
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimethylphosphite electrosynthesis from inorganic phosphorus building blocks via oxidative coupling†
Junnan
Li
a,
Hossein
Bemana
ab and
Nikolay
Kornienko
 *ab
*ab
aDepartment of Chemistry, Université de Montréal, 1375 Avenue Thérèse-Lavoie-Roux, Montréal, QC H2V 0B3, Canada
bInstitute of Inorganic Chemistry, University of Bonn, Gerhard-Domagk-Strasse 1, 53121 Bonn, Germany. E-mail: nkornien@uni-bonn.de
First published on 19th June 2024
Abstract
Organophosphorus compounds carry importance in the chemical, medical, and fertilizer industries. Their production often entails the use of white phosphorus or PCl3, which are toxic and energetically costly to produce. In this work we investigate phosphite ester formation through an electrochemical route which has the potential to serve as a greener alternative. In particular, dimethyl phosphite was electrosynthesized through oxidative coupling of an inorganic P source, H3PO2, and methanol as a model building block with high faradaic efficiencies approaching 100%. The reaction is proposed to proceed through electrooxidative phosphorus radical formation followed by coupling of this reactive species with proximal methanol molecules.
Sustainability spotlightThis work helps advance electrosynthetic routes towards societally valuable chemicals. In particular, electrosynthetic pathways towards organophosphorus compounds are an appealing route in that they can potentially be carried out under mild conditions, be powered by renewable energy, carry far less of a carbon footprint, and circumvent the need for toxic reagents. This falls in line with the UN SDGs 7, 9 and 12. Against this backdrop, this work develops an electrosynthetic route to phosphite esters, which have a diverse array of applications, using readily available H3PO2 and CH3OH building blocks. We anticipate this study will help build towards the electrification of chemical processes in industry. |
Introduction
Phosphorus is one of the essential elements of life and the chemistry of this element is consequently of interest across a wide spectrum of applications.1 Phosphorus is used in both its inorganic form, primarily as a fertilizer,2,3 and as organophosphorus compounds with a diversity of end uses including pesticides, flame retardants, plasticizers, pharmaceuticals and more.4 The industrial synthesis of organophosphorus invariably proceeds through an energy intensive thermochemical reduction of phosphate rock to form white