to the scree plot in Fig. S1(ESI),† 2 principal components already achieve a cumulated explained variance ratio of 91.31%, which is considered satisfactory to accurately represent the variance within the total data set. The resulting principal component loadings are presented in Table 1 and the resulting PCA plot for 0Cata, Al2O3 and all Pd containing catalysts is depicted in Fig. 3.
| Analysis technique | Feature | Principal component 1 loading | Principal component 2 loading |
|---|---|---|---|
| GPC-RID | M w | −0.145 | 0.294 |
| PDI | 0.146 | 0.250 | |
| GPC-UV/VIS deconvolution | Total area | −0.329 | −0.062 |
| Relative peak area of group 1 | 0.163 | −0.082 | |
| Relative peak area of group 2 | 0.079 | 0.260 | |
| Relative peak area of group 3 | −0.090 | −0.275 | |
| 31P-NMR | Aliphatic OH content | 0.086 | 0.299 |
| Phenolic OH content | 0.147 | −0.290 | |
| Carboxylic acid content | 0.156 | 0.200 | |
| Condensed phenolics | 0.097 | −0.316 | |
| Uncondensed phenolics | 0.187 | −0.228 | |
| GPC-HPLC-UV/VIS analysis | Average 1tR | 0.171 | 0.217 |
| Average 2tR | −0.141 | −0.274 | |
| V concentration | −0.171 | −0.077 | |
| DHCA concentration | 0.333 | −0.007 | |
| HMPA concentration | 0.337 | −0.005 | |
| HMCA concentration | −0.240 | 0.190 | |
| EHF concentration | 0.253 | −0.168 | |
| EUG concentration | −0.095 | −0.258 | |
| I-EUG concentration | −0.017 | −0.179 | |
| 4PS concentration | 0.245 | −0.027 | |
| MPP concentration | 0.305 | 0.055 | |
| CA concentration | −0.160 | 0.190 | |
| SA concentration | 0.316 | 0.028 |
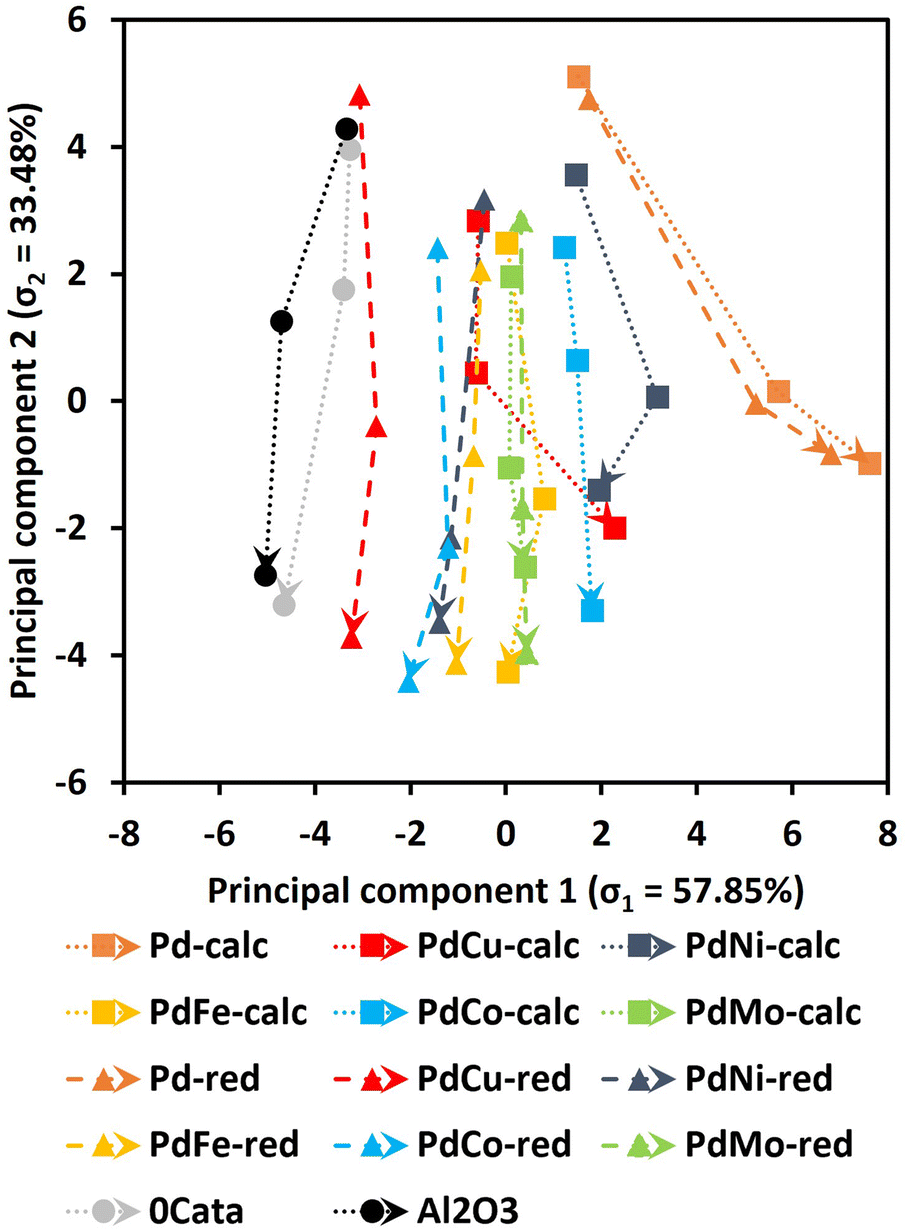 | ||
| Fig. 3 Principal component analysis for 0Cata, Al2O3 and all Pd(X)-calc/red variations as a function of reaction time (indicated as arrows between points, i.e., 2 or 3 h, 6 h and 20 h of reaction time). The explained variance ratios for principal components 1 and 2 are 57.85% and 33.48% respectively. | ||
First, the PCA results in Fig. 3 indicate that there is significant variance amongst the different experiments. However, certain pairs seem to show little to no difference. Firstly, 0Cata and Al2O3 exhibit very similar behavior, implying that the acidic functionalities within γ-Al2O3 are not contributing to the depolymerization to a substantial degree without the presence of a noble metal. Secondly, for Pd and PdMo, almost no variations are observed between the calc and red variants, implying that the preparation strategy has little to no effect on the catalytic behavior. Noteworthily, for all other combinations, i.e., PdCu, PdNi, PdFe and PdCo, albeit to varying degrees, the preparation strategy does appear to impact the catalytic behavior. Furthermore, the addition of any of the secondary metals substantially changes the behavior of a Pd catalyst.
In Fig. 3, the most prominent global trend is observed along PC1, wherein 0Cata and Al2O3 exhibit the lowest PC1 values, while Pd-calc/red display the highest values, with all PdX-calc/red variations in between. Moreover, 0Cata and Al2O3 show slightly decreasing PC1 values as a function of reaction time, whereas Pd-calc/red demonstrates a clear increase. According to the PC1 loadings in Table 1, a higher PC1 value can mainly be attributed to a higher total area in GPC-UV/VIS spectra, resulting from reduced hydrogenation and hydrodeoxygenation37,38 and higher concentrations of specific monomers, i.e., DHCA, HMPA, MPP, 4PS, SA and EHF. Additionally, lower concentrations of HMCA, CA and V will also lead to higher PC1 values. Among these, HMCA and CA have been deemed unfavorable in the literature as their para-substitution with an unsaturated bond and oxygen functionality might lead to repolymerization reactions.18 The fact that PC1, which is heavily impacted by monomer concentrations, shows the clearest trend within the entire dataset may be attributed to a size exclusion effect, where only lignin molecules below a certain hydrodynamic volume can access the active metal sites within the support material, has been noted in the literature before.40 Lower, Mw values, higher phenolic OH contents and higher uncondensed phenolic contents, all implying a higher degree of depolymerization, also contribute to higher PC1 values. This aligns with the literature, where Pd catalyzed lignin depolymerizations consistently outperform solvolysis (0Cata and Al2O3).11,12,20,23,41
The loadings indicate that the global trend along PC1 is mainly associated with selectivity, in particular towards the monomer fraction, and to a lower extent the activity. Hence, higher PC1 values should relate to overall more favorable product pools. Within this regard, the addition of a secondary metal and the preparation strategy, albeit to varying degrees, shifts the PC1 values between 0Cata/Al2O3 and Pd-calc/red. In particularly, the PC1 results position PdCu-calc as an interesting candidate as it is the only PdX variation with an increasing PC1 value between 6 h and 20 h of reaction time. In the following sections, an in-depth evaluation of the individual analyses related to selectivity will be performed aiming at a chemical understanding of the trend observed along PC1.
