
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-thin order–disorder CeO2 nanobelts as the non-carbon support of the PtCu catalyst towards methanol oxidation and oxygen reduction reactions†
Han
Zhi
a,
Boda
Dong
a,
Xingxing
Guo
b and
Feng
Xu
 *ab
*ab
aSchool of Advanced Manufacturing, Fuzhou University, Fuzhou, China. E-mail: xufeng@fzu.edu.cn
bCollege of Materials Science and Engineering, Fuzhou University, Fuzhou, China
First published on 25th September 2024
Abstract
The use of carbon supports in direct methanol fuel cells easily leads to the shedding and poisoning of the Pt catalyst and hence the decrease of catalytic activity. Non-carbon materials have been studied to enhance the metal–support interaction and the catalytic performance. Herein, we explored ultra-thin CeO2 nanobelts (2D-CeO2) with the order–disorder structure as the support of the PtCu catalyst. PtCu/2D-CeO2 shows the highest current density of 37.24 mA cm−2 toward the methanol oxidation reaction (MOR), and a limiting current density of 4.82 mA cm−2 towards the oxygen reduction reaction. The order–disorder structure of 2D-CeO2 generates a high volume of oxygen vacancies and strong metal–support interaction. The Pt0 proportion of PtCu/2D-CeO2 is much higher than that of PtCu/C which increases the active sites. The d-band center of PtCu is lowered which facilitates the adsorption and dissociation of reactants, thereby dramatically boosting the electro-catalytic performance.
Sustainability spotlightDirect methanol fuel cells (DMFCs) are widely accepted as one of the promising green energy resources to replace fossil fuels and are now on their way to commercialization. Pt/C is the commonly used catalyst of DMFCs. The carbon support leads to the deactivation during long-term operation. Moreover, carbon is a product of the petrochemical industry. In this work, ultra-thin order–disorder CeO2 was developed as the non-carbon support to load the PtCu alloy. The results showed better performance towards the methanol oxidation reaction and comparable activity towards the oxygen reduction reaction of PtCu/2D-CeO2, relative to those of Pt/C. Hence, the activity and durability of Pt-based catalysts were enhanced and the carbon consumption of DMFCs was further cut, promoting the sustainability of our world, aligning with the 7th goal of UN SDG(s), i.e., ensure access to affordable, reliable, sustainable and modern energy for all. |
Introduction
To date, platinum supported on carbon (Pt/C) remains the optimum choice as an efficient catalyst in direct methanol fuel cells (DMFCs).1–3 But Pt suffers from the durability issue and high cost as a noble metal. Both the methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR) have large reaction energy potential barriers, and the reaction rate also shows a serious impact on the overall efficiency.4 Carbon is a widely used support owing to its high conductivity and specific surface area.5,6 However, carbon materials are prone to chemical/electrochemical corrosion, which easily leads to the shedding of Pt nanoparticles and the decrease of catalytic activity.7 Moreover, carbon materials will be oxidized and generate intermediates such as CO*, which poison the Pt catalyst and degrade the overall performance.8 Therefore, developing novel catalysts to reduce the reaction potential energy barrier and improve the reaction efficiency has become the key issue for DMFCs.9One of the solutions is to explore alternative chemically and electrochemically stable supports which interact strongly with Pt nanoparticles and promote catalytic performance.10 Researchers have carried out in-depth studies on non-carbon support materials, such as oxides,11–15 nitrides,16,17etc. The oxides can effectively alter the electronic structure and prevent the agglomeration of Pt nanoparticles.18 But the critical drawback of the oxides is the inherent wide band gap which results in poor electron conductivity and severe diminishing of the reaction rate.19
It is well known that CeO2 has the advantage of switching the valence states of cerium (Ce3+ ↔ Ce4+), accompanied by charge transfer (including electrons, oxygen anions, and vacancies), which is significantly effective for regulating the ionic/electronic conductivity and the catalytic activity.20,21 The structure engineering including the morphology, size, facets, and defects is crucial to the regulation. With different nanostructures (nanorods, nanooctahedra, nanocubes, etc.), CeO2 exposes different crystal facets which facilitate certain reactions.22 The oxygen vacancy of CeO2 is commonly reckoned critical to tune the interaction with metals and the size of metallic nanoparticles, which are key issues for enhancing the catalytic performance.23 Amorphous structures have the inherent advantage of abundant vacancies relative to the crystalline counterpart.24 But amorphous structures suffer from the sluggish kinetics of electron transfer and lower electron conductivity. Therefore, the order–disorder engineering is capable of combining the separate advantages of crystalline and amorphous structures, leading to high catalytic activity.25 For example, Zhang and co-workers developed amorphous RuO2 nanosheets with well-defined amorphous-crystalline boundaries supported on carbon cloth, which displayed a low overpotential of 150 mV at 10 mA cm−2 and higher durability toward the oxygen evolution reaction.25
Herein, we explored ultra-thin two-dimensional CeO2 nanobelts (2D-CeO2) with the order–disorder structure as the support of a PtCu alloy to synthesize a dual-function catalyst. 2D-CeO2 was prepared by the sol–gel method under ambient conditions, and then the PtCu alloy was loaded on 2D-CeO2. The PtCu/2D-CeO2 catalysts exhibit better activity and durability than Pt/C towards the MOR.
