
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green and sustainable devulcanization of ground tire rubber using choline chloride–urea deep eutectic solvent†
Hesam
Ramezani
 *ab,
Fabrizio
Scarpa
b,
Qicheng
Zhang
b,
Wenfei
Ji
c,
Afifeh
Khorramshokouh
d,
Sebastien
Rochat
ce,
Jean-Charles
Eloi
c,
Robert L.
Harniman
c and
Vijay K.
Thakur
f
*ab,
Fabrizio
Scarpa
b,
Qicheng
Zhang
b,
Wenfei
Ji
c,
Afifeh
Khorramshokouh
d,
Sebastien
Rochat
ce,
Jean-Charles
Eloi
c,
Robert L.
Harniman
c and
Vijay K.
Thakur
f
aSchool of Physics, University of Bristol, Bristol, UK
bBristol Composites Institute, School of Civil, Aerospace and Design Engineering (CADE), University of Bristol, BS8 1TR Bristol, UK
cSchool of Chemistry, University of Bristol, Bristol, UK
dSchool of Civil Engineering, University of Bath, UK
eSchool of Engineering Mathematics and Technology (SEMT), University of Bristol, Bristol, UK
fBiorefining and Advanced Materials Research Center, Scotland's Rural College (SRUC), Kings Buildings, West Mains Road, EH9 3JG Edinburgh, UK
First published on 24th June 2024
Abstract
This work describes the devulcanization of ground tire rubber (GTR) with particle sizes ranging from 0.6 to 0.122 mm using a non-toxic, biodegradable, and biocompatible deep eutectic solvent (DES) based on choline chloride and urea. In addition to reducing the environmental impact of the process, other goals of this study were to minimize time and energy consumption. To meet these targets, a new de-vulcanization method has been developed. The methodology consists of using probe and bath sonication. The de-vulcanized rubber samples were then characterized using attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), electron dispersive X-ray spectroscopy (EDX) and atomic force microscopy (AFM). Flory–Rehner and Horikx analyses were also carried out to calculate the devulcanization percentage and investigate the successful devulcanization of the samples through selective crosslink scission. The results showed that rubber samples of 120 mesh (0.122 mm) were devulcanized up to 58% by using 182 W power only during a 30 minutes process.
Sustainability spotlightTo address the challenge of waste tire rubber and the imperative to recycle it, a sustainable, time and energy-efficient devulcanization process based on Deep Eutectic Solvent (DES) for ground tire rubber with four different particle sizes is introduced. This innovative approach aims to significantly reduce energy consumption, processing time, and environmental impact compared to existing devulcanization methods. It achieves a high devulcanization rate and generates larger quantities of devulcanized rubber using a green and biodegradable DES solvent. This initiative aligns with the United Nations Sustainable Development Goals, specifically SDG 7 (affordable and clean energy), SDG 9 (industry, innovation, and infrastructure), and SDG 13 (climate action). |
1 Introduction
The rubber industry has experienced consistent growth over the years, primarily driven by strong demand in the market. Natural rubber (NR) production doubled in 2015 compared to the year 2000.1 The demand for rubber is projected to remain robust in the foreseeable future as rubber products continue to play a vital role in various niche applications. However, this increased production and consumption of rubber also leads to a rise in rubber waste, which, if mishandled or processed improperly, can have detrimental effects on the environment. This casts a shadow on achieving Goal 12 of the United Nations' Sustainable Development Goals (SDG12).2Rubber products, especially synthetic types, exhibit high resistance to natural degradation due to the vulcanization process. The sulphur crosslinks formed during vulcanization enhance rubber's mechanical strength and physical and chemical properties. However, the intricate three-dimensional structure of vulcanized rubber makes it challenging to decompose through biodegradation. Additionally, additives, antioxidants, and fillers further contribute to its resistance to biodegradation.3,4
Discarded tires contribute significantly to rubber waste in the environment, primarily because the tire production sector utilizes a substantial amount of raw rubber, accounting for approximately 70% of annual natural rubber (NR) production.5 Projections suggest that the annual quantity of waste tires could reach 1.2 billion units by 2030.1,5 Additionally, the demand for rubber gloves has surged due to the pandemic, leading to widespread usage of personal protective equipment (PPE) such as masks and gloves. As of June 2020, it was estimated that around 129 billion face masks with polyisoprene rubber ear loops and 65 billion rubber gloves were disposed of monthly worldwide.6 Other significant sources of rubber waste include automotive components like shock absorbers and conveyor belts and municipal solid waste, including clothing, shoe soles, toys, electric wires, sofas, and cushions.1
Traditional approaches to rubber waste management typically involve landfill disposal or incineration. However, these methods have adverse environmental consequences, such as leaching toxic substances and heavy metals into groundwater. Rubber stockpiles can also fuel persistent fires, as demonstrated by an 18 days incident at a landfill site in Iowa City, Iowa, USA, in 2012. Such fires release hazardous air pollutants, including carbon dioxide, sulphur dioxide, fine particulate matter (PM2.5), and polycyclic aromatic hydrocarbons (PAH).7
The 4Rs waste management approach (reduce, reuse, recycle, and recover) was introduced by the European Commission in 2008 to enhance rubber waste management practices.7 Within this framework, “Reduce” takes precedence as the primary focus, aiming to minimize rubber waste generation. “Reuse” extends the lifespan of rubber products for their intended purposes. At the same time “Recycling” involves grinding rubber waste into small particles (ground rubber) or devulcanization processes that break the crosslinks between polymer chains, allowing for repurposing in other applications. Lastly, “Recover” refers to processes that decompose waste through heat, yielding fuels or valuable substances with lower molecular weights.7
Rubber recycling involves several methods. The downsizing of rubber particles can be performed via dry or wet ambient grinding or using a water jet. Other downsizing methods include Berstoff's and cryogenic methods, which are among the most commonly used techniques for downsizing rubbers.7,8 Reclaiming and devulcanizing involve the separation of crosslinks and bonds within the rubber. Reclamation and devulcanization have distinct definitions. Devulcanization refers to breaking specific crosslinks like C–S and S–S bonds, while reclamation includes breaking both the crosslinks and main chain bonds.9 Physical devulcanization methods exploit the lower bond energies of C–S bonds (310 kJ mol−1) and S–S bonds (270 kJ mol−1), whereas reclamation also involves higher bond energy C–C bonds (370 kJ mol−1).10 However, main chain scission can still occur during devulcanization due to the abundance of C–C bonds.11
Chemical methods for rubber reclamation involve using various chemicals to break the crosslinks between rubber chains or prevent the recombination of sulphur linkages. Several chemicals have been identified as effective devulcanizing agents, including sulphides, peroxides, amines, deep eutectic solvents (DESs), and ionic liquids (ILs).12–17 Walvekar et al. also investigated the use of deep eutectic solvents (DES) as devulcanizing agents. The DES comprised of zinc chloride–urea at mole ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 and 1:4, and the devulcanization was carried out via ultrasonic treatment at various temperatures. The authors concluded that DES containing ZnCl2 and urea was highly effective for the desulphurization of rubber, with sol fractions exceeding 85%.18 Thermomechanical methods are used to break crosslinks with a combination of high temperature and shear force. Different types of equipment and techniques have been considered for thermomechanical devulcanization.11,19,20 Ultrasonic methods involve the use of wave energy vibrations to induce cavities in the polymer matrix, leading to the scission of crosslinks (C–S and S–S bonds) in vulcanized rubber.21,22 Microwaves can also be used to devulcanize rubber. This method makes use of microwave heating to break crosslinks in rubber, assuming the rubber is sufficiently polar to absorb the microwave radiation. For rubbers with poor radiation absorption like NR, SBR, and ethylene-propylene-diene rubber (EPDM), polar fillers such as silica and carbon black (CB) can be added to enhance devulcanization.7 In contrast to the chemical methods, the biological degradation of rubber polymer offers a safer alternative, as it involves the use of enzymes produced by microorganisms to catalyze the devulcanization process, but the yield of the process is relatively low.1 An emerging devulcanization technique that involves the use of supercritical carbon dioxide (scCO2) as a reaction medium. The use of scCO2 offers advantages, such as non-flammability, non-toxicity, and chemical inertness, therefore ensuring process safety.1,7
:7 and 1:4, and the devulcanization was carried out via ultrasonic treatment at various temperatures. The authors concluded that DES containing ZnCl2 and urea was highly effective for the desulphurization of rubber, with sol fractions exceeding 85%.18 Thermomechanical methods are used to break crosslinks with a combination of high temperature and shear force. Different types of equipment and techniques have been considered for thermomechanical devulcanization.11,19,20 Ultrasonic methods involve the use of wave energy vibrations to induce cavities in the polymer matrix, leading to the scission of crosslinks (C–S and S–S bonds) in vulcanized rubber.21,22 Microwaves can also be used to devulcanize rubber. This method makes use of microwave heating to break crosslinks in rubber, assuming the rubber is sufficiently polar to absorb the microwave radiation. For rubbers with poor radiation absorption like NR, SBR, and ethylene-propylene-diene rubber (EPDM), polar fillers such as silica and carbon black (CB) can be added to enhance devulcanization.7 In contrast to the chemical methods, the biological degradation of rubber polymer offers a safer alternative, as it involves the use of enzymes produced by microorganisms to catalyze the devulcanization process, but the yield of the process is relatively low.1 An emerging devulcanization technique that involves the use of supercritical carbon dioxide (scCO2) as a reaction medium. The use of scCO2 offers advantages, such as non-flammability, non-toxicity, and chemical inertness, therefore ensuring process safety.1,7
Abbot et al. in 2003 showed that when hydroxyethyltrimethylammonium (choline) chloride and urea are combined, they remain in a liquid state even at normal room temperature. This is despite the melting points of choline chloride and urea, which are 302 °C and 133 °C, respectively.23 The mixture forms a eutectic system that melts at a significantly lower temperature than the melting point of its constituents. This substantial reduction in terms of melting point arises from the distribution of charges facilitated by the hydrogen bonding between the molecules of urea and the chloride ion. The resulting liquid was discovered to possess intriguing solvent properties akin to those observed in ionic liquids (ILs). To distinguish these liquids from ILs, Abbot et al. came up with the term “Deep Eutectic Solvents” (DESs), which is still being used today.23,24 DESs include a blend of organic substances involving a hydrogen-bond acceptor (HBA) and a hydrogen-bond donor (HBD), demonstrating a melting point substantially below that of either individual component. The formulation of numerous DESs is a straightforward process, achieved through the combination of an uncharged HBD (which could be an amino acid, sugar, carboxylic acid, amine, amide, or alcohol) and an HBA, for example, a quaternary ammonium salt. Choline chloride has been a frequent choice for the HBA in DESs due to its minimal toxicity.25 DESs are known for being non-flammable, maintaining high chemical and thermal stability, being recyclable, having low volatility, and displaying significant solubilisation potential. Deep eutectic solvents (DESs) offer several noteworthy advantages over ionic liquids (ILs). DESs are typically less expensive to produce than ILs due to their use of economical raw materials. DESs can also be prepared using simple procedures that do not require intricate purification steps. Significantly, DESs are often less toxic and more biodegradable than ILs. As a result, DESs are emerging as a promising category of sustainable solvents.25
Tables 1 and 2 provide a benchmark between different devulcanizing agents, and the performance and de-vulcanization yields of various methods.
