
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 hydrogenation on ruthenium: comparative study of catalyst supports†
Göran
Baade
 a,
Jens
Friedland
a,
Koustuv
Ray
b and
Robert
Güttel
*a
a,
Jens
Friedland
a,
Koustuv
Ray
b and
Robert
Güttel
*a
aInstitute of Chemical Engineering, Ulm University, Ulm, Germany. E-mail: robert.guettel@uni-ulm.de
bDepartment of Chemical Engineering, Indian Institute of Technology Kharagpur, Kharagpur, India
First published on 4th November 2024
Abstract
To achieve a significant reduction in anthropogenic CO2 in the near future, captured carbon has to be valorized. To this end, CO2 may be activated using H2 to form sustainable fuels (synthetic natural gas), platform chemicals (methanol) and higher hydrocarbons (modified Fischer–Tropsch process). In this work we synthesize Ru based catalysts from various commercially available support materials and test them under lower temperatures than usually employed at various partial pressures of CO2 and H2 using methanation as a model reaction. The results show Ru/TiO2, Ru/ZrO2 and Ru/Al2O3 as the most active catalysts with high activity, selectivity towards methane (>95%), and stability with little to no deactivation over 80 h. These most promising catalysts are further tested and kinetic parameters determined, which find reaction orders and activation energies in agreement with literature, but differing from catalyst to catalyst, hinting at complex reaction mechanisms including the support as well as the Ru. The TOF calculated for Ru/TiO2 at 190 °C is 5.7 s−1 and highlights it as the most active catalyst in this work. The study opens new and promising avenues for the valorization of CO2, as well as a basis to compare future optimizations and advances in the field of Ru-based CO2 conversion.
Sustainability spotlightIn accordance with the UN sustainable development goals regarding climate action (SDG 13), it is necessary to reduce net CO2 emissions by 2030. Since the industrial – and energy sectors cannot completely pivot away from fossil fuels as raw material until 2030, it is necessary to establish carbon-neutral fossil fuel sources. The infrastructure to use methane is already in place, which makes it a candidate for energy storage and transport. The synthesis of methane from CO2 is already well understood for centralized plants, but energy generation, H2 electrolysis and capture of CO2 are expected to be decentralized. It is therefore necessary to establish what a process at much lower temperatures may look like as foundation for optimization until 2030 and beyond. |
1 Introduction
Climate change is one of the major motivators of contemporary science and greenhouse gases are responsible for global warming, which has potentially devastating results. Among the greenhouse gases CO2 is the most notable, even when accounting for different global warming potentials, due to the large amounts that are emitted.1 Recently The Sustainable Development Goals Report of the UN has highlighted the need to drastically reduce the Greenhouse Gas emissions by 2030 and be net zero by 2050 to avoid a “Climate Calamity”.2 In accordance with these sustainability goals it is necessary to take urgent action to combat climate change and its impact.Great efforts are being made to decarbonize fossil-based industries. Among the options to reach net-zero carbon capture from exhaust gases or ambient air and the subsequent utilization (Carbon Capture and Utilization, CCU) are being investigated. As roughly half of all CO2 emissions are emitted decentralized as opposed to relatively pure point sources, a remote Direct Air Capture (DAC) and subsequent conversion without costly transport of gaseous CO2 is among the options with most potential and flexibility.3 For safe and efficient decentralized utilization of captured carbon, direct hydrogenation of CO2 at low temperatures may keep cost low and prevent potential hazards.
Ruthenium has been found to be the most active catalyst to hydrogenate CO2 with a high selectivity towards methane.4 It is widely understood that the support material of a ruthenium-based catalyst has a significant influence on the activity and stability during the reaction, which is why many studies have been conducted to determine the ideal support.5–7 However, these studies usually focus on high reaction temperatures. While studies under low-temperature conditions also exist,8–10 they usually investigate one single support material and vary the process conditions. Additionally, other works focus on the stabilization of specific active phases,11 complex catalyst geometries12 or the use of novel concepts like photo-assisted catalysis.13 The reported results, while promising, are likely far from widespread application in the short timeframe left to drastically reduce net CO2 emission. Therefore, it is necessary to close the knowledge gap in the application of different support materials in the low-temperature CO2 hydrogenation and gain a base-line understanding of what is readily achievable on Ru with commercially available materials.
In this study we investigate the activity and selectivity of a selection of ruthenium-based catalysts in the CO2 hydrogenation reaction. The catalysts are synthesized using commercial grade materials and a facile synthesis to minimize interference stemming from the procedure itself. All catalysts are tested under mild conditions by varying temperatures and partial pressures to form a quantitative understanding of the most promising candidates. These results are corroborated by subjecting the most promising catalyst materials to additional experiments to determine kinetic parameters for comparison with existing literature data and as a basis for further investigations.
2 Experimental
2.1 Catalyst synthesis and characterization
Six commercial grade support materials are used as received, among these are three reducible metal oxides: TiO2 (P25 – Evonik), ZrO2 (IBU-tec) and CeO2 (IBU-Tec) as well as two irreducible metal oxides: Al2O3 (Aeroxide Alu 130 – Evonik) and SiO2 (Aerosil 380 – Evonik) as well as one activated carbon support (IAC 402 – Infiltec). All supports are pressed into pellets, crushed and sieved to a size fraction of 150–200 μm before use to ensure comparability during the impregnation step.The catalysts are prepared by excess solvent impregnation in which the support is poured into a solution of the Ru precursor ruthenium(III) chloride hydrate (38.0–42.0% Ru basis, Sigma-Aldrich) in water to yield a nominal mass loading of 1% Ru on the final catalyst. The solution is placed in an oil bath heated to 323 K and stirred until all solvent has evaporated. The resulting powder is placed in an oven an dried at 378 K over night, followed by calcination at 473 K for 4 h in static air. After cooling down to room temperature the samples are washed three times with dilute ammonia (3 wt%) solutions and then three times with water. This was shown to facilitate the removal of residual chlorides and thus increase activity of the catalysts.14 After drying of the materials at 378 K, the dry powders are again pressed into pellets, crushed and sieved to a size fraction of 150–200 μm to prevent heat and mass transfer limitations during the reaction experiments.
The catalysts are subsequently characterized (3Flex, Micromeritics) by N2 physisorption, H2 chemisorption, H2 temperature programmed reduction (H2 TPR) and CO2 temperature programmed desorption (CO2 TPD) measurements. The samples are prepared for physisorption measurements by degassing at 1.33 mbar and up to 200 °C. The ad- and desorption isotherms are taken using a liquid nitrogen bath at relative pressures from 0.01 up to 0.99. The specific surface areas of the samples are calculated using the Brunauer–Emmett–Teller (BET) method. For the H2 TPR experiments samples are dried under flowing Ar at 120 °C for 30 min, cooled down to 50 °C and then the feed is switched to 10% H2 in Ar. After the Thermal Conductivity Detector (TCD) baseline signal stabilizes, the temperature is increased to 1000 °C with a heating rate of 10 K min−1. The H2 chemisorption experiments are conducted with the volumetric method. Circa 100 mg of each sample are reduced with H2 at 200 °C for 4 h and degassed at 300 °C for 6 h. The H2 adsorption isotherm is recorded at 50 °C. After degassing for 1 h a repeat measurement is taken. The H2 chemisorption isotherms are used to calculate the active metal surface area using the Langmuir model. The dispersion of active metal DRu on the catalyst is estimated by multiplying the H2 uptake of the catalyst QH2 with the molar mass of Ru MRuviaeqn (1).
| DRu = QH2·MRu | (1) |
The CO2 TPD experiments are conducted by in situ drying of circa 100 mg of sample at 105 °C for 10 minutes under flowing Ar. Then the sample is reduced by 10% H2 in Ar at 200 °C for 4 h. After reduction, the sample is cooled down to 50 °C and flushed with Ar until a stable TCD baseline signal is achieved. The gas flow is then switched to CO2 (4.5 purity, MTI Industriegase AG) and the sample is saturated with CO2 for 20 minutes. Afterwards the feed is switched to He (5.0 purity, MTI Industriegase AG) to remove physisorbed CO2. After a stable TCD baseline signal is achieved, the temperature is increased by 10 K min−1 up to 500 °C, where it is held for 5 min.