phosphorus (P4) which is then chlorinated to PCl3 and subsequently used as a versatile building block.5 Both the energy consumption and inherent danger of reactants (pyrophoric nature of P4 and toxicity of PCl3) present problems from a sustainability perspective, as do the high temperatures (1500 °C) used for the thermochemical reduction step.As an alternative to thermochemical routes, electrochemical methods are rapidly gaining traction. The appeal to electrosynthesis is that reactions may be carried out under mild conditions and therefore can potentially carry a much smaller carbon footprint in arriving at the same products.6,7 Against this backdrop, an ideal technological solution would be to construct organophosphorus compounds through efficient electrosynthetic pathways that utilize the most sustainable form of phosphorus and carbon, particularly PO43− and CO2 as individual building blocks (Fig. 1a).8 The direction of electrochemical CO2 reduction to small molecules like CO, CH3CH2OH and C2H4 has rapidly progressed in recent years to the point where the electrosynthesis of such products is competitive with conventional pathways.9,10
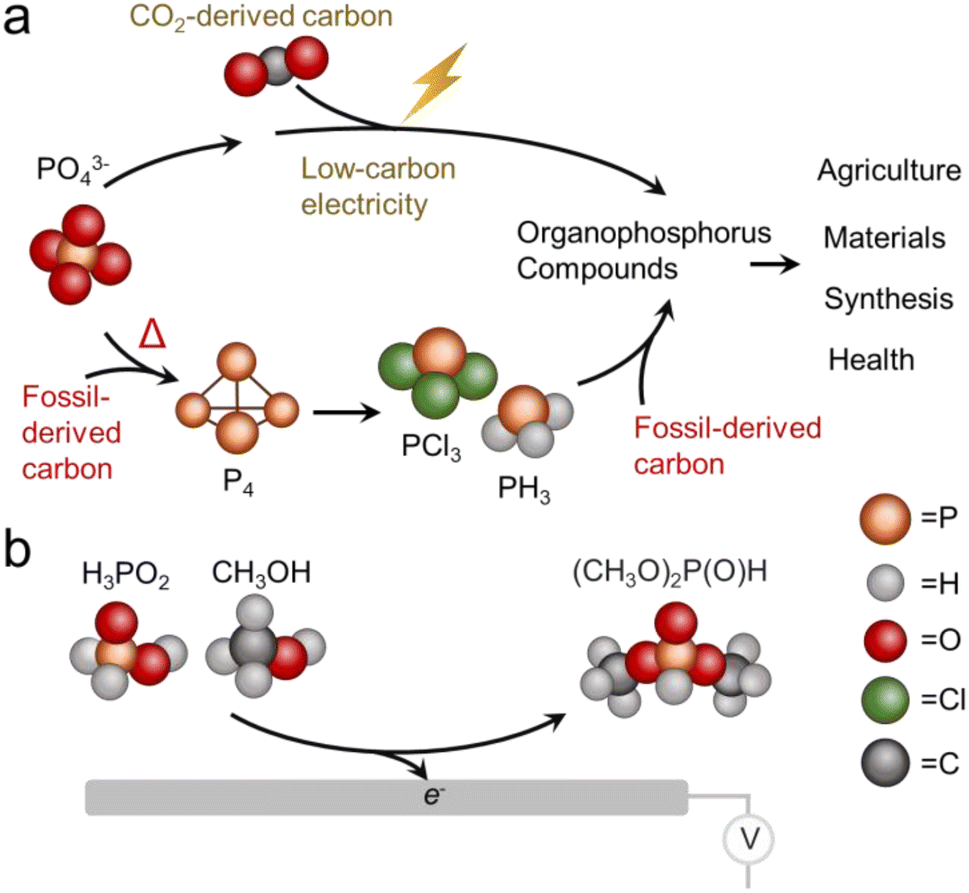 | ||
| Fig. 1 Potential production routes of organophosphorus compounds that contrast idealized electrochemical and current thermochemical pathways (a) and simplified illustration of DMP electrosynthesis from CH3OH and H3PO2 demonstrated in this work (b). | ||
Electrochemical activation of PO43− and other phosphorus building blocks on the other hand, is much less understood and consequently viable systems do not yet exist. The reduction of triphenylphosphine oxide (PV) to triphenyl phosphine (PIII) has recently been accomplished with the help of borate species in the electrolyte.11 PO43− reduction to P4 has also been carried out in a molten salt based reactor, albeit at elevated temperatures (800 °C).12 While electrochemical C–P bond formation in the realm of organic electrosynthesis is already established, these routes entail the use of pre-activated phosphorus building blocks like phosphite esters or phosphites with pre-existing C–P bonds.13 To the best of our knowledge, the electrosynthesis of organophosphorus compounds using purely inorganic phosphorus building blocks has yet to be demonstrated. Of note, mild chemical pathways to convert PO43− to C–P products14 and C–P coupling of hypophosphites with alkenes15 are emerging and another appealing direction yet are outside the scope of this work.