Experimental section
Chemicals
Chloroplatinic acid hexahydrate (H2PtCl6·6H2O, analytical grade), copper chloride dihydrate (CuCl2·2H2O, analytical grade), ethylene glycol (C2H(OH)2, analytical grade), anhydrous ethanol (CH3CH2OH, analytical grade), nitric acid (HNO3, analytical grade), sodium hydroxide (NaOH, analytical grade), sulfuric acid (H2SO4, analytical grade), and graphite (C, analytical grade) were all purchased from National Pharmaceutical Reagents. Nafion (C16H33O, analytical grade) was purchased from DuPont and deionized water (DI water) was purchased from Aquapro.Catalyst preparation
Disperse 8.68 g of cerium nitrate hexahydrate (Ce(NO3)3·6H2O) and 3.2 g of NaOH in 100 mL of deionized water (DI water), and then add the NaOH aqueous solution dropwise to the cerium nitrate aqueous solution. After the droplet addition, the suspension was filtered and washed three times with DI water. After lyophilization, the CeO2 nanobelts are obtained.The as-prepared CeO2 nanobelts were mixed with copper chloride dihydrate (CuCl2·2H2O) in ethylene glycol and the temperature was raised to 185 °C in an oil bath under a N2 atmosphere and then stirred for 10 min. The product was mixed with an appropriate amount of chloroplatinic acid (H2PtCl6) and heated at 60 °C in a water bath for 6 hours, then filtered and lyophilized again. The sample was calcined at 400 °C in a H2 atmosphere for 2 h. Finally, the calcined sample was stirred in 3 M HNO3 solution for 12 h to remove the excess Cu that was not alloyed. After filtration and lyophilization, the PtCu/2D-CeO2 catalyst was obtained. The preparation method of the PtCu/C catalyst was the same as that of the PtCu/2D-CeO2 catalyst. The difference was that CeO2 nanobelts were replaced by XC-72 carbon powder. The preparation process of the 25 wt% Pt/C catalyst was the same as the one mentioned above. The specific dosage was 30 mg of XC-72 and 6.64 mL of chloroplatinic acid glycol solution (4 mg mL−1), which was heated in an oil bath at 130 °C for 3 h, and lyophilized to obtain the 25 wt% Pt/C catalyst.
Characterization of the catalysts
The samples were characterized using an X-ray diffractometer (Ultima III X-ray diffractometer), scanning electron microscope (SUPRA 55 Scanning electron microscope), transmission electron microscope (Talos F200i high resolution transmission electron microscope), energy dispersive X-ray diffractometer (EDX), and xseries 2 inductively coupled plasma mass spectrometer.Electrochemical tests of the catalysts
The catalysts were tested by cyclic voltammetry (CV), linear voltammetry (LSV) and accelerated durability test (ADT) in a conventional three-electrode system. A conventional three-electrode system was used. The working electrodes were the glassy carbon electrode and the rotating ring-disk electrode (5 mm in diameter and 0.196 cm2 in area), the reference electrode was Ag/AgCl, and the counter electrode was a Pt wire. Electrochemical tests were performed on an Auto Lab electrochemical workstation.The MOR test was carried out by CV in the potential range of 0.00–1.20 V (vs. RHE) with a scanning rate of 50 mV s−1. Before the test, N2 was bubbled for 30 min to remove the oxygen dissolved in the electrolyte. The CV was then performed in the MOR electrolyte (0.5 M H2SO4 + 1 M methanol). The durability was also tested by CV. The ADT cycle was performed in 0.5 M H2SO4 solution in the potential range of 0.60–1.20 V (vs. RHE). The MOR performance test was conducted once every 1000 cycles to record the MOR performance.
The ORR test was carried out in 1 M HClO4 electrolyte. The LSV test was performed in the potential range of 0.00–1.20 V (vs. RHE) at the scan rate of 10 mV s−1 and different rotating speeds of 225, 400, 625, 900, 1225, 1600, and 2025 rpm. The durability test was carried out in HClO4 solution. The CV was performed at 0.56–0.96 V (vs. RHE) at a scanning rate of 100 mV s−1. A CV in the full potential range was recorded every 1000 cycles, and an ORR catalytic performance test was conducted and recorded after 5000 cycles.