Powder X-ray diffraction (PXRD) patterns of the fresh supports and finished catalysts are performed on a X'Pert MDP Pro (PANanalytical) using CuKα radiation between 5° and 80° 2θ. X-ray photoelectron spectroscopy (XPS) spectra of the as made catalysts are recorded on a PHI 5800 (Physical Electronics). Transmission electron microscopy (TEM) images of the finished catalysts are taken using a JEOL 1400 microscope.
2.2 Reaction experiments
All reaction experiments were conducted in a 1/4 inch steel tube with a concentric 1/16 inch inner steel tube to place a K-type thermocouple in the center of the packed catalyst bed. The catalyst bed is fixed with quartz wool and 400 mg of 0.5–0.75 mm silica beads before and after the bed. Previous experiments have shown no detectable blind activity of the setup. H2 (5.0 purity, MTI Industriegase AG) and CO2 (4.5 purity, MTI Industriegase AG) are dosed by mass flow controllers (Bronkhorst) at a total flow rate of 50 mLSTP min−1. A detailed flowsheet of the setup can be found in the ESI.† 100 mg of catalyst are diluted with 300 mg SiC in the size fraction 150–212 μm and filled into the reactor. The catalyst is reduced in situ under flowing H2 at ambient pressure and 473 K for 8 h.To gain insights into the (de-)activation behavior of the catalyst, reference conditions are set before and after a parameter variation. Every set of parameters is held for 8 h to ensure enough data points and stable behavior. The temperatures and partial pressures of the individual operating points are shown in Table 1. The reaction program follows seamless after reduction (A) by the first reference condition (B) of 200 °C, 5 barg and a H2/CO2 ratio of 3. For the temperature variation (C–F) the reaction temperature is set to 150 °C and in subsequent 20 K steps increased to 220 °C. After the first variation the process conditions are set to the same values as for “B”, now denoted as “G” in order to evaluate deactivation. The next reference point (H) differs from “G” by a higher pressure of 12.5 barg, at which the H2/CO2 ratio is varied between 1 and 5 in the following two operating points (I–J). Lastly the same conditions as in “H” are set in “K” again to check for deactivation.
| A | B | C | D | E | F | G | H | I | J | K | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| T/°C | 200 | 200 | 160 | 180 | 200 | 220 | 200 | 200 | 200 | 200 | 200 |
| p CO2/barg | 0 | 1.25 | 1.25 | 1.25 | 1.25 | 1.25 | 1.25 | 3.125 | 6.25 | 2.083 | 3.125 |
| P/barg | 0 | 5 | 5 | 5 | 5 | 5 | 5 | 12.5 | 12.5 | 12.5 | 12.5 |
The analysis of the dry product gas is conducted via an online gas chromatograph (GC) equipped with a TCD and a flame ionization detector (FID). One sample is taken roughly every 17.5 min and the evaluation of the results is done by taking the mean of the last three samples of the respective operating point. The GC is calibrated to calculate the percentage of C1–C6 alkanes. The corresponding unsaturated hydrocarbons can be detected, but are not calibrated.
The conversion of CO2, XCO2 is calculated from the inlet molar flow rate of CO2, ṅCO2,in and outlet molar flow rate, ṅCO2,out by assuming differential conditions (eqn (2)). With this assumption the conversion is only dependent on the set inlet molar fraction of CO2, xCO2,in, the outlet molar fractions measured by the FID, xi and the known carbon number of the corresponding hydrocarbons ni,C.
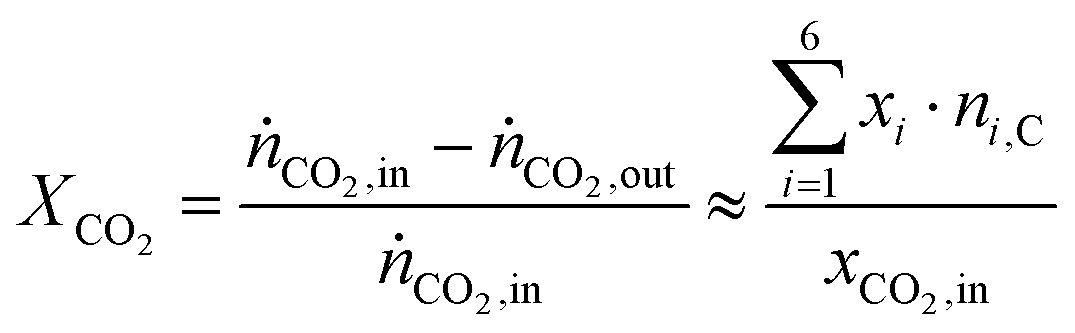 | (2) |
The CO2 consumption rate rCO2 is calculated normalized to the used catalyst mass mcat according to eqn (3).
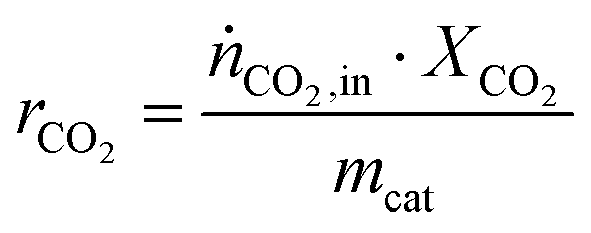 | (3) |
The selectivities towards methane and CO are calculated from the GC data by eqn (4).
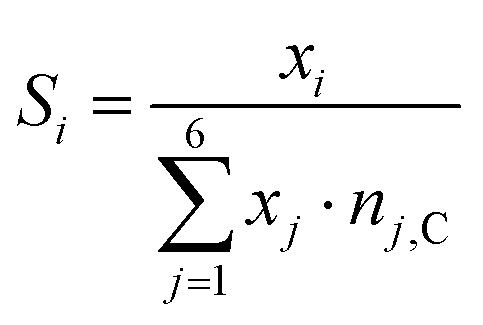 | (4) |
As no other products are detected, the selectivity to higher hydrocarbons is calculated according to eqn (5). The index C2+ denotes the lumped paraffins and olefins in the carbon number range 2–6.
| SC2+ = 1 − (SCH4 + SCO) | (5) |
Hydrocarbons higher than hexane are not detected.
With the height of the catalyst bed, hcat, the inner diameter of the reactor tube, Di and the outer diameter of the thermocouple tube, De, the bulk density of the catalysts is calculated viaeqn (6).
