
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cobalt-based metal–organic framework as a sustainable catalyst for base-free transfer hydrogenation of biomass-derived carbonyl compounds†
Aashish
,
Ruchika
Gupta
and
Rajeev
Gupta
 *
*
Department of Chemistry, University of Delhi, Delhi 110 007, India. E-mail: rgupta@chemistry.du.ac.in; Web: http://www.people.du.ac.in/%7Ergupta/
First published on 23rd September 2024
Abstract
A bifunctional cobalt-based MOF 1, offering both Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites, has been synthesized and characterized. MOF 1 presents a double-chain structure and, therefore, remarkably exposed Lewis acidic–basic and Brønsted acidic sites. MOF 1 acts as a remarkable heterogeneous catalyst for the transfer hydrogenation (TH) of carbonyl compounds using isopropanol as a green hydrogen source without the requirement of any base. MOF 1 exhibits excellent catalytic performance for the TH of assorted aldehydes and ketones, resulting in high yield and excellent selectivity. Notably, several biomass-derived substrates such as furfural, 5-methylfurfural, 5-hydroxymethylfurfural, and levulinic acid were successively converted to their corresponding products in high yield. The substrate scope further encompassed biologically relevant ones such as vanillin, cinnamaldehyde, perillaldehyde, and estrone. Subsequently, both poisoning experiments and temperature-programmed desorption studies were employed to elucidate the role of Lewis acidic–basic and Brønsted acidic sites in this MOF. To further evaluate the role of Brønsted acidic sites in TH, an ester derivative of MOF, 1-Et, was synthesized and utilized which exhibited a poor catalytic performance. Collectively, all experiments confirm a cooperative effect of Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites in TH catalysis. The entire catalytic process encompassing reagents, solvents, and operating conditions, was assessed using the CHEM21 green metrics toolkit to highlight the environmental sustainability of the present catalytic method. The MOF 1 overcomes the limitations of conventional catalysts by excluding the need for a base, offering a broad substrate scope, and achieving high yield with excellent selectivity, thus acting as a more efficient and sustainable catalyst for TH reaction.
Sustainability spotlightTransfer hydrogenation (TH) is significant due to its greener approach, operational simplicity and cost-effectiveness. The earlier-generation catalysts are not only largely based on precious metals but are mostly homogenous and suffer from recyclability challenges. Another drawback is requirement of a base which significantly limits scope for base-sensitive substrates. Such limitations demand for the development of a sustainable heterogeneous catalyst based on an earth-abundant metal. In this work, a bifunctional Co-MOF, offering Lewis acidic–basic and Brønsted acidic sites, acts as a heterogeneous catalyst for base-free TH of carbonyl compounds, including biomass-derived and biologically relevant substrates. This work emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7) and industry, innovation, and infrastructure (SDG 9). |
Introduction
Hydrogenation of carbonyl compounds is a significant and one of the most sought-after transformations in chemistry.1–5 The hydrogenation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group can be carried out using compressed hydrogen gas and metal hydride reagents (e.g., LiAlH4, NaBH4).6–9 However, such conventional methods suffer from several drawbacks. For example, compressed hydrogen gas poses considerable safety challenges while metal hydrides suffer from moisture sensitivity, non-selectivity and waste generation.6,7 As a result, focus has been shifted to explore environmentally benign chemical processes.10–14 One such strategy is transfer hydrogenation (TH) using an organic compound as a hydrogen source.4,15,16 TH has attracted significant interest in the recent years due to its greener approach, operational simplicity, cost-effectiveness and easy access to hydrogen donor sources.17–22 The in situ generated hydrogen in a TH reaction can effectively and efficiently reduce carbonyl substrates, thus, leading to a significantly improved hydrogenation rate.18,22 In this context, not only primary alcohols, such as MeOH and EtOH but also secondary alcohols, such as iPrOH, have been employed as convenient hydrogen donors for the TH of carbonyl compounds.23–26 In particular, isopropanol is a well-established hydrogen donor while also acting as a friendly solvent, thus eliminating the need for a separate solvent.25,27 Moreover, its only by-product, acetone, can be recycled and used, aligning well with the principles of green chemistry.27
O group can be carried out using compressed hydrogen gas and metal hydride reagents (e.g., LiAlH4, NaBH4).6–9 However, such conventional methods suffer from several drawbacks. For example, compressed hydrogen gas poses considerable safety challenges while metal hydrides suffer from moisture sensitivity, non-selectivity and waste generation.6,7 As a result, focus has been shifted to explore environmentally benign chemical processes.10–14 One such strategy is transfer hydrogenation (TH) using an organic compound as a hydrogen source.4,15,16 TH has attracted significant interest in the recent years due to its greener approach, operational simplicity, cost-effectiveness and easy access to hydrogen donor sources.17–22 The in situ generated hydrogen in a TH reaction can effectively and efficiently reduce carbonyl substrates, thus, leading to a significantly improved hydrogenation rate.18,22 In this context, not only primary alcohols, such as MeOH and EtOH but also secondary alcohols, such as iPrOH, have been employed as convenient hydrogen donors for the TH of carbonyl compounds.23–26 In particular, isopropanol is a well-established hydrogen donor while also acting as a friendly solvent, thus eliminating the need for a separate solvent.25,27 Moreover, its only by-product, acetone, can be recycled and used, aligning well with the principles of green chemistry.27
TH of biomass-derived carbonyl compounds, such as furfural, 5-methylfurfural, 5-hydroxymethylfurfural, and levulinic acid, has attracted considerable interest due to their significance in generating value-added organic products and biofuels.28–32 For instance, furfural has been a subject of worldwide research as its functionalization produces valuable organic derivatives.33–35 In this context, the conversion of furfural to furfuryl alcohol stands out as a significant process because the latter is extensively used for the production of synthetic fibers, resins, adhesives, as well as fuels and fuel enhancers.33,36,37 Therefore, TH of biomass-derived carbonyl compounds is a highly desirable strategy to substitute traditional fossil resources.28,31,33
For TH of carbonyl compounds, noble-metal catalysts based on Ru, Rh, Pd, and Pt have been the most successful and, therefore, widely utilized.38–41 However, their limited abundance and high cost hinder their practical employability. Similarly, most of the earlier-generation catalysts are homogenous in nature and suffer from facile separation and limited recyclability challenges.27,41 Thus, there is a critical need to develop non-precious metal-based heterogeneous catalysts that facilitate easy product separation while enhancing their recyclability.11,18,42 To date, a variety of heterogeneous catalysts, such as zeolites, mesoporous silica, metal oxides, and inorganic–organic hybrids have been employed for the TH of carbonyl compounds.3,4,28,33 However, most of these heterogeneous catalysts face challenges related to the leaching of the active species and limited catalytic efficiency.43,44 Hence, there is a crucial demand for the development of stable, reusable, sustainable, and efficient heterogeneous catalysts for the TH of carbonyl compounds.28,33
Metal–organic frameworks (MOFs) are a significant class of porous materials that have garnered extensive attention due to their well-defined structures, high crystallinity, large surface area, tunable pores, and possibilities for both pre- and post-functionalization.45–50 MOFs have been extensively investigated for a wide range of applications, including gas sorption, storage, and separation; CO2 capture and sequestration; vapor and chemical sensing; and heterogeneous catalysis.51–58 Particularly, the large surface area and high porosity of MOFs offer an extensive number of accessible active sites, thus, enhancing their catalytic performance.57,59 Furthermore, MOFs' ability to incorporate assorted metal ions and functionalized organic linkers enables them with tailorable properties, thus substantially upgrading their catalytic applications.60,61
As a result, functional MOFs hold great promise for various catalytic reactions, including oxidation, reduction, N-alkylation, C–X (X = C, N, O, and S) cross-coupling reactions, and TH.36,62–65 A few Zr-based MOFs have been employed for the TH of carbonyl compounds.66,67 For instance, Li et al. have shown the catalytic role of amorphous Zr-based bifunctional nanohybrids for the TH of biomass-derived compounds.68,69 However, most such catalysts perform inferiorly due to the presence of poor basic and Brønsted acidic sites.68 While the role of basic sites is well-established, the Brønsted acidic sites are also critical for accelerating hydrogen transfer and stabilizing key intermediates, leading to an effective TH reaction.70 Therefore, incorporating stronger basic and Brønsted acidic sites adjacent to Lewis acidic sites in MOFs is likely to enhance their catalytic performance.71,72
Considering these challenges, we assumed that integrating both Lewis acidic–basic and Brønsted acidic sites into a MOF structure would be an ideal strategy to enhance its catalytic performance towards TH of carbonyl compounds. This work presents the synthesis of a bifunctional Co-based MOF 1 offering both Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites. MOF 1 functions as an excellent heterogeneous catalyst for the TH of assorted carbonyl compounds using isopropanol as a green hydrogen source without the need for a base. MOF 1 catalyses TH of various biomass-derived as well as biologically relevant substrates. To elucidate the role of Lewis acidic–basic and Brønsted acidic sites in 1, several poisoning experiments and temperature-programmed desorption studies were carried out. To further understand the role of Brønsted acidic sites in TH, an ester derivative of MOF, 1-Et, was synthesized and utilized. Notably, in comparison to original MOF 1, 1-Et displayed lower catalytic efficiency, thus, affirming the importance of Brønsted acidic sites in TH. The greener aspects and sustainability parameters of the entire catalytic process were evaluated by using the CHEM21 green metrics toolkit.