| Devulcanizing agent | Advantages | Disadvantages |
|---|---|---|
| Diphenyl disulphide (DD) | Widely used selective crosslink scission up to 78% devulcanization | Toxic carcinogenic |
| DES | Biodegradable non-toxic biocompatible up to 97% devulcanization | Susceptible to water uptake |
| Hexadecylamine (HDA) | Biodegradable up to 59% devulcanization | The most expensive agent in the table (GBP 85.6 per 25 g) aspiration hazard (cat. 1) specific target organ toxicity (cat. 2) |
| Ionic liquids (ILs) | Simultaneous scission of backbone and crosslink up to 65% devulcanization | Non-biodegradable toxic precursors |
| scCO2 + DD | Selective crosslink scission up to 52% devulcanization | Toxic carcinogenic |
| Supercritical fluids | Selective crosslink scission up to 75% devulcanization | Produces volatile organic compounds |
| Rubber type | Devulcanizing agent | Temperature (°C) | Time (min) | Devulcanization percentage (%) | Ref. |
|---|---|---|---|---|---|
| Waste tire rubber | Benzoyl peroxide | 80 | 30 | 45.2 | 26 |
| Waste tire rubber | Diphenyl disulphide | 180 | 240 | 78.4 | 9 |
| GTR | scCO2 in the presence of diphenyl disulphide | 180 | Not available | 52 | 27 |
| GTR | DES (with sonication) | 180 | 65 | 96 | 28 |
Sustainability issues call for green chemistry-based solutions that minimize the power consumption for rubber devulcanization, especially the ones extracted from ground tyre rubber waste (GTR). Consequently, this paper describes a novel combination of chemical, thermal, and ultrasonic methods for devulcanizing GTR, which has not been described before in open literature. The proposed method achieves the target of effective devulcanization, sustainably, and with minimal environmental impact. Key features of this devulcanization process are minimizing time and energy consumption for ultrasonic and thermal methods and using a green, non-toxic, biocompatible, and biodegradable solvent for the chemical approaches. Scheme 1 in ESI† shows a schematic diagram of the devulcanization procedure proposed in this work.
2 Materials and methods
Choline chloride (ChCl) and urea were bought from Thermo Fisher Scientific – UK, and toluene was purchased from Merck. GTR samples were provided by Genan, Denmark. All the chemicals were used as purchased and without any further treatment.2.1 Rubber devulcanization procedure
The novelty of the procedure designed in this work is its focus on minimizing time and energy consumption. Due to the scarcity of information in the open literature about the use of DESs for the devulcanization of rubbers, it was decided to follow the procedure suggested by Saputra et al.28,29In the first step after the preparation of the DES (please see the ESI† for preparation method), GTR and solvent were mixed with a 1:20 mass ratio, and the mixture was sonicated at 15% power (12 W) for 10 minutes. Ultrasonic waves were applied alternatively (1 second on/1 second off) to allow the mixture to dissipate the generated heat. The heat build-up during ultrasonication can become a serious threat to the polymer chains since it can cause the backbone of the polymer to break at heightened temperatures and under the waves' pressure. Chain scission is sometimes mistaken for devulcanization, while in reality, it can be interpreted as the degradation of the polymer.19,30
In the second step, the mixture was placed in a GT Sonic D2 bath sonicator at 50 °C, operating at full power (50 W) for 15 minutes. This step complements the probe sonication and can reduce the chances of chain scission due to the reduction of applied tension to the rubber backbone and controlled temperature.
As mentioned, GTR and solvent were mixed with a 1:20 mass ratio.17 The reason for choosing this ratio was to use the minimum amount of solvent while achieving a good wetting of rubber particles and preventing the production of a sluggish mixture that would result in a reduced contact surface between GTR and DES. This would lead to a reduction in the devulcanization efficiency.1,28,29 The mixture was then added to a vial, placed on a heater/stirrer (2000 W), and heated up to 180 °C while being stirred using a magnetic bar at 1400 rpm for 5 minutes.31,32
After completing the last step, the mixture was filtered using a 500 mesh woven wire stainless steel filter. Samples were washed with ultrapure water and filtered multiple times to ensure effective solvent removal. The obtained devulcanized rubbers were dried overnight in an oven at 80 °C. Table 3 shows the coding and the mesh size of the samples.
| Rubber type | Untreated coding | Devulcanized coding |
|---|---|---|
| Super fine (0.2–0.6 mm) | S.Fu | S.Fdv |
| 40 mesh (0.411 mm) | 40 Mu | 40 Mdv |
| 80 mesh (0.178 mm) | 80 Mu | 80 Mdv |
| 120 mesh (0.122 mm) | 120 Mu | 120 Mdv |
During the 30 minutes procedure, the power usage was approximately 182 W, which is a limited amount of energy consumed. Since the work of Saputra et al.28 is the closest to this study, a comparison between the energy consumption of the two studies can provide a better understanding of the energy saved with the current method. The power of the bath sonicator used in the study by Saputra et al. was 330 W, and the sonication time was 60 minutes. Their mixture was then heated up to 180 °C; by assuming the use of a regular 2000 W heater/stirrer, the total energy consumption would have been approximately 497 W. This wattage is more than twice the amount of power needed for the procedure described in the present work. Assuming that the exact same procedures are carried out for 12 months with 20 working days and 8 hours shifts, the method proposed in this work would yield 3840 cycles, while the other procedure would lead to 1920 cycles. The energy consumption for the cycles with the proposed method would be 698.9 kW, while the other procedure would require 954.2 kW. This is a 255.3 kW save while providing twice the amount of product. However – as will be shown in later paragraphs – the percentage of devulcanization obtained with the proposed method is lower than the one provided by Saputra et al.
2.2 Density measurements using a pycnometer
The pycnometer is extensively used to measure the density of solvents and can also be utilized for detecting the density of solids. The pycnometer was weighed at room temperature when empty and after being filled with distilled water. The density of distilled water at room temperature is 0.99753 g ml−1. The actual volume of the pycnometer was measured by dividing the distilled water's mass by density. This calibration was used for all the other measurements.To find out the density of rubber, 2 g of rubber was poured into the pycnometer and then filled with distilled water. The mass of distilled water could be found by measuring the weight of the filled pycnometer. By dividing the mass of the water by its density, the volume of the water can be found. Previously, the volume of the pycnometer was calculated, and by knowing the volume of the distilled water, the volume of the rubber can be found. The density of the rubber can be finally identified by dividing the mass of the rubber (2 g) by its volume.
2.3 Characterization
536 individual data points across this region.