Results and discussion
The prepared 2D-CeO2 is a two-dimensional energy band structure with a length of about 500 nm and a width of about 100 nm (Fig. 1A). Some fragments with the size under 10 nm are observed. The thickness of 2D-CeO2 is only several nanometers. The Cu/2D-CeO2 shows Cu particles with the diameter in the range of twenty to fifty nanometers (Fig. 1B). After Pt displacement and acid leaching, the PtCu particles are intimately loaded on 2D-CeO2 (Fig. 1C). The XRD illustrates the diffraction signals of PtCu/2D-CeO2 at 2θ of 28.55°, 33.14°, 47.58°, 56.39°, and 76.70° belonging to the (1 1 1), (2 0 0), (2 2 0), (3 1 1), and (3 3 1) facets of CeO2 (PDF# 34-0394) respectively (Fig. 1D). Beside the CeO2 diffraction signals, the sharp peaks at 2θ of 39.76°, 46.24°, 67.45°, and 81.28° are ascribed to the (1 1 1), (2 0 0), (2 2 0), and (3 1 1) facets of the PtCu alloy.26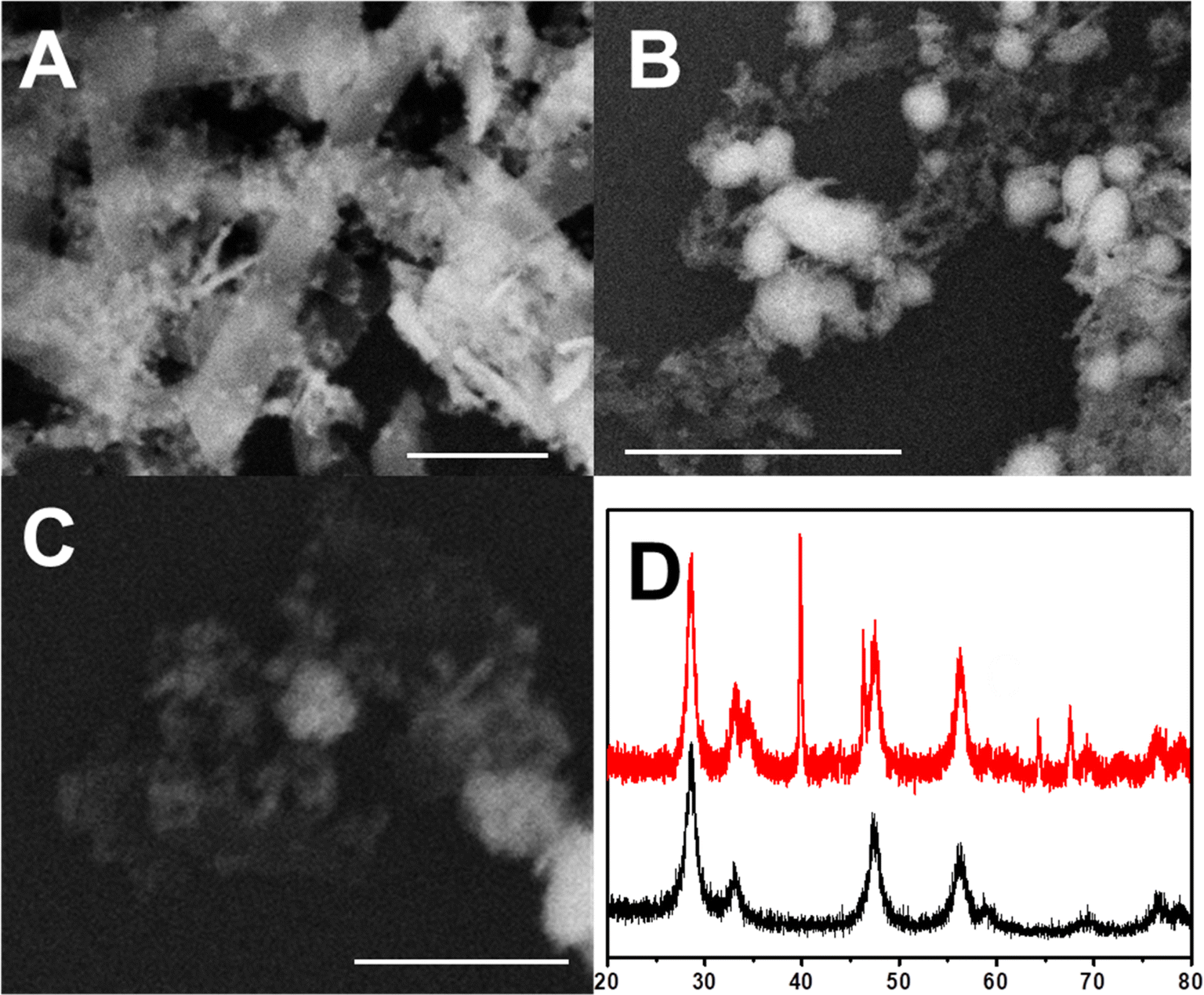 | ||
| Fig. 1 (A) The SEM images of 2D-CeO2; (B) 2D-CeO2 supported Cu; (C) 2D-CeO2 supported PtCu, and (D) the XRD of PtCu/2D-CeO2 (red line) and CeO2 (black line). The scale bars are 100 nm. | ||
The microstructure of the catalyst was further observed by TEM (Fig. 2). The Cu particles with sizes up to 100 nm can be seen (Fig. 2A). The PtCu alloy exhibits two architectures. One is hollow nanospheres with a diameter and wall thickness of about 100 and 15 nm, respectively (Fig. 2B–D). The other is solid PtCu nanoparticles with the size of 2–5 nm (Fig. 2E and F).27 The lattice spacing value of both architectures is measured to be 0.22 nm, belonging to the PtCu (1 1 1) facet.28 In the platinum displacement step of the preparation, the reaction is as follows:
| 2Cu + PtCl62− → 2Cu2+ + Pt + 6Cl− | (1) |
 | ||
| Fig. 2 TEM images of PtCu/2D-CeO2 (A–G), and high-resolution images (C–G), the EDX element distribution (H–K). | ||
When one Pt atom is reduced, two Cu atoms are oxidized to Cu2+ and a defect is formed in the surface of the particle. The defect provides a transport channel through which the internal Cu atoms migrate outward to react with PtCl6.2–29 With the migration of internal copper ions and the continuous formation of the external PtCu alloy, the hollow nanosphere finally comes into being. This architecture helps expose more active sites, leading to the improvement of catalytic activity.30
It is impressive that the 2D-CeO2 shows two phases, the amorphous and crystalline phases (Fig. 2F). The amorphous phase is the dominant one. The supersaturated nucleation in solution synthesis, because of the low solubility of CeO2 in water, can construct the amorphous structure.31 The close observation identifies the crystalline CeO2 with a size of less than five nanometers. The interplanar spacing value of 0.31 nm, corresponds to the CeO2 (1 1 1) facet.32 The well-defined crystalline–amorphous boundary is observed (Fig. 2F). The EDX images show the homogeneous distribution of Pt, Cu, and Ce elements. Therefore, the geometric structure of the catalyst consists of the PtCu nanoparticles and hollow nanospheres homogeneously loaded on the order–disorder 2D-CeO2.
ICP-AES was used to identify the content ratios of Pt, Cu, and CeO2 (Table S3†). The mass ratio of CeO2 is lower than 5%, due to the ultra-thin 2D structure. The Pt loadings of PtCu/2D-CeO2 and PtCu/C are 75.74 and 82.49 wt% respectively, and the Cu loadings are 19.37 and 17.51 wt% respectively. PtCu/2D-CeO2 exhibits the Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu atomic ratio of 1.28, lower than the 1.55 of PtCu/C, revealing that 2D-CeO2 interacts strongly with Cu and anchors Cu atoms firmly.
:Cu atomic ratio of 1.28, lower than the 1.55 of PtCu/C, revealing that 2D-CeO2 interacts strongly with Cu and anchors Cu atoms firmly.