Results and discussion
Synthesis and characterization of MOF 1
Ligand L3 (Fig. S1–S5, ESI†) was synthesized in two steps starting from L1 (Scheme 1). Ligand L3 presents three arylcarboxylic acid groups suited to coordinate with suitable metal ions. L3 on reaction with Co(OAc)2 resulted in the formation of a two-dimensional (2D) Co(II)-MOF 1 (Scheme 1). In FTIR spectrum, 1 showed strong stretches at 1672–1588 cm−1 corresponding to both νCOO and νCO groups (Fig. S6, ESI†). MOF 1 showed broad νO–H stretches at 3380–3170 cm−1 due to the presence of hydroxide moieties and ligated water molecules.73 TG analysis showed that 1 presents a good thermal stability (ca. 350 °C) after losing six ligated water molecules and two lattice DMF molecules (Fig. S7, ESI†). The elemental composition of 1 was validated using both microanalysis data and energy dispersive X-ray (EDX) spectroscopy (Fig. S8, ESI†). The PXRD patterns of as-synthezised 1 matched nicely with those simulated from the single crystal X-ray diffraction analysis, suggesting material's bulk phase purity (Fig. S9, ESI†). To evaluate its chemical stability, MOF 1 was immersed in an aqueous solution of different pH values (5, 7, and 9) and a variety of organic solvents (methanol, acetone, chloroform, and hexane). The PXRD studies nicely demonstrated that both crystallinity and framework integrity of MOF 1 remained intact in different pHs and solvents of varying polarities (Fig. S10, ESI†). These findings support that MOF 1 is chemically robust and can be employed for a variety of applications. The Brunauer–Emmett–Teller (BET) studies exhibited avg. pores of ca. 2.47 nm dimensions and a surface area of 29 m2 g−1 (Fig. S11, ESI†).74
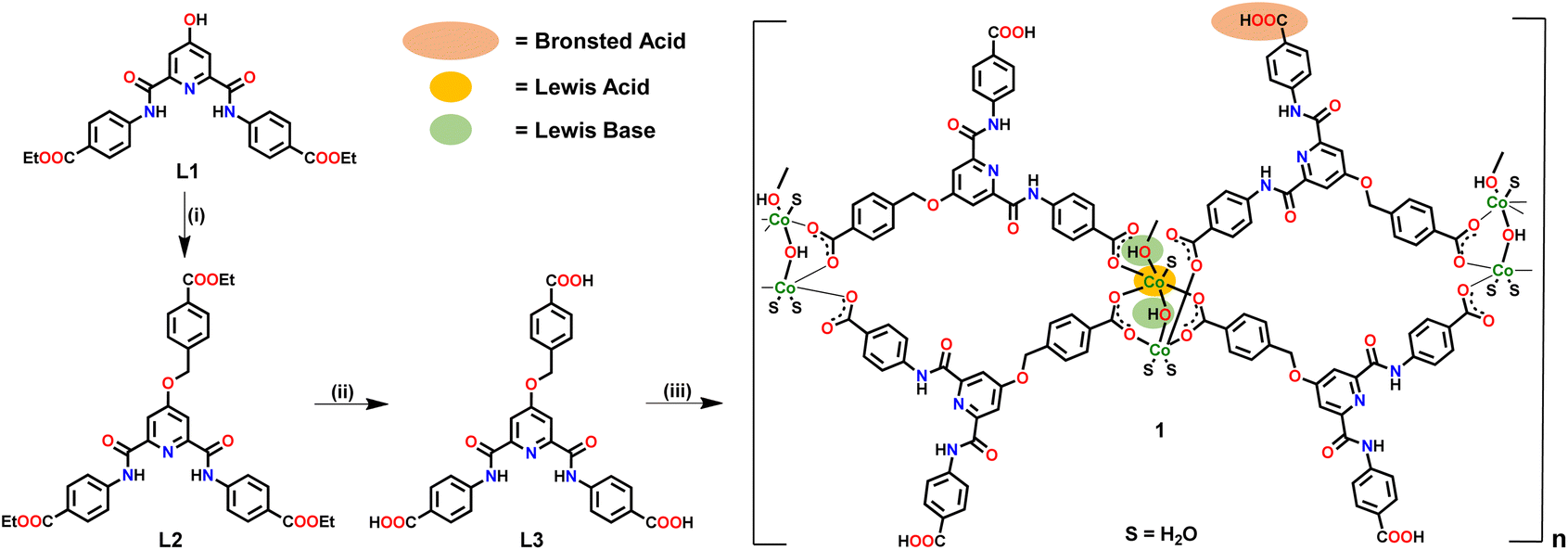 | ||
| Scheme 1 Preparative route for the synthesis of Co-MOF 1. Reaction conditions: (i) ethyl 4-(bromomethyl)benzoate, K2CO3, DMF; (ii) NaOH, THF–H2O; (iii) Co(OAc)2, DMF–H2O. | ||
MOF 1 was crystallographically characterized to gain insight into its molecular structure (Tables S1–S3, ESI†). MOF 1 crystallized in a monoclinic cell with C2/c space group. The asymmetric unit comprises two L3 ligands, two Co2+ ions, two unique bridging hydroxide groups, three coordinated water and two lattice DMF molecules (Fig. S12, ESI†). A SBU is consisted of two Co(II) ions bridged by a μ-OH group while coordinated by two bridging bidentate carboxylate groups, two monodentate carboxylate groups, and three water molecules, [Co2(μ-OH)(–COO)4(H2O)3] (Fig. 1a). In this SBU, the Co⋯Co separation is 3.47 Å. Two such SBUs are further connected via another μ-OH group to generate an overall SBU, [{[Co2(μ-OH)(–COO)4(H2O)3]}2(μ-OH)], with Co⋯Co separation of 3.97 Å. Such Co4-based SBUs are connected together by L3 ligands giving rise to a double chain structure when viewed along b-axis (Fig. 1b). One of the DMF molecules participated in H-bonding interactions within the N3 pincer cavity.75 It is important to note that the presence of Co(II) ions, μ-OH sites and free –COOH groups endows MOF 1 with a significant number of Lewis acidic, Lewis basic and Brønsted acidic sites, respectively. It is anticipated that such sites may play crucial roles in facilitating substrate activation as well as proton abstraction in catalysis (vide infra).71
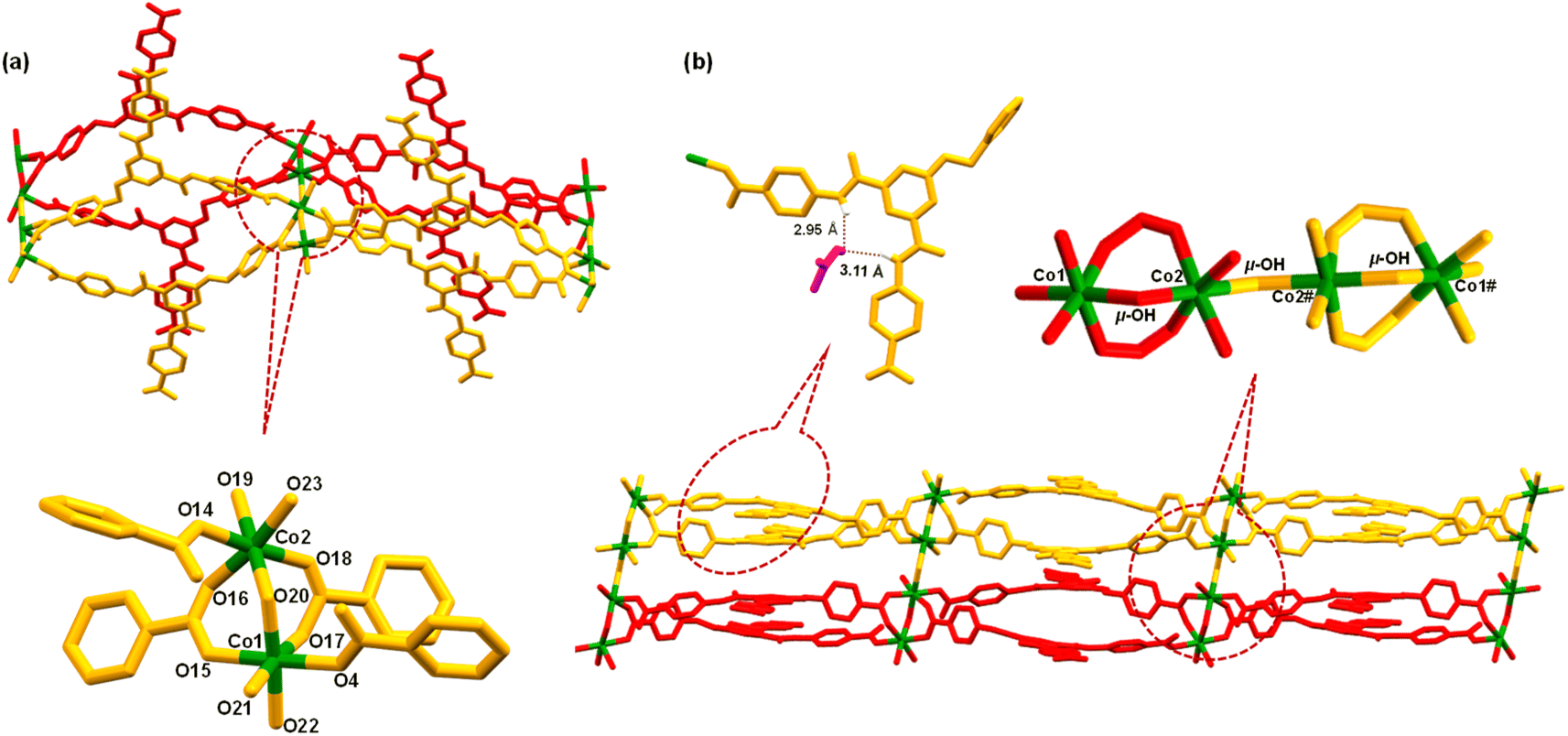 | ||
| Fig. 1 (a) Crystal structure of MOF 1, including coordination environment around the Co2+ centers. (b) A view of extended network of MOF 1, when viewed along b-axis; lattice DMF molecules have been omitted for clarity. Part of a coordinating ligand, one hydrogen bonded DMF molecule within the N3 pincer cavity, and a SBU have been zoomed. | ||
Transfer hydrogenation of carbonyl compounds
The catalytic performance of MOF 1 was investigated towards TH taking both benzaldehyde and furfural as the model substrates (Table 1). As expected, TH, in the absence of MOF 1, did not proceed (entry 1). Similarly, a few Co(II) salts, such as Co(OAc)2 and CoCl2, were largely ineffective (entries 2 and 3). To optimize the reaction conditions, several solvents such as methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, and tert-butanol were utilized as the hydrogen donors (entries 4–9). While most of the alcohols showed moderate catalytic performance, nearly quantitative product formation was observed in isopropanol (iPrOH) (entry 4). As anticipated, no benzyl alcohol was observed when tert-butanol was used as a hydrogen source (entry 9). The best reaction temperature was found to be 60 °C as comparatively lower yield was noted at both lower (30–50 °C) and higher (70 and 80 °C) temperatures (data not shown). To rule out the involvement of [Co]-nanoparticles in catalysis, both mercury and phenanthroline poisoning experiments were performed (entries 10 and 11). The catalytic performance of MOF 1 remained unaffected in their presence, thus, eliminating the involvement of [Co]-nanoparticles as the actual catalyst in TH.76,77|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Additive | Isolated yield (%) | |
| R = Phenyl | R = Furyl | ||||
| a Reaction conditions: aldehyde (1.00 mmol), MOF 1 (0.1 mol%), solvent (2 mL), time (2 h). N.R. = No reaction. | |||||
| 1 | — | iPrOH | — | N.R. | N.R. |
| 2 | Co(OAc)2 | iPrOH | 5 | N.R. | |
| 3 | CoCl2 | iPrOH | 7 | N.R. | |
| 4 | Co-MOF (1) | i PrOH | >99 | >96 | |
| 5 | Co-MOF 1 | MeOH | 40 | 32 | |
| 6 | Co-MOF 1 | EtOH | 55 | 40 | |
| 7 | Co-MOF 1 | 1-PrOH | 45 | 45 | |
| 8 | Co-MOF 1 | 1-BuOH | 25 | 15 | |
| 9 | Co-MOF 1 | t BuOH | N.R. | N.R. | |
| 10 | Co-MOF 1 | iPrOH | Hg | 97 | 96 |
| 11 | Co-MOF 1 | iPrOH | 1,10-Phenthroline | 96 | 94 |
| 12 | Co-MOF 1 | iPrOH | Acetic acid | 25 | 27 |
| 13 | Co-MOF 1 | iPrOH | 4-Methoxy pyridine | 70 | 73 |
| 14 | Co-MOF 1 | iPrOH | 2,6-Lutidine | 82 | 85 |
| 15 | 1-Et | iPrOH | — | 75 | 80 |
| 16 | L3 + Co(OAc)2 | iPrOH | — | 15 | 5 |
To elucidate the role of acidic–basic sites in MOF 1 in the TH, acetic acid, 4-methoxy pyridine, and 2,6-lutidine were employed as the poisoning additives (entries 12–14).67,78 When acetic acid was added to the reaction, a drastic decrease in the product formation was noted. Such a fact suggests that the basic sites get protonated with acetic acid and thus play a significant role in TH catalysis. The addition of 4-methoxy pyridine led to a slight decrease in the formation of both benzyl alcohol and furfuryl alcohol, implying that Lewis acidic sites also play a minor role in TH. To further examine the role of acidic sites (Lewis and Brønsted acids) on TH, 2,6-lutidine was introduced to selectively inhibit the Brønsted acidic sites. A slight decrease in the alcohol formation indicates that Brønsted acidic sites also contribute to some extent to TH catalysis.
To evaluate the role of free carboxylic acid groups on TH catalysis, an ester derivative of Co-MOF, 1-Et was synthesized by treating a crystalline sample of 1 with EtOH in the presence of a catalytic amount of conc. H2SO4. FTIR spectrum of 1-Et displayed ester stretches at 1703 cm−1, while PXRD patterns only exhibited minor changes to that of pristine 1 (Fig. S13, ESI†). Importantly, compared to MOF 1 (>99%), 1-Et exhibited a poor catalytic efficiency (75%, 80%), thus asserting a critical role of free –COOH groups on TH catalysis (entry 15; Table 1).