3 Results and discussion
3.1 Chemistry of the devulcanization
To have an improved understanding of what happens during the devulcanization process, examining the chemistry of the DES and its structure is beneficial. Fig. 1 shows the chemical structures of ChCl, urea, and ChCl–urea DES.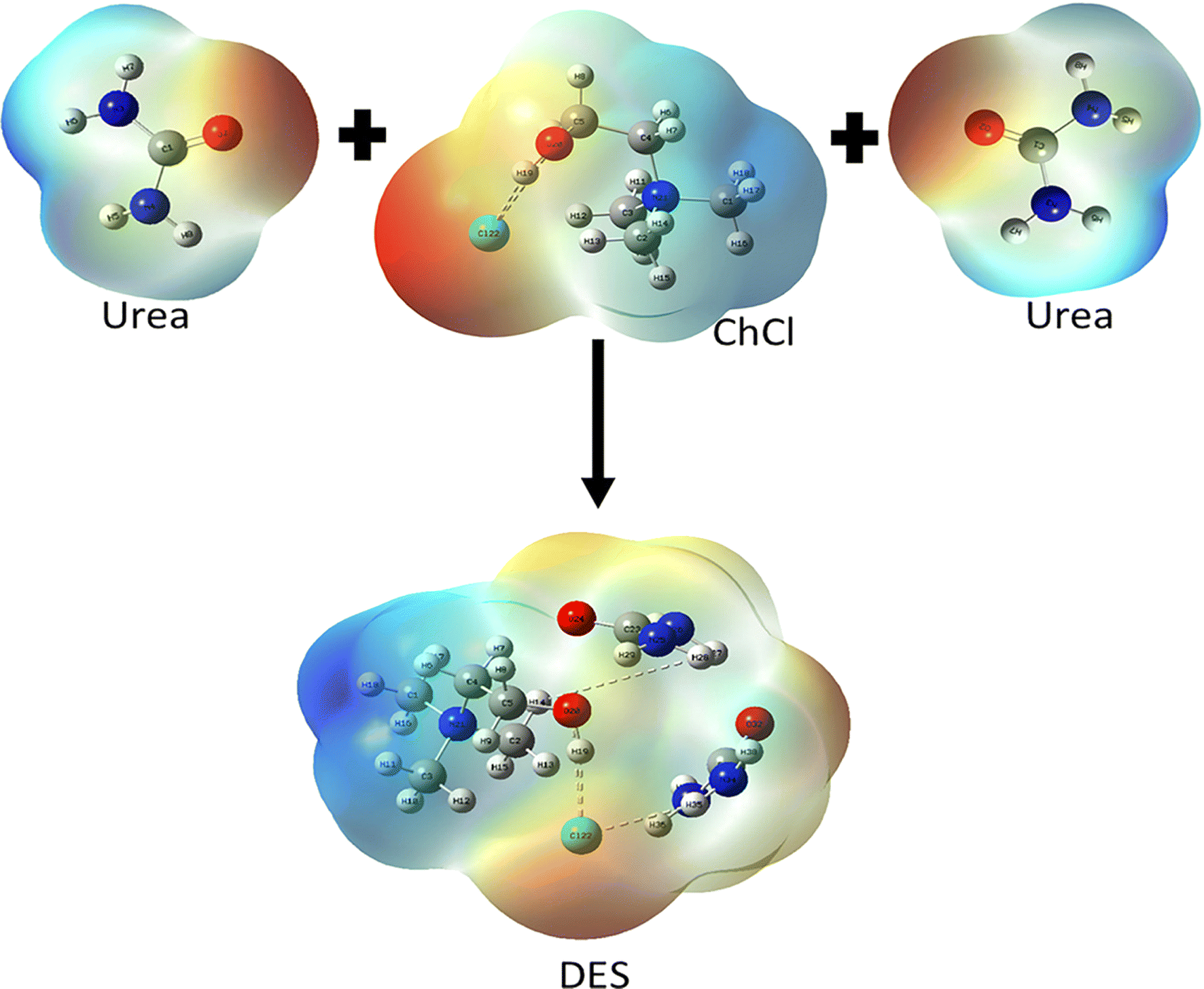 | ||
| Fig. 1 The chemical structures of ChCl, urea, and ChCl–urea DES (reprinted with permission).37,38 | ||
As shown in Fig. 1, hydrogen atoms located on the carbon atoms neighbouring the nitrogen centre (so-called α-carbons) carry a partial positive charge due to the charge distribution in the choline chloride–urea DES. This makes them prone to H-bonding with electron-rich sulphur atoms on the vulcanised rubber. The relatively acidic proton on the oxygen atom of choline will likely also be involved in the formation of the H-bonds and bring an additional contribution to the reduction of S–S bonds.
The mechanism of devulcanization using the DES is shown in Fig. 2.
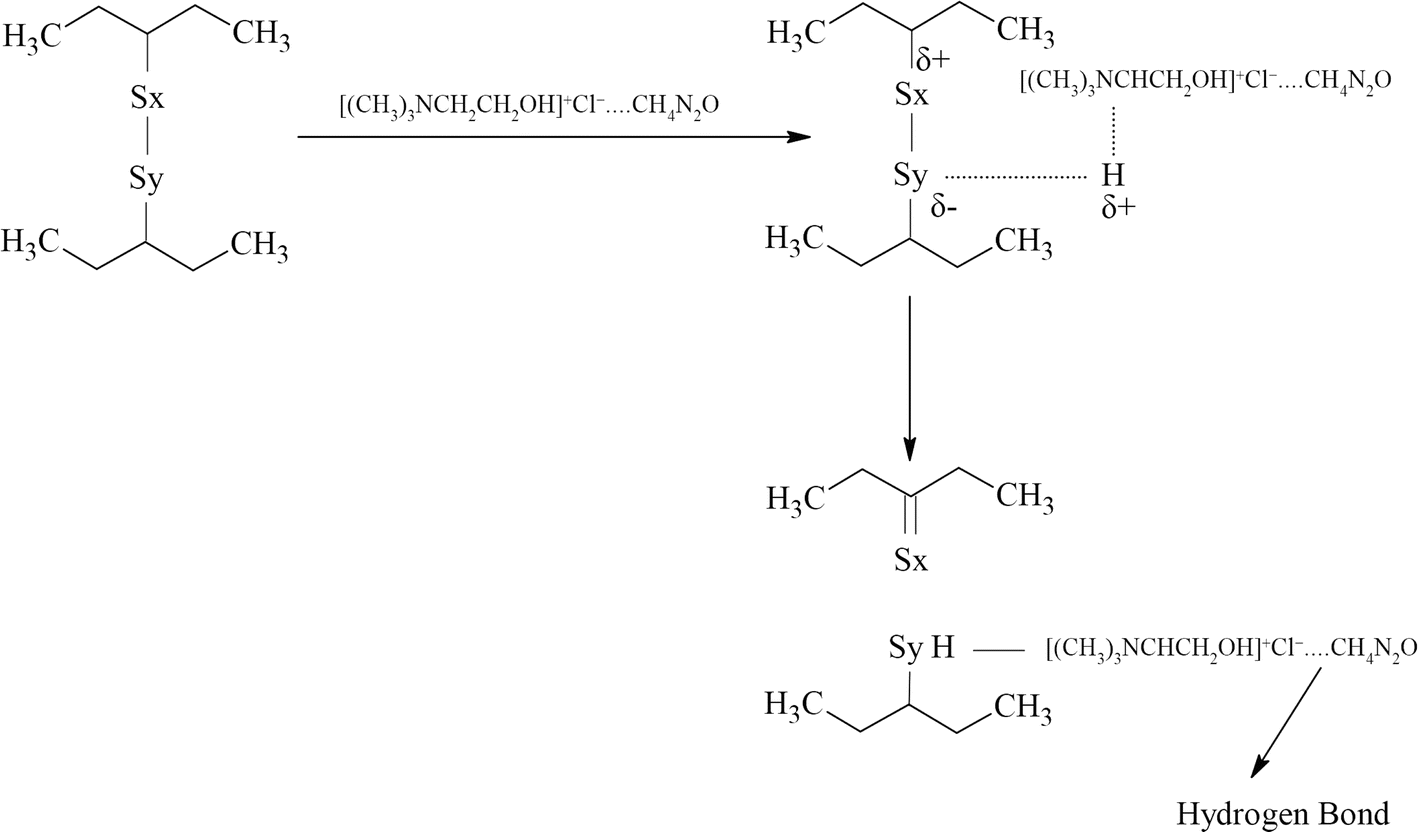 | ||
| Fig. 2 Mechanism of devulcanization using ChCl–urea DES in which hydrogen can come from the α-carbon or the OH group. | ||
During the devulcanization, the α-hydrogen atoms play a critical role. These atoms can form hydrogen bonds with sulphur which has a negative charge (δ−), and hence, the sulphur atom will be reduced, leading to the breakage of S–S bonds.29,33,39 On the other hand, hydrogens from the OH group can actively reduce S–S bonds due to their positive charge. Hence, the hydrogen atom that reduces the S–S bond can come from the α-carbons or the OH group. The other sulphur atom can form a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and get neutralized after the S–S bond cleavage.
S and get neutralized after the S–S bond cleavage.
3.2 Scanning electron microscopy (SEM)
The microstructure and morphology of the samples before and after devulcanization are shown in Fig. 3–6. Notably, in the images, the left column belongs to backscattered electrons, and the right column shows the secondary electron scans, which were detected using a backscattered electron detector (BED) and a secondary electron detector (SED). Furthermore, there are the difference between these modes is that backscattered electrons are the results of an elastic interaction after the beam reaches the sample. As a result, backscattered electrons are reflected from deeper regions and they are also highly sensitive to the atomic number of the elements they are reflected from. Hence, the brighter areas in the backscattered electron images correspond to areas containing species with higher atomic numbers.40,41 | ||
| Fig. 3 SEM images of the S.Fu (top) and S.Fdv (bottom) samples, left column: SED and right column: BED. | ||
 | ||
| Fig. 4 SEM images of the 40 Mu (top) and 40 Mdv (bottom) samples, left column: SED and right column: BED. | ||
 | ||
| Fig. 5 SEM images of the 80 Mu (top) and 80 Mdv (bottom) samples, left column: SED and right column: BED. | ||
 | ||
| Fig. 6 SEM images of the 120 Mu (top) and 120 Mdv (bottom) samples, left column: SED and right column: BED. | ||
On the other hand, the secondary electrons' source is the rubber atoms. When the beam reaches the sample, secondary electrons are reflected because of an inelastic interaction between the beam and the rubber. Hence, secondary electrons come from the surface and give an insight into the surface morphology of the samples.40,42
As can be observed from the SEM images, the untreated GTR samples feature more impurities and inhomogeneity in terms of particle size. A closer look at the images shows that GTR samples have a slightly smoother surface than devulcanized specimens. As the devulcanization percentage increases, the surface roughness shows an increase, which can be a sign of successful devulcanization, according to Saputra et al.28,29
Another observation from the SEM images is the availability of bright points in backscattered electron images. These points dispersed throughout the rubber matrix can be silica, zinc, magnesium, iron, calcium, or other additives used in the rubber compound. Normally, the most abundant additives should be silica and zinc. In the next section, the composition of the rubber is discussed in detail using the EDX method.
To have a better investigation of the surface morphology after devulcanization, high-magnification images can be very helpful. As can be seen from Fig. 3, the surface morphology of the S.Fdv sample is slightly rougher than that of the S.Fu. This has increased for the 40 Mdv, 80 Mdv, and 120 Mdv. When the devulcanization percentage increases, the surface of the polymer becomes like a molten plastic's surface.28
This plasticization results from breaking crosslinks that limit the polymer chain movements. When the crosslinks are broken and because of high temperature, polymer chains can move with a higher degree of freedom; this, in turn, leads to changes in the surface morphology and the adoption of plasticized surface morphology.