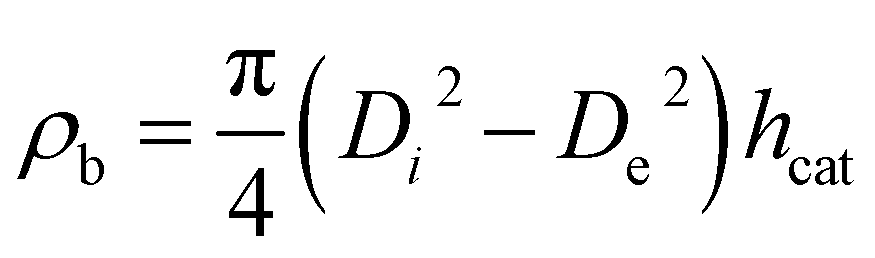 | (6) |
With the bulk density the consumption rate of CO2 is related to the volume of the catalyst bed by eqn (7).
| rCO2,V = rCO2·ρb | (7) |
Due to the low conversion associated with the assumption of differential conditions, the carbon balance is implicitly assumed as closed.
2.3 Kinetic experiments
To gain insight into the reaction network, the results from the reaction experiments are used to design an experiment to determine apparent reaction orders of CO2 and H2, as well as the apparent activation energy of the reaction. The three most promising candidates are chosen for this kinetic study. 80 mg of catalyst are used and the total volumetric flow of gas is set to 100 mLSTP min−1. The H2 and CO2 flow rates are varied with Argon (5.0 purity, MTI Industriegase AG) as balance. The pressure is set to 5 barg. The kinetic test program is shown in Table 2. First the flow rates are set to 50/10/40 mLSTP min−1 H2/CO2/Ar and the temperature is varied from 150 to 190 °C in 20 K steps to determine the activation energy. The first temperature is held for 8 h to ensure stable operation; the other two temperatures are held 4 h. Afterwards the reactant inlet flow composition is varied at 190 °C with 2 h of reaction time as the optimum between stable operation and deactivation of the catalyst. For every operating point the last sample from the GC is chosen for evaluation. Measured reaction rates are evaluated by assuming that the power law expression in eqn (8) sufficiently describes the reaction rate r with the partial pressures of CO2, pCO2 and H2, pH2 as well as the respective reaction orders, nCO2 and nH2.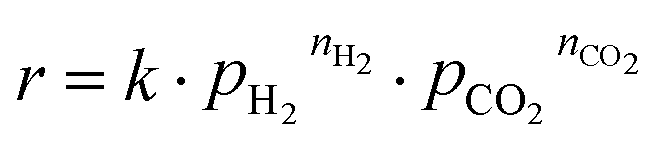 | (8) |
| A | T-variation | p i -variation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T/°C | 200 | 150 | 170 | 190 | 190 | 190 | 190 | 190 | 190 | 190 | 190 | 190 |
| H2/CO2 | 50/0 | 50/10 | 50/10 | 50/10 | 45/10 | 45/15 | 50/15 | 60/10 | 60/15 | 60/20 | 50/20 | 45/20 |
| Duration/h | 8 | 8 | 4 | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
The values for the reaction orders and the kinetic constant, k are estimated by fitting the experimentally obtained reaction rates and known partial pressures to eqn (9). With the measured temperatures of the catalyst bed and the determined rate constants, the apparent activation energy is estimated based on the linearized Arrhenius equation, while the Arrhenius plot is established, as well. The turn over frequency (TOF) is calculated by eqn (9) using the H2 sorption capacity, QH2 and the mass fraction of Ru, ωRu to estimate the number of active sites on the catalyst.
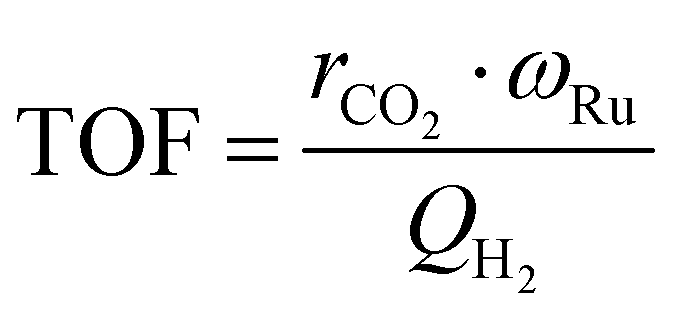 | (9) |
3 Results and discussion
3.1 Catalyst characterization
The physisorption measurements were conducted on the pure supports and on the final catalyst and both measured BET surface areas are compared with the manufacturer specifications in Table 3. The results show that the pure supports are within the manufacturer specifications, with the exception of the activated carbon, and the BET surface area of the catalyst is only slightly below that of the pure support. This shows that the pore structure remains intact after the synthesis procedure and that accessibility of the active material is provided.| Material | Specification/m2 g−1 | Pure support/m2 g−1 | Ru/support/m2 g−1 |
|---|---|---|---|
| TiO2 | 35–65 | 55.3 ± 0.3 | 46.3 ± 0.2 |
| ZrO2 | 25–35 | 32.7 ± 0.1 | 22.3 ± 0.4 |
| CeO2 | 38–42 | 39.4 ± 0.3 | 35.9 ± 0.2 |
| SiO2 | 350–410 | 346.7 ± 3.7 | 232.3 ± 2.0 |
| Al2O3 | 110–150 | 122.0 ± 0.7 | 98.7 ± 1.1 |
| C | 1100 | 890.9 ± 28.5 | 808.1 ± 24.7 |
Results of H2 TPR experiments for all catalyst and supports are shown in Fig. 1 for the relevant temperature range. The Ru/C and Ru/CeO2 catalysts exhibit a much larger TCD-signal than the other samples and are therefore scaled down by a factor of 2 and 3, respectively. Ru/C measurements show a constant and steady increase in H2 consumption, possibly indicating hydrogenation of the carbon support, making a stable operation of this catalyst in reaction experiments unlikely. Ru/CeO2 shows two pronounced peaks between 101–119 °C and 141–163 °C indicating the reduction of two different Ru species. All other catalysts show H2 consumption over a broader range of temperatures, indicating no separation between strongly- and weakly interacting Ruthenium. The silica-supported catalyst shows little H2 consumption which may indicate strong interaction between the silica and the ruthenium resulting in little reduction at the used temperature or a loss of active material throughout the synthesis procedure. All other catalysts are reducible below 200 °C, which is why this temperature is chosen as the reduction temperature for the chemisorption and reaction experiments. Metallic surface areas and H2 uptake measured by H2-chemisorption, as well as estimated dispersions provided in Table 4 show a higher Ru surface area on reducible metal oxides, indicating a better stabilization of the Ru precursor or the metal throughout the synthesis and pretreatment. The silica-supported catalyst shows an especially small metallic surface area again indicating a insufficient stabilization of the active material. By comparing the BET surface area and the Ru surface area and dispersion, it becomes clear that a higher surface area of the support does not automatically result in a higher active surface area and better dispersion.