The temperature-programmed desorption (TPD) studies were performed using NH3 and CO2 as probe molecules to quantitatively evaluate the Lewis acidic–basic and Brønsted acidic sites in 1 (Fig. S14, ESI†).79,80 The NH3-TPD profile reveals two distinct desorption peaks at 150 and 220 °C, indicating the presence of abundant acidic sites within this MOF. The primary peak at 150 °C is associated with the interaction of NH3 to that of metal nodes, while the shoulder at 220 °C suggests its interaction with Brønsted acidic groups (–COOH). On the other hand, the CO2-TPD profile exhibits a desorption peak at 250 °C, corresponding to the interaction of CO2 with –OH− groups, confirming the presence of basic sites. The higher desorption temperature for CO2 compared to NH3 highlights MOF's strong basicity. These findings not only validate the presence of Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites but also underscore their crucial role in the enhanced catalytic performance of MOF 1.79,80
Finally, to support the importance of well-defined Lewis acidic–basic catalytic sites offered by MOF 1, a mixture of ligand L3 and Co(OAc)2 was employed as a catalyst. Notably, only a negligible amount of product was formed (15%; entry 16). Such a fact convincingly illustrates the significance of well-organized catalytic sites in a crystalline sample of MOF 1. Collectively, a synergistic interplay of Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites is operational in controlling the outcome of heterogeneous TH catalysis.66,71
Subsequently, MOF 1 was employed as a heterogeneous catalyst to explore the substrate scope for the TH of assorted carbonyl compounds. A variety of aldehydes and ketones, bearing both electron-withdrawing and electron-donating groups, were efficiently converted to their respective alcohols as exclusive products (Schemes 2 and 3). For instance, benzaldehyde (1) and its derivatives were converted to their corresponding benzyl alcohols in nearly quantitative yields. However, it was noted that benzaldehydes containing electron-withdrawing groups, such as 4-nitro (2), 3-nitro (3), 2-nitro (4), 4-bromo (5), and 3-bromo (6) underwent faster reduction as compared to the benzaldehydes having electron-donating groups such as 4-methyl (7), 3-methyl (8), 2-methyl (9), 4-methoxy (10), 4-amino (11), 4-hydroxy (12) and 2,4-dimethyl (13). Further, 1-naphthaldehyde (14), 9-anthraldehyde (15) and piperonyl aldehyde (16) were also reduced to their corresponding alcohols in very high yield.
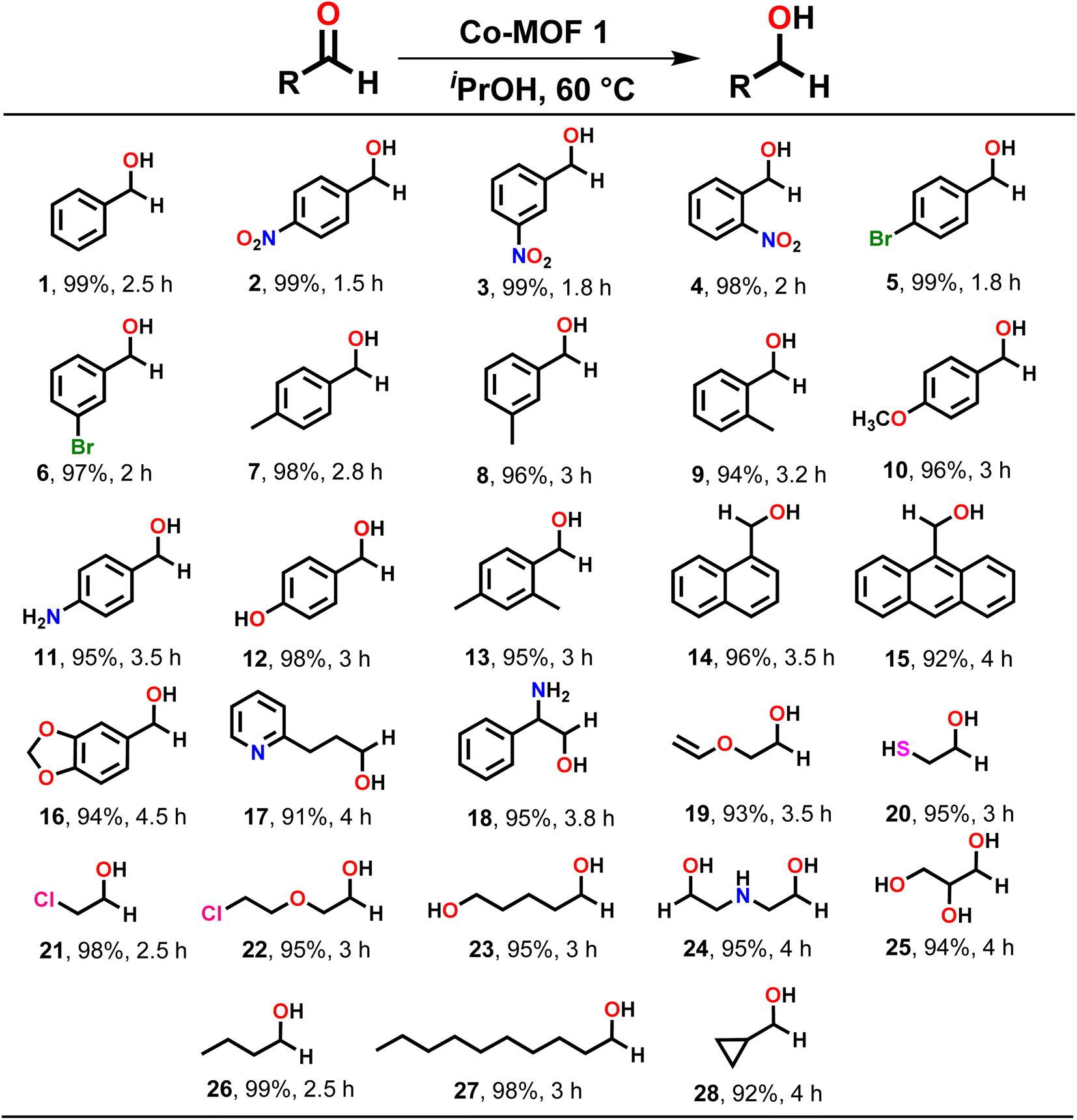 | ||
| Scheme 2 Substrate scope for the TH of assorted aldehydes. Reaction conditions: aldehyde (1.00 mmol), MOF 1 (0.1 mol%), iPrOH (2 mL), temp. (60 °C). | ||
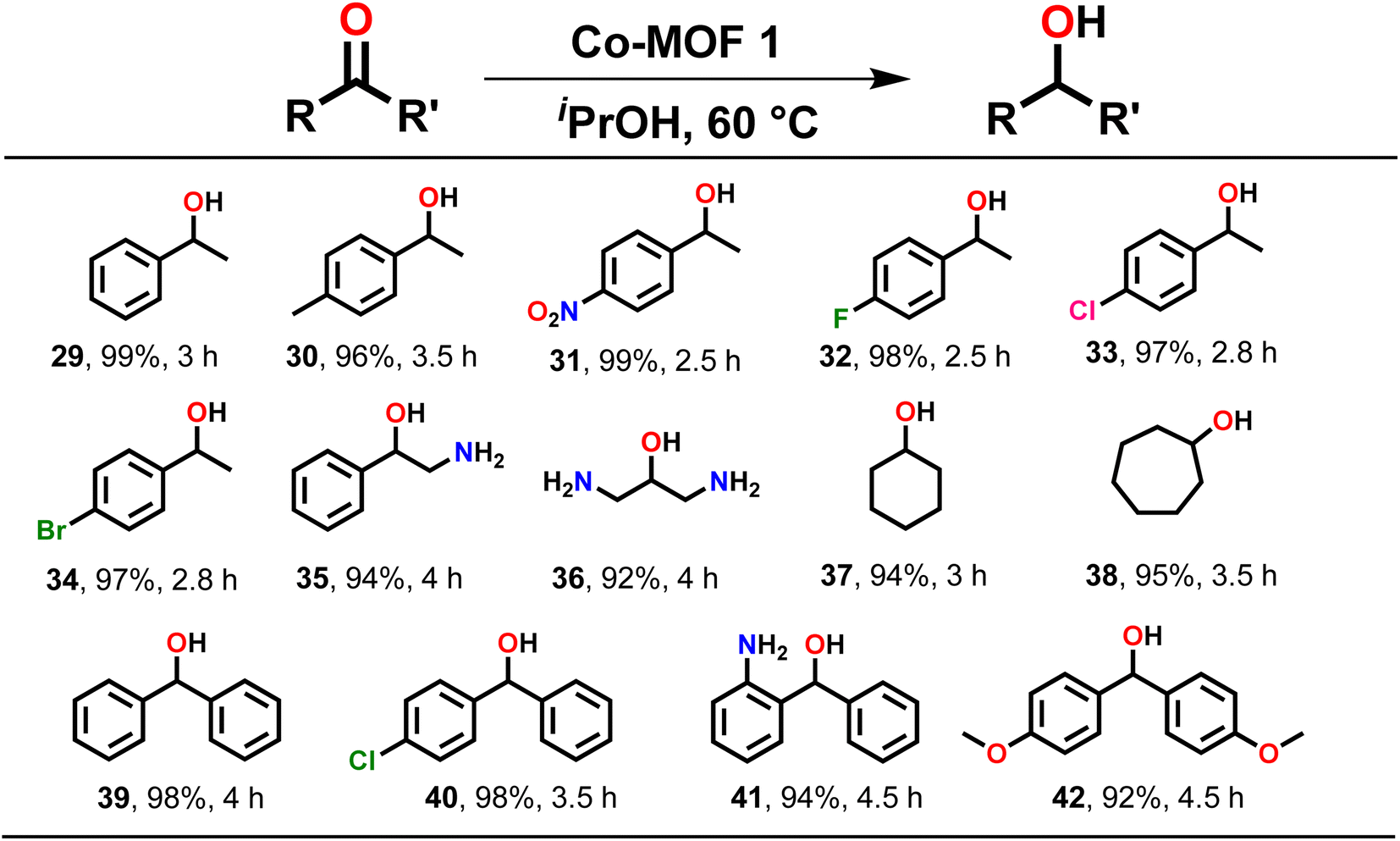 | ||
| Scheme 3 Substrate scope for the TH of assorted ketones. Reaction conditions: ketone (1.00 mmol), MOF 1 (0.1 mol%), iPrOH (2 mL), temp. (60 °C). | ||
Subsequently, assorted aliphatic substrates such as 3-(pyridin-2-yl)propanal (17), 2-amino-2-phenyl-acetaldehyde (18), 2-(vinyloxy)acetaldehyde (19), 2-mercapto-acetaldehyde (20), 2-chloro-acetaldehyde (21), 2-(2-chloroethoxy)acetaldehyde (22), 5-hydroxypentanal (23), 2,2′-azanediyldiacetaldehyde (24), 2,3-dihydroxy-propanal (25), butanal (26), and decanal (27) were efficiently converted to their respective alcohols in high yield. Furthermore, TH of a cyclic aldehyde such as cyclopropane aldehyde (28) also provided respective alcohol in excellent yield.