XPS was used to analyze the valence and chemical states of the Pt, Cu, and Ce elements (Fig. 3). The peaks at 71.0 and 74.2 eV correspond to Pt0 4f7/2 and Pt0 4f5/2 (Fig. 3A). After splitting, the signals of Pt0 (p and t), Pt2+ (p′ and t′), and Pt4+ (p′′ and t′′) are obtained. Relative to the binding energy (B.E.) of peak p of PtCu/C (71.52 eV), the one of PtCu/2D-CeO2 (70.97 eV) shifts negatively by 0.55 eV. Through the integrated areas of peaks corresponding to different valence states, the proportion of atoms with different valence states is calculated as follows:
| Pt0% = SPt0/ (SPt0 + SPt2+ + SPt4+) × 100% | (2) |
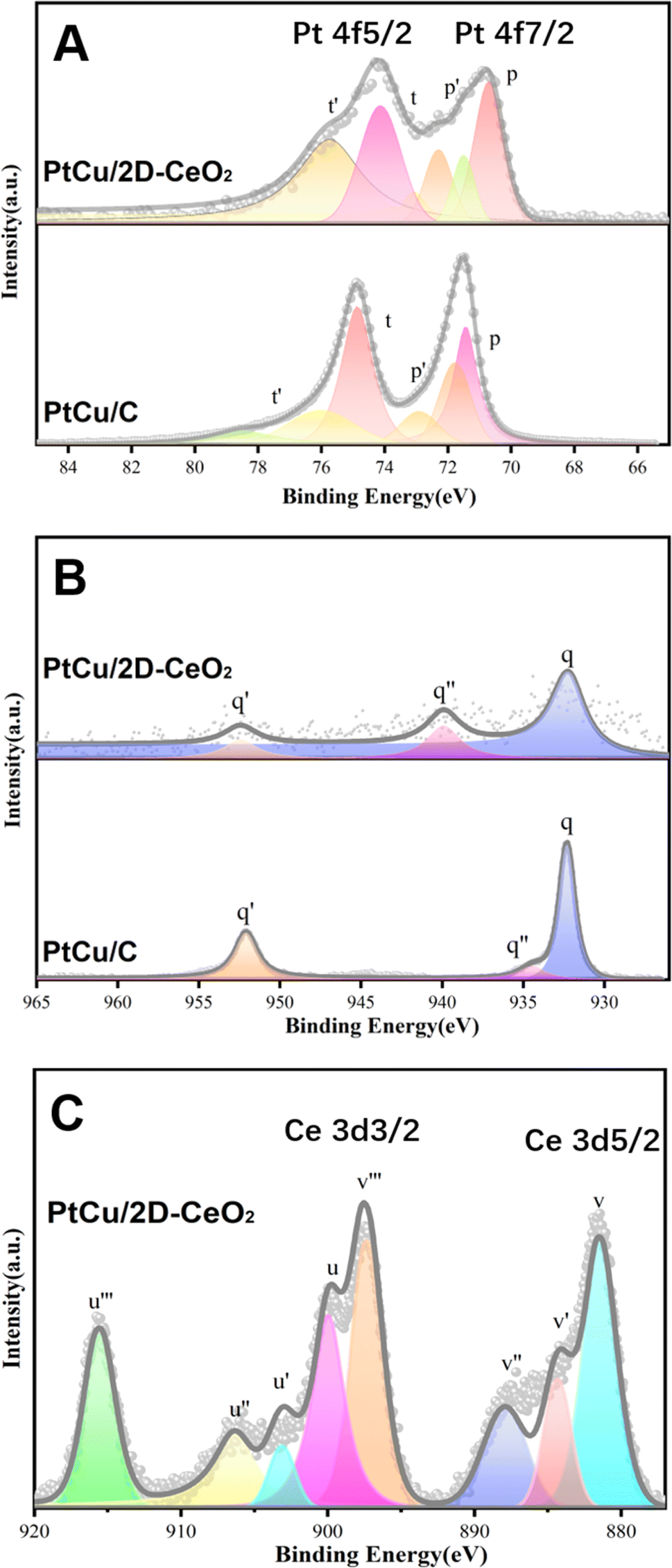 | ||
| Fig. 3 XPS spectra of Pt 4f (A), Cu 2p (B), and Ce 3d of PtCu/2D-CeO2 (C) orbitals. | ||
The Pt0 proportion (Pt0%) of PtCu/2D-CeO2 is 89.34%, much higher than 32.83% of PtCu/C, which indicates the impressive inhibition on the oxidation of the PtCu alloy, due to the Ce3+/Ce4+ pairs in CeO2 which can flexibly adjust their valence states.33
Fig. 3B shows the spectrum of the Cu 2p orbital. The q and q′ peaks represent the Cu0 2p3/2 and Cu0 2p1/2, respectively.34 Similar to the scenario of the Pt 4f orbital, the Cu0 peak of PtCu/2D-CeO2 (931.14 eV) shifts negatively by 1.12 eV relative to the one of PtCu/C (932.26 eV). The q′′ peak corresponding to Cu2+ 2p3/2 is located at 934.8 eV, representing the presence of non-zero valence copper atoms on the catalyst surface, due to the oxygenation of surficial Cu atoms during the catalyst preparation and preservation.35 The negative B. E. shifts of Pt 4f and Cu 2p orbitals of PtCu/2D-CeO2 indicate the lower d-band center of the PtCu alloy.36
The Ce 3d spectrum shows the peaks at near 882.1 and 902.5 eV representing Ce 3d2/3 (labeled series c) and Ce 3d2/5 (labeled series e), respectively. The v, v′′, u, and u′ are the characteristic peaks of Ce3+, and the other peaks (v′, v′′, v′′′, u′′, and u′′′) corresponded to the characteristic peaks of Ce4+. The presence of Ce3+ indicates the existence of oxygen vacancy defects in 2D-CeO2. The Ce3+ proportion in CeO2 preliminarily determines the concentration of oxygen vacancies. The Ce3+ proportion (Ce3+%) is determined by comparing the integral of the characteristic peak of Ce3+ with the integral of the full peak as follows:37
| Ce3+% = ΣSCe3+/(ΣSCe3+ + ΣSCe4+) × 100% | (3) |
The Ce3+% of PtCu/2D-CeO2 is calculated to be 24.91%. The disordered structure results in the high Ce3+% and the related concentration of oxygen vacancy defects, which is important to the interaction with PtCu nanoparticles and the catalytic activity.38