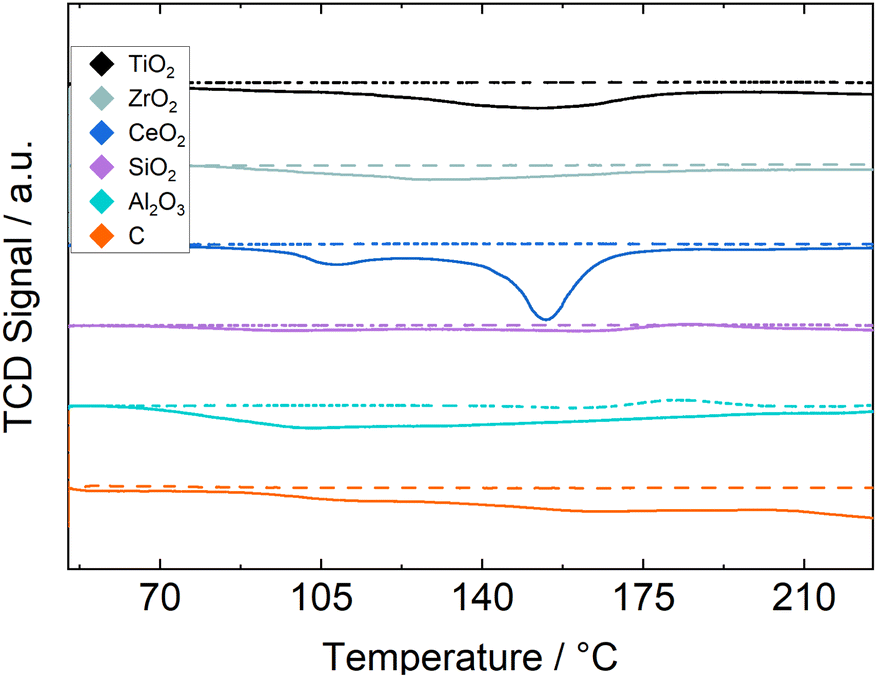 | ||
| Fig. 1 TCD-signal during H2 TPR measurements, for pure support (dashed lines) and Ru/support (solid lines). | ||
| Catalyst | Metal surface area/m2 gcat−1 | H2 uptake/μmol g−1 | Dispersion/% |
|---|---|---|---|
| Ru/TiO2 | 0.96 | 7.51 | 7.59 |
| Ru/ZrO2 | 5.82 | 72.6 | 73.4 |
| Ru/CeO2 | 6.57 | 80.4 | 81.2 |
| Ru/Al2O3 | 0.24 | 4.70 | 4.75 |
| Ru/SiO2 | 0.04 | 0.63 | 0.64 |
| Ru/C | 0.59 | 4.43 | 4.47 |
The CO2-TPD results for both the catalysts and fresh supports are shown in Fig. 2 to distinguish between desorption peaks from the support and changes due to the supported Ru. Oxygen vacancies on the support enhance the adsorption of CO2 (ref. 15 and 16) and therefore the addition of Ru might increase the number of adsorption sites on the support due to H2 spillover. In general the difference between the two signals is not large, as the Ru loading is low. Especially for the Ru/ZrO2 catalyst the two lines barely deviate, indicating either very little adsorption of CO2 on both support and active material or a strong bonding of CO2 to ZrO2 and little bonding to the supported Ru. The only noticeable deviation is detected between 270 and 380 °C. The deviation between the signals is similarly small for the Ru/SiO2 catalyst with a step change for both catalyst and support at 260 °C. This indicates a large adsorption capacity for CO2 on SiO2 and little to no influence from the supported Ru. A similar step-change at 260 °C is seen for the Ru/C catalyst, but not the pure C support, this means that a significant amount of CO2 adsorbs due to the Ru. The Ru/Al2O3 catalyst shows a broad but more defined peak from 250 to 480 °C with a smaller peak at 100–160 °C. The Ru/TiO2 catalyst shows two distinct peaks at 100–250 °C and 300–390 °C with the former being larger, which is contrary to the other catalysts. The Ru/CeO2 catalyst shows a large deviation from the signal of the support with two noticeable peaks, the smaller between 80–170 °C and the larger being a step change starting from 260 °C. Here the CeO2 support also shows a noticeable peak from 350 to 470 °C indicating that much of the large CO2 capacity is due to the support and the addition of Ru results in more partial reduction of the CeO2 and thus an increase in CO2 accepting sites. The two defined H2 consumption peaks in Fig. 1 support the reduction of more than one surface species and therefore significant reduction of the support as well.
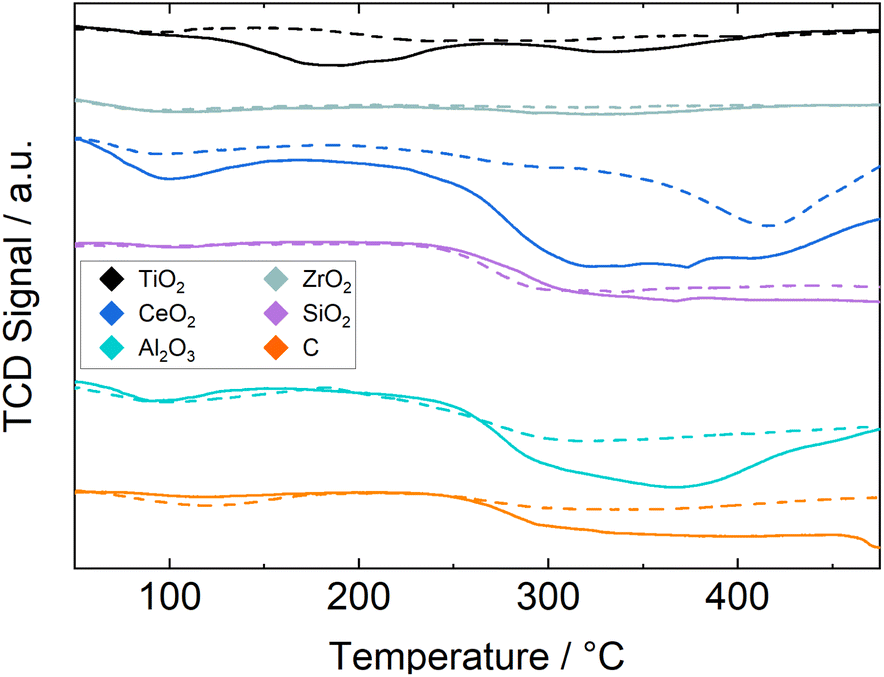 | ||
| Fig. 2 TCD-signal during CO2-TPD experiments, for pure support (dashed lines) and Ru/support (solid lines). | ||
The XPS results (Table 5) are used to analyze the surface composition of the catalysts. The results for the C percentage is unreliable, as the sample holder is a carbon pad and it is shown only for the sake of completion. The XPS results confirm the support as the main component of the catalysts with the respective metal and oxygen exhibiting the largest percentage. The oxygen/metal ratio indicates oxygen excess with respect to the expectations from stoichiometry, which may be explained by the presence of H2O. The low fraction of Cl for most catalysts shows the efficacy of the ammonia washing, while the low percentage of N shows that NH3 can subsequently be almost completely removed easily. The comparatively large Cl percentage on Ru/CeO2 might cause the poisoning of active sites and thus a decrease in activity. The measurements show a low fraction of Ru on the Ru/SiO2 catalyst, which indicates either a loss of active material, or a significant influence of other effects like wettability or capillary forces, which might lead to migration of Ru to the bulk of the support particles. This would cause Ru being outside of the surface sensitive XPS technique. The Ru amount on Ru/C is much higher than expected, which shows an uneven distribution of Ru on the Ru/C catalyst. Both observations may explain the low dispersion despite the large BET surface area for both the Ru/SiO2 and Ru/C catalyst.