After TH of assorted aldehydes, substrate scope was extended to various ketones, which are generally more challenging to reduce (Scheme 3). Acetophenone was quantitatively reduced to 1-phenylethanol (29). Similarly, acetophenone derivatives, such as 4-methylacetophenone (30), 4-nitroacetophenone (31), 4-fluoroacetophenone (32), 4-chloroacetophenone (33), 4-bromoacetophenone (34) and 2-aminoacetophenone (35) were reduced to their respective products in generally high yield. Notably, the TH of substrates having electron-withdrawing groups required less time as compared to the ones having electron-donating groups. An aliphatic substrate, 1,3-diaminoacetone (36), also produced the respective secondary alcohol in high yield. Subsequently, cyclic ketones such as cyclohexanone (37) and cycloheptanone (38) were conveniently converted to their respective secondary alcohols in high yield. Moreover, benzophenone (39) and its derivatives such as 4-chlorobenzophenone (40), 2-aminobenzophenone (41), and 4,4′-dimethoxy-benzophenone (42) also provided the corresponding secondary alcohols in excellent yield (Scheme 3). It is important to note that ketones needed a longer reaction time for reduction when compared to aldehydes.3
To further elaborate substrate scope, TH catalysis was extended to biomass-derived carbonyl compounds such as furfural, 5-methylfurfural, 5-hydroxymethylfurfural (HMF) and levulinic acid (Scheme 4). Importantly, TH of furfural led to a nearly complete conversion to furfuryl alcohol (43). Similarly, 5-methylfurfural was reduced to 5-methylfurfuryl alcohol (44) in high yield. Likewise, 5-hydroxymethylfurfural (45) and levulinic acid (46) were also converted to their respective products in good yield. Subsequently, a few biologically relevant substrates, such as vanillin (47), cinnamaldehyde (48), perillaldehyde (49) and estrone (50) were employed (Scheme 4). Notably, such biologically relevant substrates presented an intriguing challenge for attempting TH as all of them featured a carbonyl group alongside other functional groups. Gratifyingly, MOF 1, as a heterogeneous catalyst, effectively converted these substrates to their respective alcohols in excellent yield. However, both perillaldehyde and estrone required a higher catalyst loading of 0.5 mol% to drive the TH catalysis effectively. It is noteworthy that MOF 1 facilitated TH of all substrates without the requirement of a base (vide infra).41
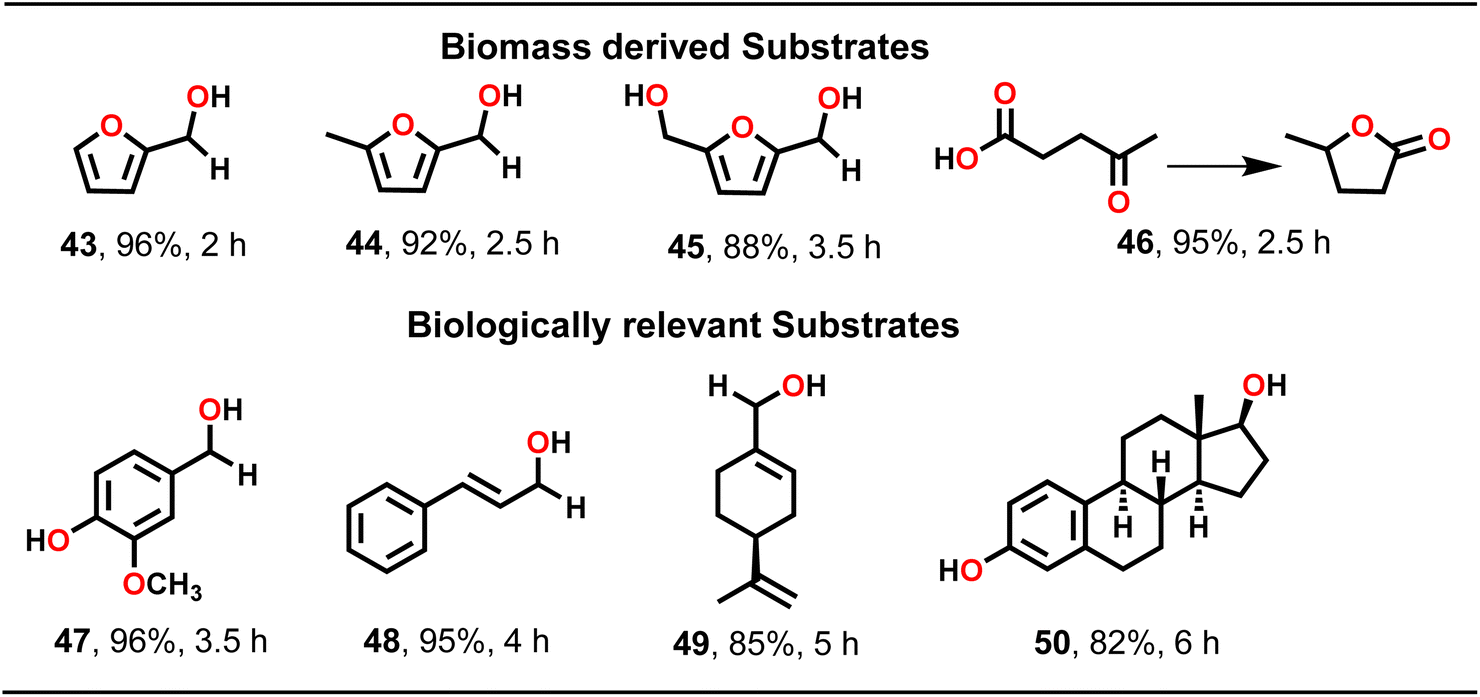 | ||
| Scheme 4 Substrate scope for the TH of a few biomass-derived and biologically relevant substrates. Reaction conditions: substrate (1.00 mmol), MOF 1 (0.1 mol%), iPrOH (2 mL), temp. (60 °C). | ||
To study the kinetics of the TH reaction, time-dependent NMR spectra were recorded, taking furfural as a model substrate.81,821H NMR spectral analysis displayed that TH of furfural proceeded efficiently in the presence of MOF 1 (Fig. 2a). New signals were identified at 4.60, 6.28, 6.34, and 7.40 ppm that are consistent with the formation of furfuryl alcohol replacing the original peaks of furfural at 6.57, 7.22, 7.66 and 9.61 ppm. The same reaction was utilized to monitor the formation of furfuryl alcohol, which exhibited a first-order kinetics with the associated rate constant of 0.0194 min−1 at 60 °C. Subsequently, kinetic experiments were performed at different temperatures (303, 313, 323, and 333 K) to determine the activation energy (Ea) of the TH reaction (Fig. 2b).83 The Ea, calculated from the Arrhenius equation, was found to be 15.75 kJ mol−1 (Fig. 2c).
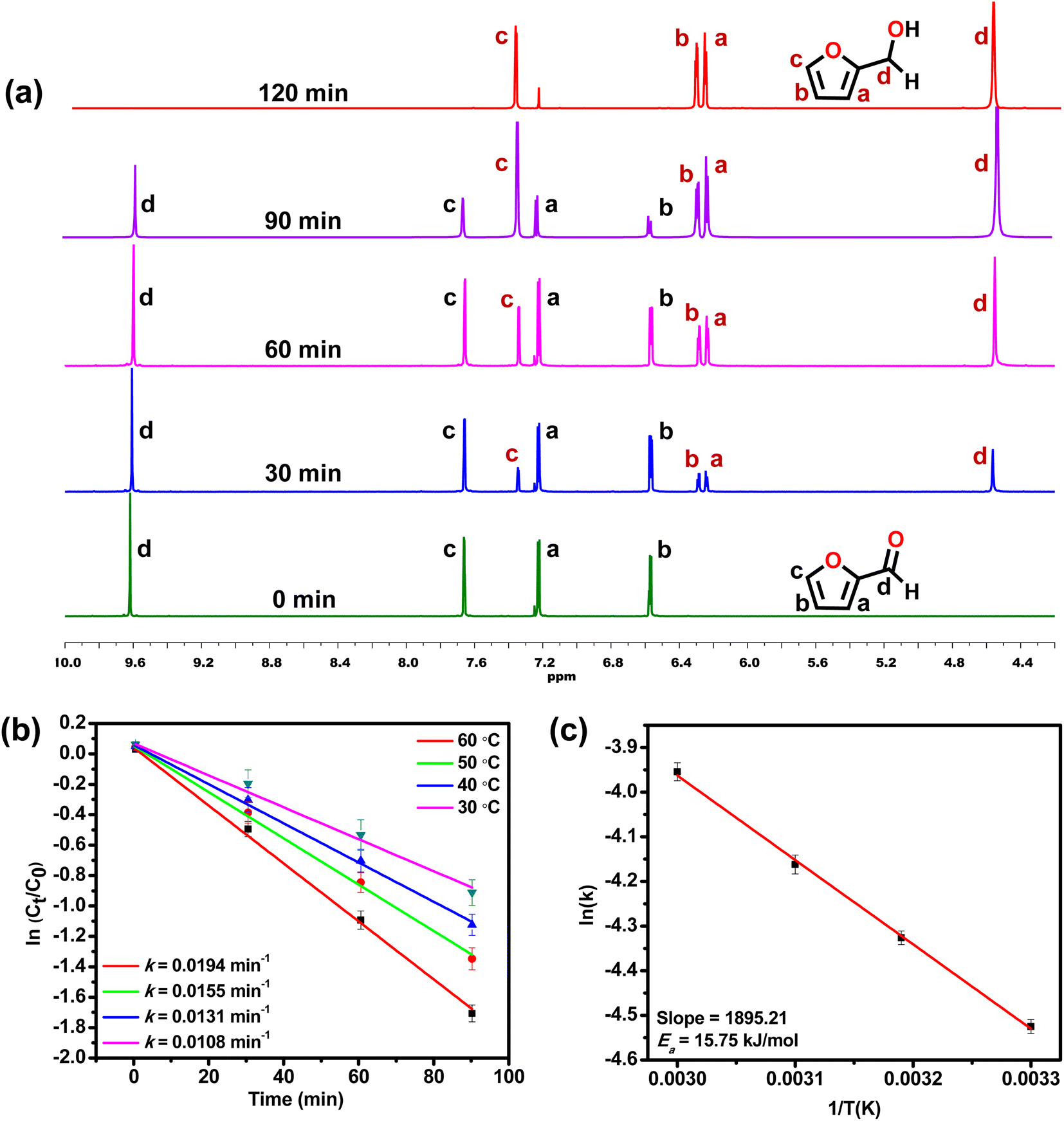 | ||
| Fig. 2 (a) Progress of the TH of furfural monitored by time-dependent 1H NMR spectroscopy. (b) Plots of ln(Ct/Co) versus time presenting the first-order kinetics at four different temperatures, and (c) the corresponding Arrhenius plot for the TH of furfural in the presence of MOF 1 as a catalyst. | ||
Mechanistic insights
TH of carbonyl compounds typically follows two distinct mechanistic pathways: metal-mediated hydrogenation and Meerwein–Ponndorf–Verley (MPV) hydrogenation pathway.33,84,85 In first case, a [M–H] species serves as an active catalyst followed by the transfer of hydride to the carbonyl carbon atom of a substrate.41 In latter pathway, β-H of isopropanol is directly transferred to the carbonyl carbon of a substrate through the formation of a six-membered transition state.84,86 Notably, in MPV pathway, generation of a [M–H] species is not necessary. Based on literature reports, control and poisoning experiments,66,71,84 a plausible reaction mechanism is proposed for the TH of carbonyl substrates, taking furfural as a representative example, catalyzed by Co-MOF 1 (Scheme 5).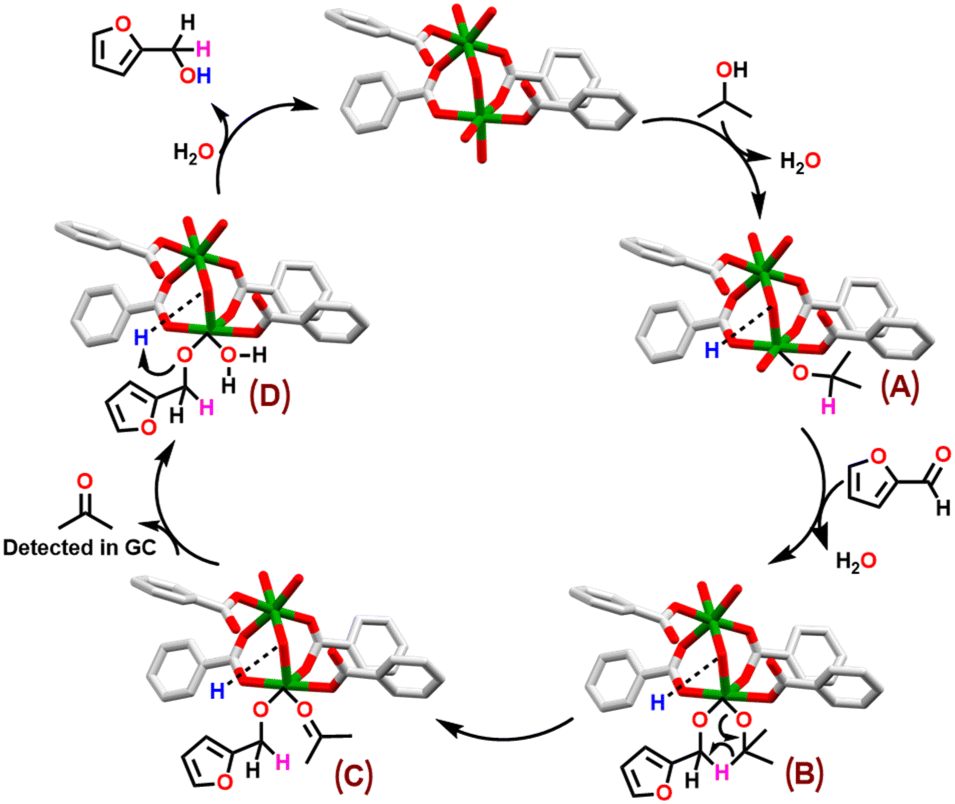 | ||
| Scheme 5 Proposed reaction mechanism for the TH of a carbonyl substrate, taking furfural as a representative case, catalyzed by MOF 1. | ||
The MPV mechanistic cycle starts with the dissociative coordination of a molecule of isopropanol resulting in the formation of species A. Herein, while basic bridging hydroxide group (Lewis basic site) abstracts proton of the isopropanol, the resulting isopropoxide anion is coordinated to the cobalt ion (Lewis acidic site). The second step involves the insertion of furfural to the active site of MOF 1, leading to the formation of a six-membered transition state B. The next step involves direct transfer of hydrogen to the carbonyl carbon atom through β-H elimination, affording species C. This is succeeded by the formation of species D, while releasing a molecule of acetone. It is important to mention that acetone was detected in both GC and proton NMR spectra of the reaction mixture (Fig. S15, ESI†). In the final step, furfuryl alcohol is released, as the exclusive product, with hydrogen atom being transferred from the protonated basic site (–O(H)–H+). This is followed by the coordination of a water molecule to the Co(II) ion, thus, regenerating the catalyst.