Against this backdrop, this work aimed to develop electrosynthetic pathways to organophosphorus compounds (here defined as compounds containing both P and C atoms that also includes esters) from inorganic phosphorus building blocks. While the long-term goal is to directly use PO43−, we opted to first use hypophosphorous acid (H3PO2) as a model reagent with higher reactivity. Likewise, we turned to CH3OH as a model carbon-based building block with higher reactivity than CO2. Again, the aim would be to either directly use CO2 or use building blocks like CH3OH that were electrochemically derived from CO2 in a previous step.16 Using a Pd catalyst, we demonstrate an ambient temperature/pressure and air/moisture-tolerant electrosynthesis of dimethylphosphite (DMP) from H3PO2 and CH3OH co-oxidation (Fig. 1b). DMP is a phosphite ester with applications as an intermediary compound in the manufacture of pesticides, pharmaceuticals and fireproof materials, The route developed in this work is an alternative to its current production method, carried out via PCl3 and CH3OH starting reagents at a scale of approx. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons annually.17 Under optimized conditions, the faradaic efficiency reaches nearly 100%, such that almost all electrons are being directed towards the intended reaction pathway at the anode and side reactions there are minimized. The proof-of-principle here stands to open new pathways to electrochemical C/O–P coupling and take a step towards the sustainable synthesis of organophosphorus compounds, with the ultimate goal being the electrochemical activation of phosphate and CO2 en route to an array of organic phosphorus compounds. We must also note that while electrochemical approaches are just emerging, there is a substantial push towards sustainable organophosphorus chemistry with thermochemical routes.18 For example, H-phosphonate diesters were prepared from alcohols and H3PO2 over Ni/SiO2 catalysts.19
000 tons annually.17 Under optimized conditions, the faradaic efficiency reaches nearly 100%, such that almost all electrons are being directed towards the intended reaction pathway at the anode and side reactions there are minimized. The proof-of-principle here stands to open new pathways to electrochemical C/O–P coupling and take a step towards the sustainable synthesis of organophosphorus compounds, with the ultimate goal being the electrochemical activation of phosphate and CO2 en route to an array of organic phosphorus compounds. We must also note that while electrochemical approaches are just emerging, there is a substantial push towards sustainable organophosphorus chemistry with thermochemical routes.18 For example, H-phosphonate diesters were prepared from alcohols and H3PO2 over Ni/SiO2 catalysts.19
Results and discussion
To begin investigating the electrochemical activation of H3PO2, we used a Pd foil catalyst. Pd was chosen as a model metal starting catalyst that is straightforward to work with in both aqueous and non-aqueous environments.20,21 The surface of the Pd was roughened through electrodepositing a Pd layer in an aqueous acidic solution.22 This procedure resulted in a flower-like hierarchical structure, as noted by scanning electron microscopy (SEM) imaging (Fig. 2). An advantage of this route was that, unlike that with typical wet-chemical nanoparticle synthesis, there were no surface-bound ligands remaining that could interfere with the catalytic process, a high surface area with large density of active sites was attained, and that there was a direct electronic connection to the current collector. | ||
| Fig. 2 Morphology and roughness of the Pd electrode used in this study under low (a) and high (b) magnifications. | ||
We first probed the electrochemical oxidation of H3PO2 and CH3OH reactants individually using cyclic voltammetry (CV) measurements. Dimethylformamide (DMF) was used as a solvent with Tetrabutylammonium hexafluorophosphate (TBA PF6) supporting electrolyte in a standard 3-electrode configuration with an Ag wire as a quasi-reference electrode (Fig. S1†), whose potential was calibrated to be at −0.75 V vs. a standard Fc/Fc+ redox couple (Fig. S2†). We noted that the oxidation of DMF began at around 1 V (Fig. 3a). When 10% vol. CH3OH was added to the system, additional irreversible oxidation current initiated at 0.7 V. This likely stemmed from the oxidation of CH3OH to species like CO, HCOOH and CO2.
 | ||
| Fig. 3 Electrochemical oxidation of CH3OH and H3PO2 reactants in a DMF solvent. | ||
However, a DMF solution with 0.2 M H3PO2 resulted in first, a large oxidation wave initiating at 0.4 V and on the reverse sweep, an asymmetric wave that often occurs when there is a deactivation process occurring at high potentials and these inhibiting species are removed again once the potential returns to 1.1 V. This is seen, for example, in electrochemical alcohol oxidation when *CO intermediates formed at highly oxidizing potentials block poison catalytic sites and get reductively desorbed.23 Alternatively, this phenomenon, also occurs when less active surface oxides form under oxidizing potentials, but we believe this is less likely because we did not see this in the case of CH3OH oxidation. We therefore attribute this to the site-blocking behavior of H3PO2-derived species. A possible chemical explanation of this is that the positive current entails the 1 e− oxidation of H3PO2 to [˙PH2O2]+via cleavage of the weak P–H bond.15,24 This radical species can then presumably further react with other species in the electrolyte (e.g. CH3OH) or dimerize with proximal H3PO2 species to form pyrophosphate-like products, the latter of which we tentatively attribute to the site-blocking inactive species. With both CH3OH and H3PO2 present, the catalytic current increased and the stripping feature slightly decreased, providing an initial layer of evidence that a new reaction pathway involving both reactants was emerging.