On the one hand, the PC2 loadings in Table 1 imply a heavy correlation between lower PC2 values and high depolymerization degrees, i.e., product pools with low Mw and PDI values, low condensed phenolics, high phenolic OH content and low concentrations of the unstable monomers HMCA and CA.18 Additionally, the aliphatic OH content is anticipated to decrease during depolymerization, as the most energetically favorable cleavage pathways of the β-O-4 linkage, the most abundant linkage within the miscanthus lignin39 and most native-like lignins, facilitates the transformation and/or removal of the aliphatic OH function on the α-position.42 Moreover, even if not removed through β-O-4 cleavage (i.e., hydrogenolysis occurred), the OH group on the α-position and the potential OH group on the γ-position can still be transformed or removed in various modification reactions under the mild reductive environment.43,44 Previous research has also related higher degrees of depolymerization to increased average 1tR in the GPC-HPLC-UV/VIS analysis.37,38 Indeed, PC2 values decrease as a function of reaction time for all experiments as shown in Fig. 3. On the other hand, no clear trend can be observed along the PC2 axis amongst the samples. Moreover, Pd-calc/red display higher PC2 values at 20 h of reaction time than 0Cata/Al2O3 while, based on the literature, they should achieve substantially higher degrees of depolymerization.11,12,20 Hence, PC2 appears not to be exclusively correlated with the reaction activity. The latter is substantiated by the fact that the relative peak areas of groups 2 and 3 and average 2tR, which are primarily related to selectivity, also have high PC2 loadings.37,38 Additionally, higher condensed phenolic contents, which are generally unwanted as they imply a lot or repolymerization, would also lower PC2 values. The lack of a clear trend along PC2, despite the strong correlation with the degree of depolymerization, implies that the differences amongst the various experiments are also relatively small.
Therefore, to obtain a more comprehensive understanding of the origins of the trends observed in PCA, or the lack thereof, in-depth analyses of the catalyst activities and selectivities are deemed crucial.
As a benchmark to allow a fair assessment of the catalyst performances, the next section starts with an investigation of the effects of potential interfering factors, i.e., outside of the intrinsic catalyst performances. As organic solvents and pristine γ-Al2O3 have been reported to cause varying degrees of depolymerization,11,24 their effects on the depolymerization process are studied first. Subsequently, the varying Pd content in the different catalysts is taken into account in the catalyst activity evaluation. Then, the effect of the addition of a non-noble metal as well as the catalyst preparation strategy on the catalyst activity is mapped. Finally, to gain a chemical understanding of the PC1 trend, mainly associated with selectivity, especially in the monomer fraction, the selectivity differences of the catalysts are thoroughly investigated.
3.2. Depolymerization activity and selectivity without a Pd containing catalyst
For the situation without any catalytic material (0Cata) and with pristine γ-Al2O3 (Al2O3), depolymerization was performed and evaluated after 3 h, 6 h and 20 h of reaction. Due to the absence of Pd in the reactor for these experiments, the results are presented as a function of reaction time instead of batch time (τ).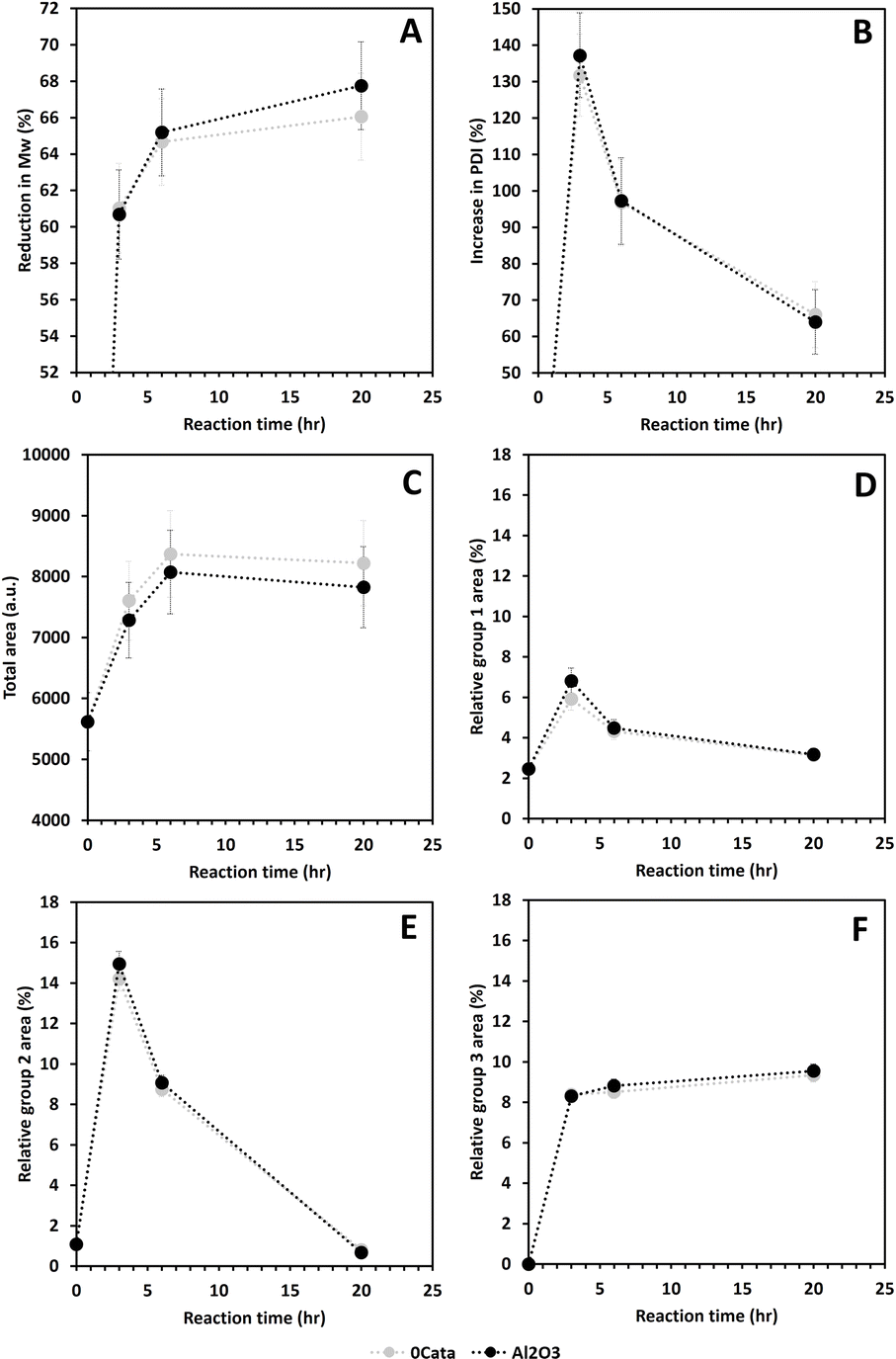 | ||
| Fig. 4 Reduction in Mw (A) and increase in PDI (B) determined through GPC-RID and total area under the GPC-UV/VIS chromatogram (C), and relative peak 1 area (E), relative peak 2 area (D) and relative peak 3 area (F) determined through deconvolution of the GPC-UV/VIS chromatogram (F), all as a function of reaction time for 0Cata and Al2O3. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg lignin and 50 mg catalyst if used. | ||
3.3. Depolymerization activity and selectivity in the presence of a Pd containing catalyst
| Catalyst | Pd content (wt%) (yield (%)) | M2 content (wt%) (yield (%)) |
|---|---|---|
| Pd-calc | 3.18 (64) | Not applicable |
| PdCu-calc | 1.96 (63) | 1.44 (77) |
| PdNi-calc | 2.38 (74) | 1.59 (89) |
| PdFe-calc | 1.84 (56) | 1.32 (77) |
| PdCo-calc | 1.86 (58) | 1.50 (84) |
| PdMo-calc | 1.91 (73) | 2.03 (86) |
| Pd-red | 3.09 (62) | Not applicable |
| PdCu-red | 2.17 (69) | 1.51 (81) |
| PdNi-red | 1.86 (58) | 1.65 (93) |
| PdFe-red | 2.10 (64) | 1.48 (86) |
| PdCo-red | 1.64 (51) | 1.63 (91) |
| PdMo-red | 1.82 (69) | 1.97 (83) |
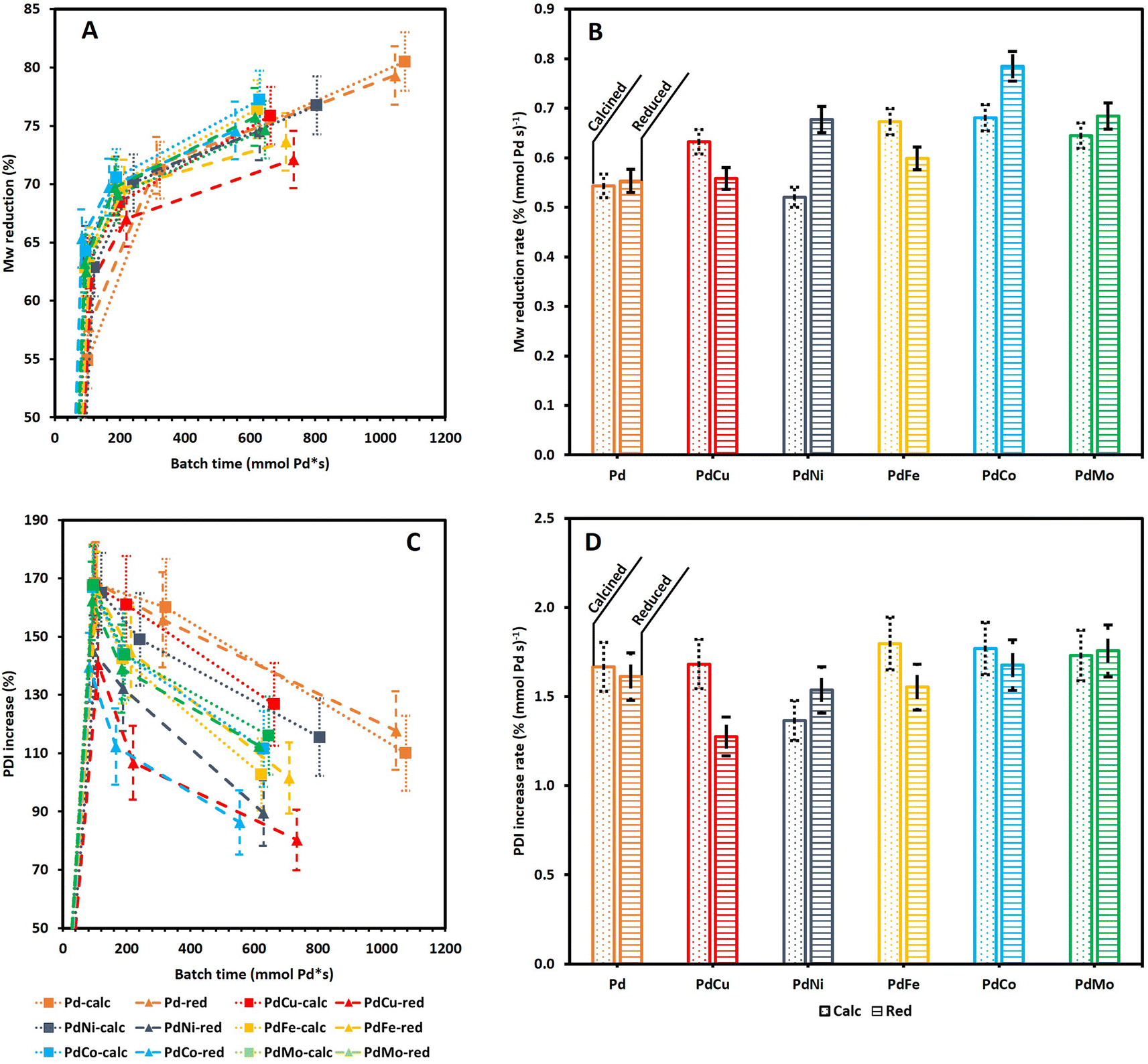 | ||
| Fig. 5 M w reduction (A) and PDI increase (C) as a function of batch time and the resulting Mw reduction rate (B) and PDI increase rate (D) at 3 h (2 h for Pd-calc/red) for all Pd(X)-calc/red catalysts. Dotted lines/bars are used for calcined catalysts; striped lines/bars indicate reduced catalysts. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 259 mg lignin and 50 mg catalyst. | ||
Fig. 5A shows a continuous reduction in Mw as a function of τ, for all catalysts. Moreover, Fig. S3A† clearly indicates that all Pd containing catalysts achieve a higher Mw reduction than 0Cata and Al2O3 which is in line with the literature.23,41,50 Notably, Fig. S3† illustrates that the difference in Mw reduction between the Pd containing catalysts and 0Cata/Al2O3 becomes more pronounced at longer reaction times, which further substantiates the presence of a size exclusion effect.40 However, repolymerization at longer reaction times for 0Cata and Al2O3 and the successful inhibition thereof by the Pd containing catalysts could also explain this observation. Regardless of the cause, the addition of a Pd containing catalyst significantly enhances the Mw reduction over 0Cata and Al2O3 as seen in Fig. S3.† Moreover, the bimetallic catalysts, with the exception of PdCu-red, exhibit Mw reductions similar to the monometallic ones as a function of batch time, despite their lower Pd content, as indicated in Section 3.3.1.