Stability and recyclability of MOF 1
For a promising heterogeneous catalyst, both stability and reusability are considered to be the significant factors.87,88 To evaluate a true heterogeneous nature of MOF 1 and its stability in catalysis, hot filtration test was employed taking TH of furfural as a model reaction (Fig. 3a). The said reaction was allowed to continue in the presence of 1 for 75 min, and thereafter MOF 1 was removed by centrifugation. Removal of 1 led to a complete stop of the catalytic reaction. Remarkably, re-addition of the same catalyst revived the catalytic activity, leading to the formation of product in a quantitative yield. Such a filtration test unveiled a true heterogeneous nature of MOF 1. Upon completion of the said catalytic reaction, MOF 1 was separated via centrifugation, washed with water, dried and subsequently employed for five consecutive cycles. As shown in Fig. 3b, only a marginal drop in its catalytic activity was observed during the five consecutive cycles. In fact, furfuryl alcohol formation only decreased from 99% to 95% in such five reusability cycles. We believe that a slight drop in the catalytic performance is due to the potential loss of MOF 1 during the isolation and re-addition steps. Importantly, FTIR spectral, PXRD and SEM studies of the recovered MOF 1, after five catalytic runs, revealed that both structural integrity and morphology of MOF remain preserved during the catalytic cycles (Fig. S16–S18, ESI†). These findings illustrate a robust nature of MOF 1 as a true heterogeneous catalyst.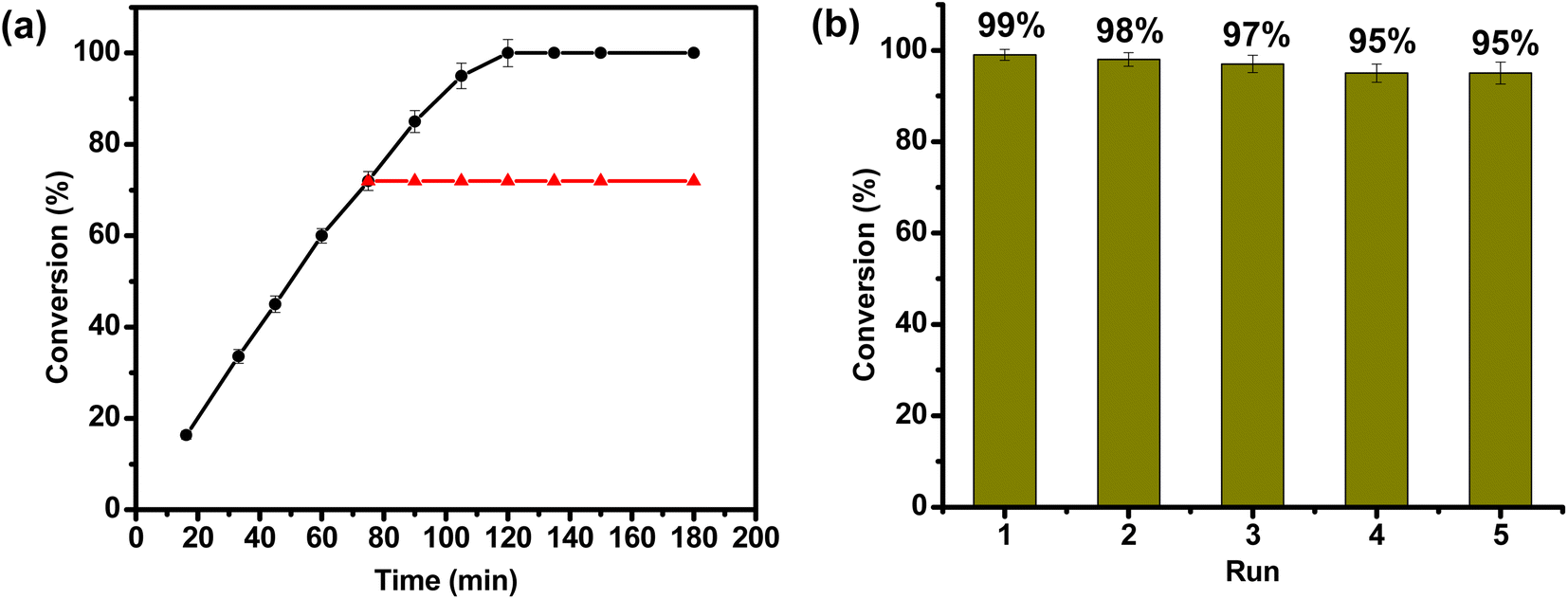 | ||
| Fig. 3 (a) Hot filtration test for the TH of furfural in the presence of MOF 1 as a catalyst. MOF 1 was filtered off at 75 min (red triangles) which led to the cease of catalysis; re-addition of MOF 1 led to the formation of furfuryl alcohol in a quantitative yield (black circles). (b) A bar graph displaying recyclability of MOF 1 for five consecutive cycles. | ||
Green chemistry metrics
The environmental sustainability of one-step TH was assessed by using the publicly available CHEM21 green metrics toolkit developed by Clark and co-workers (Scheme 6 and Tables S4 and S5, ESI†).89,90 This toolkit encompasses 12 principles of green chemistry by offering a quantitative method to evaluate, measure, and compare the sustainability of a chemical reaction. The said toolkit includes four levels of assessment with each one with increasing complexity: zero pass, first pass, second pass and third pass. These levels are used to check the green credentials of a chemical reaction.89 In CHEM21 green metrics toolkit, an initial evaluation of various methods is carried out using the zero pass. Herein, the assessment focuses on the significant use of hazardous solvents and chemicals, taking into consideration health and safety aspects.91,92 Subsequently, efficacy of the reaction is examined on the basis of various factors like conversion, yield, selectivity, atom economy (AE), and reaction mass efficiency (RME). Promising methods identified in zero pass are further examined using the first pass where process mass intensity (PMI), hazards to health, safety and ecology are investigated in an elaborated manner.92 The second and third passes are relevant for industrial-scale processes and are beyond the scope of the present laboratory-scale research.41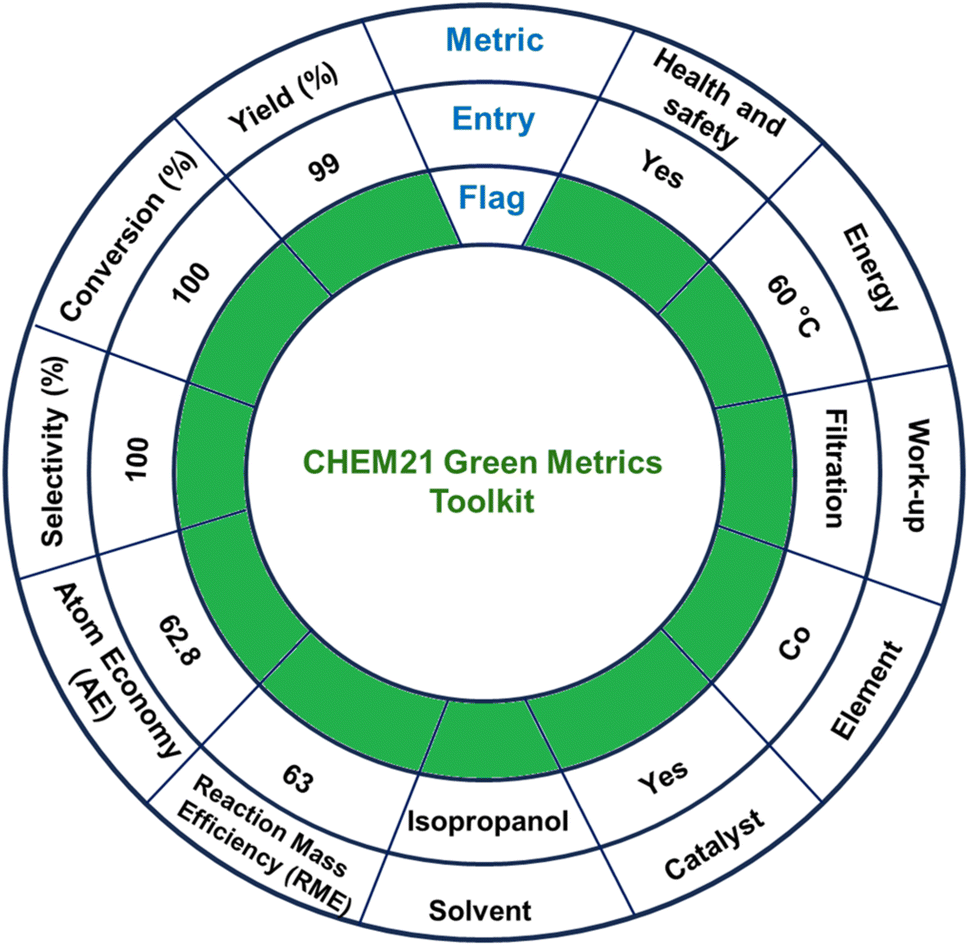 | ||
| Scheme 6 CHEM21 green metrics toolkit calculations for TH of furfural catalyzed by MOF 1. | ||
A gram–scale reaction was carried out using furfural (1.00 g, 0.01 mol) as a model substrate, to assess the catalytic performance, sustainability, and robustness of MOF 1 (0.1 mol%, 0.031 g) as a catalyst. The reaction was carried out at 60 °C while a complete conversion was attained in 2 h, yielding furfuryl alcohol (0.99 g, 0.01 mol), as the sole product, and acetone as the only by-product. The CHEM21 green metrics toolkit was employed to investigate the entire catalytic process. This method provided green flags for yield, conversion, selectivity, AE, and RME.89 The use of isopropanol as an environmentally friendly solvent further earned a green flag. The utilization of cobalt as a catalytic metal, which is earth-abundant, inexpensive, environmentally benign and nontoxic, received a green flag. The work-up procedure was straightforward and thus further earned a green flag. As TH of furfural was carried out at 60 °C, the reaction temperature also received a green flag. Furthermore, the entire catalytic process showed good health and safety concerns, with acetone being the only by-product, thus further earning a green flag. Collectively, the aforementioned catalytic results effectively support CHEM21 green metrics toolkit as a reliable method and provide both qualitative and quantitative information for the TH of carbonyl compounds.89,92
Conclusions
In conclusion, a bifunctional Co-based MOF 1 providing both Lewis acidic–basic (Co and –OH−) and Brønsted acidic (–COOH) sites was synthesized. MOF 1 offered a double-chain structure and, therefore, remarkably exposed Lewis acidic–basic and Brønsted acidic sites. MOF 1 was utilized as a heterogeneous catalyst for the TH of assorted carbonyl compounds using isopropanol as a green hydrogen donor. MOF 1 illustrated a remarkable catalytic efficiency and reduced a large number of aldehydes and ketones to their respective alcohols in excellent yield. Several biomass-derived as well as biologically relevant substrates further illustrated remarkable TH results of MOF 1. Various poisoning experiments and temperature-programmed desorption studies substantiated the significant roles of both Lewis acidic–basic and Brønsted acidic sites in catalysis. To nail, an ester derivative of MOF 1, 1-Et, was utilized to elucidate the role of Brønsted acidic sites in catalysis. These experiments asserted a synergistic role of Lewis acidic–basic and Brønsted acidic sites offered by MOF 1 in efficiently promoting TH without the use of any external base. The CHEM21 green metrics toolkit provided ‘green flags’ for the entire catalytic system. This work not only overcomes the limitations of conventional catalysts but also opens up new avenues for the development of earth-abundant MOFs as sustainable yet effective heterogeneous catalysts for the conversion of biomass-derived compounds to biofuels and value-added chemicals.Experimental section
Materials
All chemicals and reagents were procured from commercial companies and were used without further purification. The solvents were purified utilizing the standard literature methods.93 The precursor L1 was synthesized according to our earlier report.50Syntheses
O). Elemental analysis for C35H33N3O9: C, 65.72; H, 5.20; N, 6.57. Found: C, 65.79; H, 5.29; N, 6.51.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) and treated with 5 equiv. of NaOH (0.15 g, 3.90 mmol). This reaction mixture was stirred for 12 h at room temperature. To this reaction mixture, an aqueous solution of 4 N HCl was added until pH reached to 3.5–4.0. The resulting solution was vacuum evaporated to remove THF which led to the precipitation of a product which was filtered, washed with water, and air dried. Yield: 0.36 g (85%). 1H NMR spectrum (400 MHz, DMSO-d6) δ 12.96 (s, 2H), 11.22 (s, 2H), 8.37–7.80 (m, 12H), 7.66 (d, J = 6.8 Hz, 2H), 5.55 (s, 2H). 13C NMR spectrum (100 MHz, DMSO-d6) δ 167.41, 162.17, 151.17, 142.47, 141.01, 130.81, 130.08, 127.99, 126.78, 120.62, 112.49, 70.06. FTIR spectrum (selected peaks, ν/cm−1): 3187 (N–H), 2962 (C–H), 1680 (COOH), 1589 (CO). Elemental analysis for C29H21N3O9: C, 62.70; H, 3.81; N, 7.56. Found: C, 62.62; H, 3.76; N, 7.55.