We carried out a series of constant potential electrolysis runs but with only 10% CH3OH, we were not able to reliable detect any accumulated products in the electrolyte using 31P-NMR as our detection method. Modifying the H3PO2 concentration did help either, suggesting that under these conditions H3PO2 is not the limiting reagent. However, once we increased the CH3OH concentration to 50%, we began to see a new peak arise around 12 ppm. This peak was more evident when CH3OH concentration was increased to 100% (e.g. CH3OH completely replaced DMF as the solvent) (Fig. 4a). This gave a first indication that CH3OH was the limiting reagent in our system. The peak matched that of DMP and we could further verify its identity using 2D NMR and mass spectrometry (Fig. S4 and 5†).
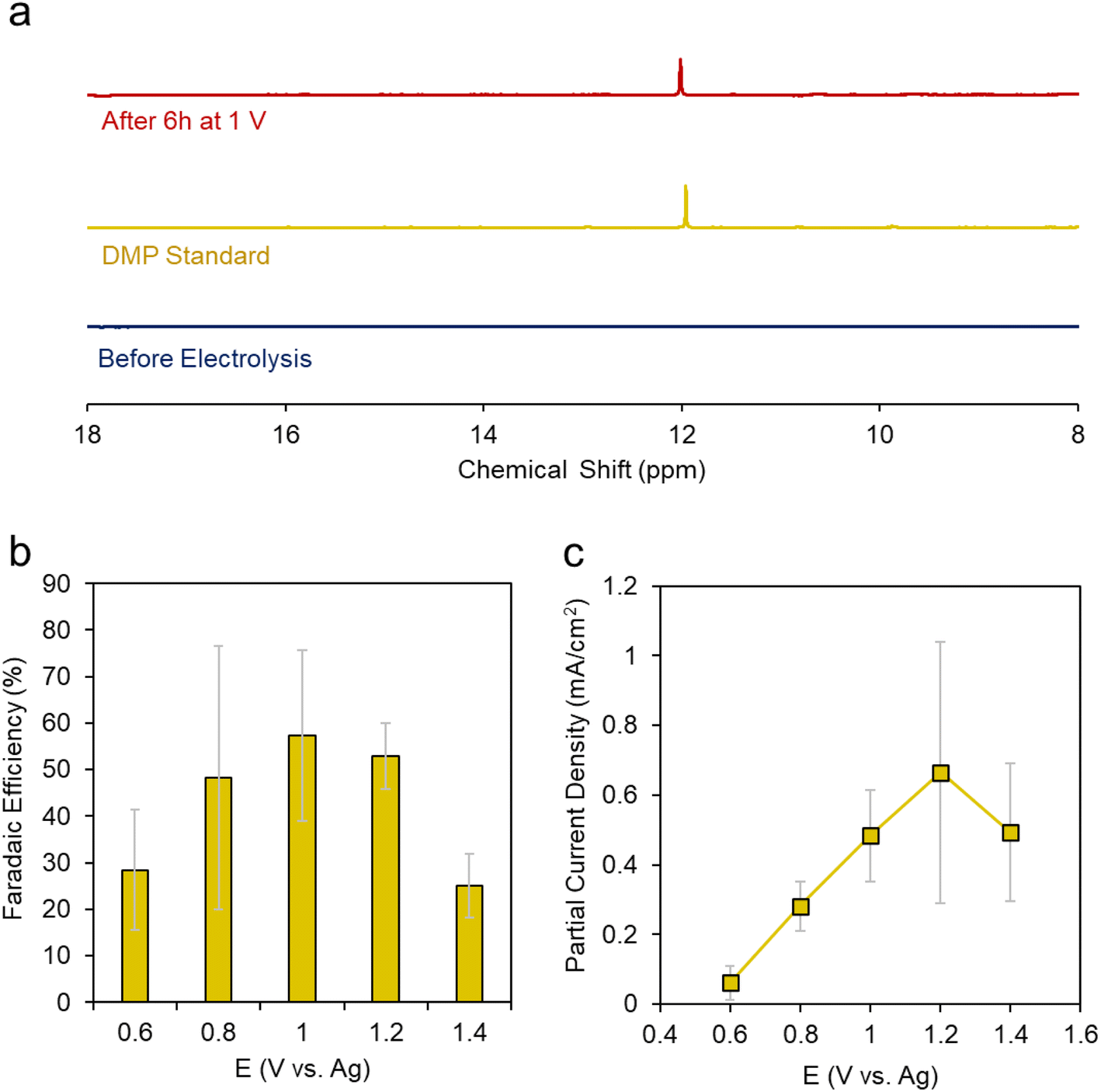 | ||
| Fig. 4 NMR spectra of DMP and our pre/post electrolysis solutions (a). Faradaic efficiency (b) and partial current density (c) for DMP production using the roughened Pd electrode as a function of voltage in CH3OH solvent with 0.2 M TBA PF6 supporting electrolyte and 0.1 M H3PO2 reactant. | ||
We then carried out a series of constant potential electrolysis runs to quantify the selectivity and formation rate of DMP using CH3OH as the solvent with 0.02 M TBA PF6 supporting electrolyte and 0.1 M H3PO2 reactant. We noted that DMP began to form around 0.6 V, slightly positive of its oxidation potential as approximated through CV measurements. The faradaic efficiency (FE), a measure of selectivity