The CV and MOR plots were recorded, shown in Fig. 4. The as-prepared PtCu/2D-CeO2 catalyst shows electrocatalytic activity comparable to that of Pt/C (Fig. 4A). PtCu/2D-CeO2 exhibits a larger thickness of the double-layer in the potential range of 0.4–0.6 V vs. RHE, owing to the large surface of CeO2 nanobelts which can adsorb quantity charge, illustrating that CeO2 nanobelts possess high conductivity.39 The electrochemical active surface area (ECSA) diagram was drawn by integrating the hydrogen region in the potential range of 0–0.4 V (Fig. 4B). The ECSA of PtCu/2D-CeO2, PtCu/C, and Pt/C is 8.91, 11.39, and 8.36 m2 gPt−1 respectively. Both PtCu/2D-CeO2 and PtCu/C exhibit higher ECSA than Pt/C, due to the higher activity of the PtCu alloy than Pt.40 Better electron conductivity of carbon than CeO2 leads to a higher ECSA of PtCu/C than PtCu/2D-CeO2. It is still noteworthy that PtCu/2D-CeO2 illustrates higher ECSA than Pt/C, revealing higher electrochemical activity. Two factors are crucial to the high activity: (i) the sufficient active sites on which the CH3OH molecule adsorbs and dissociates, (ii) the high conductivity of the support due to which the electrons coming from the CH3OH dissociation can be transported to the circuit.41 The PtCu alloy provides abundant active sites owing to the structure of porous and hollow nanospheres. Moreover, the high Pt0% as calculated by XPS represents abundant active sites exposed for reactant adsorption, resulting in the high ECSA.42 The electron can be transported through oxygen vacancies, and hence, a high volume of oxygen vacancies facilitates electron transportation, leading to the high conductivity.43 It has been revealed that the 2D materials with the thickness of several nanometers can boost the charge transfer between the support and metal, which promotes the catalytic activity.44
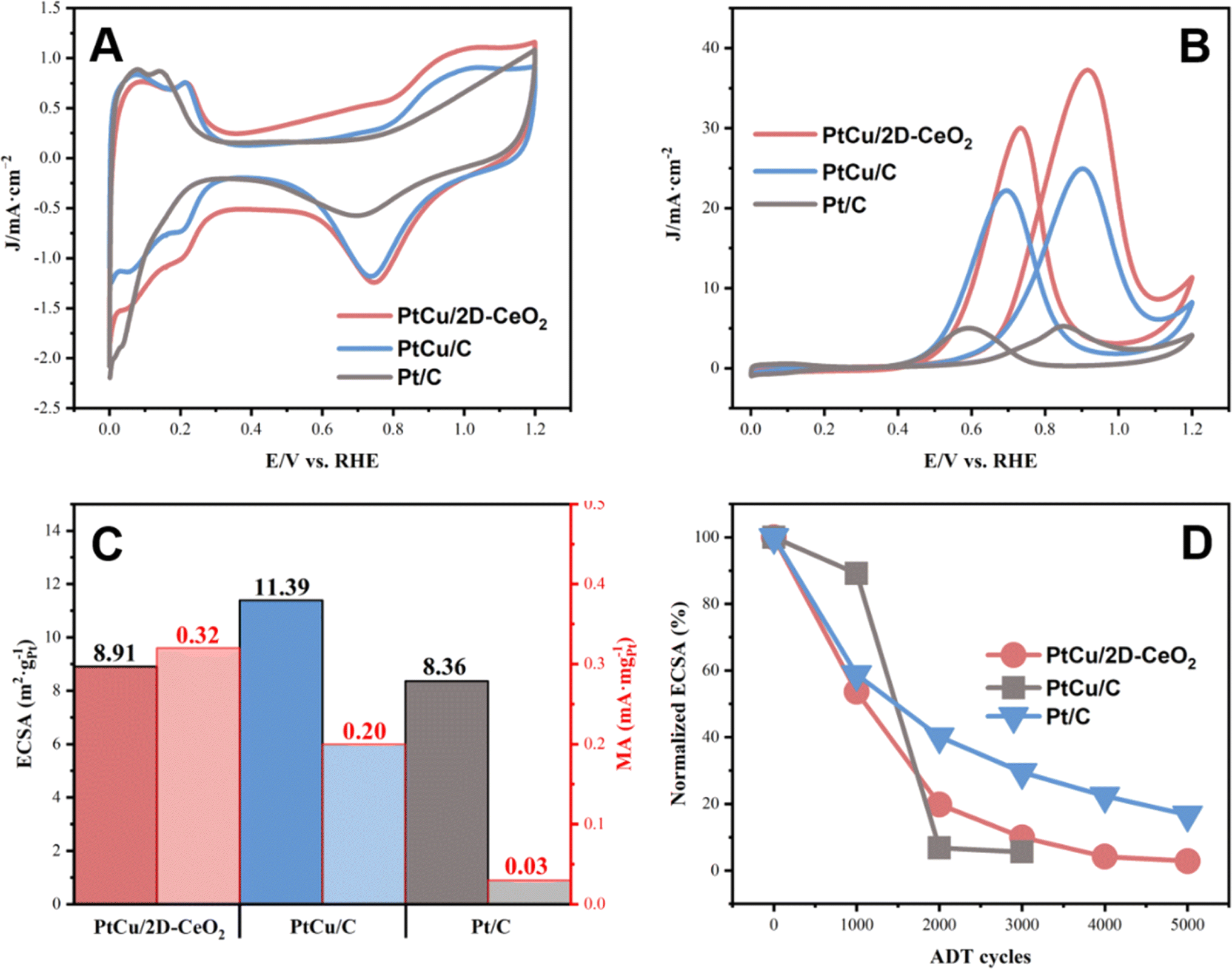 | ||
| Fig. 4 The CV (A) and MOR (B) plots of PtCu/2D-CeO2, PtCu/C, and Pt/C; ECSA and MA diagrams of PtCu/2D-CeO2, PtCu/C, and Pt/C (C); the normalized ECSA after ADT cycling (D). | ||
Fig. 4B shows the MOR performance of the catalysts. The peak current densities of forward scanning of PtCu/2D-CeO2, PtCu/C, and Pt/C are 37.24, 24.93, and 5.23 mA cm−2, respectively. The value of PtCu/2D-CeO2 is 7.08 times that of Pt/C, revealing the very high activity toward the MOR. The MA was calculated based on the limiting current density (Fig. 4C). The MA of PtCu/2D-CeO2, PtCu/C, and Pt/C is 0.32, 0.20, and 0.03 mA mgPt−1 respectively. The MA of PtCu/2D-CeO2 is 1.6 and 10.67 times those of PtCu/C and Pt/C, respectively. The specific activity (SA) of the catalyst was calculated from ECSA and MA (Table S4†). The SA of PtCu/C (0.18 mA cm−2) is much higher than that of Pt/C (0.04 mA cm−2), confirming the high activity of the PtCu alloy. PtCu/2D-CeO2 shows the SA of 0.36 mA cm−2, which is double the value of PtCu/C, owing to the high Pt0%. The current density ratio of peak values of the forward scanning (If) to backward scanning (Ib) is related to the Faraday efficiency, i.e., the oxidation capacity of methanol oxidation and its intermediate products. The If/Ib ratios of