O). Elemental analysis calculated for C64H58N8O23Co2: C, 53.94; H, 4.10; N, 7.86; Co, 8.27%. Found: C, 53.89; H, 4.06; N, 7.82; Co, 8.36%. The Co content was measured by inductively coupled plasma mass spectrometry (ICP-MS).
:1, v/v) and treated with 5 equiv. of NaOH (0.15 g, 3.90 mmol). This reaction mixture was stirred for 12 h at room temperature. To this reaction mixture, an aqueous solution of 4 N HCl was added until pH reached to 3.5–4.0. The resulting solution was vacuum evaporated to remove THF which led to the precipitation of a product which was filtered, washed with water, and air dried. Yield: 0.36 g (85%). 1H NMR spectrum (400 MHz, DMSO-d6) δ 12.96 (s, 2H), 11.22 (s, 2H), 8.37–7.80 (m, 12H), 7.66 (d, J = 6.8 Hz, 2H), 5.55 (s, 2H). 13C NMR spectrum (100 MHz, DMSO-d6) δ 167.41, 162.17, 151.17, 142.47, 141.01, 130.81, 130.08, 127.99, 126.78, 120.62, 112.49, 70.06. FTIR spectrum (selected peaks, ν/cm−1): 3187 (N–H), 2962 (C–H), 1680 (COOH), 1589 (CO). Elemental analysis for C29H21N3O9: C, 62.70; H, 3.81; N, 7.56. Found: C, 62.62; H, 3.76; N, 7.55.
O). Elemental analysis calculated for C64H58N8O23Co2: C, 53.94; H, 4.10; N, 7.86; Co, 8.27%. Found: C, 53.89; H, 4.06; N, 7.82; Co, 8.36%. The Co content was measured by inductively coupled plasma mass spectrometry (ICP-MS).
1-Et. MOF 1 (100.00 mg, 0.07 mmol) was taken in 20 mL EtOH followed by the addition of a few drops of conc. H2SO4. The reaction mixture was stirred for 12 h at room temperature. After completion of the reaction, the product was filtered, thoroughly washed with water, followed by diethyl ether, and dried under the vacuum. Yield: 98 mg (95%). FTIR spectrum (selected peaks, ν/cm−1): 3326 (N–H), 2977 (C–H), 1662, 1703 (COOCH2CH3), 1672, 1588 (CO). Elemental analysis calculated for C68H66N8O23Co2: C, 55.14; H, 4.49; N, 7.57. Found: C, 55.09; H, 4.48; N, 7.51.
Typical procedure for TH
A 20 mL round bottom flask was charged with catalyst (0.1 mol %), substrate (1.0 mmol), and isopropanol (2 mL). The reaction mixture was stirred for 2 h at 60 °C. The progress of the reaction was monitored with the thin layer chromatography (TLC). Upon completion, the reaction mixture was subjected to centrifugation, which led to the separation of the catalyst, which was isolated. The recovered catalyst was characterized by FTIR spectral as well as PXRD studies. The organic product(s) were extracted using 10 mL EtOAc. The EtOAc solution was then passed through a Celite plug to eliminate any remaining inorganic impurities. The resulting solution was concentrated to obtain a pure TH product, which was subsequently characterized using both 1H and 13C NMR spectroscopic studies. Fig. S19 and S118† in ESI contain 1H and 13C NMR spectra of all isolated TH products.1H and 13C NMR spectral data of TH products:
(1) Benzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.35–7.23 (m, 2H), 4.58 (s, 2H), 2.64 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 140.92, 128.56, 127.61, 127.04, 65.13.
(2) 4-Nitrobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.21 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.9 Hz, 2H), 4.83 (s, 2H), 2.23 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 148.36, 147.19, 127.01, 123.70, 63.92.
(3) 3-Nitrobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.17 (s, 1H), 8.06 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 4.77 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 148.21, 143.01, 132.74, 129.42, 122.35, 121.36, 63.67.
(4) 2-Nitrobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.07 (d, J = 8.2 Hz, 1H), 7.74 (d, J = 7.3 Hz, 1H), 7.66 (t, J = 7.0 Hz, 1H), 7.46 (t, J = 7.7 Hz, 1H), 4.96 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 147.49, 136.88, 134.14, 129.73, 128.43, 124.96, 62.35.
(5) 4-Bromobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.47 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 4.62 (s, 2H), 2.05 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 139.76, 131.63, 128.60, 121.45, 64.53.
(6) 3-Bromobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.46 (s, 1H), 7.38 (d, J = 5.1 Hz, 1H), 7.23–7.14 (m, 2H), 4.56 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 143.08, 130.60, 130.13, 129.87, 125.37, 122.61, 64.24.
(7) 4-Methylbenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.21 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 7.9 Hz, 2H), 4.57 (s, 2H), 2.33 (s, 3H), 2.21 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 137.96, 137.34, 129.24, 127.15, 65.12, 21.18.
(8) 3-Methylbenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.21 (t, J = 7.5 Hz, 1H), 7.10 (dd, J = 16.4, 7.3 Hz, 3H), 4.56 (s, 2H), 2.53 (s, 1H), 2.33 (s, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 140.90, 138.21, 128.47, 128.35, 127.81, 124.09, 65.17, 21.42.
(9) 2-Methylbenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.34–7.26 (m, 1H), 7.21–7.12 (m, 3H), 4.61 (s, 2H), 2.31 (s, 3H), 2.15 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 138.73, 136.10, 127.76, 127.54, 126.06, 63.37, 18.75.
(10) 4-Methoxybenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.23 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 4.52 (s, 2H), 3.76 (s, 3H), 2.62 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 159.09, 133.21, 128.65, 113.90, 64.75, 55.29.
(11) 4-Aminobenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.07 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 8.0 Hz, 2H), 4.44 (s, 2H), 4.09 (s, 1H), 3.64 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 146.10, 131.08, 128.81, 115.18, 65.27.
(12) 4-Hydroxybenzyl alcohol. 1H NMR spectrum (DMSO-d6, 400 MHz) δ 9.30 (s, 1H), 7.11 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 4.96 (s, 1H), 4.36 (s, 2H). 13C NMR spectrum (DMSO-d6, 100 MHz) δ 156.68, 133.11, 128.51, 115.25, 63.25.
(13) 2,4-Dimethylbenzyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.20 (d, J = 8.1 Hz, 2H), 6.99 (d, J = 4.4 Hz, 2H), 4.62 (s, 2H), 2.31 (d, J = 3.6 Hz, 6H). 13C NMR spectrum (CDCl3, 100 MHz) δ 137.55, 136.14, 135.78, 131.24, 127.90, 126.63, 63.39, 21.05, 18.60.
(14) 1-Napthalenemethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.10 (d, J = 8.1 Hz, 1H), 7.87 (d, J = 9.4 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.57–7.48 (m, 3H), 7.46–7.39 (m, 1H), 5.11 (d, J = 4.4 Hz, 2H), 1.92 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 136.28, 133.81, 131.24, 128.69, 128.61, 126.37, 125.91, 125.42, 125.36, 123.67, 63.69.
(15) 9-Anthracenemethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.46 (s, 1H), 8.40 (d, J = 9.4 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 7.62–7.52 (m, 2H), 7.53–7.42 (m, 2H), 5.66 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 131.56, 131.02, 130.26, 129.16, 128.41, 126.48, 125.12, 123.89, 57.43.
(16) 1,3-Benzodioxol-5-ylmethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 6.84 (s, 1H), 6.81–6.73 (m, 2H), 5.94 (s, 2H), 4.54 (s, 2H), 2.09 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 147.80, 147.07, 134.90, 120.52, 108.21, 107.90, 101.02, 65.17.
(17) 2-Pyridinepropanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.48 (d, J = 4.9 Hz, 1H), 7.61 (td, J = 7.7, 1.8 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.12 (ddd, J = 7.4, 5.0, 1.0 Hz, 1H), 4.67 (s, 1H), 3.70 (t, J = 6.0 Hz, 2H), 2.95 (t, J = 7.1 Hz, 2H), 2.06–1.93 (m, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 161.51, 148.65, 136.81, 123.18, 121.16, 61.96, 35.11, 31.88.
(18) 2-Amino-2-phenylethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.36–7.24 (m, 5H), 4.01 (dd, J = 8.3, 4.2 Hz, 1H), 3.70 (dd, J = 10.9, 4.3 Hz, 1H), 3.53 (dd, J = 10.9, 8.3 Hz, 1H), 2.65 (s, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 142.49, 128.62, 127.52, 126.58, 67.88, 57.41.
(19) 2-(Vinyloxy)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 6.51 (dd, J = 14.3, 6.8 Hz, 1H), 4.23 (dd, J = 14.3, 2.2 Hz, 1H), 4.05 (dd, J = 6.8, 2.2 Hz, 1H), 3.82 (dd, J = 7.7, 3.7 Hz, 2H), 2.40 (t, J = 5.2 Hz, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 151.55, 87.12, 69.15, 61.18.
(20) 2-Mercaptoethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.73 (t, J = 5.3 Hz, 2H), 2.72 (dd, J = 10.2, 4.3 Hz, 2H), 1.44 (t, J = 8.5 Hz, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 63.98, 27.67.
(21) 2-Chloroethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.92–3.84 (m, 2H), 3.72–3.62 (m, 2H), 2.48 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 62.94, 46.82.
(22) 2-(2-Chloroethoxy)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.77 (dd, J = 10.1, 4.6 Hz, 4H), 3.69–3.61 (m, 4H), 2.38 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 72.39, 71.13, 61.67, 42.93.
(23) 1,5-Pentanediol. 1H NMR spectrum (D2O, 400 MHz) δ 3.50 (t, J = 6.6 Hz, 4H), 1.54–1.40 (m, 4H), 1.37–1.19 (m, 2H). 13C NMR spectrum (D2O, 100 MHz) δ 61.62, 30.97, 21.39.
(24) 2,2′-Azanediylbis(ethan-1-ol). 1H NMR spectrum (CDCl3, 400 MHz) δ 4.18 (s, 2H), 3.75–3.63 (m, 4H), 2.82–2.64 (m, 4H). 13C NMR spectrum (CDCl3, 100 MHz) δ 60.60, 51.19.
(25) 1,2,3-Propanetriol. 1H NMR spectrum (D2O, 400 MHz) δ 3.73–3.63 (m, 1H), 3.50 (ddd, J = 18.2, 11.7, 5.4 Hz, 4H). 13C NMR spectrum (D2O, 100 MHz) δ 72.03, 62.45.
(26) 1-Butanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.64 (t, J = 6.6 Hz, 2H), 2.07 (s, 1H), 1.62–1.48 (m, 2H), 1.42–1.37 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 62.64, 34.81, 18.87, 13.83.