of the fraction of electrons being used to generate this product, increased up to 57% at 1.2 V, then decreased when moving more positive to 1.4 V (Fig. 4b). The same trend was observed for the partial current density, which peaked at 0.66 mA cm−2 at 1.2 V (Fig. 4c). Interestingly, the voltage range in which DMP is effectively produced corresponds to what in which H3PO2 is oxidized but before substantial CH3OH oxidation begins. At 1.4 V and higher, we believe that CH3OH oxidation begins to dominate, and this is responsible for the decreased selectivity and production rate of DMP.
We did not observe any notable decrease in performance throughout a 12 h stability measurement (Fig. S6†) or obvious changes to the Pd catalyst (Fig. S7†).
We next sought to gain a further level of mechanistic insight into the reaction mechanism by first modulating the concentration of H3PO2. We hypothesized that increasing the H3PO2 concentration may promote the deactivating dimerization mechanism while decreasing this would instead promote the reaction of the active species, presumably [˙PH2O2]+, with CH3OH. This was confirmed as increasing the H3PO2 concentration from 0.1 M to 0.3 M while keeping other parameters constant (roughened Pd electrode, CH3OH solvent + 20 mM TBA PF6 supporting electrolyte) led to a decrease of both the faradaic efficiency (Fig. 5a) and partial current density (Fig. 5b) for DMP production. Conversely, decreasing the H3PO2 concentration to 0.05 M resulted in DMP production with 98% faradaic efficiency while the partial current density was also raised from 0.48 to 1.18 mA cm−2.