Specifically, as the reduction in Mw represents the degree to which the linkages within the original lignin structure are cleaved, the Mw reduction rate per mmol of Pd, measured at 3 h (2 h for Pd-calc/red) of reaction time, can serve as an equivalent to TOF in traditional catalyst screening and an indicator for the catalyst activity in lignin depolymerization. Fig. 5B supports the conclusion that the addition of a secondary metal to the Pd catalyst does not diminish the catalyst activity, as the Mw reduction rate for all bimetallic catalysts is comparable to that of Pd-calc/red and, in most cases, even significantly higher. The lower Pd content of the PdX catalysts, compared to Pd-calc/red, could potentially affect the Pd dispersion, which, in turn, might impact the activity. However, as aforementioned, there is no clear relationship between the dispersion of Pd and the perceived activity of the catalyst based on the literature.13,24 Furthermore, the secondary metal could also induce additional depolymerization through its own catalytic activity. To investigate the latter, monometallic Cu, Ni, Fe, Co and Mo catalysts, both only calcined (calc) and thermally reduced after calcination (red), were evaluated at 6 h of reaction time. The GPC-RID results (see Fig. S6, S7A and B (ESI)†) reveal significantly lower reduction in Mw than Pd-calc/red. Furthermore, both the calc and red variations of the Cu, Ni, Fe, Co and Mo catalysts show statistically indistinguishable reduction in Mw and an increase in PDI, along with molecular weight distributions nearly identical to those of 0Cata and Al2O3. Hence, it can be concluded that the secondary metals, on their own, are inactive in the mild reductive depolymerization of lignin, which agrees with the literature that base metal catalysts require harsher conditions.11,12 Therefore, the increased activity of the bimetallic catalysts is likely (partly) due to synergetic relationships between Pd and Cu, Ni, Fe, Co or Mo. Moreover, these results also validate the calculation for batch time, i.e., only including Pd content, used throughout this study.
The Mw reduction rate is the highest for PdCo-red, followed by PdMo-red, PdCo-calc, PdNi-red, PdFe-calc, PdMo-calc, PdCu-calc, PdFe-red, PdCu-red, Pd-red, Pd-calc and PdNi-calc. However, as indicated in Fig. 5B, there is statistical overlap between several of the Mw reduction rate values. Nonetheless, PdCo-red clearly demonstrates the highest activity, i.e., the Mw reduction rate, and, most notably, PdNi-calc shows the lowest, even significantly lower than Pd-calc and Pd-red. Additionally, when comparing all calcined catalysts to their respective reduced counterparts, only the Pd-calc/red and PdMo-calc/red pairs show no significant difference in Mw reduction rates, which is in line with the PCA results in Section 3.1. Moreover, PdCu-calc and PdFe-calc achieve a significantly higher Mw reduction rate than their reduced counterparts while an opposite trend is observed for PdNi and PdCo. This implies that the preferred preparation strategy also depends on the secondary metal. The fact that PdMo-calc/red shows no significant differences in the Mw reduction rate is likely due to the necessity for 2 separate precursor solutions during incipient wetness impregnation, where all other Pd(X) catalysts can be impregnated with one precursor solution.
Fig. 5C suggests substantial differences in the PDI increase as a function of batch time among the Pd containing catalysts, yet all display a continuously decreasing trend. Fig. S3B (ESI)† confirms that except for PdCu-red, the PDI is higher for the Pd containing catalysts than for 0Cata and Al2O3. Similar to the Mw reduction (Fig. S3A (ESI)†), the differences become more pronounced at longer reaction times, likely due to the aforementioned size exclusion effect. However, as all PdX catalysts overlap with Pd-calc/red as shown in Fig. 5D, the differences amongst the catalysts in terms of PDI increase rate seem to be rather insignificant at low reaction times. The only exception is PdCu-red, which exhibits a significantly lower PDI increase rate than PdCu-calc.
Finally, Fig. 5A and C indicate that the PDI decreases with an increase in Mw reduction. However, as indicated by Fig. S2A and B (ESI),† except for PdCu-red, all Pd(X)-calc/red variations demonstrate a significantly higher increase in PDI, yet also a larger reduction in Mw than 0Cata/Al2O3 at all reaction times. Therefore these results suggest that, in addition to achieving higher degrees of depolymerization, the Pd(X)-calc/red variations also exhibit differences in product selectivity compared to 0Cata and Al2O3. This is also evident when comparing the molecular weight distributions of 0Cata and Al2O3 in Fig. S2 (ESI)† to those of the catalysts in Fig. S4 (ESI).† Moreover, the molecular weight distributions of the various catalysts at all batch times, presented in Fig. S4 (ESI),† clearly demonstrate differences between all 12 catalysts throughout the molecular weight range. The only exception is monometallic Pd as the calcined and reduced variants only show a noticeable difference at 20 h of reaction time, which is more explicitly displayed in Fig. S5 (ESI).†
To conclude, the results in Fig. 5A and C indicate that the lack of a clear activity related trend in the PCA results is most likely related to the fact that all variations show relatively small differences in Mw and PDI. However, inclusion of the catalyst loadings, i.e., batch times, results in more substantial and significant differences, allowing for a more in-depth and accurate assessment of the catalyst activities. As a result, PdCu and PdFe are the most interesting catalysts with regard to activity as Cu and Fe are cheaper than Ni, Co or Mo, and they perform best when calcination, which is cheaper than thermal reduction, is used as the preparation strategy and still provide increased activity over Pd-calc/red.