| Material | C | O | Al | Si | Ti | Zr | Ce | N | Cl | Other impurities | Ru |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ru/TiO2 | 34.4 | 46.5 | 0 | 0 | 17.9 | 0 | 0 | 0.57 | 0.15 | 0 | 0.57 |
| Ru/ZrO2 | 22.1 | 53.8 | 0 | 0 | 0 | 20.4 | 0 | 1.09 | 0.27 | 0.74 | 1.55 |
| Ru/CeO2 | 14.0 | 54.3 | 0 | 0 | 0 | 0 | 25.0 | 0.63 | 2.37 | 2.21 | 1.55 |
| Ru/SiO2 | 1.38 | 72.0 | 0.25 | 26.2 | 0 | 0 | 0 | 0 | 0 | 0 | 0.11 |
| Ru/Al2O3 | 17.7 | 57.5 | 23.3 | 0 | 0 | 0 | 0 | 0.28 | 0.27 | 0.67 | 0.34 |
| Ru/C | 61.3 | 25.9 | 0 | 0.94 | 0 | 0.31 | 0 | 2.74 | 0.31 | 1.2 | 7.26 |
3.2 Variation of process parameters
The consumption rates of all synthesized catalysts under the process conditions described in Table 1 are shown in Fig. 3. Ru/TiO2 and Ru/ZrO2 are by an order of magnitude the most active catalysts, followed by Ru/Al2O3. Among the other catalysts Ru/C is the least active catalyst with activity circa two orders of magnitude lower than the Ru/ZrO2 catalyst. Generally, the activity of the synthesized catalysts is in the order of Ru/TiO2 ≈ Ru/ZrO2 ≫ Ru/Al2O3 > Ru/SiO2 > Ru/CeO2 > Ru/C. The calculated CO2 conversion of all catalysts at all operating points is shown in the ESI.† Vannice et al.17 investigated the influence of the support on the CO hydrogenation. In their experiments only Ru/TiO2 and Ru/Al2O3 showed enough activity to produce measurable quantities of methane, which is in line with our results. However, they did not test Ru/ZrO2 or Ru/CeO2 catalysts. While the high activity of Ru/TiO2 has been extensively studied and accredited to a range of metal-support-interaction mechanisms like lattice-matching18 or the formation of encapsulating overlayers,19 investigations into Ru/ZrO2 are scarce. One such study done by Alves et al.20 investigated various ZrO2 supports and found a high activity, but they did not compare their results with Ru/TiO2 catalysts. Our results thus show that Ru/ZrO2 is indeed of comparable or even higher activity than Ru/TiO2, especially at temperatures above 200 °C. Ru/CeO2, another reducible metal oxide, displays much lower activity, which is more comparable to Ru supported on the irreducible metal oxides SiO2 and Al2O3. This may be due to the availability of partially reduced sites, which is beneficial to facilitate the adsorption and bond-cleavage of carbon oxides. This bond-cleavage has been proposed before by works like that of Xu et al.19 The low activity of Ru/CeO2 indicates that the reduction of Ru/CeO2 catalysts requires careful tuning as noted by Dreyer et al.6 with their experiments using Ru/CeO2. The low activity here is therefore ascribed to the CeO2 not being reduced to a sufficient degree under the chosen conditions.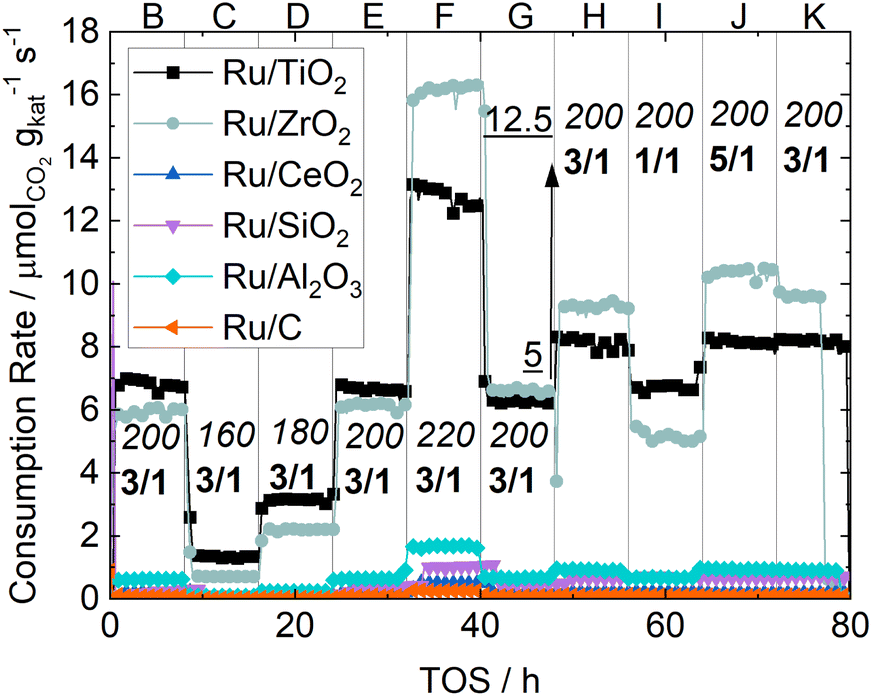 | ||
| Fig. 3 CO2 consumption rates of all catalysts under various process conditions as function of TOS. Reduction phase (A) not shown. Points C–F indicate different temperature at 5 barg and H2/CO2 ratio of 3 with B and G as reference measurements for deactivation due to temperature variation. Points H–J indicate the variation of the H2/CO2 ratio at 12.5 barg and 200 °C with H and K as reference measurements for deactivation due to composition variation. Italic numbers indicate the temperature in degree Celsius, bold numbers the H2/CO2 ratio and underlined numbers the switch from 5 barg to 12.5 barg. | ||
The comparison of the reference points B and G shows that in B Ru/TiO2 is the most active catalyst and in point G Ru/ZrO2 is more active than Ru/TiO2. This steady deactivation of Ru/TiO2 and a steady activation of Ru/ZrO2 is most notable at 220 °C, where the consumption rate of the Ru/TiO2 catalyst noticeable decreases and stays below that of the Ru/ZrO2 catalyst upon returning to 200 °C. It is also worth noting that for both these catalysts the consumption rate decreases noticeably when the syngas ratio is switched from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1. No effect on conversion is visible for Ru/TiO2 between H2/CO2 ratios of 3 and 5. The influence of the gas composition is much larger for Ru/ZrO2 when switching to the lowest ratio, while there is a noticeable difference between H2/CO2-ratios of 5 and 3. Since the active material is identical, this shows that the support material may change the reaction mechanism through hetero-contacts between the support and active metal in addition to the already studied oxygen-vacancies, which merely accelerate the rate-limiting C–O-bond cleavage.
:1 to 1:1. No effect on conversion is visible for Ru/TiO2 between H2/CO2 ratios of 3 and 5. The influence of the gas composition is much larger for Ru/ZrO2 when switching to the lowest ratio, while there is a noticeable difference between H2/CO2-ratios of 5 and 3. Since the active material is identical, this shows that the support material may change the reaction mechanism through hetero-contacts between the support and active metal in addition to the already studied oxygen-vacancies, which merely accelerate the rate-limiting C–O-bond cleavage.