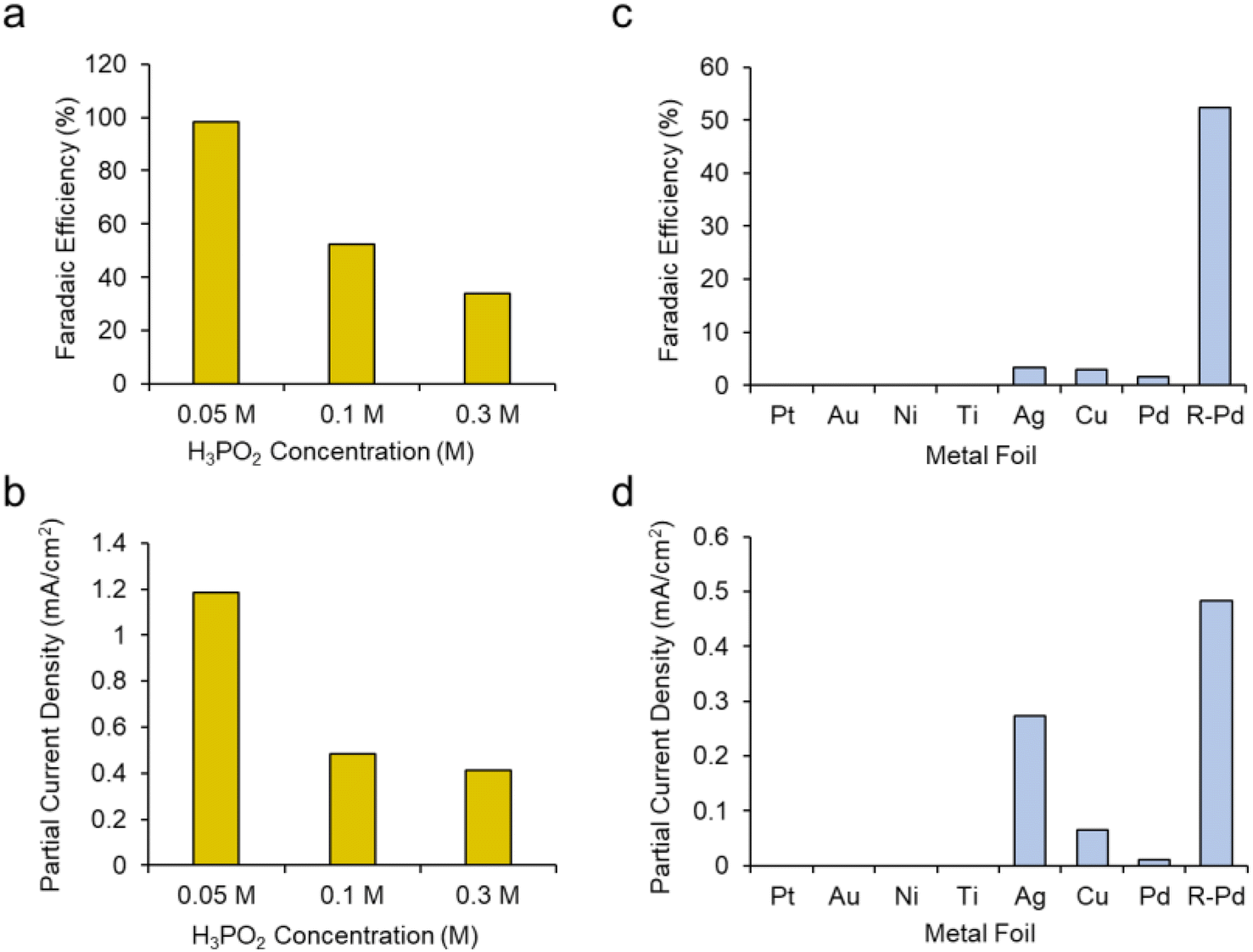 | ||
| Fig. 5 The faradaic efficiency (a) and partial current density (b) for DMP production with roughened Pd as a function of H3PO2 concentration. Planar electrodes comprised of different metals were also tested for their faradaic efficiency (c) and partial current density (d) for DMP formation and compared to the roughened Pd. All tests were run in CH3OH + 20 mM PBA PF6 at 1 V. | ||
Our next series of experiments entailed the use of different electrodes (catalysts) for this reaction. We tested planar, polycrystalline Pt, Au, Ni, Ti, Cu and Pd electrodes for their DMP production capacity using a standard set of conditions (CH3OH + 20 mM PBA PF6 + 0.1 M H3PO2 at 1 V). We noted that only Ag, Cu and Pd produced DMP, albeit with low FE (Fig. 5c). Even the planar Pd pared poorly in comparison with the roughened Pd. Interestingly, the planar Ag electrode produced DMP at comparable rates relative to the roughened Pd (Fig. 5d). This indicates that both the catalyst identity, as well as the surface roughness was key in this reaction mechanism. A potential explanation here could be the varying propensities of each catalyst to oxidize H3PO2 and CH3OH at the selected 1 V set potential. Further, we tentatively believe that the surface roughness of the Pd worked to ‘dilute’ the phosphorus radical intermediates and similarly facilitate their reaction with CH3OH rather than dimerizing.