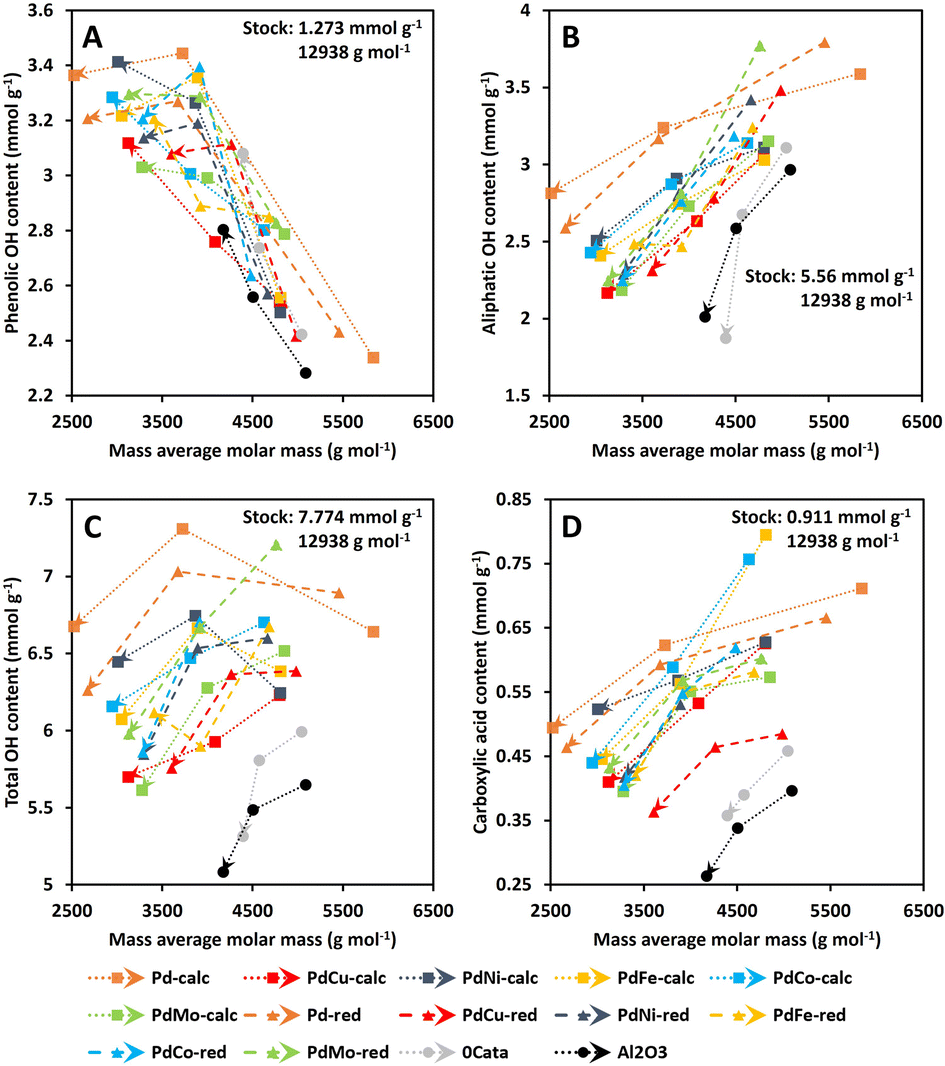 | ||
| Fig. 6 Average phenolic OH content (A), average aliphatic OH content (B), total OH content (C) and average carboxylic acid content (D) as a function of weight average molar mass for the lignin stock solution, and 0Cata, Al2O3 and all Pd(X)-calc/red catalysts as a function of reaction time. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg lignin and 50 mg catalyst if used. | ||
Firstly, on the one hand, the phenolic OH content is expected to increase during depolymerization.51 On the other hand, the presence of Pd can lead to unwanted hydrogenation of the aromatic system within the lignin-derived compounds, leading to a decrease in phenolic OH functions.52,53 Both effects are confirmed in Fig. 6A as the average phenolic OH content continuously increases for 0Cata and Al2O3 with reaction time, yet displays a maximum for the Pd-calc and Pd-red catalysts. Moreover, as mentioned before, the presence of a secondary metal can inhibit the hydrogenation of aromatic systems over Pd catalysts,27,28 leading to a continuously increasing phenolic OH content. This is observed more prominently for calcined variations (PdCu-calc, PdNi-calc, PdCo-calc and PdMo-calc) than for reduced variations (PdNi-red and PdFe-red). Therefore, these results suggest that the calcined PdX catalysts may be more effective in inhibiting the undesired hydrogenation of the aromatic systems than the reduced variants.
Secondly, as mentioned in Section 3.1, a continuous decrease in aliphatic OH-content as a function of reaction time (decreasing Mw) is noted for all catalysts, 0Cata and Al2O3, in Fig. 6B.42–44 However, it can be noted that Pd-calc/red maintains, overall, the highest aliphatic OH content, 0Cata/Al2O3 the lowest and all Pd(X)-calc/red catalysts fall somewhere in between. This implies that the stabilization of aliphatic OH groups requires a Pd containing catalyst and that the addition of the secondary metals, to varying degrees, increases selectivity towards dehydration of these groups, when compared to Pd-calc/red.
Thirdly, the total OH content, i.e. the sum of the average aliphatic and phenolic OH contents, is an important parameter for valorization of the product pool in the polymer industry.6–8 On the other hand, lower aliphatic OH content, along with low heterogeneity and molecular weight, has been noted to enhance the antioxidant activity of lignin, which is beneficial for the pharmaceutical industry.5 Hence, depending on the intended purpose, e.g., low molecular weights with a high or low total OH content, points within the top left or bottom left corner of Fig. 6C, respectively, would represent interesting product pools.
Finally, the carboxylic acid content is expected to decrease during depolymerization as the reductive environment leads to modification of this functionality and the esterification of this group with ethanol is also possible. This is, indeed, observed in Fig. 6D for all catalyst variations, 0Cata and Al2O3.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H functionalities as para-substitutions.37,38 The resulting total area and relative peak areas for groups 1, 2 and 3, i.e., total peak area of the respective groups divided by the total area underneath the chromatogram, are presented in Fig. 7B–E as a function of batch time and Fig. S8B–E (ESI)† as a function of reaction time, respectively.
O)H functionalities as para-substitutions.37,38 The resulting total area and relative peak areas for groups 1, 2 and 3, i.e., total peak area of the respective groups divided by the total area underneath the chromatogram, are presented in Fig. 7B–E as a function of batch time and Fig. S8B–E (ESI)† as a function of reaction time, respectively.