The surface roughness for the 120 Mdv sample is much higher than other devulcanized samples, and it is shown in Fig. 6. The rubber particle has a highly rough surface, and there are signs of stretched rubber in the middle of the particle. Furthermore, there are droplet-like points on the surface, which show that the rubber behaved very similarly to a molten polymer during the devulcanization procedure.
3.3 Atomic force microscopy (AFM)
AFM was utilised to investigate the morphology and adhesive properties of the 120 M sample, which had the highest percentage of devulcanization. Fig. 7 and 8 show the topography of the samples and the concurrent map of adhesive interaction (in blue). The left-most images of each figure are without any flattening; this shows the tilt in the sample and its true heights. The central images provide a higher contrast for identifying fine surface structure through applying a second-order polynomial fitting, removing the inherent tilt of the sample. | ||
| Fig. 7 AFM images of 120 Mu sample, blue the concurrent map of the adhesive interaction. | ||
 | ||
| Fig. 8 AFM images of 120 Mdv sample, blue the concurrent map of adhesive interaction. | ||
The surface of the 120 Mu sample presents small grains/platelets of material in a stepped layer structure, while areas of higher adhesive interaction are largely at the interface between grains.
The 120 Mdv sample appears to contain larger flatter platelets than the 120 Mu, therefore forming large regions protruding from a film of amorphous material. This amorphous material is highly adhesive and poorly resolved, suggesting the presence of a potentially semi-liquid state. This confirms the changes of the rubber after devulcanization and the plasticization observed in the 120 Mdv sample observed from SEM (Fig. 9).
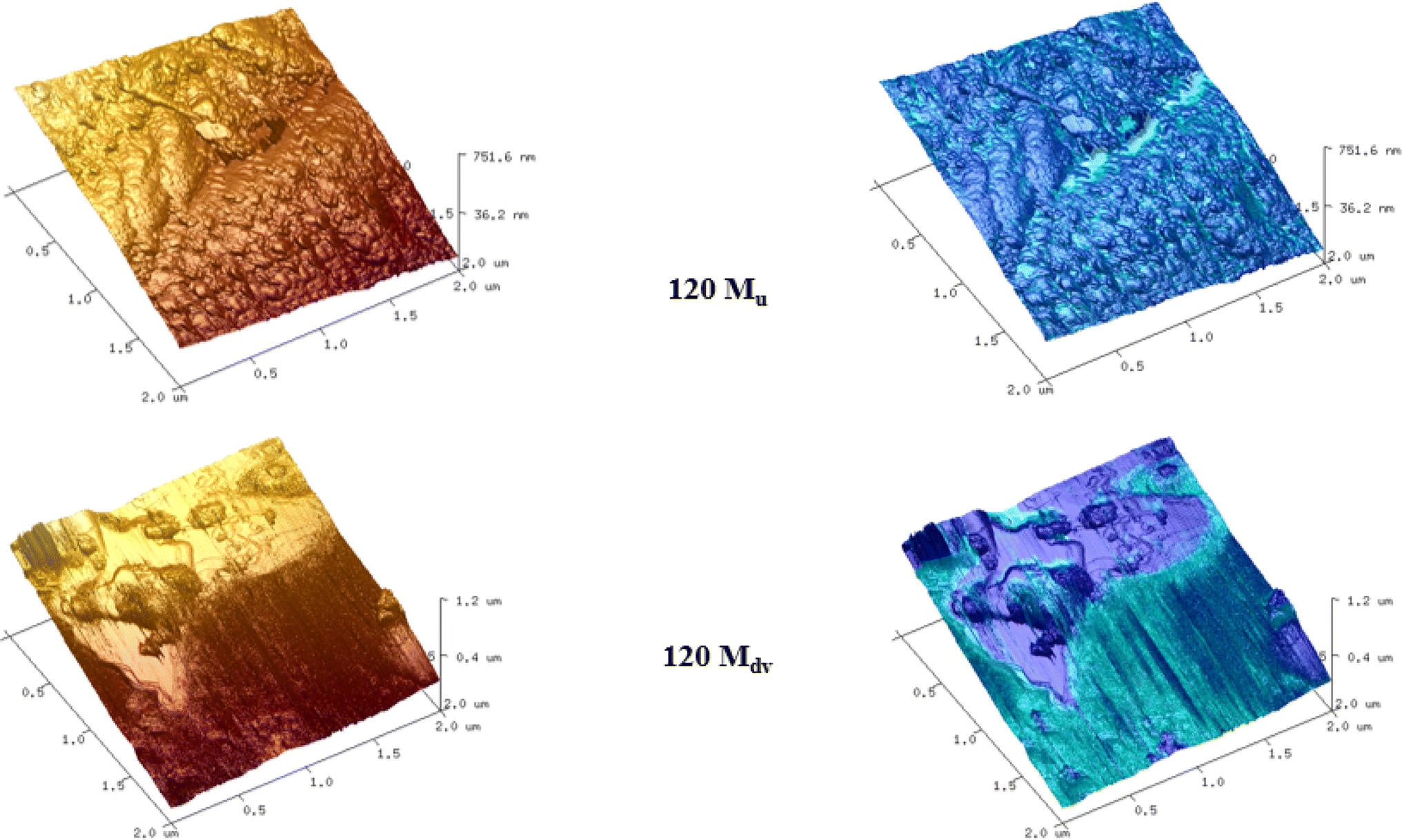 | ||
| Fig. 9 AFM images of 120 Mu and 120 Mdv samples, blue the concurrent map of adhesive interaction. | ||
Fig. 10 shows the average force of the adhesive interaction for the 120 Mu and 120 Mdv. The values of the force are 1.19 ± 0.73 nN and 9.84 ± 2.52 nN, respectively, as averaged from 65536 measurements per sample over 2 μm × 2 μm regions. This clearly indicates an approximately 8-fold increase in adhesive interaction after devulcanization. These observations confirm the potential of the devulcanized rubber to form blends and compounds with other polymers and additives for various applications.
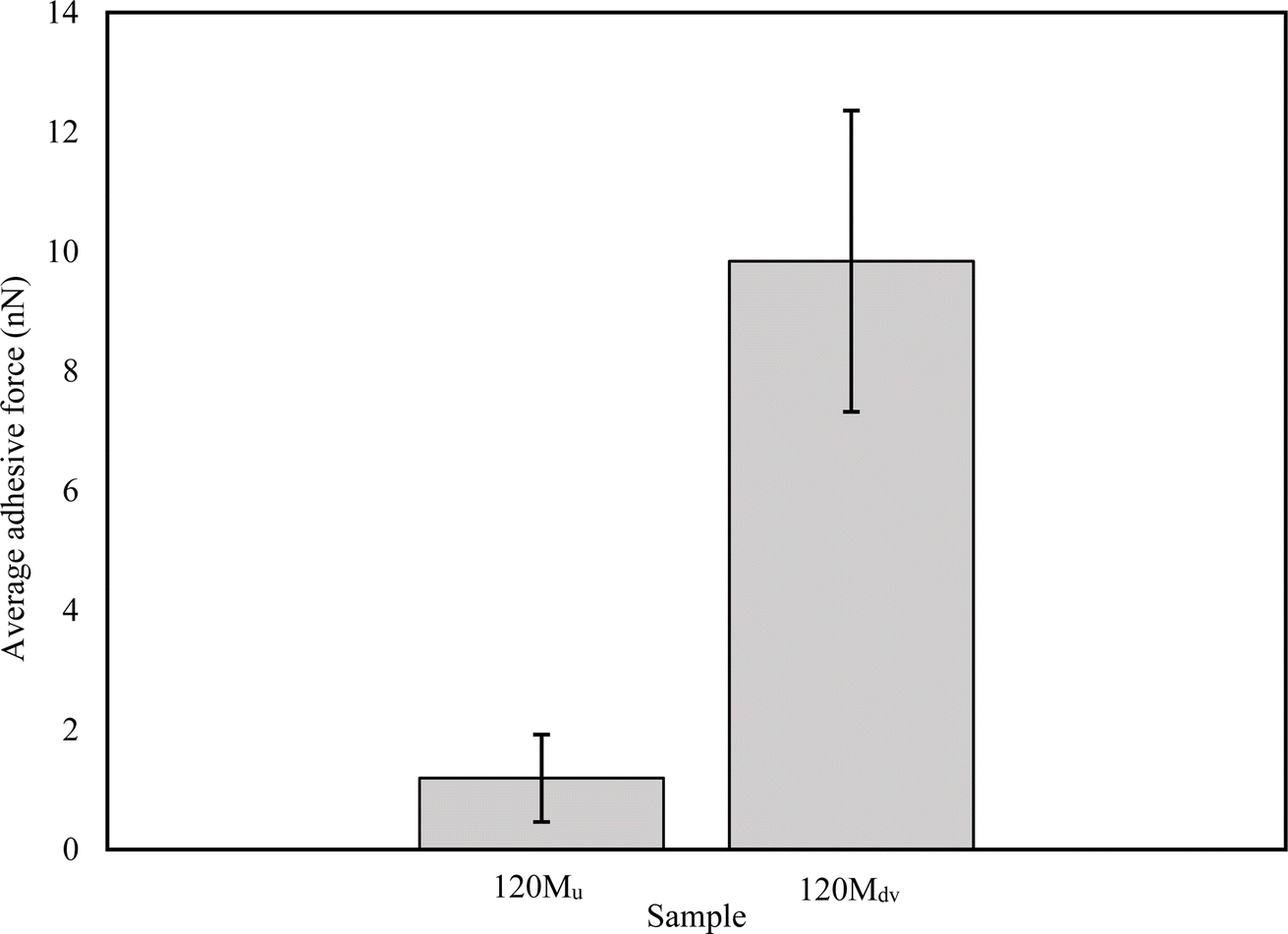 | ||
| Fig. 10 The average adhesive force of the untreated and devulcanized 120 M samples (averaged from 65536 measurements per sample). | ||
3.4 Energy dispersive X-ray spectroscopy (EDX)
The rubber samples were analysed before and after treatment to investigate the changes in composition. The reported percentages are the average values of five different spectra taken from each sample to consider the difference in the dispersion of the elements. Tables 4 and 5 show the composition of the samples before and after devulcanization, respectively. The available elements in the GTR samples included C, O, S, Si, Zn, Fe, Al, Mg, Na, and Ca. For this study, C, O, S, Si, and Zn were recorded and investigated, which are shown in Table 4.| C | O | S | Si | Zn | |
|---|---|---|---|---|---|
| S.Fu | 91.8 | 6.1 | 0.6 | 0.2 | 0.8 |
| 40 Mu | 93.4 | 4.8 | 0.8 | 0.3 | 0.4 |
| 80 Mu | 90.7 | 7.3 | 0.5 | 0.8 | 0.3 |
| 120 Mu | 93 | 5.4 | 0.5 | 0.5 | 0.3 |
| C | O | S | Si | Zn | N | Cl | |
|---|---|---|---|---|---|---|---|
| S.Fdv | 90.4 | 5.9 | 0.8 | 0.4 | 0.4 | 1.0 | 0.3 |
| 40 Mdv | 85.5 | 8.1 | 0.9 | 0.7 | 0.4 | 3.1 | 0.7 |
| 80 Mdv | 86.9 | 7.1 | 0.8 | 1.2 | 0.4 | 2.4 | 0.6 |
| 120 Mdv | 85.9 | 6.5 | 0.9 | 1.2 | 0.4 | 3.6 | 0.6 |
Apart from the carbon black fillers, the highest additive contents belong to the sulphur, silicon, and zinc, which are the curing agent, reinforcing and wear-resistant filler, and vulcanization activator which were also mentioned in previous studies.43,44
As shown in Table 5, the devulcanization process affects the atomic percentage of all the elements. During vulcanization, sulphur forms bonds between the polymer chains as crosslinking agent. These bonds are the targets of the devulcanization process. One can notice a slight increase in the atomic percentage of sulphur in every sample. Sulphur content cannot be directly correlated to the available crosslinks but, residual sulphur on the surface of the rubber from the vulcanization process and sulphur free radicals after crosslink cleavage can all lead to the formation or rearrangement of bonds, including new crosslinks that are reflected in the atomic percentage. In addition, the possible formation of H2S and CS2 could also affect the sulphur content.28,45
Another interesting fact is the availability of N and Cl after devulcanization. This shows that during devulcanization, the ChCl–urea molecules are attached to the sulphur atom through a hydrogen bond. That also proves the incorporation of the DES in the bulk of the material, which confirms the suggested mechanism shown in Fig. 2.