(27) 1-Decanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.63 (t, J = 6.6 Hz, 2H), 1.60–1.54 (m, 2H), 1.39–1.21 (m, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 63.04, 32.79, 31.90, 29.62, 29.56, 29.44, 29.32, 25.75, 22.68, 14.10.
(28) Cyclopropylmethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.22 (d, J = 7.0 Hz, 2H), 1.74 (s, 1H), 0.96–0.81 (m, 1H), 0.31 (dt, J = 5.7, 4.5 Hz, 2H), 0.06–0.02 (m, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 67.61, 13.39, 2.68.
(29) 1-Phenylethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.38–7.31 (m, 4H), 7.27 (dd, J = 6.0, 2.7 Hz, 1H), 4.87 (q, J = 6.2 Hz, 1H), 1.48 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 145.84, 128.52, 127.49, 125.42, 70.41, 25.16.
(30) 1-(4-Methylphenyl)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.26 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 7.9 Hz, 2H), 4.86 (q, J = 6.4 Hz, 1H), 2.34 (s, 3H), 1.90 (s, 1H), 1.48 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 142.88, 137.17, 129.18, 125.37, 70.26, 25.08, 21.10.
(31) 1-(4-Nitrophenyl)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 8.18 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 5.01 (q, J = 6.5 Hz, 1H), 1.51 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 153.25, 147.10, 126.14, 123.72, 69.42, 25.44.
(32) 1-(4-Fluorophenyl)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.40–7.28 (m, 2H), 7.02 (dd, J = 9.8, 7.7 Hz, 2H), 4.88 (q, J = 6.4 Hz, 1H), 2.04 (s, 1H), 1.47 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 163.33, 160.90, 141.53, 141.50, 127.09, 127.01, 115.36, 115.15, 69.77, 25.28.
(33) 1-(4-Chlorophenyl)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.33–7.27 (m, 4H), 4.86 (q, J = 6.4 Hz, 1H), 2.07 (s, 1H), 1.46 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 144.26, 133.06, 128.61, 126.81, 69.74, 25.26.
(34) 1-(4-Bromophenyl)ethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.46 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 4.85 (q, J = 6.3 Hz, 1H), 2.04 (s, 1H), 1.46 (d, J = 6.5 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 144.78, 131.56, 127.17, 121.16, 69.78, 25.24.
(35) 2-Amino-1-phenylethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.37–7.15 (m, 5H), 4.54 (dd, J = 7.6, 4.1 Hz, 1H), 2.85 (s, 2H), 2.80–2.67 (m, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 142.90, 128.37, 127.44, 125.93, 74.24, 49.24.
(36) 1,3-Diaminopropanol. 1H NMR spectrum (D2O, 400 MHz) δ 3.55–3.43 (m, 1H), 2.54 (ddd, J = 21.0, 13.4, 6.0 Hz, 4H). 13C NMR spectrum (D2O, 100 MHz) δ 73.77, 44.03.
(37) Cyclohexanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.69–3.51 (m, 1H), 1.98 (s, 1H), 1.95–1.84 (m, 2H), 1.73 (t, J = 6.4 Hz, 2H), 1.54 (dd, J = 11.4, 7.1 Hz, 1H), 1.27 (dd, J = 15.7, 7.2 Hz, 4H). 13C NMR spectrum (CDCl3, 100 MHz) δ 70.25, 35.49, 25.45, 24.16.
(38) Cycloheptanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 3.62–3.55 (m, 1H), 2.11 (s, 1H), 1.89 (d, J = 5.4 Hz, 2H), 1.73 (d, J = 4.2 Hz, 2H), 1.45 (d, J = 4.5 Hz, 1H), 1.37–1.07 (m, 6H). 13C NMR spectrum (CDCl3, 100 MHz) δ 71.26, 41.93, 27.15, 25.65.
(39) Diphenylmethanol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.28–7.24 (m, 5H), 7.38–7.34 (m, 3H), 7.34–7.30 (m, 2H), 5.83 (d, J = 3.3 Hz, 1H), 2.30 (d, J = 3.5 Hz, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 413.82, 128.53, 127.60, 126.56, 76.28.
(40) 4-Chlorobenzhydrol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.34 (d, J = 4.4 Hz, 4H), 7.31–7.26 (m, 5H), 5.80 (d, J = 3.1 Hz, 1H), 2.32 (d, J = 3.4 Hz, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 143.45, 142.23, 133.29, 128.67, 128.62, 127.89, 126.54, 75.63.
(41) 2-Aminobenzhydrol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.41–7.32 (m, 4H), 7.29 (t, J = 9.3 Hz, 1H), 7.11 (td, J = 7.7, 1.6 Hz, 1H), 7.01 (dd, J = 7.6, 1.5 Hz, 1H), 6.79–6.60 (m, 2H), 5.82 (s, 1H), 3.93 (s, 2H), 2.79 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 144.84, 141.93, 128.69, 128.52, 127.63, 126.57, 118.40, 117.02, 74.95.
(42) 4,4′-Dimethoxybenzhydrol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.27 (d, J = 8.5 Hz, 4H), 6.86 (d, J = 8.8 Hz, 4H), 5.76 (d, J = 3.2 Hz, 1H), 3.78 (s, 6H), 2.20 (d, J = 3.5 Hz, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 158.96, 136.38, 127.76, 113.83, 75.39, 55.30.
(43) Furfuryl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.40 (d, J = 2.6 Hz, 1H), 6.38–6.31 (m, 1H), 6.29 (d, J = 3.0 Hz, 1H), 4.60 (s, 2H), 2.06 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 154.00, 142.59, 110.37, 107.78, 57.44.
(44) 5-Methylfurfuryl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 6.34 (dd, J = 3.2, 1.9 Hz, 1H), 6.29 (d, J = 3.0 Hz, 1H), 4.60 (s, 2H), 2.46 (s, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 154.01, 142.60, 110.37, 107.78, 57.44, 15.32.
(45) 5-(Hydroxymethyl)furfuryl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 6.52 (d, J = 3.5 Hz, 2H), 4.72 (s, 4H). 13C NMR spectrum (CDCl3, 100 MHz) δ 152.35, 109.99, 57.58.
(46) Gamma-valerolactone. 1H NMR spectrum (CDCl3, 400 MHz) δ 4.75–4.56 (m, 1H), 2.62–2.49 (m, 2H), 2.44–2.30 (m, 1H), 1.91–1.80 (m, 1H), 1.42 (d, J = 6.2 Hz, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 177.31, 77.29, 29.68, 29.08, 21.04.
(47) Vanillyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 6.76–6.58 (m, 2H), 6.30 (d, J = 8.5 Hz, 1H), 4.47 (s, 2H), 3.58 (s, 3H). 13C NMR spectrum (CDCl3, 100 MHz) δ 151.71, 147.17, 129.89, 118.14, 114.40, 108.79, 60.34, 56.14.
(48) Cinnamyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 7.37 (d, J = 7.2 Hz, 2H), 7.34–7.28 (m, 2H), 7.23 (dd, J = 10.8, 3.6 Hz, 1H), 6.60 (d, J = 15.9 Hz, 1H), 6.35 (dt, J = 15.9, 5.7 Hz, 1H), 4.30 (d, J = 5.7 Hz, 2H), 2.00 (s, 1H). 13C NMR spectrum (CDCl3, 100 MHz) δ 136.71, 131.11, 128.62, 128.54, 127.71, 126.50, 63.66.
(49) Perillyl alcohol. 1H NMR spectrum (CDCl3, 400 MHz) δ 5.92 (s, 1H), 4.94 (s, 2H), 4.22 (s, 2H), 2.35 (d, J = 10.0 Hz, 4H), 2.14 (dd, J = 46.4, 12.9 Hz, 2H), 1.96 (s, 3H), 1.88–1.64 (m, 2H). 13C NMR spectrum (CDCl3, 100 MHz) δ 149.82, 137.24, 122.45, 108.66, 67.22, 41.14, 30.40, 27.46, 26.10, 20.79.
(50) Estradiol. 1H NMR spectrum (DMSO-d6, 400 MHz) δ 9.03 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.50 (dd, J = 8.4, 2.6 Hz, 1H), 6.43 (d, J = 2.5 Hz, 1H), 4.49 (d, J = 4.2 Hz, 1H), 3.52 (dd, J = 11.4, 8.3 Hz, 1H), 2.70 (t, J = 7.5 Hz, 2H), 2.23 (dd, J = 13.2, 3.1 Hz, 1H), 2.06 (t, J = 8.6 Hz, 1H), 1.95–1.72 (m, 3H), 1.66–1.50 (m, 1H), 1.44–1.05 (m, 7H), 0.66 (s, 3H). 13C NMR spectrum (DMSO-d6, 100 MHz) δ 155.38, 137.59, 130.87, 126.49, 115.39, 113.18, 80.53, 50.00, 44.00, 43.28, 37.06, 30.37, 29.63, 27.42, 26.55, 23.25, 11.74.
Physical measurements
The FTIR spectra were recorded using a Cary 630 spectrometer having a diamond ATR. NMR spectral measurements were done with a Jeol 400 MHz spectrometer. Elemental analysis was carried out using an Elementar Analysen Systeme GmbH Vario EL-III instrument. Powder X-ray diffraction (PXRD) patterns were obtained either with a Rigaku Table-Top XRD or a Bruker AXS D8 Discover instrument (CuKα radiation, λ = 1.54184 Å). The samples were ground and subjected to a range of θ = 5–60° at a slow scan rate at room temperature. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed with a Shimadzu DTG-60 and TA-DSC Q200 instruments, respectively, under a N2 atmosphere with a heating rate of 10 °C per min. The nitrogen sorption isotherms were collected using a Quantachrome gas sorption analyzer. Barrett–Joyner–Halenda (BJH) model was used for the analysis of pore size based on N2 adsorption. Gas chromatographic studies were performed with a PerkinElmer Clarus 580 equipped with an RTX-5SIL-MS column. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was conducted using Agilent 8900 Triple Quadrupole equipment. Temperature-programmed desorption (TDP) using NH3 and CO2 gases was carried out using a BELCAT II Catalyst Analyzer from Microtrac Corp. The samples were degassed at 150 °C for 2 h and then cooled to room temperature for 30 min in a He flow. After the adsorption of CO2 and NH3 at 50 °C, the temperature was increased to 400 °C with 10 °C min−1. Given the thermal stability of MOF 1 up to 400 °C, the maximum desorption temperature for both NH3 and CO2 was set at 400 °C.X-ray diffraction studies
Single crystal X-ray diffraction data for MOF 1 was collected on a Bruker SMART APEX-II CCD diffractometer equipped with a graphite monochromatic MoKα radiation (λ = 0.71073 Å).94 For MOF 1, SMART95 was used for collecting frames of data, indexing reflections and determining the lattice parameters; SAINT95 for integration of the intensity of reflections and scaling; and SADABS96 for absorption correction. The frames were collected at 293(2) K. The structure was solved by the direct methods using SIR-97 (ref. 97) and refined by the full-matrix least-squares refinement techniques on F2 using SHELXL-97 (ref. 98) incorporated in Olex2 crystallographic package.99 All calculations and structure refinements were performed using the Olex2 program. The hydrogen atoms were fixed at the calculated positions using the isotropic thermal parameters, whereas non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the coordinated as well as uncoordinated water molecules could not be located from the Fourier map; however, their contributions are included in the empirical formulae. The crystallographic data collection and structure solution parameters are presented in Table S1,† whereas selected bond distances and bond angles are presented in Tables S2 and S3.† CCDC 2334280 (1) contains the supplementary crystallographic data for this paper.Kinetic studies
A calibration curve was generated for the TH reaction of furfural using benzene as an internal standard. The kinetics for the TH of furfural was studied under first-order conditions and monitored by gas chromatography with respect to the concentration of a substrate. The rate constant (k) was calculated from the slope of the linear plot of ln(Ct/Co) against time, where (Ct/Co) is the ratio of the concentration of substrate at a certain time ‘t’ to that of initial time ‘0’.100The activation energy was calculated by using the following equation:
| lnk = lnA – Ea/RT | (1) |
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for MOF 1 has been deposited at the CCDC under accession number 2334280 and can be obtained from [https://www.ccdc.cam.ac.uk/].Author contributions
A. A. and Ru. G. data curation, formal analysis, methodology, software. R. G. conceptualization, editing, funding, supervision.Conflicts of interest
There is no conflict of interest involved in this work and authors declare no competing financial interest.Acknowledgements
Rajeev Gupta gratefully acknowledges financial support from the Science & Engineering Research Board (SERB), New Delhi (CRG/2021/001700). Aashish and Ruchika Gupta thank CSIR, New Delhi, for their SRF fellowships. The authors thank USIC at this university for the instrumental facilities and Ms. Upma for the technical assistance.References
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- A. Suárez, Phys. Sci. Rev., 2018, 3, 20170028 Search PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- R. A. Grey, G. P. Pez and A. Wallo, J. Am. Chem. Soc., 1981, 103, 7536–7542 CrossRef CAS.