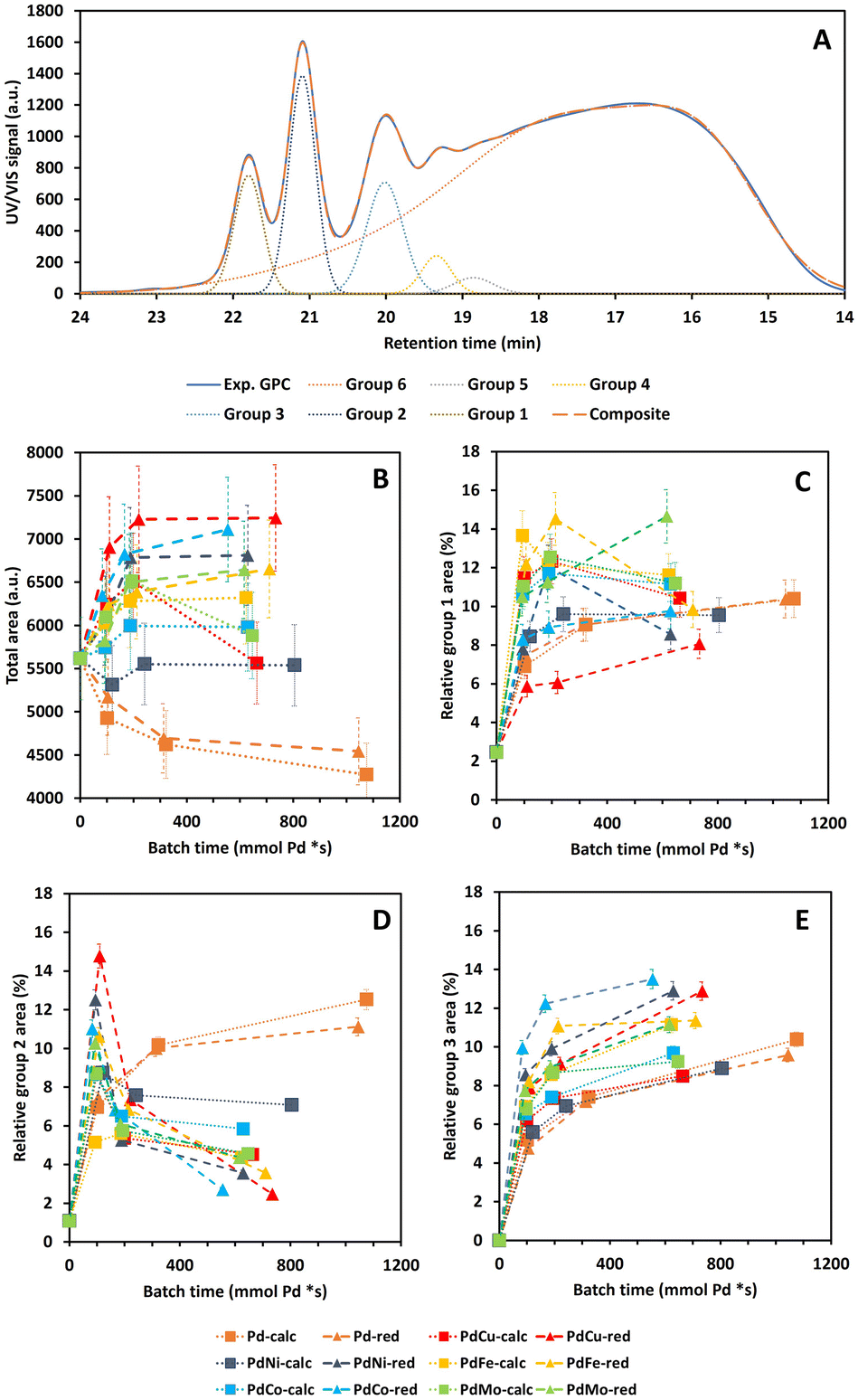 | ||
| Fig. 7 Example deconvolution of the GPC-UV/VIS chromatogram with indication of the chromatogram, deconvoluted groups and the composite curve after deconvolution (A), total area under the GPC-UV/VIS chromatogram (B) and relative areas for groups 1–3 (C–E), as determined through deconvolution of GPC-UV/VIS, as a function of batch time for all Pd(X)-calc/red variations. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg lignin and 50 mg catalyst. | ||
The main trend along PC1 in Section 3.1 is only clearly observed for the total area in Fig. S8B,† in agreement with the respective PC1 loading. More specifically, the total area is significantly the largest for the indistinguishable 0Cata/Al2O3 pair and the smallest for the Pd-calc/red pair, with all PdX-calc/red samples in between. Despite literature reports indicating Pd to be a performant hydrogenation catalyst, even under these mild conditions,52,53 the 31P-NMR results, see Section 3.3.3, suggest that the Pd-calc/red catalysts do not exhibit significantly lower phenolic OH contents compared to PdX-calc/red catalysts, and even significantly higher than 0Cata and Al2O3. Therefore, the lower total area of the PdX-calc/red and, particularly, the Pd-calc/red catalysts, is unlikely caused by hydrogenation of aromatic centres. However, this might be related to the hydrogenation of conjugated aliphatic double bonds and/or hydrodeoxygenation of the native functionalities. Moreover, the suppression of Pd hydrogenation activity through addition of a secondary metal, caused by the ensemble effect, can explain the increased total areas for the PdX catalysts compared to Pd itself.27–29 Additionally, while not always significant, the PdX-red catalysts display larger total areas than their PdX-calc counterpart, which again substantiates the importance of the preparation strategy.
Fig. S8 (ESI)† demonstrates that the relative peak areas of groups 1 and 2, the groups with the lowest molecular weight range, decrease after a maximum at 3 h of reaction time for 0Cata and Al2O3. In contrast, all Pd(X)-calc/red catalysts display higher relative peak areas for group 1 at all reaction times and at 20 h of reaction time for group 2. This clearly indicates that the Pd and PdX catalysts also strongly steer the selectivity within the product pool. When considering the relative peak area of group 1, as shown in Fig. 7C, some catalysts, i.e., Pd-calc, Pd-red, PdCu-red, PdNi-calc, PdCo-red and PdMo-red, exhibit a continuous increase, while others reach a maximum. This suggests that some or all of the constituents of this group are compounds that can convert further during the reaction and are stabilized to varying degrees by Pd-containing catalysts. This is supported by 0Cata and Al2O3 exhibiting significantly lower relative peak areas for group 1 at all reaction times. However, as aforementioned, previous research has indicated that group 1 primarily contains monomers with propyl/propenyl as para-substitution.38 Due to conjugation, the UV/VIS response of a monomer with a propenyl substitution, e.g., isoeugenol, is drastically higher than that of a monomer with a propyl substitution. Hence, a larger relative area for group 1 can most likely be attributed to prominent hydrodeoxygenation, leading to propenyl and propyl chains, and a lack of hydrogenation of conjugated aliphatic double bonds. All bimetallic catalysts, besides PdCu-red and PdNi-red, appear, in general, to display more hydrodeoxygenation and/or less hydrogenation than Pd-calc/red. This agrees with the 31P-NMR results in Section 3.3.3 as all PdX-calc/red catalysts displayed lower aliphatic OH contents compared to Pd-calc/red.
While Pd-calc and Pd-red exhibit a continuously increasing relative peak area for group 2 as shown in Fig. 7D and S8D,† 0Cata, Al2O3 and all PdX-calc/red catalysts reach a maximum at 3 h (6 h for PdFe-calc) of reaction time. However, unlike 0Cata and Al2O3, the PdX-calc/red catalysts retain a relative peak area for group 2 significantly larger than 0, albeit to varying degrees, at 20 h of reaction time. In conclusion, the loss in relative area for group 2 for the PdX-catalysts, combined higher relative areas for group 1, compared to Pd-calc/red, implies that the PdX catalysts display more hydrodeoxygenation and/or less hydrogenation than Pd-calc/red. The latter is important as, depending on the intended goal, the presence of aliphatic double bonds and/or oxygen containing functionalities is crucial for valorization of bio-aromatic compounds.5–8,54,55 The fact that the relative peak areas of both groups 1 and 2 display a continuous decrease for 0Cata and Al2O3 implies that the monomers are not stabilized and (in part) repolymerize.
Finally, it is worth noting that, although the differences amongst catalysts, and with 0Cata and Al2O3, are smaller, the relative peak area for group 3, see Fig. 7E, is significantly larger for the PdX-red catalysts than their PdX-calc counterparts. In particular, PdCo-red shows significantly larger relative peak areas for group 3 at all batch times than all other variations. Furthermore, the constituents of group 3, which are mainly small fragments yet larger than monomers,38 are likely not intermediates but rather end products as the relative peak area of this group continuously increases as a function of batch time (or reaction time) for all variations.