3.5 Attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR)
The samples were characterized using ATR-FTIR spectroscopy before and after devulcanization. The data obtained is shown in Fig. 11 and the observed peaks are summarized in Table 6.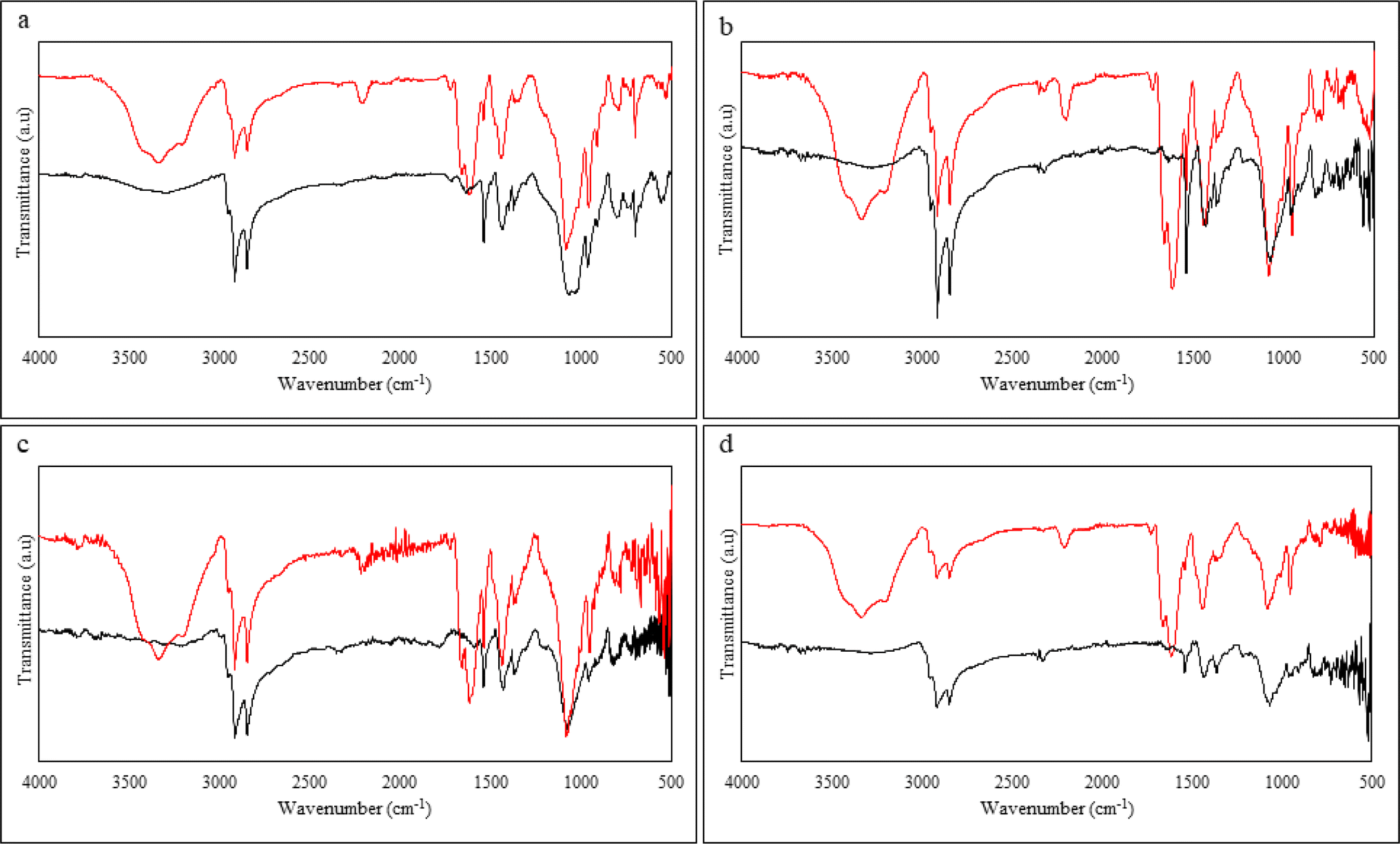 | ||
| Fig. 11 ATR-FTIR spectra of (a) S.F sample, (b) 40 M sample, (c) 80 M sample, (d) 120 M sample, the black line belongs to untreated, and the red line belongs to the devulcanized rubber. | ||
| Wavelength (cm−1) | Description | References |
|---|---|---|
| 3400–3500 | Stretching of N–H, –OH from water uptake, and Si–OH | 32, 46 and 47 |
| 3100–3400 | Stretching of N–H | 28 and 32 |
| 2910–2950 | Stretching of –CH from the backbone | 29 and 48 |
| 2850 | Stretching of –CH from the backbone | 29 and 49 |
| 1608–1666 | Stretching of C–N, coupling of symmetric and asymmetric N–H | 28 and 50 |
| 1500–1540 | Related to Zn compounds and crosslinks | 28 and 45 |
| 1444 | Bending vibration of –CH2 | 49 and 51 |
| 1370 | Bending of –CH3 | 46 |
| 1030–1110 | Carbon black, stretching of Si–O–Si | 28 and 46 |
| 827 | Bending of CC |
46 |
| 669–938 | Representing mono, di, and polysulphidic bonds | 28, 29, 43 and 52 |
Starting from the 3400 to 3500 cm−1, in this specific range, there are multiple stretching related to N–H in primary amide, stretching of –OH from moisture uptake, and stretching of Si–OH, which is between the silica, water, and –OH functionality of the DES.29,32,47
In addition to that, the wavenumbers at 2910 to 2950, 2850, 1444, 1370, and 827 cm−1 belong to the symmetric and asymmetric stretching of –CH in –CH3, stretching of –CH in –CH2– bending of –CH2– and CH3, and bending of CC, respectively. These are characteristic peaks of natural rubber present in the GTR and did not show any reduction in any of the samples after devulcanization. It is likely that the backbone scission was minimal during the devulcanization process and the process did not harm the polymer structure in a way observable via FTIR technique. This also agrees with other observations of this study and also available in open literature.28,29,49,51
The stretching of N–H that occurs at 3100 to 3400 cm−1 alongside the C–N stretching and the coupling of symmetric and asymmetric N–H bend at 1608 to 1666 cm−1 confirms the grafting of the ammonium group from the solvent onto the chains of the polymer. This is further proof of the suggested mechanism of devulcanization.25,28,50
The product of the reaction between ZnO and stearic acid (the vulcanization activators) is Zn(COO)2 and can be seen at the wavenumber around 1500 to 1540 cm−1. There is another sign of the available additives in the rubber compound, which can be seen at 1030 to 1110 cm−1; this belongs to the carbon black fillers and stretching of Si–O–Si. Observing this band can show that the devulcanization reaction did not affect the carbon black fillers.28
The band between 669 and 938 cm−1 shows the presence of the C–S bonds. Fig. 3–6 in the ESI† show the FTIR spectra in the mentioned wavenumbers. All the spectra show a slight decrease in intensity when comparing GTR and the devulcanized rubber. This indicates that the mono, di, and polysulphidic bonds were broken, and selective crosslink scission occurred during the devulcanization. This observation can be the most important data extracted from the ATR-FTIR spectroscopy to prove the successful devulcanization of the GTR samples.28,29,33,43 There are signs of peaks, especially in the 80 Mdv sample. These peaks can be attributed to impurities originating from the cellulose-based filter paper used after the devulcanization and the rearrangement or reformation of sulphur bonds, which can also be in the form of new crosslinks.