PtCu/2D-CeO2, PtCu/C, and Pt/C are 1.24, 1.12, and 1.05 respectively. The higher If/Ib ratio PtCu/2D-CeO2 exhibits better oxidation capability for the intermediates.45 The durability is also an important issue. Fig. 4D shows the MOR curves of PtCu/2D-CeO2, PtCu/C, and Pt/C after 5000 cycles and their MA losses. The forward scan current of Pt/C after 1000 cycles was already lower than the negative scanning current value, which indicated that the Faraday efficiency of Pt/C decreased dramatically after 1000 cycles. However the forward scan current of PtCu/C and PtCu/2D-CeO2 is always higher than negative ones, illustrating the high Faraday efficiency. The degradation of catalytic activity was directly observed by the MA loss. The activity of the Pt/C catalyst decreases sharply after 2000 ADT cycles. The MA of PtCu/C after 5000 cycles retained 14.21%; due to the better durability of the PtCu alloy, the PtCu/2D-CeO2 gives a slight improvement in durability. The negative shift of the d-band center of Pt 4f and Cu 2p caused by the strong interaction between PtCu and 2D-CeO2 represents the lower adsorption energy of the reactant and intermediates, which facilitates the MOR. Due to the unique redox behavior between Ce3+ and Ce4+, the dissolved Ce ions could also participate in the radical scavenging reaction, which prevents the chemical degradation of polymeric components in the cell.46
The ORR activity was also tested (Fig. 5). The onset potentials of PtCu/2D-CeO2, and PtCu/C catalysts are very close at around 0.93 V. PtCu/2D-CeO2 shows lower limiting current density (4.8 mA cm−2) than PtCu/C (6.0 mA cm−2). The Tafel slopes of PtCu/2D-CeO2 and PtCu/C are 242.90 and 268.20 mV dec−1 respectively, and the lower value represents that 2D-CeO2 is of great help to improve the ORR performance. There are two- and four-electron paths in the ORR. The two-electron path generates H2O2 which damages the proton exchange membrane;47 therefore, it is necessary to calculate the number of transferred electrons in the reaction. The K–L equation was used for the diffusion-restricted region to calculate the number of transferred electrons. The electron transfer number of PtCu/2D-CeO2 is calculated to be 2.66, indicating that the ORR of PtCu/CeO2 is more inclined to the four-electron transfer process, while the latter is more inclined to the two-electron reaction. To evaluate the durability of the catalysts, we conducted the accelerated durability tests (ADTs) to test their ORR performance after 10000 cycles (Fig. 5D–F). In the first 1000 cycles, the ECSA of both PtCu/2D-CeO2 and PtCu/C increases by over 20%, because the active sites are gradually exposed. The performance decreases as the cycling goes on. After 5000 ADT cycles, PtCu/C (108.35%) retains higher ECSA than PtCu/2D-CeO2 (85.48%). The onset potentials of PtCu/2D-CeO2, and PtCu/C remain consistent with the original ones.