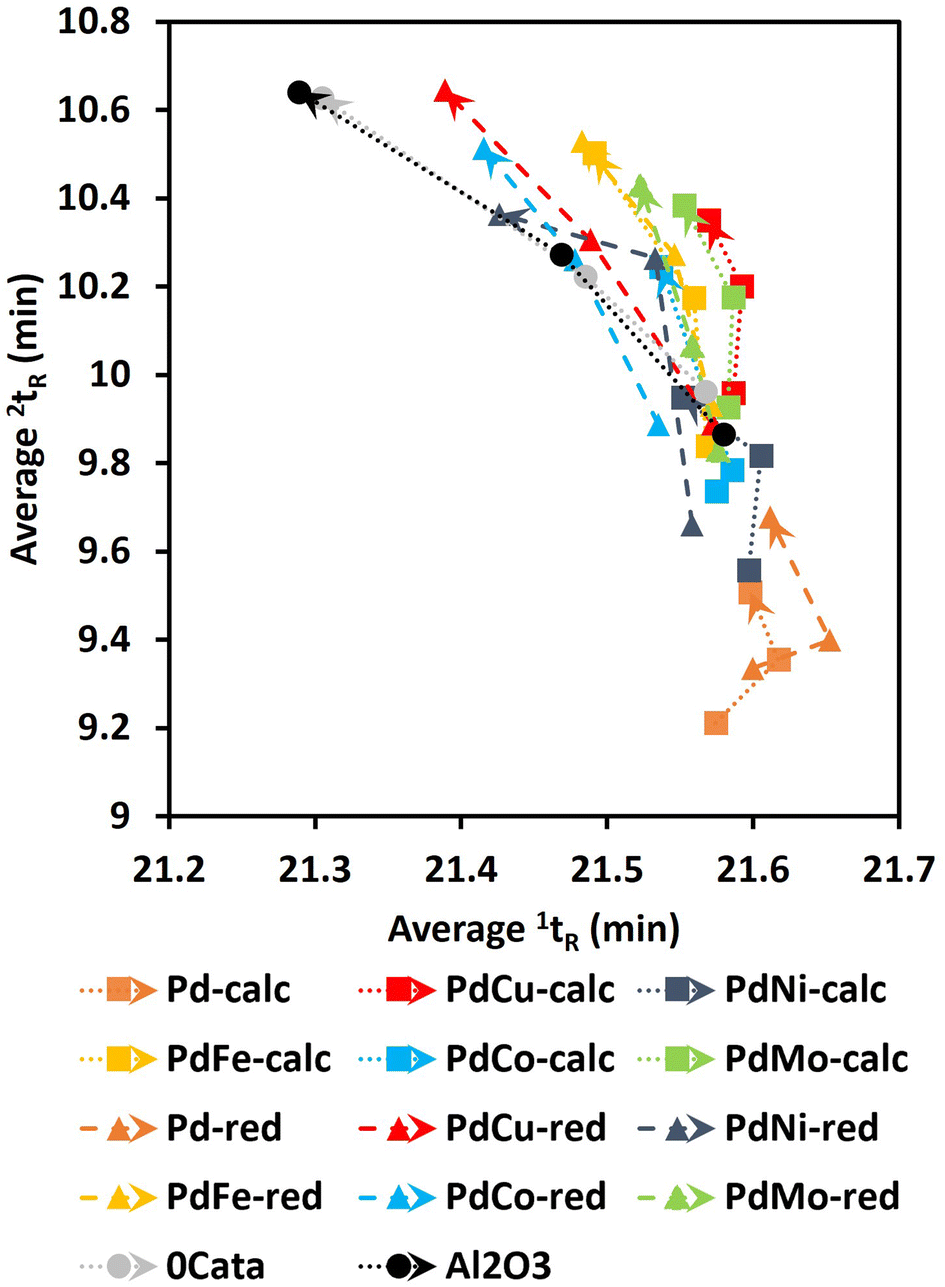 | ||
| Fig. 8 The center points, as determined through GPC-HPLC-UV/VIS analysis, for 0Cata, Al2O3 and all Pd(X)-calc/red catalysts as a function of reaction time. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg lignin and 50 mg catalyst if used. | ||
The mass yields of V, DHCA, HMPA, HMCA, EHF, EUG, I-EUG, 4PS, MPP, CA and SA as well as the total monomer mass yield as a function of batch time for all Pd containing catalysts are presented in Fig. 9. Fig. S12 (ESI)† further details the mass yields as a function of reaction time, allowing comparison with 0Cata and Al2O3. The selectivity differences between the different variations are more substantial for the monomer yields than for the 31P-NMR of the entire product pools and GPC-UV/VIS deconvolution results in Sections 3.3.3 and 3.3.4, substantiating the presence of a size exclusion effect.40
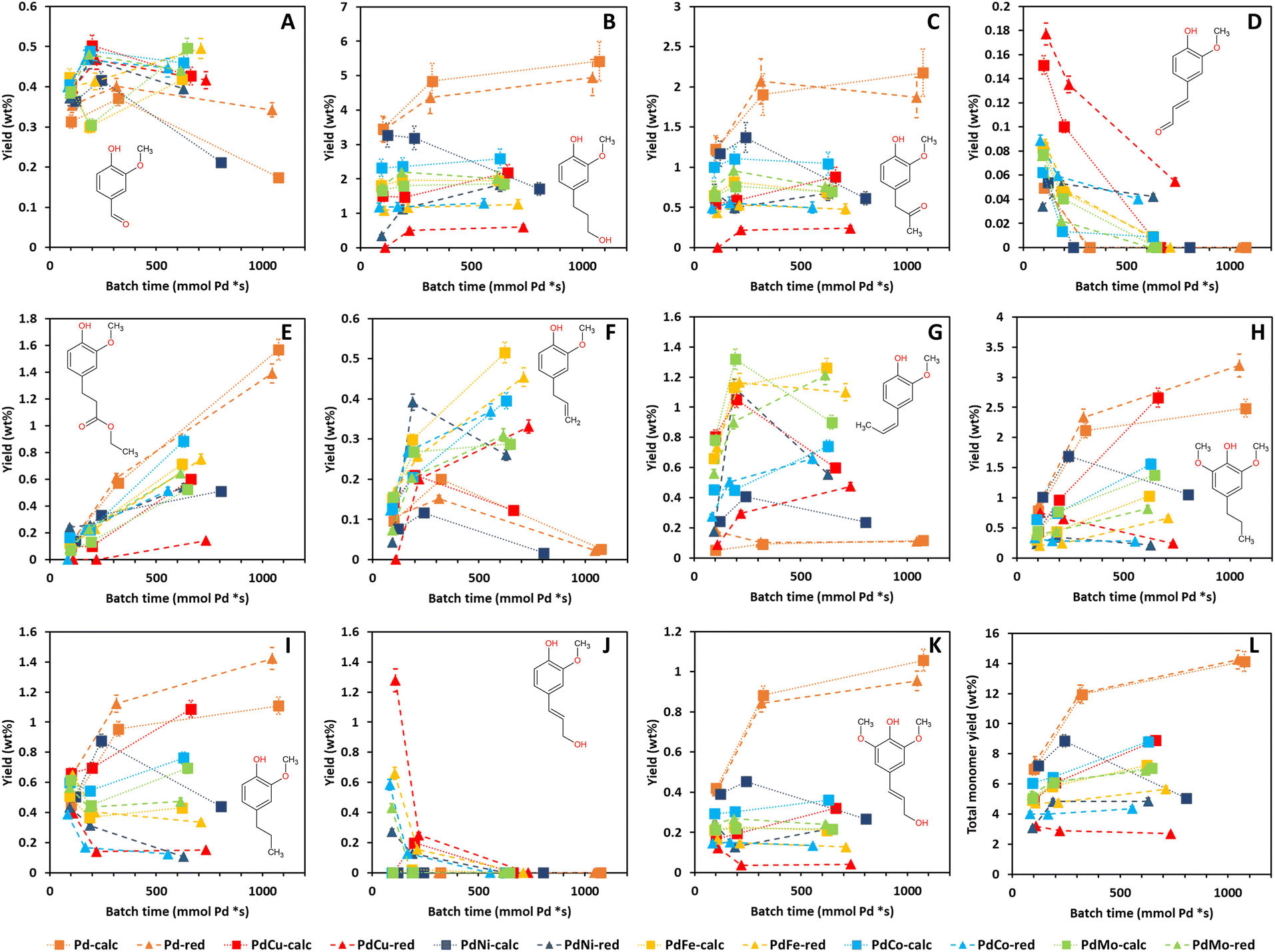 | ||
| Fig. 9 Mass yields of V (A), DHCA (B), HMPA (C), HMCA (D), EHF (E), EUG (F), I-EUG (G), 4PS (H), MPP (I), CA (J), and SA (K) and the total monomer mass yield (L) as a function of batch time for all Pd(X)-calc/red variations. Reaction conditions: 10 bar H2 initial, 180 °C, 10 mL 70/30 EtOH/H2O (vol%), 265 mg lignin and 50 mg catalyst. | ||
The global PC1 trend from Section 3.1, i.e., the 0Cata/Al2O3 and Pd-calc/red pairs showing the most different results with all PdX catalysts in between, is clearly observed for the mass yields of DHCA (B), HMPA (C), HMCA (D), EHF (E), MPP (I), CA (J) and SA (K) and the total mass yield (L) in Fig. 9 and S12.† Moreover, Pd-calc/red achieves the highest overall mass yields for DHCA, HMPA, EHF, MPP, and SA, all of which have a strongly positive PC1 loading, and the lowest overall mass yields for HMCA and CA, both of which have a strongly negative PC1 loading. As mentioned in Section 3.1, HMCA and CA, for which 0Cata and Al2O3 show the highest concentrations, have been associated with repolymerization.18 In contrast, SA, which also possesses an aliphatic OH group and a double bond in the para-substitution, not only shows a continuously increasing concentration for Pd-calc/red but is also present in significantly higher concentrations for Pd-calc/red, compared to PdX-calc/red catalysts. This difference between CA and SA is likely due to the influence of the additional methoxy group in SA compared to CA, which has been noted to drastically alter the adsorption energy of the compound on Pd containing catalysts.56 CA, if present, is likely converted to compounds such as DHCA, EUG and/or MPP by the Pd containing catalysts through modification reactions, evading potential repolymerization. However, the same is most likely not happening for 0Cata and Al2O3 as DHCA is absent and EUG and MPP are only present in relatively low concentrations. Furthermore, Pd-calc/red also achieves the highest total monomer yields with 0Cata/Al2O3 exhibiting the lowest. Hence, high PC1 values within the PCA results are heavily related to more stabilization of monomers and/or the absence of monomers that might induce repolymerization.
Inhibiting repolymerization as a result of different catalyst selectivities also influences the activity for the mild reductive depolymerization. The latter is in line with the literature and substantiated by the fact that the chemical structure of most quantified monomeric compounds can be related to a specific cleavage pathway of β-O-4 linkages. More specifically, dehydration of the OH group on the α-carbon is followed by either hydrogenolysis or hydrolysis of the ether linkage and subsequent modifications of the para-substituted alkyl and alkenyl chains, as demonstrated in Fig. S13 (ESI).†42
However, the presence of dehydration, hydrogenation of aliphatic double bonds and other modification reactions causes the monomers to be chemically related to each other, as seen in Fig. S14 (ESI).† Therefore, the relative abundance of the monomers cannot be unanimously related to differences in β-O-4 cleavage pathways. Nonetheless, these results can be utilized to study the variations in selectivity for hydrogenation and hydrodeoxygenation reactions in depth. Additionally, it is important to note that an iso-conversion point cannot be defined for lignin when comparing different catalysts due to the absence of a clearly definable conversion.