CO formation is only detected for Ru/CeO2 and Ru/C. While the general activity of Ru/CeO2 is low, the selectivity to CO is reliably detected to be around 50% under all process conditions. The activity of Ru/C is so low that the detected amounts of CO are close to the detection limit and the resulting error in selectivity is too large to provide a reliable number. The methane selectivities of all catalyst at the reference points are shown in Table 6. A complete list of selectivities can be found in the ESI.† The catalysts with higher activity show a high selectivity towards methane under all process conditions, while the least active catalyst, Ru/CeO2 and Ru/C, show a much lower selectivity.
| Ru/TiO2 | Ru/ZrO2 | Ru/CeO2 | Ru/SiO2 | Ru/Al2O3 | Ru/C | |
|---|---|---|---|---|---|---|
| B | 96.8 | 96.4 | 44.1 | 98.9 | 97.4 | 83.4 |
| G | 97.1 | 96.6 | 41.9 | 99.3 | 97.7 | 62.4 |
| H | 97.3 | 96.7 | 50.8 | 99.0 | 96.0 | 77.3 |
| K | 97.8 | 96.8 | 52.7 | 99.1 | 97.4 | 74.3 |
As expected the relationship between temperature and selectivity to methane correlates. Increasing the pressure from reference G to reference H does not influence selectivity towards higher hydrocarbons for the Ru/TiO2, Ru/ZrO2 and Ru/CeO2 catalysts. Temperature measurements show no hot-spot formation which could explain this behavior by thermocatalytic effects. A comparison of the references H and K shows an increase in selectivity towards methane over time for most catalysts, while the activity remains basically the same, even after exposure to a feed composition of H2/CO2 of 1. This shows that carbon-rich synthesis gas does not lead to deactivation of any of the catalysts. These results prove that the Ru catalysts used in this study are robust under a broad range of temperatures and CO2/H2 ratios.
The small difference in both activity and selectivity when varying the H2/CO2 ratio and the moderate influence of increased pressure, shows that an elevated hydrogen partial pressures is not necessary for low-temperature CO2 methanation when using ruthenium-based catalysts. This may significantly reduce operating cost as hydrogen and its compression are expected to be at 2 € per kg (ref. 21) and CO2 at 50 € per tonne in 2040.22 The significant consumption rates at low temperatures may also allow the use of much more cost-effective and safe heat-management like medium-pressure steam, possibly allowing efficient process-windows inaccessible to other catalysts due to low activity or rapid deactivation. Longer studies and even lower syngas-ratios are necessary to rule out carbon-deposition as a significant deactivation mechanism over extended periods of time.
The consumption rates related to the catalyst bed volume are calculated for the first reference point B via the eqn (6) and (7) and shown in Table 7. Due to its higher bulk density, Ru/ZrO2 has a higher volumetric consumption rate than Ru/TiO2. This is an important consideration for reactors, where the heat generated per volume is a crucial parameter for design of the thermal management.
| Catalyst | Bulk density/g cm−3 | Consumption rate/μmol gcat−1 s−1 | Volumetric consumption rate/μmol cm−3 s−1 |
|---|---|---|---|
| Ru/TiO2 | 0.34 | 6.75 | 2.28 |
| Ru/ZrO2 | 0.44 | 6.07 | 2.70 |
| Ru/CeO2 | 0.34 | 0.20 | 0.07 |
| Ru/Al2O3 | 0.30 | 0.63 | 0.19 |
| Ru/SiO2 | 0.23 | 0.31 | 0.07 |
| Ru/C | 0.35 | 0.09 | 0.03 |
3.3 Reaction kinetics
The CO2 conversion is below 5% for all operating points, which ensures differential conditions. The apparent reaction orders with respect to CO2, shown in Fig. 4, are practically zero for the Ru/ZrO2 and Ru/Al2O3 catalysts, which is in line with the results from Mansour and Iglesia, who found that the reaction of surface CO is rate-limiting and therefore the reaction rate to be independent of CO2 pressure.23 The reaction order for the Ru/TiO2 catalyst is non-zero, which indicates a different reaction mechanism compared to the other two catalysts, even though the difference is small. The apparent reaction orders with respect to H2, shown in Fig. 5, are close to 0.5 for the Ru/TiO2 and Ru/ZrO2 catalyst, which is in line with those determined by Wang et al.24 in a much more in-depth analysis of the kinetics of a Ru/Al2O3 catalyst. Their results found a reaction order of 0.3–0.5 for H2. The reaction order of H2 for the Ru/Al2O3 catalyst in this study is 0.25 and therefore below that expected from literature. The different H2 reaction order of the Ru/Al2O3 catalyst compared to the other catalysts may indicate a different reaction mechanism when comparing Ru/Al2O3 to Ru/TiO2 and Ru/ZrO2.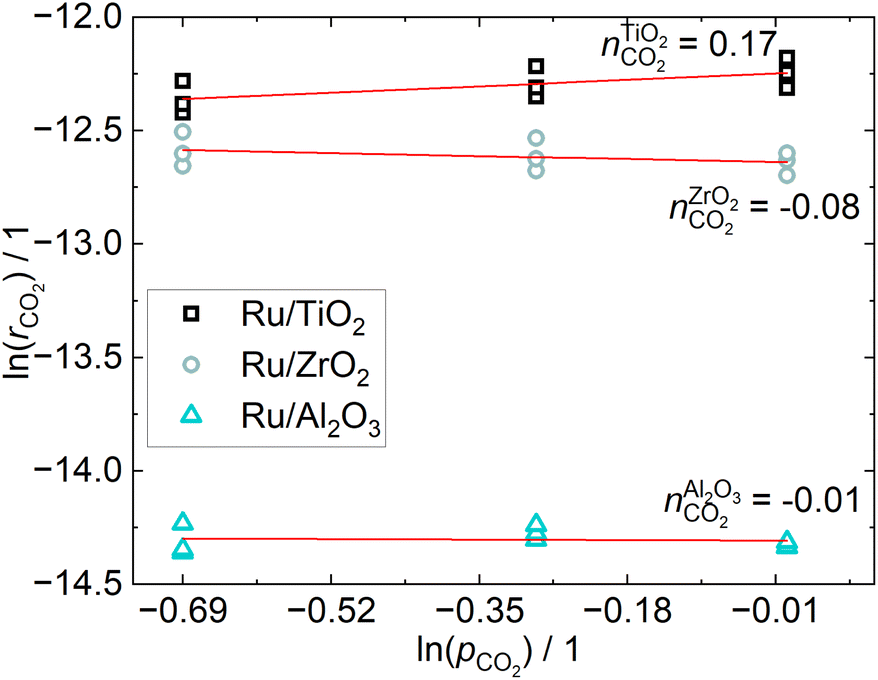 | ||
| Fig. 4 Reaction orders of CO2 obtained from a linear fit of the consumption rates calculated according to 9 at three different constant H2 pressures. | ||
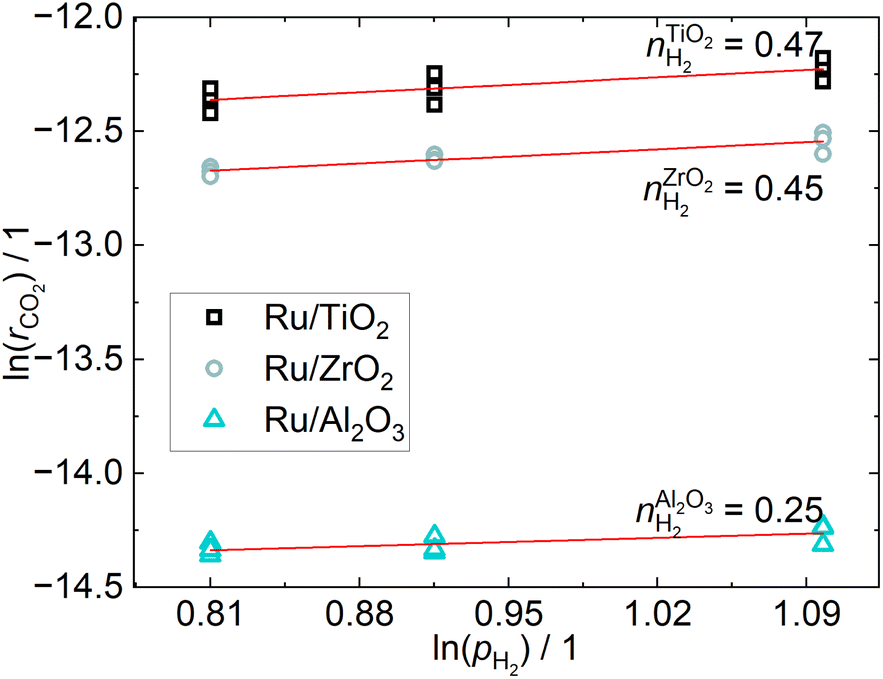 | ||
| Fig. 5 Reaction orders of H2 resulting from a linear fit through the consumption rates calculated from varying the H2-partial pressure at three different constant CO2 pressures. | ||
The Arrhenius-plot for the investigated catalysts is shown in Fig. 6. The experiments of Wang et al.24 found activation energies between 57 and 80 kJ mol−1 depending on the H2/CO2 ratio. At a ratio of 5, as was used here, the resulting activation energy was 78 kJ mol−1 for their 5% Ru/Al2O3 catalyst, while it is 58 kJ mol−1 for 0.5% Ru/Al2O3, which is well in line with the Ru/TiO2 and Ru/Al2O3 catalysts in this study. The activation energy for our Ru/ZrO2 catalyst is much higher than the TiO2 and Al2O3 supported Ru in this study, indicating a different reaction mechanism on Ru/ZrO2 compared to the other two catalysts. This solidifies the indication of different reaction mechanisms depending on the chosen support.