From the whole of our data, we then come to an initial set of insights and tentative catalytic mechanism (Fig. 6). We believe that the first reaction step entails the 1 e− oxidation and P–H cleavage of H3PO2 to [˙PH2O2]. Under high local concentrations of H3PO2, this species can react with H3PO2 to form unreactive dimers. However, under low local H3PO2 concentrations, promoted by both the low bulk H3PO2 concentration and high catalyst surface area, as well as under high CH3OH concentrations, the initial reactive species of [˙PH2O2] can react with CH3OH to form a monomethyl phosphite. This species is readily oxidized once more and a subsequent coupling with CH3OH results in the DMP main product. However, we cannot rule out reaction mechanisms that involve, for example, a chemical reaction of the monomethyl ester that takes place before the 2nd oxidation step. This would be an interesting point to follow up on in greater depth via in situ techniques like vibrational spectroscopy25 in subsequent works. Further, we cannot completely identify each dimerization or deactivation pathway but we do see pyrophosphate/pyrophosphite byproducts that are likely the result of this (Fig. S8†). The existence of the radical-based reaction pathway is further supported by the fact that we were able to generate the DMP product using a sodium thiosulfate radical initiator in the same reaction media (Fig. S9†). Beyond DMP, additional products detected include the monomethyl phosphite product that is thought to be an intermediary species en route to DMP as well as mono and dimethyl phosphate species (Fig. S10†). Finally, we attempted to use other alcohols instead of methanol to extend the scope of the synthetic route.
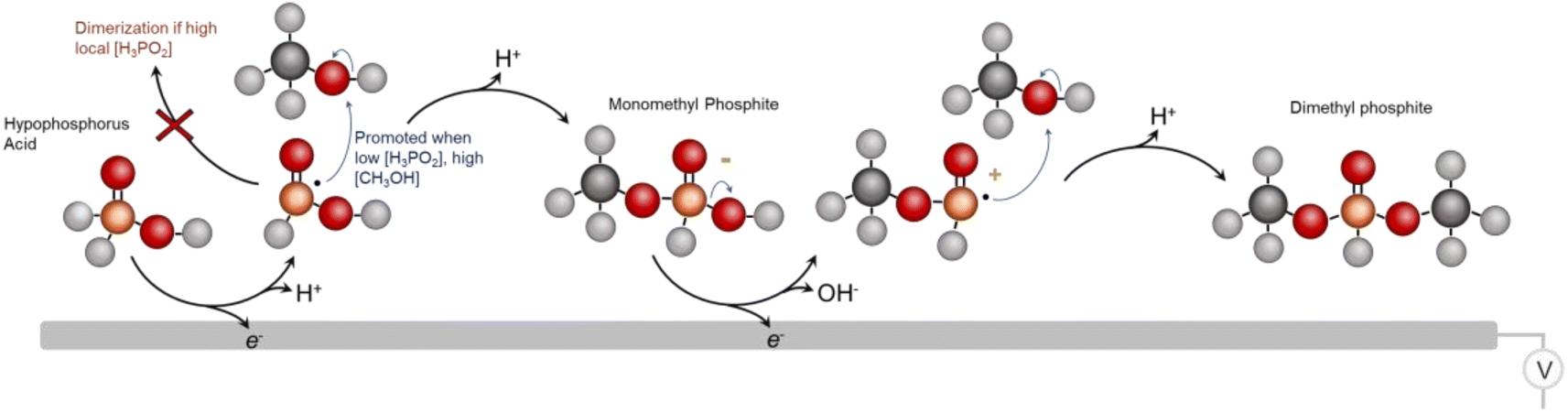 | ||
| Fig. 6 The proposed mechanism for DMP electrosynthesis from H3PO2 and CH3OH reactants. | ||
We note that the use of phosphonic acid or phosphoric acid would be a worthy follow-up in this overall direction. However, the use of these reagents under similar reaction conditions did not reliably yield new phosphorus-containing products in high concentrations.