The monomer yields reflect that Pd-calc/red prefer hydrogenation of aliphatic double bonds over hydrodeoxygenation while PdX-calc/red, albeit to varying degrees, prefers hydrodeoxygenation over hydrogenation of aliphatic double bonds. As a result, compounds such as HMPA and, especially, DHCA, both having oxygen functionalities and no aliphatic double bonds in the para-substitution, are the most prominent monomers for the Pd containing catalysts yet nearly absent for 0Cata and Al2O3 with all PdX catalysts somewhere in between these two extremes. The vanillin yields appear to contradict the notion that the PdX-calc/red catalysts exhibit higher hydrodeoxygenation activities than the Pd-calc/red catalysts as, in general, besides PdNi-calc, the former demonstrate significantly higher V yields. However, this can be explained by the fact that the first step in hydrodeoxygenation of (CO)H groups, such as the one in the para-substitution of V, is hydrogenation of the (CO)H group to an CH2OH group.44,57 The latter should be, according to this study, facilitated more by the Pd-calc/red catalysts. Moreover, the increased activity of the Pd-calc/red catalysts in hydrogenation of both aliphatic double bonds and (CO)H groups is likely also the reason why HMCA is absent from the Pd-calc/red product pool at 6 h and 20 h of reaction time. As an additional result of the increased hydrogenation activity of Pd-calc/red, they demonstrate higher MPP and 4PS yields, compounds with no aliphatic double bonds in the para-substitution, yet lower EUG and, especially, I-EUG yields, with propenol substitutions, than most PdX catalysts, besides EUG for PdCu-calc and, especially, PdNi-calc.
Furthermore, PdCu-calc displays an atypical behavior. Namely, while PdCu-calc demonstrates lower hydrogenation of aliphatic double bonds and higher HDO activity at 3 h and 6 h of reaction time than Pd-calc/red, akin to the other PdX-calc/red catalysts, increased hydrogenation activity and decreased hydrodeoxygenation activity between 6 h and 20 h of reaction time can be observed. More specifically, the 4PS and MPP concentrations increase, while the EUG and I-EUG concentrations decrease drastically between 6 h and 20 h of reaction time for PdCu-calc. This shift in behavior between 6 h and 20 h of reaction time by PdCu-calc was also identified using a shift towards higher PC1 values in the PCA results, see Section 3.1.
EHF is an interesting compound as the carboxylic acid group on the γ-carbon has been esterified with ethanol. Notably however, Fig. 8E and S12E (ESI)† indicate that all Pd containing catalysts, albeit to varying degrees, show a continuously increasing EHF concentration while EHF is not present in the product pool for 0Cata/Al2O3. This implies that either the esterification of dihydroferulic acid or the hydrogenation of ferulic acid to dihydroferulic acid, prior to or after esterification with ethanol, can only occur in the presence of a Pd containing catalyst. However, as Fig. S12H and I† clearly indicate that MPP and 4PS are present for 0Cata/Al2O3, the former hypothesis is likely more accurate.
In conclusion, while some monomer yields strongly reflect the global PC1 trend within the PCA results in Section 3.1, others do not, in agreement with the PC1 loadings. In-depth analysis of the results strongly suggests that Pd containing catalysts are required to stabilize monomers and, hence, avoid repolymerization. Additionally, all PdX-calc/red catalysts, albeit to varying degrees, demonstrate increased hydrogenation activities and decreased hydrodeoxygenation activities compared to Pd-calc/red. PdCu-calc is, again, selected as the most interesting candidate as its catalytic behavior shifts more towards that of Pd-calc/red between 6 h and 20 h of reaction time, which was also identified in the PCA results in Section 3.1. As a result, PdCu-calc, along with PdCo-calc, achieves the highest total monomer yield outside of Pd-calc/red.
4. Conclusions
The effects of the incorporation of a secondary metal, i.e., Cu, Ni, Fe, Co or Mo, and the catalyst preparation strategy, i.e., calcination or thermal reduction, on the performance, i.e., activity and selectivity, of Pd nanoparticle catalysts, supported on γ-Al2O3, in the mild reductive depolymerization of lignin were evaluated using a novel and systematic approach. To this end, 2 supported monometallic Pd catalysts and 10 supported bimetallic PdX catalysts were tested at three different reaction times and compared to experiments without the presence of a catalyst and with the presence of pristine γ-Al2O3. To address the complexity of lignin depolymerization and its inherent incompatibility with conventional evaluation parameters such as conversion, turnover frequency (TOF), or turnover number (TON), innovative and comprehensive strategies for assessing both activity and selectivity were developed and implemented.Principal component analysis of the entire dataset had a cumulative explained variance ratio of 91.31% with 2 PC. A clear global trend was identified along PC1 with Pd-calc/red achieving the highest PC1 values and 0Cata/Al2O3 achieving the lowest PC1 values, with all PdX catalysts in between. Analysis of the PC1 loadings implied that the values are heavily driven by differences in monomer yields and that, in general, more favorable product pools should achieve higher PC1 values. The PCA results identified that the addition of pristine γ-Al2O3 has little to no effect and that the preparation strategy also doesn't impact the behavior of Pd and PdMo. PdCu-calc demonstrates a substantial shift towards higher PC1 values between 6 h and 20 h of reaction time, denoting it as the most interesting bimetallic combination. Although the PC2 loadings are heavily, yet not exclusively, driven by parameters related to the degree of depolymerization, no clear trend in line with the literature could be observed in the PCA results, implying that activity differences are relatively small and require more in-depth analysis. The latter was confirmed as the differences in Mw reduction amongst the Pd and PdX catalysts are mostly not significant if the differences in Pd loading are not taken into account. However, evaluation of the Mw reduction rate, a novel parameter representing the percentual reduction in Mw per batch time, revealed significant differences amongst the Pd(X)-calc/red catalysts. It was concluded that addition of Cu, Ni, Fe, Co and Mo, which are inactive catalysts on their own, to the Pd based catalyst significantly increased the Pd activity. Additionally, the preparation strategy has a significant effect on the activity of the PdCu, PdNi, PdFe and PdCo catalysts but not the Pd and PdMo catalysts. Moreover, while calcination is the preferred preparation strategy for PdCu and PdFe, thermal reduction is preferred for PdNi and PdCo.
In-depth analysis of the differences in selectivity was performed at an increasing depth, often agreed with the global PC1 trend while also elucidating the chemical reason behind this trend. Additionally, the selectivity related data strongly indicate the presence of a size exclusion effect. More specifically, the 31P-NMR, GPC-UV/VIS deconvolution and GPC-HPLC-UV/VIS quantification of the monomers, with increasingly substantial differences, consistently indicate reduced hydrogenation activities and increased hydrodeoxygenation activities, especially dehydration of aliphatic OH groups, at varying degrees, for the PdX catalysts compared to Pd-calc/red. Furthermore, these results also substantiate the PCA derived conclusion that the reaction selectivity of PdCu-calc shifts towards Pd-calc/red between 6 h and 20 h of reaction time. Additionally, the results regarding selectivity, especially the monomer quantifications, substantiate that without the presence of a Pd containing catalyst, i.e., for 0Cata and Al2O3, repolymerization occurs (partly) due to a lack of stabilization of the monomers.
Hence, this study demonstrates that the performance, i.e., activity and selectivity, of Pd based catalysts in the mild reductive depolymerization of lignin can be altered through addition of a secondary non-noble metal and variation of the preparation strategy. Moreover, novel methods for the assessment of differences in activity or selectivity were successfully implemented for a large number of samples to select interesting catalysts. Hence, this study improves on the state of the art, offering a more refined method for catalyst screening while simultaneously demonstrating the potential of PdX catalysts to reduce catalyst synthesis costs and improve activity without steering selectivity towards undesired products. Specifically, the systematic evaluation of activity and selectivity identified PdCu-calc as the most interesting catalyst for further research as, when compared to Pd-calc/red, it achieves a strong reduction in synthesis cost, significantly improved activity and a comparable and favorable selectivity towards functionalized bio-aromatics. Consequently, future efforts should prioritize elucidating relationships between the catalyst's properties and its performance, with a goal to further refine and optimize them.
Author contributions
Conceptualization TDS, JDC, AV, JL; data curation TDS, BA; formal analysis TDS; funding acquisition TDS, KVG, JDC, AV, JL; investigation TDS, BA; methodology TDS, JDC, AV, JL; project administration AV; resources PV, KVG, JDC, AV, JL; software TDS; supervision KVG, JDC, AV, JL; validation TDS; visualization TDS, JL; writing – original draft TDS; writing – review & editing TDS, BA, PV, KVG, JDC, AV, JL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. D. S. and B. A. are doctoral (1SD8721N and 1S15123N) fellows of the Research Foundation Flanders. Additionally, J. L. also acknowledges the Research Foundation Flanders for financial support through grant number 12Z2221N.References
- T. De Saegher, J. Lauwaert, J. Hanssen, E. Bruneel, M. Van Zele, K. Van Geem, K. De Buysser and A. Verberckmoes, Materials, 2020, 13, 691 CrossRef CAS PubMed.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- I. Van Nieuwenhove, T. Renders, J. Lauwaert, T. De Roo, J. De Clercq and A. Verberckmoes, ACS Sustain. Chem. Eng., 2020, 8, 18789–18809 CrossRef CAS.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- J. Domínguez-Robles, Á. Cárcamo-Martínez, S. A. Stewart, R. F. Donnelly, E. Larrañeta and M. Borrega, Sustainable Chem. Pharm., 2020, 18, 100320 CrossRef.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Polymers, 2019, 11, 1176 CrossRef PubMed.