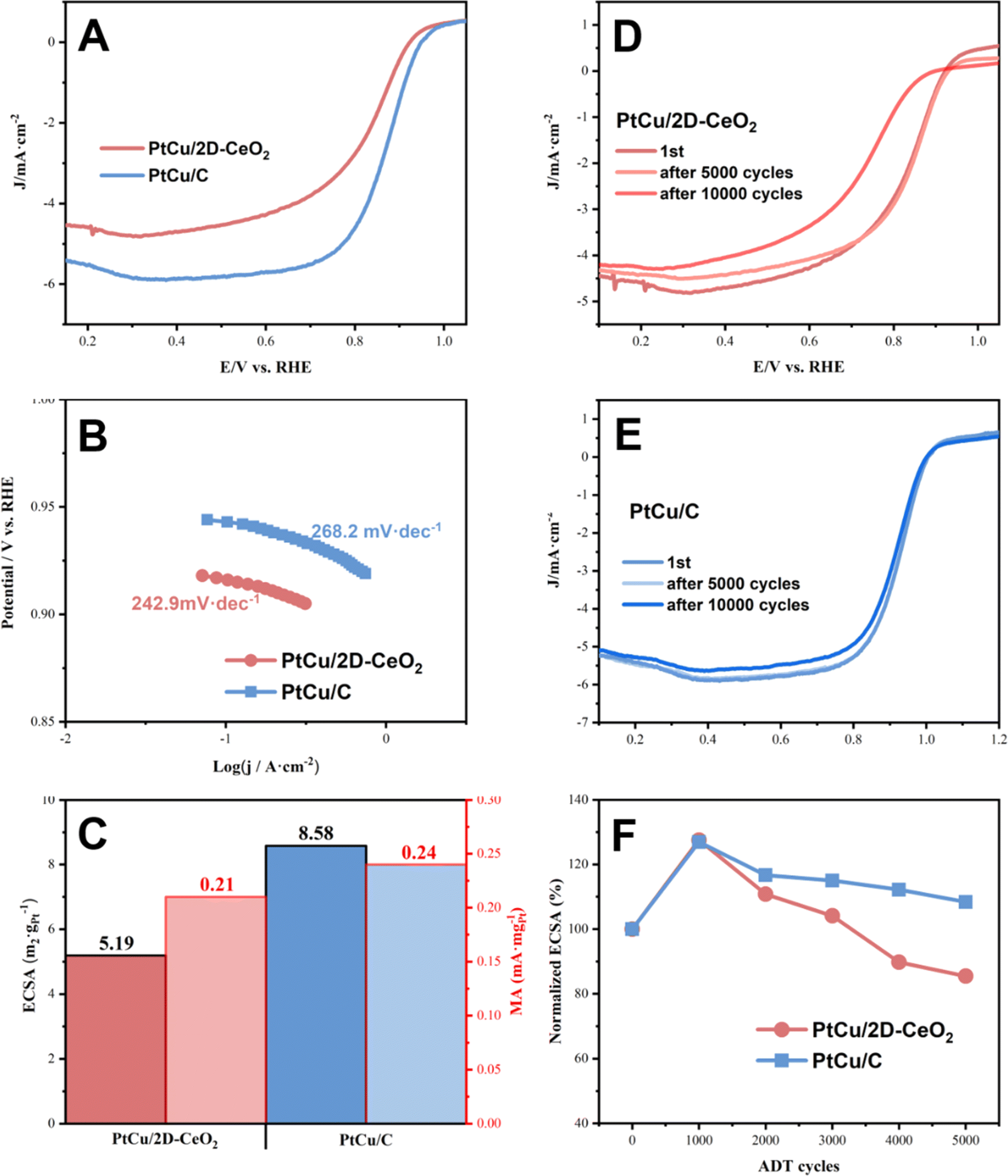 | ||
| Fig. 5 The LSV curves (A), Tafel slopes (B), and ECSA and MA (C) of PtCu/C and PtCu/2D-CeO2; LSV curves of PtCu/2D-CeO2 (D) and PtCu/C (E) 10000 ADT cycles; and normalized ECSA (F). | ||
Conclusions
In this paper, a catalyst with the geometry of order–disorder two-dimensional ceria nanobelt (2D-CeO2) supported PtCu nanoparticles and hollow nanospheres was prepared. Owing to the order–disorder structure, the 2D-CeO2 shows a high concentration of oxygen vacancies, leading to the strong interaction with the PtCu alloy. The Pt0 proportion of PtCu/2D-CeO2 is much higher than that of PtCu/C, and both Pt 4f and Cu 2p orbitals shift negatively which indicates the lower d-band center of the PtCu alloy. The PtCu/2D-CeO2 exhibits specific activity which is 10.67 times that of Pt/C toward the methanol oxidation reaction and comparable activity toward the oxygen reduction reaction. The high performance of PtCu/2D-CeO2 mainly comes from two aspects: (i) the high volume of oxygen vacancies of 2D-CeO2 facilitates electron transportation, leading to high conductivity. (ii) The sufficient active sites and the lower d-band center of the PtCu alloy facilitate the adsorption and dissociation of reactants. This work offers an attractive avenue to design order–disorder oxide supports to achieve high performance and long-term durability of catalysts.Data availability
Data are available on request from the authors. The data that support the findings of this study are available from the corresponding author, Feng Xu, upon reasonable request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the Natural Science Foundation of Fujian Province (No. 2022J01089).References
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao, M. Zeng, J. Zhong, C. Jin, Y. Li, S.-T. Lee and H. Dai, Nat. Commun., 2015, 6, 10035 CrossRef CAS PubMed.
- Q. Fang, H. Wang, X. Lv, X. Wei, X. Luo, J. Huang, L. Jiao, W. Gu, W. Song and C. Zhu, ACS Sustain. Chem. Eng., 2021, 9, 13039–13046 CrossRef CAS.
- J. Wang, B. Zhang, W. Guo, L. Wang, J. Chen, H. Pan and W. Sun, Adv. Mater., 2023, 35, 2211099 CrossRef CAS PubMed.
- X. Fan, M. Zhao, T. Li, L. Y. Zhang, M. Jing, W. Yuan and C. M. Li, Nanoscale, 2021, 13, 18332–18339 RSC.
- M. Li, F. Yang, J. Chang, A. Schechter and L. Feng, Acta Phys.-Chim. Sin., 2023, 2301005 Search PubMed.
- A. Riese, D. Banham, S. Ye and X. Sun, J. Electrochem. Soc., 2015, 162, F783–F788 CrossRef CAS.
- G. Bai, C. Liu, Z. Gao, B. Lu, X. Tong, X. Guo and N. Yang, Small, 2019, 15, 1902951 CrossRef PubMed.
- W. Qiao, L. Yu, J. Chang, F. Yang and L. Feng, Chin. J. Catal., 2023, 51, 113–123 CrossRef CAS.
- B. Cai, S. Henning, J. Herranz, T. J. Schmidt and A. Eychmüller, Adv. Energy Mater., 2017, 7, 1700548 CrossRef.
- H. C. Song, G. R. Lee, K. Jeon, H. Lee, S. W. Lee, Y. S. Jung and J. Y. Park, ACS Nano, 2020, 14, 8335–8342 CrossRef CAS PubMed.
- A. Kakoria, B. Devi, A. Anand, A. Halder, R. R. Koner and S. Sinha-Ray, ACS Appl. Nano Mater., 2018, 2, 64–74 CrossRef.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruña, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317–9332 CrossRef CAS.
- Z. Yan, W. Wei, J. Xie, S. Meng, X. Lü and J. Zhu, J. Power Sources, 2013, 222, 218–224 CrossRef CAS.