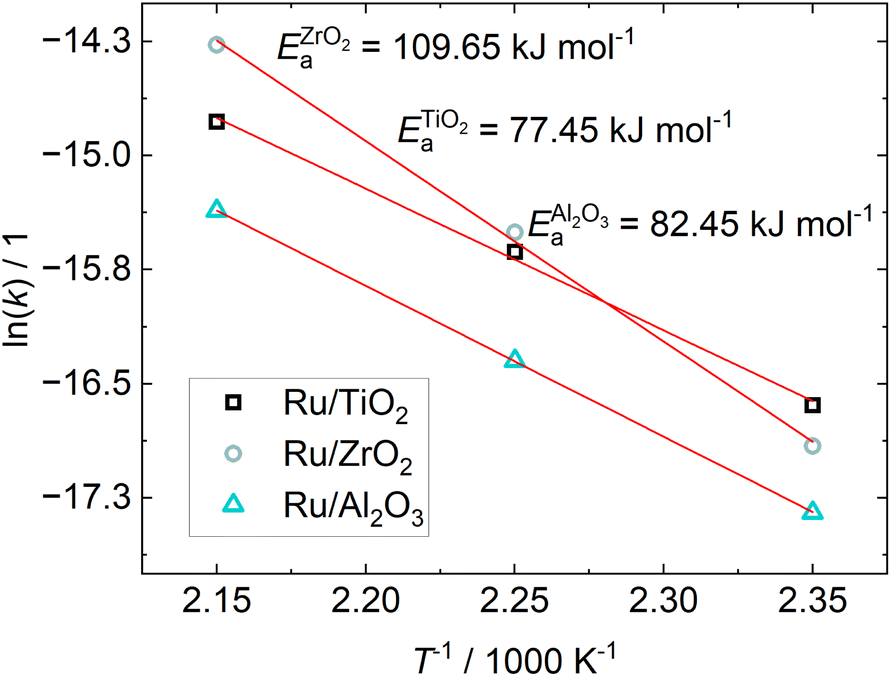 | ||
| Fig. 6 Arrhenius-plot from the kinetic constant obtained from parameter fitting viaeqn (8) and the resulting apparent activation energy for different catalysts. | ||
The TOF during temperature variation calculated viaeqn (9), shown in Fig. 7, highlights Ru/TiO2 as the most active catalyst.
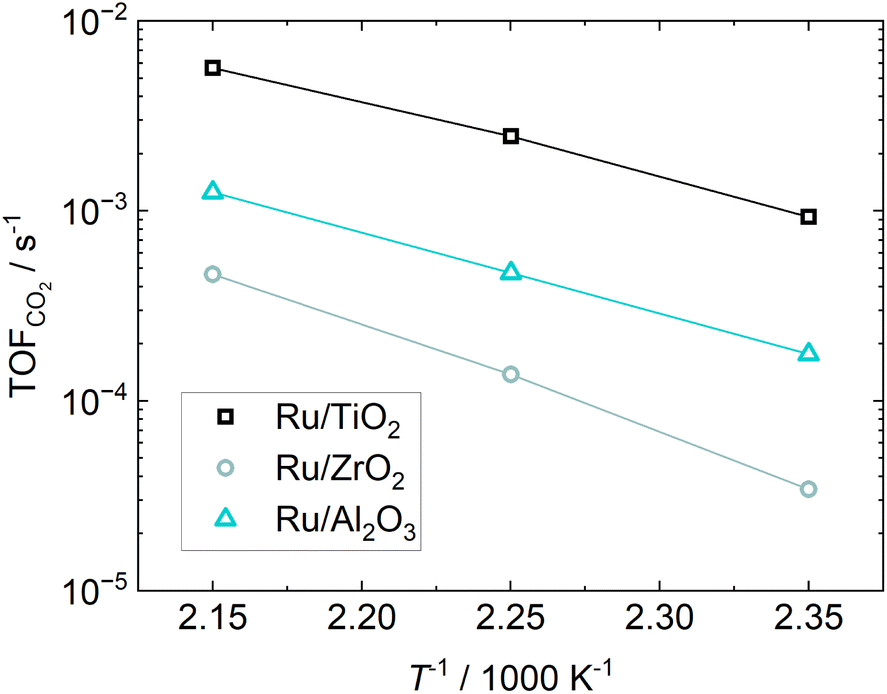 | ||
| Fig. 7 Arrhenius-plot based on the TOF from CO2 consumption rate. | ||
The TOF of 5.7 × 10−3 s−1 for the Ru/TiO2 catalyst at 190 °C is well in line with our previous results13 and slightly below the value of 5 to 10 × 10−3 s−1 measured by Abdel-Mageed et al.10 Chen et al.25 investigated a 2.3 wt% Ru/Al2O3 catalyst and at 190 °C found TOFs of 4 × 10−4 s−1 and 6.8 × 10−3 s−1, depending on the pretreatment of the catalyst. The TOF of 1.3 × 10−3 s−1 at 190 °C of the Ru/Al2O3 catalyst in this study is in good agreement with the TOF achieved by Chen et al. The same group also investigated a 2.1 wt% Ru/ZrO2 catalysts26 and at 190 °C found a TOF of 3.4 × 10−3 s−1, which is much higher than the 4.6 × 10−4 s−1 measured in this study. This may be due to the assumption that H2 only chemisorbs on Ru. In particular, the spillover of hydrogen atoms onto the support is a known phenomenon,27 which may lead to H2 at both the Ru and support surface. This might explain the large measured H2 capacity of the catalysts using partially reducible metal oxides as supports, which would result in overestimating the active surface area and dispersion and consequently to the underestimation of the TOF. The reported TOFs and literature data are summarized in Table 8.