3.6 Flory–Rehner analysis
Flory–Rehner's analysis provides a better understanding of the efficiency of the devulcanization procedure by using equations 5 and 6 from the ESI.† The density of the crosslinks can be measured before and after devulcanization; this is used as a criterion to determine the success rate of the devulcanization method. The devulcanization percentage for each sample was calculated (Fig. 12) after the soluble content of the samples was obtained (Fig. 13). | ||
| Fig. 12 Devulcanization percentage calculated from the Flory–Rehner equation. | ||
 | ||
| Fig. 13 The soluble content of the untreated and devulcanized samples. | ||
Results showed that as the particle size decreases, the efficiency of the devulcanization increases. The reduction of the particle size allows the solvent molecules to penetrate the polymer network more easily and more surface area is available. Smaller particle sizes are also likely to provide an effective energy transfer from ultrasonic waves to the C–S and S–S bonds. The resulting network would be smaller in dimension, and the applied energy would affect the sulphur bonds instead of the backbone of the polymer. This explanation can also be applied to other samples; however, the energy transfer is less effective due to the particle and network sizes.1,28,46
The C–C bonds of the polymer's backbone are broken if the sol content increases during devulcanization. The measurements of the sol content showed that the least amount of sol was generated during the devulcanization procedure, which confirms the selective scission of sulphur bonds in the polymer network.53–55 This can also be investigated using the Horikx analysis.
3.7 Horikx analysis
Horikx analysis provides an evaluation of the devulcanization procedure by using equations 7 and 8 in the ESI.† Horikx analysis determines if the broken bonds belong to the polymer's backbone or the crosslinks. In Fig. 14, if the devulcanization percentage (the black triangle) of a sample falls below the selective crosslink scission curve (red curve), it indicates that the least amount of sol content was generated during the process. Consequently, it can be concluded that during the procedure no or a minimum number of C–C chains were broken.28,33 Furthermore, this shows that the intensity of the method was lower than the intensity needed to inflict damage on the backbone of the polymer which is regarded as degradation.19,30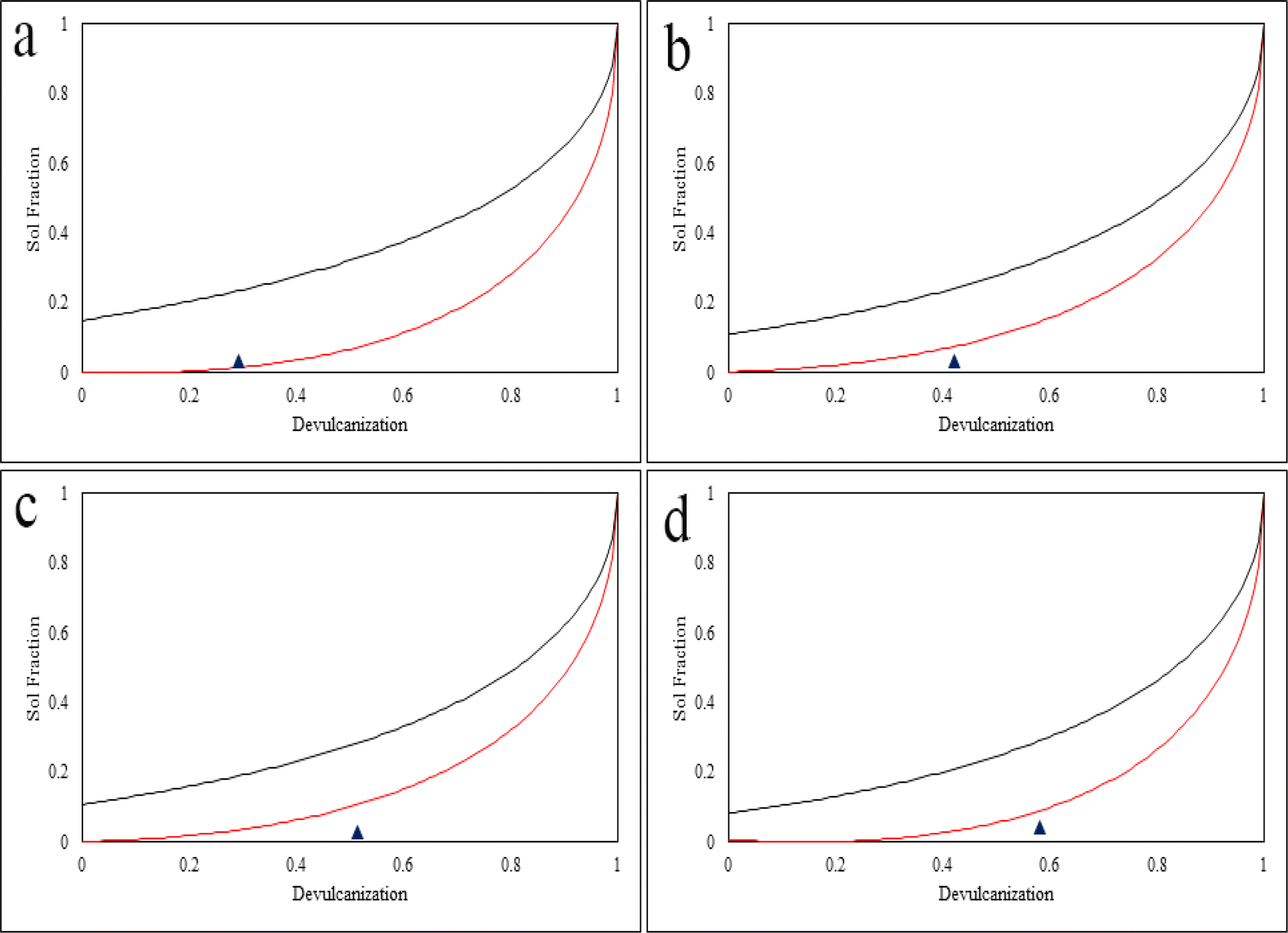 | ||
| Fig. 14 Horikx plot for (a) S.Fdv, (b) 40 Mdv, (c) 80 Mdv, and (d) 120 Mdv samples. Below the red curve, selective crosslink scission is happening, the area between the red and black curves shows that crosslink and backbone scission are coinciding, and above the black curve is the region in which the backbone scission is dominant. | ||
As can be observed in Fig. 14, the black triangles show the devulcanization percentage and are also an indication of the amount of sol fraction. These triangles either fall below the selective crosslink scission curve or – in the case of the S.Fdv – on the curve. This shows that during the devulcanization procedure, a minimal amount of energy was applied to the backbone of the polymer which was unable to damage it.21,28 It was previously observed in open literature that devulcanization occurs at the same time as reclamation.30,33 During this process, many studies show that the dominant type of scission belongs to the C–C bonds of the backbone.1,28,46 As a result, any procedure that can reduce the amount of main chain scission or minimize it can be considered successful.19,30,33
Another interesting observation from the Horikx plots goes back to the effect of particle size on the efficiency of the devulcanization. Although the procedure was similar for every sample, the 120 Mdv has the highest efficiency. This shows that smaller rubber particles make it more feasible for the solvent to penetrate through the bulk of the rubber and break the C–S or S–S bonds with the least amount of energy and time. The results of the Horikx analysis can prove that the devulcanization process was carried out successfully. This was achieved using a green, biodegradable, biocompatible, and non-toxic DES.56–58
3.8 Thermogravimetric analysis
TGA curves of the untreated and devulcanized samples are presented in Fig. 15 and 16, respectively. As can be observed, the untreated rubber samples have the same behaviour. The onset of mass loss falls between 340 to 345 °C, while their final mass loss falls between 59 to 61%. The samples' thermal stability and degradation behaviour are also similar; this shows that all the samples have the same origin and additive contents with slight differences in filler content.28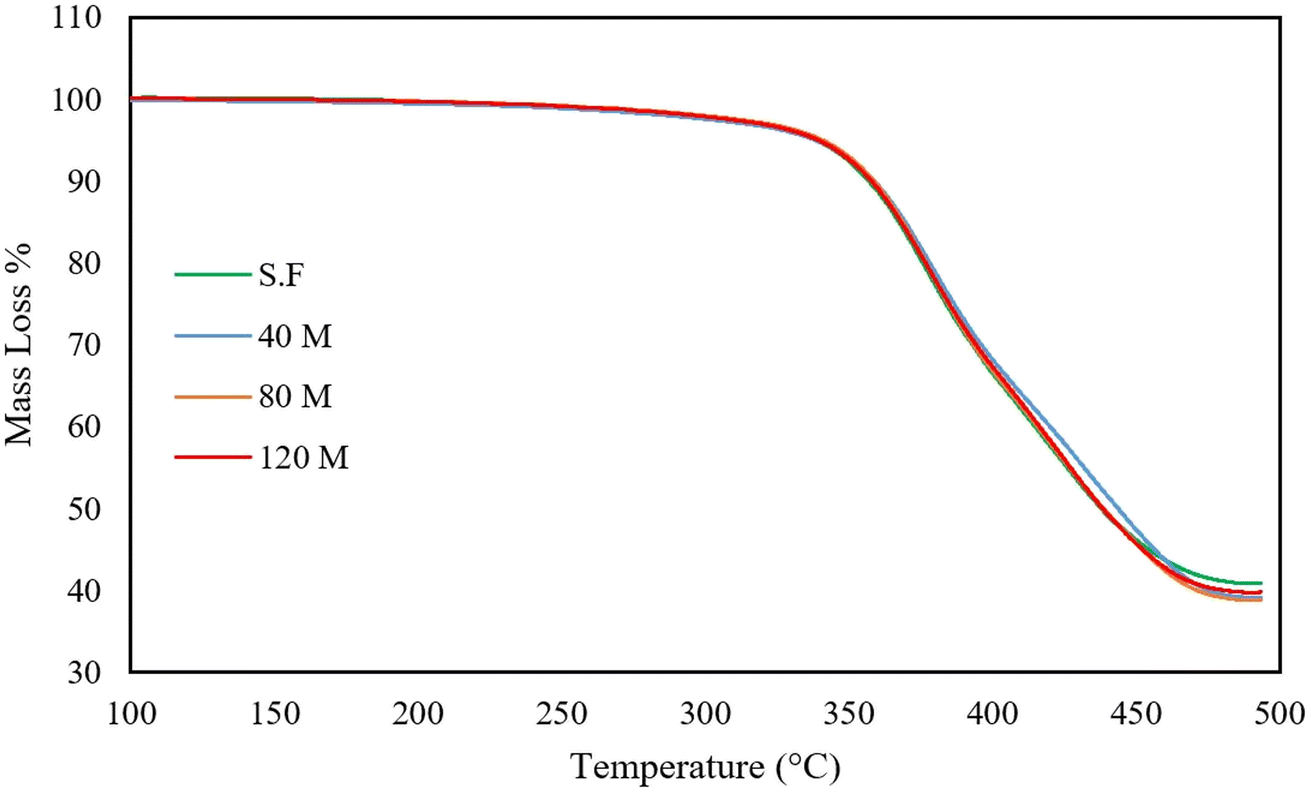 | ||
| Fig. 15 TGA curves of the untreated samples. | ||
 | ||
| Fig. 16 TGA curves of the devulcanized samples. | ||
Fig. 16 shows four different thermal regions attributed to volatilization, degradation of isoprene, degradation of styrene-butadiene and char residue. Within the first region (from 100 to 250 °C), low molecular weight materials, any absorbed moisture, oils, and waxes are degraded.28 In the following region, natural rubber (the major constituent of GTR) degrades at approximately 250 to 400 °C.4,29
In the next region, styrene-butadiene, which has higher thermal stability than natural rubber and is another major constituent of the GTR, is degraded at a temperature range of 400 to 480 °C.28
The char residue after 500 °C is attributed to carbon black, silica, and degraded natural rubber.29 Normally, char residue remains unchanged after devulcanization, as also mentioned in previous studies.28,33,46 This is the case for S.F, 40 M, and 120 M samples, but the 80 M has a different char residue. This can be attributed to cleavage of the polymer backbone, existing impurities, or differences in composition. According to the Horikx diagram (Fig. 14C), the 80 Mdv sample sits below the selective crosslink scission curve which shows that the assumption of backbone cleavage is false. The TGA plots for the untreated samples show that they have similar compositions but, there can be differences in filler content since the GTR is sourced from various types of rubber. This can be confirmed by the slight char residue differences in the untreated TGA curves (refer to Table 7 for details). In addition, the SEM images in the ESI† clearly show that fibrous impurities are available in the GTR. These impurities can also affect the char yield since they have various sources such as polyester cords that are used as ply fabric.