- K. Sasaki, L. Zhang and R. R. Adzic, Phys. Chem. Chem. Phys., 2008, 10, 159–167 RSC.
- X. Tian, H. Tang, J. Luo, H. Nan, T. Shu, L. Du, J. Zeng, S. Liao and R. R. Adzic, ACS Catal., 2017, 7, 3810–3817 CrossRef CAS.
- Y. Kuang, W. Qiao, S. Wang, F. Yang and L. Feng, ACS Mater. Lett., 2024, 6, 1722–1731 CrossRef CAS.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC.
- J. Yu, D. Wang, G. Wang, Y. Cui and S. Shi, Adv. Mater., 2022, 35, 2209210 CrossRef PubMed.
- T. A. Zepeda, R. Ponce-Pérez, A. Solis-Garcia, J. Guerrero-Sanchez, S. Fuentes and S. A. Gomez, Appl. Catal., B, 2023, 336, 122936 CrossRef CAS.
- W. Chen, J. Xue, Y. Bao and L. Feng, Chem. Eng. J., 2020, 381, 122752 CrossRef CAS.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28 CrossRef CAS.
- Y. Zhou, A. Chen, J. Ning and W. Shen, Chin. J. Catal., 2020, 41, 928–937 CrossRef CAS.
- B. Lu, R. Dun, W. Wang, J. Huang, J. Wu, Z. Hua and J. Shi, Appl. Catal., B, 2024, 342, 123343 CrossRef CAS.
- Y. Zhang, Y. Zhang, Z. Zeng and D. Ho, Mater. Horiz., 2023, 10, 2904–2912 RSC.
- F. Xu, S. Cai, B. Lin, L. Yang, H. Le and S. Mu, Small, 2022, e2107387 CrossRef PubMed.
- F. Wang, X. Wang, Z. Guo, J. Yu and H. Zhu, Energy Fuels, 2021, 35, 3368–3375 CrossRef CAS.
- T. Zou, Y. Wang and F. Xu, ACS Appl. Mater. Interfaces, 2023, 15, 58296–58308 CrossRef CAS PubMed.
- M. Guo, Q. Tu, L. Wang, Y. Tang, H. Song, J. Zhou, Z. Zhang, Y. Wang and C. Liu, Int. J. Hydrogen Energy, 2019, 44, 6886–6895 CrossRef CAS.
- X. Qiao, J. Jin, H. Fan, Y. Li and S. Liao, J. Mater. Chem. A, 2017, 5, 12354–12360 RSC.
- Z. Guo, Z. Liu and R. Tang, Mater. Chem. Front., 2024, 8, 1703–1730 RSC.
- N. K. Eswar, V. V. Katkar, P. C. Ramamurthy and G. Madras, Ind. Eng. Chem. Res., 2015, 54, 8031–8042 CrossRef CAS.
- A. Chen, X. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. Jin, C. Yang, A. Nefedov, C. Wöll, Y. Wang and W. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS.
- Y. Liu, A. Zhang, L. Xue, H. Zhang, Y. Hao, Y. Wang, J. Wu and S. Zeng, ACS Appl. Energy Mater., 2021, 5, 604–614 CrossRef.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- M. Jin, Y. Liu, X. Zhang, J. Wang, S. Zhang, G. Wang, Y. Zhang, H. Yin, H. Zhang and H. Zhao, Appl. Catal., B, 2021, 298, 120545 CrossRef CAS.
- F. Xu, K. Cheng, Y. Yu and S. Mu, Electrochim. Acta, 2017, 229, 253–260 CrossRef CAS.
- Y. Yu, X. Wang, W. Gao, P. Li, W. Yan, S. Wu, Q. Cui, W. Song and K. Ding, J. Mater. Chem. A, 2017, 5, 6656–6663 RSC.
- B. Feng, Z. U. Khan and W. U. Khan, Environ. Sci.: Nano, 2023, 10, 1163–1176 RSC.
- J. Wang, J. Zhang, G. Liu, C. Ling, B. Chen, J. Huang, X. Liu, B. Li, A.-L. Wang, Z. Hu, M. Zhou, Y. Chen, H. Cheng, J. Liu, Z. Fan, N. Yang, C. Tan, L. Gu, J. Wang and H. Zhang, Nano Res., 2020, 13, 1970–1975 CrossRef CAS.
- H. Hu, F. Ding, H. Ding, J. Liu, M. Xiao, Y. Meng and L. Sun, Adv. Compos. Hybrid Mater., 2020, 3, 498–507 CrossRef CAS.
- G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. K. Kirkensgaard, T. E. L. Smitshuysen, S. Kadkhodazadeh, M. Juelsholt, K. M. Ø. Jensen, K. Anklam, H. Wan, J. Schäfer, K. Čépe, M. Escudero-Escribano, J. Rossmeisl, A. Quade, V. Brüser and M. Arenz, Nat. Mater., 2020, 20, 208–213 CrossRef PubMed.
- Y. Fang, H. Li, Q. Zhang, C. Wang, J. Xu, H. Shen, J. Yang, C. Pan, Y. Zhu, Z. Luo and Y. Guo, Environ. Sci. Technol., 2022, 56, 3245–3257 CrossRef CAS PubMed.
- K. Zhang, W. Guo, Z. Liang and R. Zou, Sci. China Chem., 2019, 62, 417–429 CrossRef CAS.
- L. Zhang, L.-X. Ding, H. Chen, D. Li, S. Wang and H. Wang, Small, 2017, 13, 1604000 CrossRef PubMed.
- K. R. Yoon, J. M. Kim, K. A. Lee, C.-K. Hwang, S. G. Akpe, Y. J. Lee, J. P. Singh, K. H. Chae, S. S. Jang, H. C. Ham and J. Y. Kim, J. Power Sources, 2021, 496, 229798 CrossRef CAS.
- Y. Duan, Y. Sun, S. Pan, Y. Dai, L. Hao and J. Zou, ACS Appl. Mater. Interfaces, 2016, 8, 33572–33582 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00449c |
| This journal is © The Royal Society of Chemistry 2024 |