Conclusion
In all, we developed an electrochemical route to DMP synthesis from purely inorganic H3PO2 building blocks. We believe that the reaction first proceeds through the oxidation of H3PO2 to the corresponding [˙PH2O2] species. We propose that the selectivity in this mechanism is determined through the competition between dimerization pathways and this can be steered through modifying local reactant concentrations and catalyst surface area. Carefully selecting reaction conditions here led to a nearly 100% faradaic efficiency for DMP synthesis. While H3PO2 is still produced from P4 and this does not entirely circumvent the thermochemical reduction of phosphates, this route at least takes place under ambient and air/moisture tolerant conditions instead of the conventional route to DMP synthesis involving PCl3 reagents. We anticipate this work helping advance the community's efforts in developing green pathways to organophosphorus synthesis and eventually finding viable routes to use phosphate as the building block.Data availability
Data will be available free of charge following reasonable requests to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. K., J. L. and H. B. acknowledge NSERC Discovery Grant RGPIN-2019-05927.References
- A. Sharpley, H. Jarvie, D. Flaten and P. Kleinman, J. Environ. Qual., 2018, 47, 774–777 CrossRef CAS PubMed.
- J. J. Weeks Jr and G. M. Hettiarachchi, J. Environ. Qual., 2019, 48, 1300–1313 CrossRef PubMed.
- W. Schipper, Eur. J. Inorg. Chem., 2014, 2014, 1567–1571 CrossRef CAS.
- V. Iaroshenko, Organophosphorus Chemistry: from Molecules to Applications, John Wiley & Sons, 1 edn, 2019 Search PubMed.
- M. B. Geeson and C. C. Cummins, ACS Cent. Sci., 2020, 6, 848–860 CrossRef CAS PubMed.
- T. Gao, B. Xia, K. Yang, D. Li, T. Shao, S. Chen, Q. Li and J. Duan, Energy Fuels, 2023, 37, 17997–18008 CrossRef CAS.
- D. Rojas Sánchez, K. Khalilpour and A. F. A. Hoadley, Sustainable Energy Fuels, 2021, 5, 5866–5880 RSC.
- J. Li, H. Heidarpour, G. Gao, M. McKee, H. Bemana, Y. Zhang, C.-T. Dinh, A. Seifitokaldani and N. Kornienko, Nat. Synth., 2024 DOI:10.1038/s44160-024-00530-8.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. S. Elias, C. Costentin and D. G. Nocera, J. Am. Chem. Soc., 2018, 140, 13711–13718 CrossRef CAS PubMed.
- J. F. Melville, A. J. Licini and Y. Surendranath, ACS Cent. Sci., 2023, 9, 373–380 CrossRef CAS PubMed.
- Y. H. Budnikova, E. L. Dolengovsky, M. V. Tarasov and T. V. Gryaznova, Front. Chem., 2022, 10, 1054116 CrossRef CAS PubMed.
- T. Schneider, K. Schwedtmann, J. Fidelius and J. J. Weigand, Nat. Synth., 2023, 2, 972–979 CrossRef.
- Z. Huang, Y. Chen and M. W. Kanan, Chem. Commun., 2022, 58, 2180–2183 RSC.
- S. Kong, X. Lv, X. Wang, Z. Liu, Z. Li, B. Jia, D. Sun, C. Yang, L. Liu, A. Guan, J. Wang, G. Zheng and F. Huang, Nat. Catal., 2023, 6, 6–15 CrossRef CAS.
- OECD, Screening Information Dataset: (SIDS) Initial Assessment Report (SIAR) for Dimethyl Phosphonates, 2014 Search PubMed.
- S. P. M. Ung and C.-J. Li, RSC Sustainability, 2023, 1, 11–37 RSC.
- H. C. Fisher, L. Prost and J.-L. Montchamp, Eur. J. Org Chem., 2013, 2013, 7973–7978 CrossRef CAS.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Nano Energy, 2016, 29, 198–219 CrossRef CAS.
- C. Liu, Y. Wu, B. Zhao and B. Zhang, Acc. Chem. Res., 2023, 56, 1872–1883 CrossRef CAS PubMed.
- Y. Zhang and N. Kornienko, Chem Catal., 2022, 2, 499–507 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- J. E. Nycz, Molecules, 2021, 26(23), 7286 CrossRef CAS PubMed.
- N. Heidary, K. H. Ly and N. Kornienko, Nano Lett., 2019, 19, 4817–4826 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00134f |
| This journal is © The Royal Society of Chemistry 2024 |