4 Conclusions
In this work we tested various Ru-based catalysts using commercial support materials under mild process conditions to find the most promising catalyst and process conditions for a safe and sustainable CH4 synthesis from CO2. The results highlight Ru/TiO2 as the most active catalyst with high activity even at low temperatures. The catalyst remains stable even at elevated CO2 concentrations with high CH4 selectivity. The kinetic parameters determined for the Ru/TiO2, Ru/ZrO2 and Ru/Al2O3 catalysts give an indication that the reaction mechanism for each of these catalysts may differ from the others to a certain extent. Since the main difference between these catalysts is the support material, the significant influence of metal-support interactions in governing the activity of the catalyst is revealed. The activity of the TiO2, ZrO2 and Al2O3 supported catalysts are not just a morphological effect due to high metal surface areas, but possibly a mechanistic effect originating from the interaction of Ruthenium with the support materials.The results obtained for the unexplored low temperatures and H2/CO2 ratios indicates potentially attractive process windows by reducing the cost of thermal management and H2 from electrolysis by leveraging sub-stoichiometric H2/CO2 ratios. The results of kinetic experiments show that the reaction mechanism must be influenced by the support material, highlighting the need for catalyst specific optimization strategies for metal-support interactions. The low temperatures employed during calcination and reduction of the catalyst show that the higher temperatures often employed for both steps may not be necessary.
The comparative nature of the materials and wide process window tested in this study makes it an ideal basis for future studies of more sophisticated catalyst materials or for CO2 hydrogenation at mild conditions. Finally, the results also show that the most promising Ru-based catalysts for CO2 hydrogenation are supported on either ZrO2 or TiO2.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Göran Baade: conceptualization, investigation, methodology, visualization, writing – original draft preparation. Jens Friedland: methodology, writing – reviewing and editing. Koustuv Ray: writing – reviewing and editing. Robert Güttel: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Vector Stiftung for funding this project (P2022-0013) and the Deutsche Forschungsgemeinschaft for funding the experimental setup within the Major Research Instrumentation Programme (INST 40/516-1 FUGG) as well as IBU-tec advanced materials AG for providing the ZrO2 and CeO2 support, Evonik Industries AG for providing the TiO2, Al2O3 and SiO2 support and Infiltec GmbH for providing the C support. Additionally we would like to thank Samuel Blessing of the Institute of Inorganic Chemistry II at Ulm University for conducting the PXRD measurements and Konstantin Schüttler of the Institute of Surface Chemistry and Catalysis at Ulm University for conducting the XPS measurements.Notes and references
- D. A. Lashof and D. R. Ahuja, Nature, 1990, 344, 529–531 CrossRef
.
- UN-DESA, The Sustainable Development Goals Report 2023: Special Edition, 2023, https://unstats.un.org/sdgs/report/2023/ Search PubMed.
- L. Jiang, W. Liu, R. Wang, A. Gonzalez-Diaz, M. Rojas-Michaga, S. Michailos, M. Pourkashanian, X. Zhang and C. Font-Palma, Prog. Energy Combust. Sci., 2023, 95, 101069 CrossRef
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef
- J. Ilsemann, M. M. Murshed, T. M. Gesing, J. Kopyscinski and M. Bäumer, Catal. Sci. Technol., 2021, 11, 4098–4114 RSC
- J. A. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H.-L. Sit and W. Y. Teoh, Appl. Catal., B, 2017, 219, 715–726 CrossRef
- S. Scirè, C. Crisafulli, R. Maggiore, S. Minicò and S. Galvagno, Catal. Lett., 1998, 51, 41–45 CrossRef
- C. Wang, H. Sun, X. Liu, X. Jin, Y. Feng, H. Shi, D. Wang, Y. Zhang, Y. Wang and Z. Yan, Fuel, 2023, 345, 128238 CrossRef
- Z. Zhao, Q. Jiang, Q. Wang, M. Wang, J. Zuo, H. Chen, Q. Kuang and Z. Xie, ACS Sustain. Chem. Eng., 2021, 9, 14288–14296 CrossRef
- A. M. Abdel-Mageed, K. Wiese, M. Parlinska-Wojtan, J. Rabeah, A. Brückner and R. J. Behm, Appl. Catal., B, 2020, 270, 118846 CrossRef
- C. Tébar-Soler, V. M. Diaconescu, L. Simonelli, A. Missyul, V. Perez-Dieste, I. Villar-García, D. Gómez, J.-B. Brubach, P. Roy, A. Corma and P. Concepción, ACS Catal., 2024, 14, 4290–4300 CrossRef
- A. Aitbekova, E. D. Goodman, L. Wu, A. Boubnov, A. S. Hoffman, A. Genc, H. Cheng, L. Casalena, S. R. Bare and M. Cargnello, Angew. Chem., Int. Ed., 2019, 58, 17451–17457 CrossRef
- H. Becker, D. Ziegenbalg and R. Güttel, Preprint: Photoassisted amplification of chain-growth in CO2 hydrogenation: Switching selectivities of heterogeneously catalyzed reactions with light, 2024, https://www.chemrxiv.org/engage/chemrxiv/article-details/65e822589138d23161c69590.
- J. M. Crawford, B. E. Petel, M. J. Rasmussen, T. Ludwig, E. M. Miller, S. Halingstad, S. A. Akhade, S. H. Pang and M. M. Yung, Appl. Catal., A, 2023, 663, 119292 CrossRef
- J. Niu, C. Zhang, H. Liu, Y. Jin and R. Zhang, Front. Energy, 2023, 17, 545–554 CrossRef
- X. Yan, C. Duan, S. Yu, B. Dai, C. Sun and H. Chu, J. CO2 Util., 2024, 79, 102648 CrossRef
- M. Vannice, J. Catal., 1982, 74, 199–202 CrossRef CAS
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CrossRef
- J. Xu, X. Su, H. Duan, B. Hou, Q. Lin, X. Liu, X. Pan, G. Pei, H. Geng, Y. Huang and T. Zhang, J. Catal., 2016, 333, 227–237 CrossRef
- L. M. Alves, M. P. Almeida, M. Ayala, C. D. Watson, G. Jacobs, R. C. Rabelo-Neto, F. B. Noronha and L. V. Mattos, Chem. Eng. Sci., 2021, 239, 116604 CrossRef
- T. Terlouw, C. Bauer, R. McKenna and M. Mazzotti, Energy Environ. Sci., 2022, 15, 3583–3602 RSC
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef
- H. Mansour and E. Iglesia, J. Am. Chem. Soc., 2021, 143, 11582–11594 CrossRef
- X. Wang, Y. Hong, H. Shi and J. Szanyi, J. Catal., 2016, 343, 185–195 CrossRef
- S. Chen, A. M. Abdel-Mageed, M. Dyballa, M. Parlinska-Wojtan, J. Bansmann, S. Pollastri, L. Olivi, G. Aquilanti and R. J. Behm, Angew. Chem., Int. Ed., 2020, 59, 22763–22770 CrossRef
- S. Chen, A. M. Abdel-Mageed, M. Li, S. Cisneros, J. Bansmann, J. Rabeah, A. Brückner, A. Groß and R. J. Behm, J. Catal., 2021, 400, 407–420 CrossRef CAS
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00469h |
| This journal is © The Royal Society of Chemistry 2024 |