| Sample | T 5% (°C) | T 10% (°C) | T 20% (°C) | T 50% (°C) | Residual mass (%) |
|---|---|---|---|---|---|
| S.Fu | 338.3 | 356.8 | 375.6 | 437.8 | 40.94 |
| S.Fdv | 319.3 | 352.3 | 373.5 | 439.5 | 39.88 |
| 40 Mu | 338.2 | 358.7 | 378.1 | 443.6 | 39.1 |
| 40 Mdv | 295.9 | 347.6 | 371.8 | 440.4 | 38.73 |
| 80 Mu | 340.7 | 358.5 | 376.7 | 438.5 | 38.81 |
| 80 Mdv | 293.6 | 345.4 | 369.4 | 428.1 | 33.38 |
| 120 Mu | 339.2 | 357.7 | 376.6 | 438.3 | 39.79 |
| 120 Mdv | 239.4 | 313.8 | 364 | 431.9 | 37.94 |
Furthermore, the FTIR curve for the 80 Mdv shows peaks in the range between 600–1000 cm−1 that normally should be less intense after devulcanization. This again can be due to impurities from the filter paper used for extracting the devulcanized rubber which may also affect the char residue. Table 7 represents the thermal decomposition temperature of the samples at 5, 10, 20, and 50% mass loss in addition to the residual mass.
3.9 Efficiency and scalability of the procedure
An efficient and scalable procedure to obtain a product with the desired characteristics is essential for manufacturing and synthesis processes. To better understand the efficiency of the proposed method, the devulcanization percentages from Table 2 are compared to the highest percentage achieved in this study. Considering both time consumption and devulcanization percentage, the proposed method can be considered an efficient and environmentally friendly procedure, with a 58% devulcanization percentage achieved in a 30 minutes procedure with low energy consumption.Given the availability of industrial scale sonicators, sonication is a feasible method for scaling up this procedure. In addition, many industries already use highly efficient heated reactors, which means heating the mixture to 180 °C is also easily achievable. The most important part is the product yield of 1 g per 20 g of DES on the lab scale. Assuming a linear scale-up of the process (which should be confirmed by further investigations), if the capacity of a reactor is 1000 kg, the amount of solvent needed is 952 kg, and the amount of obtained devulcanized rubber after a 30 minutes procedure will be 48 kg (neglecting the possible losses). This means that 96 kg of devulcanized rubber can be produced each hour using a 1000 kg reactor, and after 8 hours (daily shift), 768 kg of devulcanized rubber can be produced. This assumption is made based on a linear scale-up and has to be investigated and evaluated in the future.
This procedure can be even more efficient if the DES can be used multiple times with the same devulcanization efficiency. To confirm this, DES should be thoroughly studied before and after the recycling procedure, and the devulcanization percentage should be investigated. The suggested procedure can be highly effective on the condition of using one batch of DES for multiple devulcanization procedures.
Finally, the DES constituents' price, urea and choline chloride should also be considered when assessing the performance of this devulcanization approach. At the time of writing this paper, urea (99%) from Sigma-Aldrich was sold for GBP 57.80 and choline chloride (98%) for GBP 80.60 per kg resulting in a GBP 65.39 for a kilogram of the ChCl–urea DES. For better insight, DD (99%), HDA (98%), and benzoyl peroxide are priced at GBP 147 per 250 g, GBP 85 per 25 g, and GBP 154 per kg, respectively.
In conclusion, this procedure is a scalable method with the potential to be highly efficient and profitable if DES proves to be reusable. This claim should be investigated more thoroughly from financial and chemical perspectives in future studies.
4 Conclusion
In this study, four different GTR samples were successfully devulcanized using ChCl–urea DES and a combination of probe and bath sonicators, followed by heating. The 120 M ground tire rubber samples had the highest devulcanization percentage of 58.13%, while the devulcanization for 80 M, 40 M, and S.F samples were 51.48, 42.1 and 29.3%, respectively.The morphology of the devulcanized rubber was similar to plasticized polymers with a rough surface that increased with the percentage of devulcanization. The change of morphology in the devulcanized rubber was observed both by SEM and AFM. The average adhesion force measured via AFM in the 120 M devulcanized sample was almost 8 times larger than the vulcanized counterpart. ATR-FTIR analysis confirmed that multiple disulphide bonds were selectively broken during devulcanization with minimal or no damage to the C–C bonds of the polymer backbone. EDX analysis revealed that there was no backbone scission during devulcanization and that N and Cl from the DES had formed chemical bonds with the polymer chains. TGA analysis showed the thermal behaviour and stability of the samples before and after devulcanization.
Flory–Rehner analysis calculated the devulcanization percentage using various formulations and measurement data. Horikx analysis and plots were used to evaluate the results of the Flory–Rehner analysis. Horikx analysis showed that the samples fell below the selective crosslink chain scission curve, confirming the accuracy and credibility of the data from the characterization techniques and Flory–Rehner analysis.
One of the goals of this study was to devulcanize GTR using the least amount of time and energy consumption and minimizing the environmental impacts of using a solvent. This method achieved a 58% devulcanization of the 120 M sample after 30 minutes and consuming only 182 W. We suggest that future research investigate various green DESs and optimize devulcanization parameters to achieve even higher devulcanization percentages, which could have significant scientific and technological importance.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The GREEN-LOOP project has received funding from the European Union's Horizon Europe, grant number 101057765. This includes funds from the UK Research and Innovation (UKRI) under the UK Government's Horizon Europe funding guarantee, grant number 10038028. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them. Hesam Ramezani also acknowledges the support of the Bristol Centre for Functional Nanomaterials (BCFN) of the University of Bristol.References
- H. Chittella, et al., Rubber waste management: A review on methods, mechanism, and prospects, Polym. Degrad. Stab., 2021, 194, 109761 CrossRef CAS.
- T. Cernev and R. Fenner, The importance of achieving foundational Sustainable Development Goals in reducing global risk, Futures, 2020, 115, 102492 CrossRef.
- M. G. Aboelkheir, et al., Biodegradation of Vulcanized SBR: A Comparison between Bacillus subtilis, Pseudomonas aeruginosa and Streptomyces sp, Sci. Rep., 2019, 9(1), 19304 CrossRef CAS PubMed.
- A. Fazli and D. Rodrigue, Waste rubber recycling: A review on the evolution and properties of thermoplastic elastomers, Materials, 2020, 13(3), 782 CrossRef CAS PubMed.
- M. Schmidt, et al., Sustainable Global Value Chains, Springer, vol. 730, 2019 Search PubMed.
- K. Selvaranjan, et al., Environmental challenges induced by extensive use of face masks during COVID-19: A review and potential solutions, Environ. Challenges, 2021, 3, 100039 CrossRef CAS PubMed.
- S.-Y. Leong, et al., 4R of rubber waste management: current and outlook, J. Mater. Cycles Waste Manage., 2023, 25(1), 37–51 CrossRef PubMed.
- S. Ramarad, et al., Waste tire rubber in polymer blends: A review on the evolution, properties and future, Prog. Mater. Sci., 2015, 72, 100–140 CrossRef CAS.
- J. Shi, et al., Structure and performance of reclaimed rubber obtained by different methods, J. Appl. Polym. Sci., 2013, 129(3), 999–1007 CrossRef CAS.