- X. Gong, Y. Meng, J. Lu, Y. Tao, Y. Cheng and H. Wang, Macromol. Chem. Phys., 2022, 223, 2100434 CrossRef CAS.
- X. Zhen, H. Li, Z. Xu, Q. Wang, S. Zhu, Z. Wang and Z. Yuan, Int. J. Biol. Macromol., 2021, 182, 276–285 CrossRef CAS.
- H. Yang, X. Zhu, H. W. Amini, B. Fachri, M. Ahmadi, G. H. ten Brink, P. J. Deuss and H. J. Heeres, Appl. Catal., A, 2023, 654, 119062 CrossRef CAS.
- B. Liu and M. M. Abu-Omar, in Adv. Inorg. Chem., ed. P. C. Ford and R. van Eldik, Academic Press, 2021, pp. 137–174 Search PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. Beckham and B. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- D. Bourbiaux, J. Pu, F. Rataboul, L. Djakovitch, C. Geantet and D. Laurenti, Catal. Today, 2021, 373, 24–37 CrossRef CAS.
- J. Park, H. Setiadi, U. Mushtaq, D. Verma, D. Han, K.-W. Nam, S. K. Kwak and J. Kim, ACS Catal., 2020, 10, 12487–12506 CrossRef CAS.
- X. Zhang, W. Tang, Q. Zhang, T. Wang and L. Ma, Appl. Energy, 2018, 227, 73–79 CrossRef CAS.
- X. Wang, S. Feng, Y. Wang, Y. Zhao, S. Huang, S. Wang and X. Ma, Green Energy Environ., 2021, 8, 927–937 CrossRef.
- A. Bjelić, M. Grilc, M. Huš and B. Likozar, Chem. Eng. J., 2019, 359, 305–320 CrossRef.
- M. Hou, H. Chen, Y. Li, H. Wang, L. Zhang and Y. Bi, Energy Fuels, 2022, 36, 1929–1938 CrossRef CAS.
- S. den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- G. Yao, G. Wu, W. Dai, N. Guan and L. Li, Fuel, 2015, 150, 175–183 CrossRef CAS.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- Q. Li, Q. Liang, Y. Fu and J. Chang, Ind. Eng. Chem. Res., 2023, 62, 6005–6015 CrossRef CAS.
- T. Ročnik, B. Likozar, E. Jasiukaitytė-Grojzdek and M. Grilc, Chem. Eng. J., 2022, 448, 137309 CrossRef.
- I. Van Nieuwenhove, J. Lauwaert, T. De Saegher, J. Gracia-Vitoria, K. Vanbroekhoven, T. Renders, T. De Roo, J. De Clercq and A. Verberckmoes, Waste Biomass Valorization, 2022, 14, 1447–1460 Search PubMed.
- A. Karnitski, J.-W. Choi, D. J. Suh, C.-J. Yoo, H. Lee, K. H. Kim, C. S. Kim, K. Kim and J.-M. Ha, Catal. Today, 2023, 411–412, 113844 CrossRef CAS.
- J. Wang, S. Hong, B. Wang, X. Shen, J.-L. Wen and T.-Q. Yuan, Chem Catal., 2023, 3, 100797 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS.
- G. Warner, T. Hansen, A. Riisager, E. Beach, K. Barta and P. Anastas, Bioresour. Technol., 2014, 161, 78–83 CrossRef CAS PubMed.
- C. Espro, B. Gumina, E. Paone and F. Mauriello, Catalysts, 2017, 7, 78 CrossRef.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustain. Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- S. Huang, N. Mahmood, Y. Zhang, M. Tymchyshyn, Z. Yuan and C. C. Xu, Fuel, 2017, 209, 579–586 CrossRef CAS.
- H. Luo, L. Wang, G. Li, S. Shang, Y. Lv, J. Niu and S. Gao, ACS Sustain. Chem. Eng., 2018, 6, 14188–14196 CrossRef CAS.
- T. Rinesch and C. Bolm, ACS Omega, 2018, 3, 8386–8392 CrossRef CAS.
- K. Gao, M. Xu, C. Cai, Y. Ding, J. Chen, B. Liu and Y. Xia, Org. Lett., 2020, 22, 6055–6060 CrossRef CAS.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef CAS.
- M. Chen, X. Ma, R. Ma, Z. Wen, F. Yan, K. Cui, H. Chen and Y. Li, Ind. Eng. Chem. Res., 2017, 56, 14025–14033 CrossRef CAS.
- T. De Saegher, J. Lauwaert, J. Vercammen, K. M. Van Geem, J. De Clercq and A. Verberckmoes, ChemistryOpen, 2021, 10, 740–747 CrossRef CAS.
- T. De Saegher, J. Vercammen, B. Atanasova, K. M. Van Geem, J. De Clercq, A. Verberckmoes and J. Lauwaert, Anal. Chim. Acta, 2023, 1278, 341738 CrossRef CAS PubMed.
- J. Lauwaert, I. Stals, C. S. Lancefield, W. Deschaumes, D. Depuydt, B. Vanlerberghe, T. Devlamynck, P. C. A. Bruijnincx and A. Verberckmoes, Sep. Purif. Technol., 2019, 221, 226–235 CrossRef CAS.
- L. Chen, A. P. van Muyden, X. Cui, Z. Fei, N. Yan, G. Laurenczy and P. J. Dyson, JACS Au, 2021, 1, 729–733 CrossRef CAS PubMed.
- M. V. Galkin, C. Dahlstrand and J. S. M. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS.
- B. Zhang, Z. Qi, X. Li, J. Ji, L. Zhang, H. Wang, X. Liu and C. Li, Green Chem., 2019, 21, 5556–5564 RSC.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- P. Mäki-Arvela and D. Y. Murzin, Catalysts, 2017, 7, 265 CrossRef.
- P. D. Kouris, D. J. G. P. van Osch, G. J. W. Cremers, M. D. Boot and E. J. M. Hensen, Sustainable Energy Fuels, 2020, 4, 6212–6226 RSC.
- S. F. Hashmi, H. Meriö-Talvio, K. Ruuttunen and H. Sixta, Fuel Process. Technol., 2020, 197, 106200 CrossRef CAS.
- D. M. d. A. Fragoso, F. P. Bouxin, J. R. D. Montgomery, N. J. Westwood and S. D. Jackson, Bioresour. Technol. Rep., 2020, 9, 100400 CrossRef.
- P. Velin, C.-R. Florén, M. Skoglundh, A. Raj, D. Thompsett, G. Smedler and P.-A. Carlsson, Catal. Sci. Technol., 2020, 10, 5460–5469 RSC.
- A. S. G. G. Santos, J. Restivo, C. A. Orge, M. F. R. Pereira and O. S. G. P. Soares, Appl. Catal., A, 2022, 643, 118790 CrossRef CAS.
- X. Ouyang, X. Huang, B. M. S. Hendriks, M. D. Boot and E. J. M. Hensen, Green Chem., 2018, 20, 2308–2319 RSC.
- X. Dou, W. Li, C. Zhu, X. Jiang, H. Chang and H. Jameel, RSC Adv., 2020, 10, 43599–43606 RSC.
- P. Zhang, T. Wu, M. Hou, J. Ma, H. Liu, T. Jiang, W. Wang, C. Wu and B. Han, ChemCatChem, 2014, 6, 3323–3327 CrossRef CAS.
- D. Poondi and M. A. Vannice, J. Catal., 1996, 161, 742–751 CrossRef CAS.
- W. Yang, H. Ding, D. Puglia, J. M. Kenny, T. Liu, J. Guo, Q. Wang, R. Ou, P. Xu, P. Ma and P. J. Lemstra, SusMat, 2022, 2, 535–568 CrossRef CAS.
- R. Morales-Cerrada, S. Molina-Gutierrez, P. Lacroix-Desmazes and S. Caillol, Biomacromolecules, 2021, 22, 3625–3648 CrossRef CAS PubMed.
- I. Kumaniaev and J. S. M. Samec, Ind. Eng. Chem. Res., 2019, 58, 6899–6906 CrossRef CAS.
- A. Sulman, P. Mäki-Arvela, L. Bomont, V. Fedorov, M. Alda-Onggar, A. Smeds, J. Hemming, V. Russo, J. Wärnå, M. Käldström and D. Murzin, Catal. Lett., 2018, 148, 2856–2868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00054d |
| This journal is © The Royal Society of Chemistry 2024 |