- L. H. Slaugh, J. Org. Chem., 1967, 32, 108–113 CrossRef CAS.
- H.-P. Jia and E. Alessandra Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- D. L. Reger, M. M. Habib and D. J. Fauth, J. Org. Chem., 1980, 45, 3860–3865 CrossRef CAS.
- M. V. Lototskyy, V. A. Yartys, B. G. Pollet and R. C. Bowman, Int. J. Hydrogen Energy, 2014, 39, 5818–5851 CrossRef CAS.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- D. Spasyuk, C. Vicent and D. G. Gusev, J. Am. Chem. Soc., 2015, 137, 3743–3746 CrossRef CAS PubMed.
- Y.-M. He and Q.-H. Fan, ChemCatChem, 2015, 7, 398–400 CrossRef CAS.
- Z. Guo, P. Zhou, L. Jiang, S. Liu, Y. Yang, Z. Li, P. Wu, Z. Zhang and H. Li, Adv. Mater., 2024, 36, 2311149 CrossRef CAS PubMed.
- G. Shen, J. Chen, D. Xu, X. Zhang, Y. Zhou and B. Fan, Org. Lett., 2019, 21, 1364–1367 CrossRef CAS PubMed.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750–1770 CrossRef CAS PubMed.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS.
- R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., 2015, 127, 5260–5263 CrossRef.
- C. G. Santana and M. J. Krische, ACS Catal., 2021, 11, 5572–5585 CrossRef CAS PubMed.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS.
- R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed.
- B. Ding, Z. Zhang, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 3690–3693 CrossRef CAS PubMed.
- Á. M. Pálvölgyi, J. Bitai, V. Zeindlhofer, C. Schröder and K. Bica, ACS Sustainable Chem. Eng., 2019, 7, 3414–3423 CrossRef.
- P. Weingart and W. R. Thiel, ChemCatChem, 2018, 10, 4844–4848 CrossRef.
- S. Yadav, P. Vijayan, S. Yadav and R. Gupta, Dalton Trans., 2021, 50, 3269–3279 RSC.
- W. Fang and A. Riisager, Green Chem., 2021, 23, 670–688 RSC.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370–11419 CrossRef CAS PubMed.
- R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang, X. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS.
- Y. Meng, Y. Jian, J. Li, H. Wu, H. Zhang, S. Saravanamurugan, S. Yang and H. Li, Chem. Eng. J., 2023, 452, 139477 CrossRef CAS.
- Z. An and J. Li, Green Chem., 2022, 24, 1780–1808 RSC.
- Y. Fan, C. Zhuang, S. Li, Y. Wang, X. Zou, X. Liu, W. Huang and G. Zhu, J. Mater. Chem. A, 2021, 9, 1110–1118 RSC.
- S. Li, Y. Fan, C. Wu, C. Zhuang, Y. Wang, X. Li, J. Zhao and Z. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 8507–8517 CrossRef CAS PubMed.
- J. Wu, D. Liang, X. Song, T. Liu, T. Xu, S. Wang and Y. Zou, J. Energy Chem., 2022, 71, 411–417 CrossRef CAS.
- K. Zhang, Q. Meng, H. Wu, M. He and B. Han, ACS Sustainable Chem. Eng., 2022, 10, 10286–10293 CrossRef CAS.
- W. Baratta, G. Chelucci, E. Herdtweck, S. Magnolia, K. Siega and P. Rigo, Angew. Chem., Int. Ed., 2007, 46, 7651–7654 CrossRef CAS PubMed.
- D. S. Matharu, D. J. Morris, A. M. Kawamoto, G. J. Clarkson and M. Wills, Org. Lett., 2005, 7, 5489–5491 CrossRef CAS PubMed.
- N. Pannetier, J.-B. Sortais, J.-T. Issenhuth, L. Barloy, C. Sirlin, A. Holuigue, L. Lefort, L. Panella, J. G. de Vries and M. Pfeffer, Adv. Synth. Catal., 2011, 353, 2844–2852 CrossRef CAS.
- S. Yadav and R. Gupta, ACS Sustainable Chem. Eng., 2023, 11, 8533–8543 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee and K. Wilson, J. Chem. Technol. Biotechnol., 2017, 92, 1125–1135 CrossRef CAS.
- A. M. Hengne, B. S. Kadu, N. S. Biradar, R. C. Chikate and C. V. Rode, RSC Adv., 2016, 6, 59753–59761 RSC.
- Z. Yu, F. Meng, Y. Wang, Z. Sun, Y. Liu, C. Shi, W. Wang and A. Wang, Ind. Eng. Chem. Res., 2020, 59, 7416–7425 CrossRef CAS.
- H.-C. “Joe” Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- M. Bosch, S. Yuan, W. Rutledge and H.-C. Zhou, Acc. Chem. Res., 2017, 50, 857–865 CrossRef CAS PubMed.
- Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403 RSC.
- Aashish, R. Gupta and R. Gupta, Sens. Diagn., 2023, 2, 1585–1596 RSC.
- C. Jiang, X. Wang, Y. Ouyang, K. Lu, W. Jiang, H. Xu, X. Wei, Z. Wang, F. Dai and D. Sun, Nanoscale Adv., 2022, 4, 2077–2089 RSC.
- K. Nath, A. Ahmed, D. J. Siegel and A. J. Matzger, J. Am. Chem. Soc., 2022, 144, 20939–20946 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, W. Zhou and B. Chen, J. Phys. Chem. Lett., 2014, 5, 3468–3479 CrossRef CAS PubMed.
- A. A. Olajire, Renewable Sustainable Energy Rev., 2018, 92, 570–607 CrossRef CAS.
- D. J. Wales, J. Grand, V. P. Ting, R. D. Burke, K. J. Edler, C. R. Bowen, S. Mintova and A. D. Burrows, Chem. Soc. Rev., 2015, 44, 4290–4321 RSC.
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- G. Kumar, F. Hussain and R. Gupta, Dalton Trans., 2017, 46, 15023–15031 RSC.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- S. Ali Akbar Razavi and A. Morsali, Coord. Chem. Rev., 2019, 399, 213023 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. G. Iii, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- N. Van Velthoven, M. Henrion, J. Dallenes, A. Krajnc, A. L. Bugaev, P. Liu, S. Bals, A. V. Soldatov, G. Mali and D. E. De Vos, ACS Catal., 2020, 10, 5077–5085 CrossRef CAS.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- Y. Liu, J. Sun, L. Fan and Q. Xu, Front. Chem., 2022, 10, 845274 CrossRef CAS PubMed.
- B. Bohigues, S. Rojas-Buzo, M. Moliner and A. Corma, ACS Sustainable Chem. Eng., 2021, 9, 15793–15806 CrossRef CAS PubMed.
- A. H. Valekar, M. Lee, J. W. Yoon, J. Kwak, D.-Y. Hong, K.-R. Oh, G.-Y. Cha, Y.-U. Kwon, J. Jung, J.-S. Chang and Y. K. Hwang, ACS Catal., 2020, 10, 3720–3732 CrossRef CAS.
- L. Hu, A. He, X. Shen, Q. Gu, J. Zheng, Z. Wu, Y. Jiang, X. Wang, J. Xu, Y. Kan and F. Xu, Green Chem., 2022, 24, 6931–6944 RSC.
- H. Li, J. He, A. Riisager, S. Saravanamurugan, B. Song and S. Yang, ACS Catal., 2016, 6, 7722–7727 CrossRef CAS.
- H. Li, X. Liu, T. Yang, W. Zhao, S. Saravanamurugan and S. Yang, ChemSusChem, 2017, 10, 1761–1770 CrossRef CAS PubMed.
- F. Lin, Y. Yang and Y.-H. Chin, ACS Catal., 2017, 7, 6909–6914 CrossRef CAS.
- F. Wang, Z. Chen, H. Chen, T. A. Goetjen, P. Li, X. Wang, S. Alayoglu, K. Ma, Y. Chen, T. Wang, T. Islamoglu, Y. Fang, R. Q. Snurr and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 32090–32096 CrossRef CAS PubMed.
- P. Ji, K. Manna, Z. Lin, A. Urban, F. X. Greene, G. Lan and W. Lin, J. Am. Chem. Soc., 2016, 138, 12234–12242 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Ltd, 2008 Search PubMed.
- F. Ambroz, T. J. Macdonald, V. Martis and I. P. Parkin, Small Methods, 2018, 2, 1800173 CrossRef.
- G. Kumar, S. Pachisia, P. Kumar, V. Kumar and R. Gupta, Chem.–Asian J., 2019, 14, 4594–4600 CrossRef CAS PubMed.
- K.-N. T. Tseng, A. M. Rizzi and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 16352–16355 CrossRef CAS PubMed.
- S. Pachisia, R. Kishan, S. Yadav and R. Gupta, Inorg. Chem., 2021, 60, 2009–2022 CrossRef CAS PubMed.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- G. V. A. Martins, G. Berlier, C. Bisio, S. Coluccia, H. O. Pastore and L. Marchese, J. Phys. Chem. C, 2008, 112, 7193–7200 CrossRef CAS.
- M. Lusardi, T. Struble, A. R. Teixeira and K. F. Jensen, Catal. Sci. Technol., 2020, 10, 536–548 RSC.
- R. Gupta, G. Kumar and R. Gupta, Inorg. Chem., 2022, 61, 7682–7699 CrossRef CAS.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed.
- Y. Bedjanian, P. Szabó and G. Lendvay, J. Phys. Chem. A, 2023, 127, 6916–6923 CrossRef CAS PubMed.
- R. S. Assary, L. A. Curtiss and J. A. Dumesic, ACS Catal., 2013, 3, 2694–2704 CrossRef CAS.
- K. Liu, R. Qin and N. Zheng, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS PubMed.
- F. Li, Z. Li, L. J. France, J. Mu, C. Song, Y. Chen, L. Jiang, J. Long and X. Li, Ind. Eng. Chem. Res., 2018, 57, 10126–10136 CrossRef CAS.
- G. Kumar, G. Kumar and R. Gupta, Inorg. Chem., 2015, 54, 2603–2615 CrossRef CAS PubMed.
- S. Pachisia and R. Gupta, Cryst. Growth Des., 2019, 19, 6039–6047 CrossRef CAS.
- C. Robert McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- M. Prieschl, J. García-Lacuna, R. Munday, K. Leslie, A. O'Kearney-McMullan, C. A. Hone and C. Oliver Kappe, Green Chem., 2020, 22, 5762–5770 RSC.
- T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703–15715 CrossRef CAS.
- M. A. Droesbeke and F. E. Du Prez, ACS Sustainable Chem. Eng., 2019, 7, 11633–11639 CrossRef CAS.
- W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, 4 edn, reprint, Butterworth-Heinemann, Oxford, 2002 Search PubMed.
- CrysAlisPro, Oxford Diffraction Ltd, Yarnton Engl., 2009 Search PubMed.
- SMART & SAINT Software Reference Manual, Version 6.45, Bruker Analytical X-Ray Systems, Inc., Madison, 2003 Search PubMed.
- G. M. Sheldrick, SADABS, Version 2.05. A Software for Empirical Absorption Correction, University of Göttingen, Göttingen, 2002 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, SIR92 – a Program for Automatic Solution of Crystal Structures by Direct Methods, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, A Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A Complete Structure Solution, Refinement and Analysis Program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. Yadav and R. Gupta, Inorg. Chem., 2022, 61, 15463–15474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis, characterization data, figures and tables. CCDC 2334280. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4su00368c |
| This journal is © The Royal Society of Chemistry 2024 |