- R. Diaz, et al., Thermo-mechanical recycling of rubber: Relationship between material properties and specific mechanical energy, J. Mater. Process. Technol., 2018, 252, 454–468 CrossRef CAS.
- K. Formela, M. Cysewska and J. T. Haponiuk, Thermomechanical reclaiming of ground tire rubber via extrusion at low temperature: Efficiency and limits, J. Vinyl Addit. Technol., 2016, 22(3), 213–221 CrossRef CAS.
- B. Vega, et al., Control of vulcanizing/devulcanizing behavior of diphenyl disulfide with microwaves as the heating source, J. Appl. Polym. Sci., 2008, 108(3), 1969–1975 CrossRef CAS.
- P. Thaicharoen, P. Thamyongkit and S. Poompradub, Thiosalicylic acid as a devulcanizing agent for mechano-chemical devulcanization, Korean J. Chem. Eng., 2010, 27, 1177–1183 CrossRef CAS.
- Z. Zhang, et al., Understanding H2O2-induced thermo-oxidative reclamation of vulcanized styrene butadiene rubber at low temperatures, ACS Sustain. Chem. Eng., 2021, 9(5), 2378–2387 CrossRef CAS.
- M. Sabzekar, et al., Influence of process variables on chemical devulcanization of sulfur-cured natural rubber, Polym. Degrad. Stab., 2015, 118, 88–95 CrossRef CAS.
- R. Walvekar, et al., Parametric study for devulcanization of waste tire rubber utilizing deep eutectic solvent (DES), in MATEC Web of Conferences, EDP Sciences, 2018 Search PubMed.
- S. Seghar, et al., Devulcanization of styrene butadiene rubber by microwave energy: Effect of the presence of ionic liquid, eXPRESS Polym. Lett., 2015, 9(12), 1076–1086 CrossRef CAS.
- R. Walvekar, et al., Devulcanization of waste tire rubber using amine based solvents and ultrasonic energy, in MATEC Web of Conferences, 2018 Search PubMed.
- S. Seghar, et al., Thermo-mechanical devulcanization and recycling of rubber industry waste, Resour., Conserv. Recycl., 2019, 144, 180–186 CrossRef.
- Ł. Piszczyk, et al., Effect of ground tire rubber on structural, mechanical and thermal properties of flexible polyurethane foams, Iran. Polym. J., 2015, 24, 75–84 CrossRef.
- A. I. Isayev, T. Liang and T. M. Lewis, Effect of particle size on ultrasonic devulcanization of tire rubber in twin-screw extruder, Rubber Chem. Technol., 2014, 87(1), 86–102 CrossRef CAS.
- X. Sun and A. Isayev, Continuous ultrasonic devulcanization: Comparison of carbon black filled synthetic isoprene and natural rubbers, Rubber Chem. Technol., 2008, 81(1), 19–46 CrossRef CAS.
- A. P. Abbott, et al., Novel solvent properties of choline chloride/urea mixtures, Chem. Commun., 2003,(1), 70–71 RSC.
- A. P. Abbott, et al., Deep eutectic solvents formed between choline chloride and carboxylic acids: versatile alternatives to ionic liquids, J. Am. Chem. Soc., 2004, 126(29), 9142–9147 CrossRef CAS PubMed.
- S. Arriaga and A. Aizpuru, Innovative non-aqueous phases and partitioning bioreactor configurations, in Advances in Chemical Engineering, Elsevier, 2019, pp. 299–348 Search PubMed.
- S. Rooj, New Route for Devulcanization of Natural Rubber and the Properties of Devulcanized Rubber, J. Polym. Environ., 2011, 19(2), 382–390 CrossRef CAS.
- I. Mangili, et al., Characterization and supercritical CO2 devulcanization of cryo-ground tire rubber: Influence of devulcanization process on reclaimed material, Polym. Degrad. Stab., 2014, 102, 15–24 CrossRef CAS.
- R. Saputra, et al., Effective devulcanization of ground tire rubber using choline chloride-based deep eutectic solvents, J. Environ. Chem. Eng., 2019, 7(3), 103151 CrossRef.
- R. Saputra, et al., Devulcanisation of ground rubber tyre by novel ternary deep eutectic solvents, J. Mol. Liq., 2020, 306, 112913 CrossRef CAS.
- H. Lian, et al., Processing of lignin in urea–zinc chloride deep-eutectic solvent and its use as a filler in a phenol-formaldehyde resin, RSC Adv., 2015, 5(36), 28778–28785 RSC.
- A. Berlin, et al., Tire-rubber-waste recycling and active powder of discretely devulcanized rubber, Polym. Sci., Ser. D, 2018, 11, 323–329 CrossRef CAS.
- C. Jiang, et al., Tailoring the properties of ground tire rubber/high-density polyethylene blends by combining surface devulcanization and in situ grafting technology, Mater. Chem. Phys., 2018, 220, 161–170 CrossRef CAS.
- M. Molanorouzi and S. O. Mohaved, Reclaiming waste tire rubber by an irradiation technique, Polym. Degrad. Stab., 2016, 128, 115–125 CrossRef CAS.
- W. Dierkes and S. Saiwari, Regeneration and devulcanization, Tire Waste and Recycling, 2021, p. 97 Search PubMed.
- M. Verbruggen, et al., Mechanisms involved in the recycling of NR and EPDM, Rubber Chem. Technol., 1999, 72(4), 731–740 CrossRef CAS.
- M. Horikx, Chain scissions in a polymer network, J. Polym. Sci., 1956, 19(93), 445–454 CrossRef CAS.
- L. Liu, et al., Synthesis of N-unsubstituted cyclic imides from anhydride with urea in deep eutectic solvent (DES) choline chloride/urea, Chem. Pap., 2020, 74, 1351–1357 CrossRef CAS.
- M. Santra, D. Kunzru and D. Rabari, A stability analysis of choline chloride: urea deep eutectic solvent using density functional theory, Comput. Theor. Chem., 2022, 1217, 113921 CrossRef CAS.
- K. Formela, et al., Microwave treatment in waste rubber recycling–recent advances and limitations, eXPRESS Polym. Lett., 2019, 13(6), 565–588 CrossRef CAS.
- M. A. Saghiri, et al., Back-scattered and secondary electron images of scanning electron microscopy in dentistry: a new method for surface analysis, Acta Odontol. Scand., 2012, 70(6), 603–609 CrossRef CAS PubMed.
- L. Reimer and L. Reimer, Emission of backscattered and secondary electrons, Scanning Electron Microscopy: Physics of Image Formation and Microanalysis, 1998, pp. 135–169 Search PubMed.
- Z.-J. Ding and R. Shimizu, Monte Carlo study of backscattering and secondary electron generation, Surf. Sci., 1988, 197(3), 539–554 CrossRef CAS.
- Ł. Zedler, et al., Synergistic effects of bitumen plasticization and microwave treatment on short-term devulcanization of ground tire rubber, Polymers, 2018, 10(11), 1265 CrossRef PubMed.
- J. Araujo-Morera, et al., On the use of mechano-chemically modified ground tire rubber (GTR) as recycled and sustainable filler in styrene-butadiene rubber (SBR) composites, J. Compos. Sci., 2021, 5(3), 68 CrossRef CAS.
- X. Colom, et al., Structural and physico-mechanical properties of natural rubber/GTR composites devulcanized by microwaves: Influence of GTR source and irradiation time, J. Compos. Mater., 2018, 52(22), 3099–3108 CrossRef CAS.
- R. Saputra, et al., Current progress in waste tire rubber devulcanization, Chemosphere, 2021, 265, 129033 CrossRef CAS PubMed.
- H. Ramezani, T. Behzad and R. Bagheri, Synergistic effect of graphene oxide nanoplatelets and cellulose nanofibers on mechanical, thermal, and barrier properties of thermoplastic starch, Polym. Adv. Technol., 2020, 31(3), 553–565 CrossRef CAS.
- A. Zanchet, et al., Activator from sugar cane as a green alternative to conventional vulcanization additives, J. Cleaner Prod., 2018, 174, 437–446 CrossRef CAS.
- S. Saiwari, W. K. Dierkes and J. W. Noordermeer, Devulcanization of whole passenger care tire material, Kautsch. Gummi, Kunstst., 2013, 66(7–8), 20–25 CAS.
- Q. Zhang, et al., Deep eutectic solvents: syntheses, properties and applications, Chem. Soc. Rev., 2012, 41(21), 7108–7146 RSC.
- F. D. De Sousa, et al., Devulcanization of waste tire rubber by microwaves, Polym. Degrad. Stab., 2017, 138, 169–181 CrossRef CAS.
- K. Shahbaz, et al., Prediction of the surface tension of deep eutectic solvents, Fluid Phase Equilib., 2012, 319, 48–54 CrossRef CAS.
- P. Song, et al., Vegetable derived-oil facilitating carbon black migration from waste tire rubbers and its reinforcement effect, Waste Manage., 2018, 78, 238–248 CrossRef CAS PubMed.
- S. Ghorai, et al., Reclaiming of waste guayule natural rubber vulcanizate—reclaim rubber for green tire applications: An approach for sustainable development, J. Elastomers Plast., 2019, 51(3), 193–210 CrossRef CAS.
- P. Song, X. Wu and S. Wang, Effect of styrene butadiene rubber on the light pyrolysis of the natural rubber, Polym. Degrad. Stab., 2018, 147, 168–176 CrossRef CAS.
- M. Protti, et al., Co-solvents and mobile phase additives in HPLC, in Liquid Chromatography, Elsevier, 2023, pp. 283–321 Search PubMed.
- A. Si and A. K. Misra, Perspective on the transformation of carbohydrates under green and sustainable reaction conditions, in Recent Trends in Carbohydrate Chemistry, Elsevier, 2020, pp. 3–71 Search PubMed.
- U. Alshana and M. Soylak, Deep eutectic solvents in microextraction, in Analytical Sample Preparation with Nano-And Other High-Performance Materials, Elsevier, 2021, pp. 471–512 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00079j |
| This journal is © The Royal Society of Chemistry 2024 |